
Imidazoles as Potential Anticancer Agents: An Update on Recent Studies

 ,
, 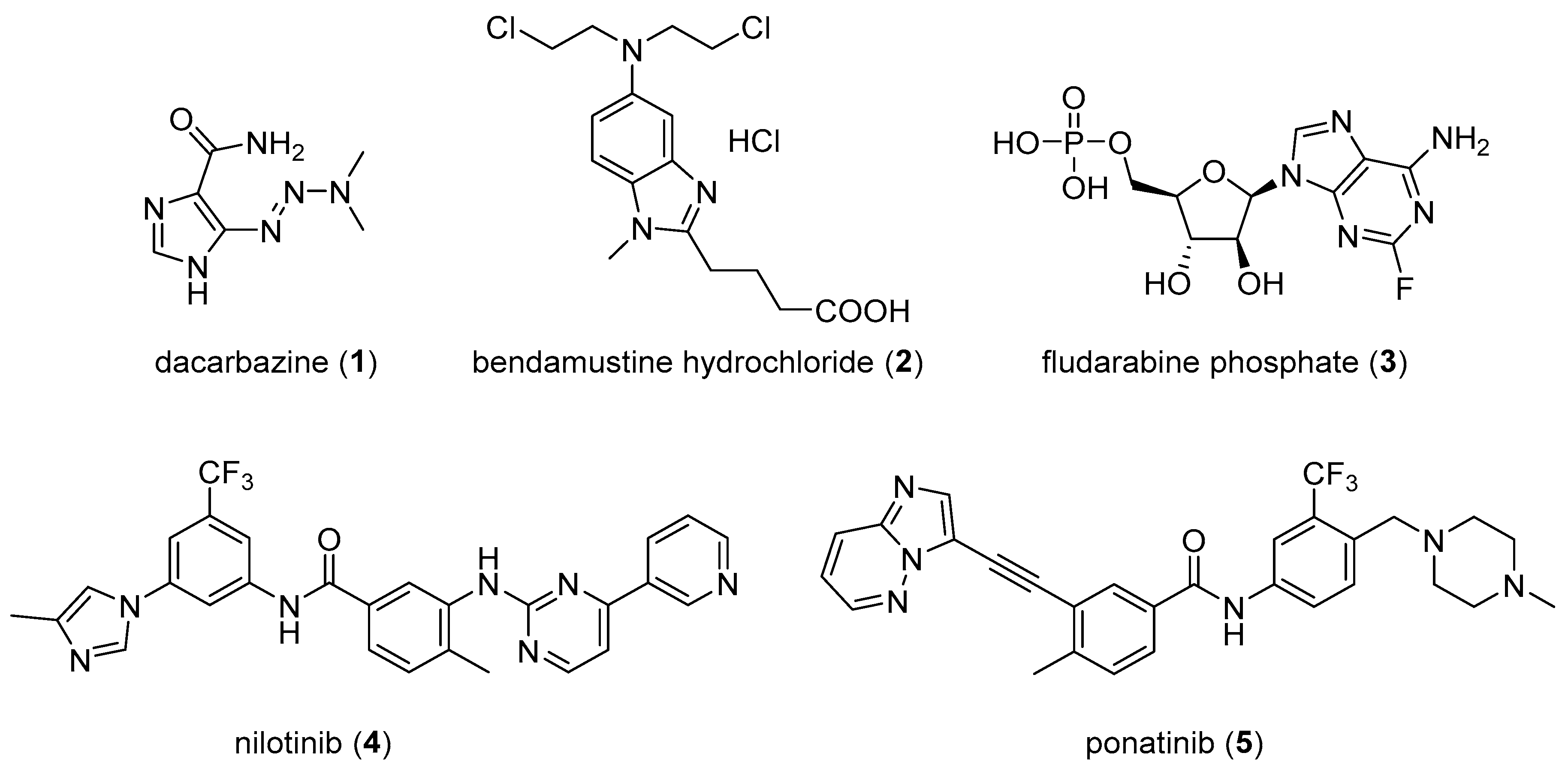{kind=link}
{kind=link}
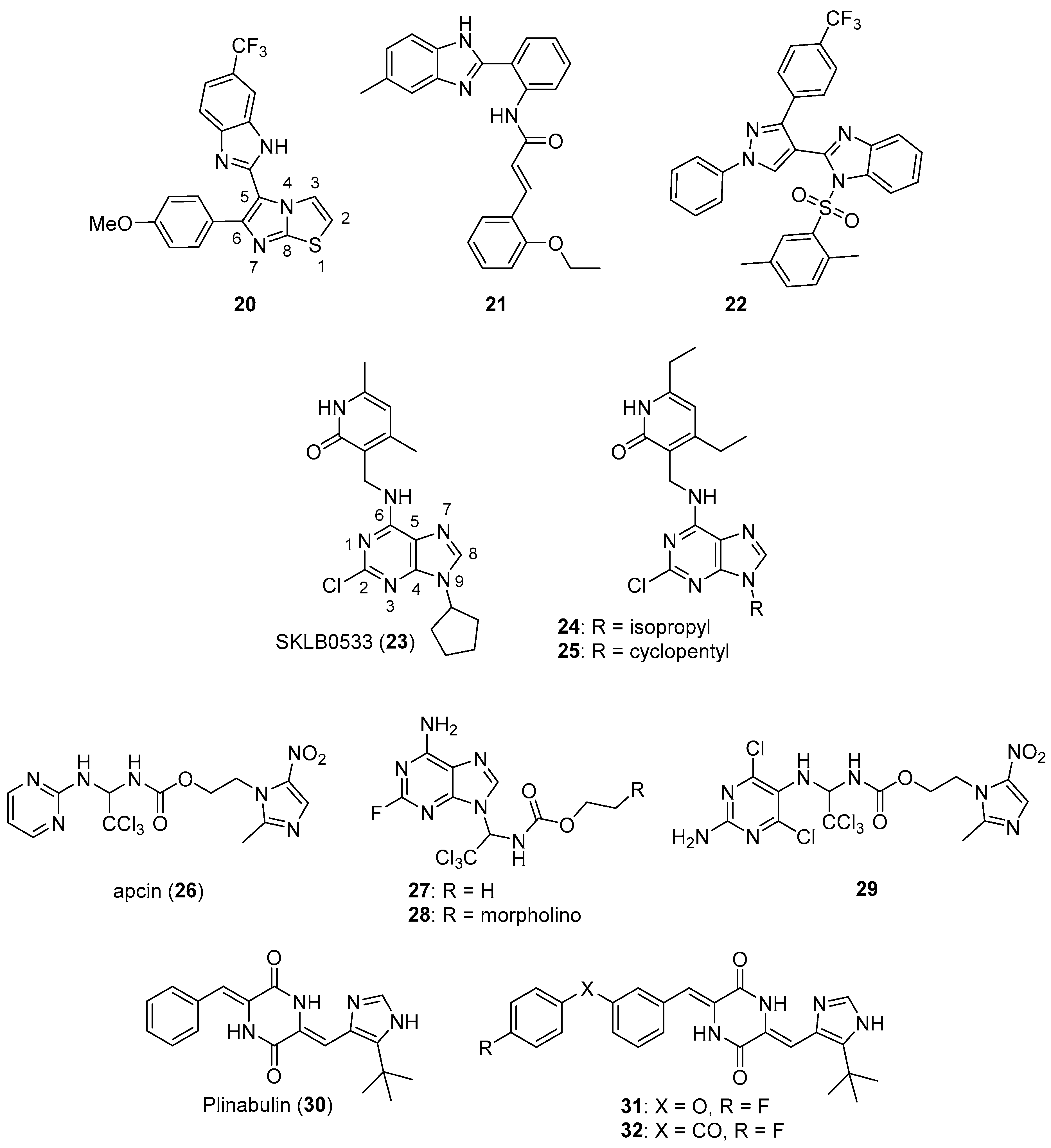{kind=link}
{kind=link}
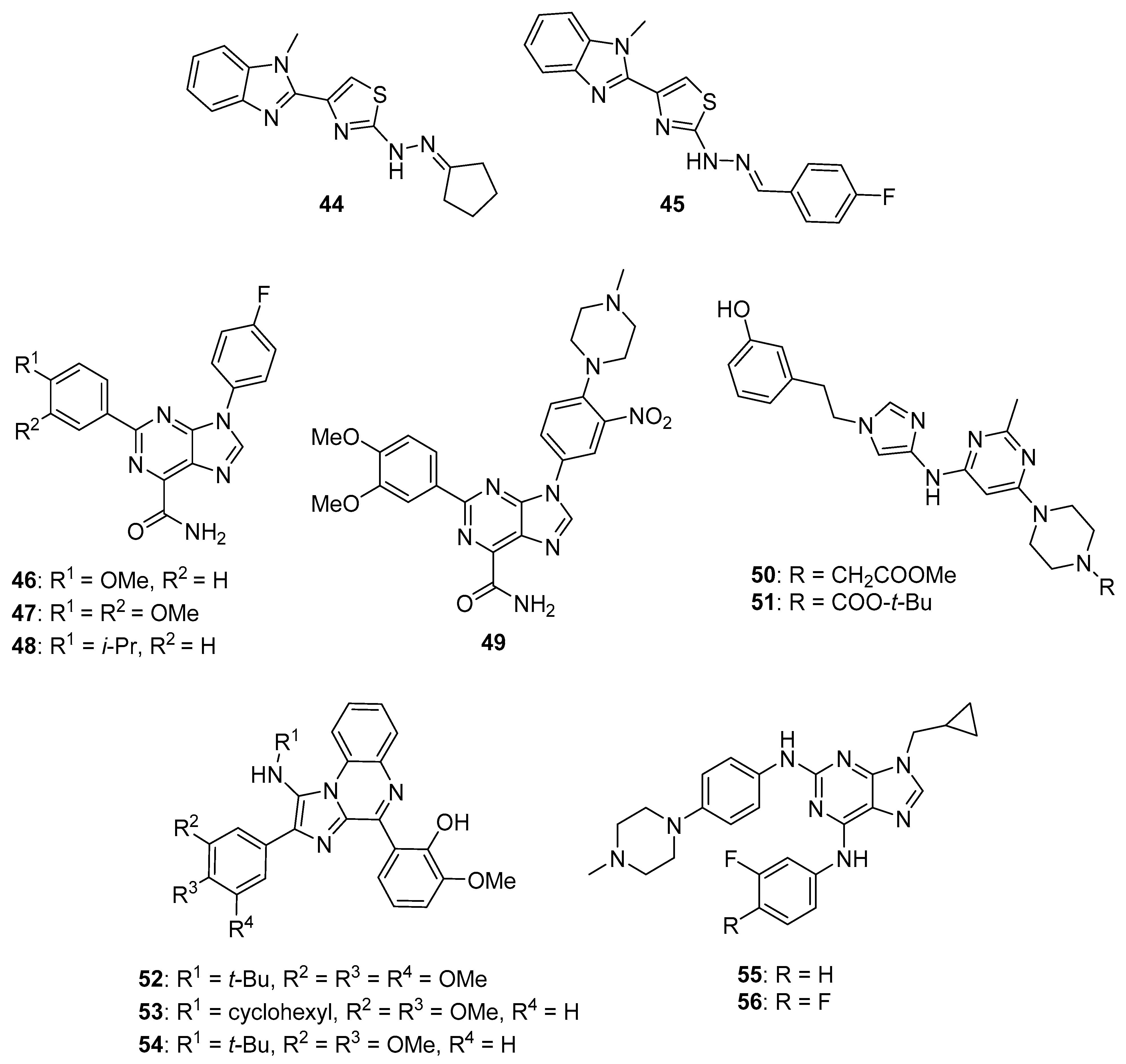{kind=link}
{kind=link}
{kind=link}
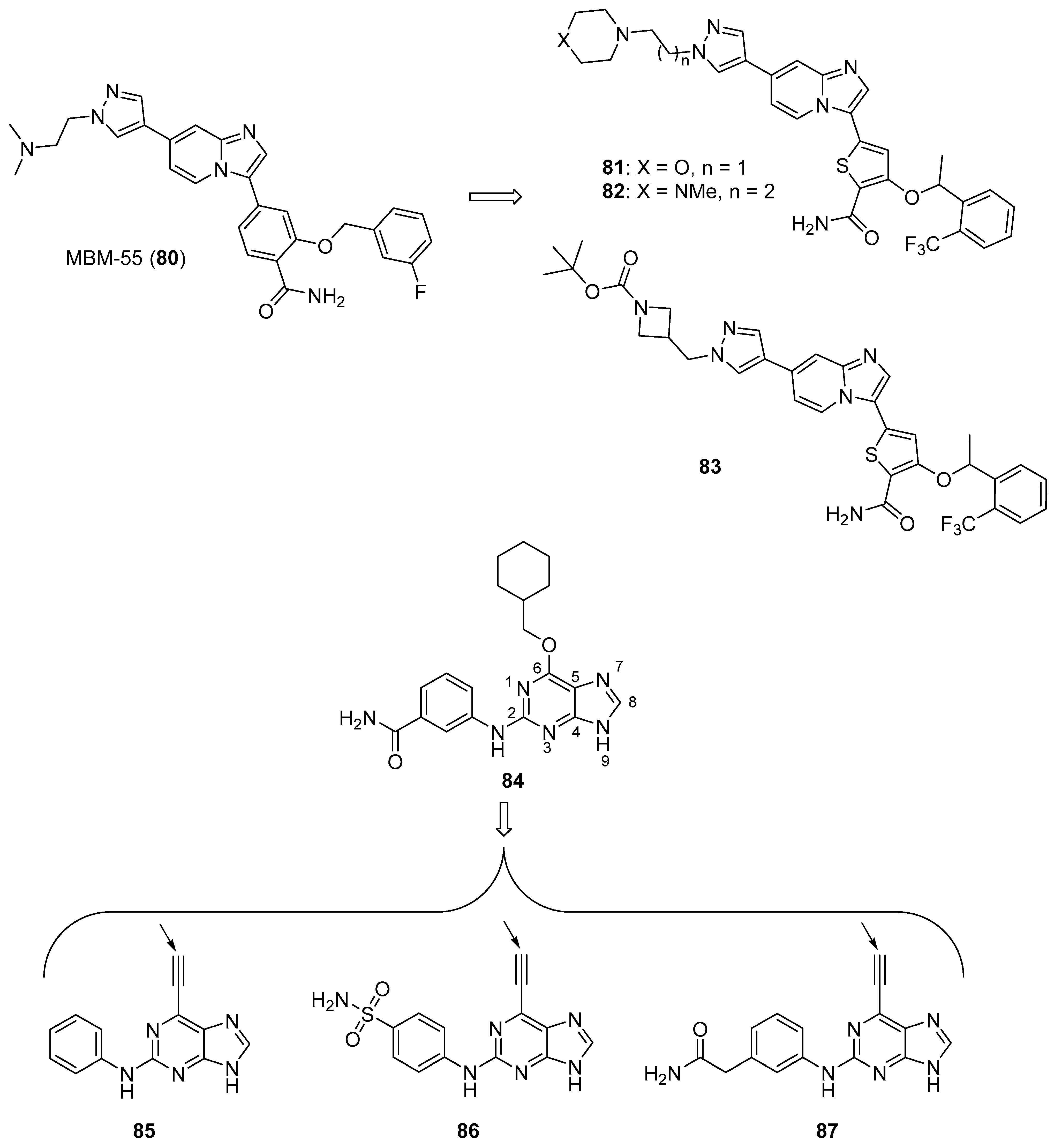{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Imidazoles as Tubulin Polymerization Inhibitors
3. Imidazoles as Kinase Inhibitors
3.1. Tyrosine Kinase Inhibitors
3.1.1. Vascular Endothelial Growth Factor Receptor (VEGFR) Inhibitors
3.1.2. Epidermal Growth Factor Receptor (EGFR) Inhibitors
3.1.3. Src Family Kinase (SFK) Inhibitors
3.1.4. Bcr-Abl and Bruton’s Tyrosine Kinase (BTK) Inhibitors
3.2. Serine-Threonine Kinase Inhibitors
3.2.1. Activin Receptor-Like Kinase 5 (ALK5) Inhibitors
3.2.2. Inhibitors of Checkpoint Kinases
3.2.3. Rapidly Accelerated Fibrosarcoma (RAF) Kinase Inhibitors
3.2.4. Cyclin-Dependent Kinase (CDK) Inhibitors
3.2.5. Aurora Kinase (AURK) Inhibitors
3.2.6. Nek2 Kinase Inhibitors
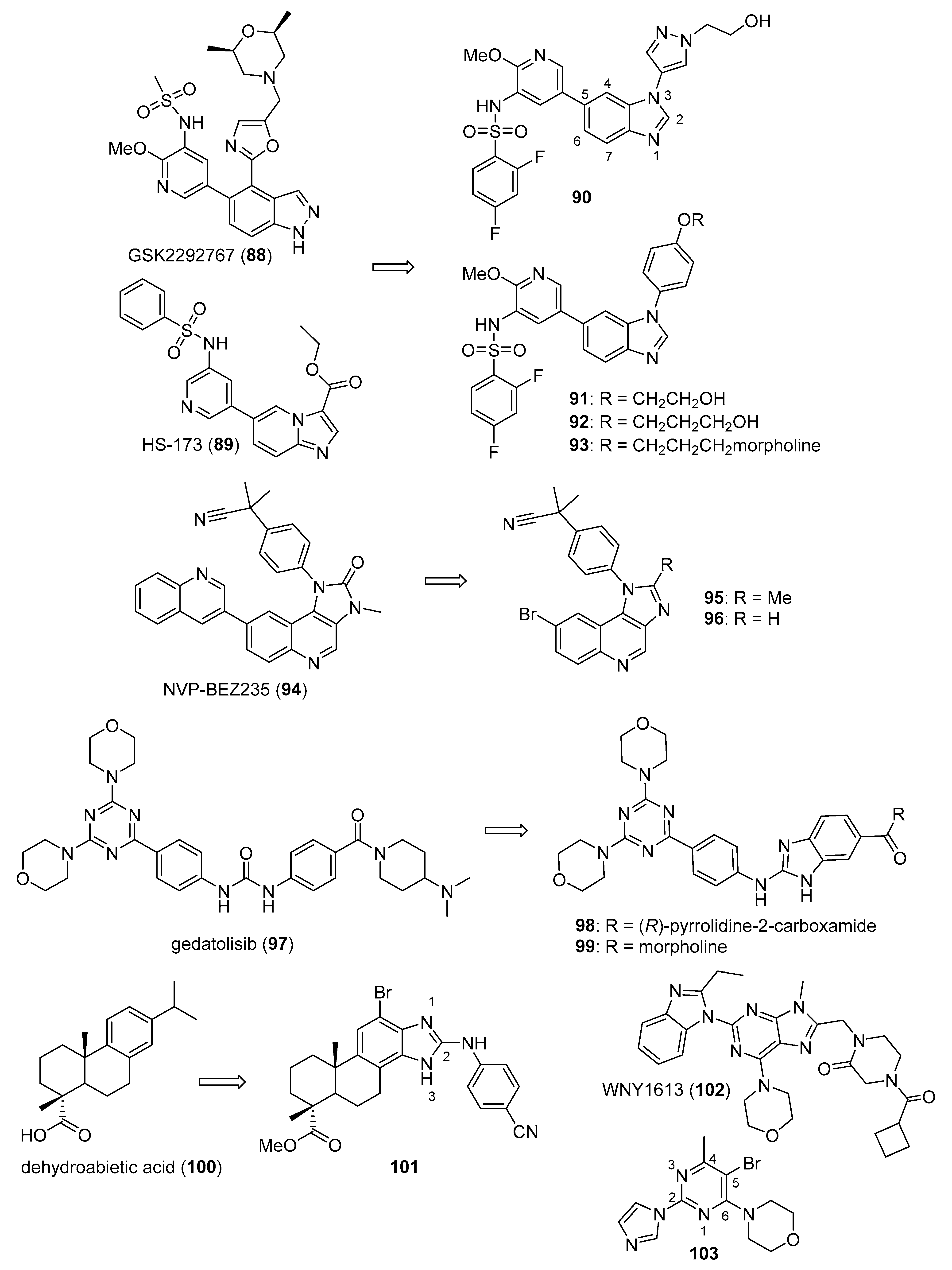
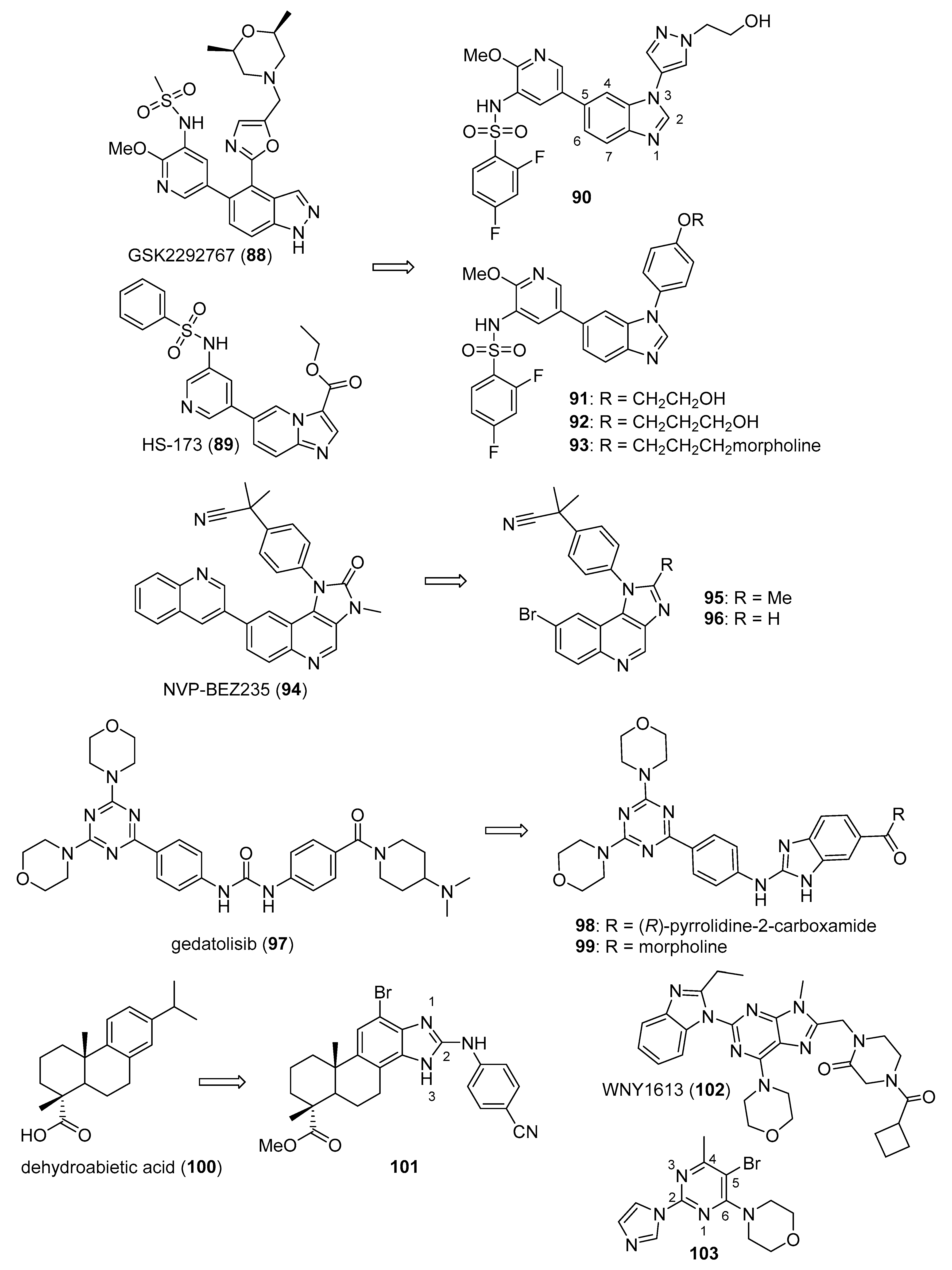
3.3. Phosphatidylinositol-3-Kinase (PI3K)/AKT/mTOR Inhibitors
4. Imidazoles as Inhibitors of Other Targets
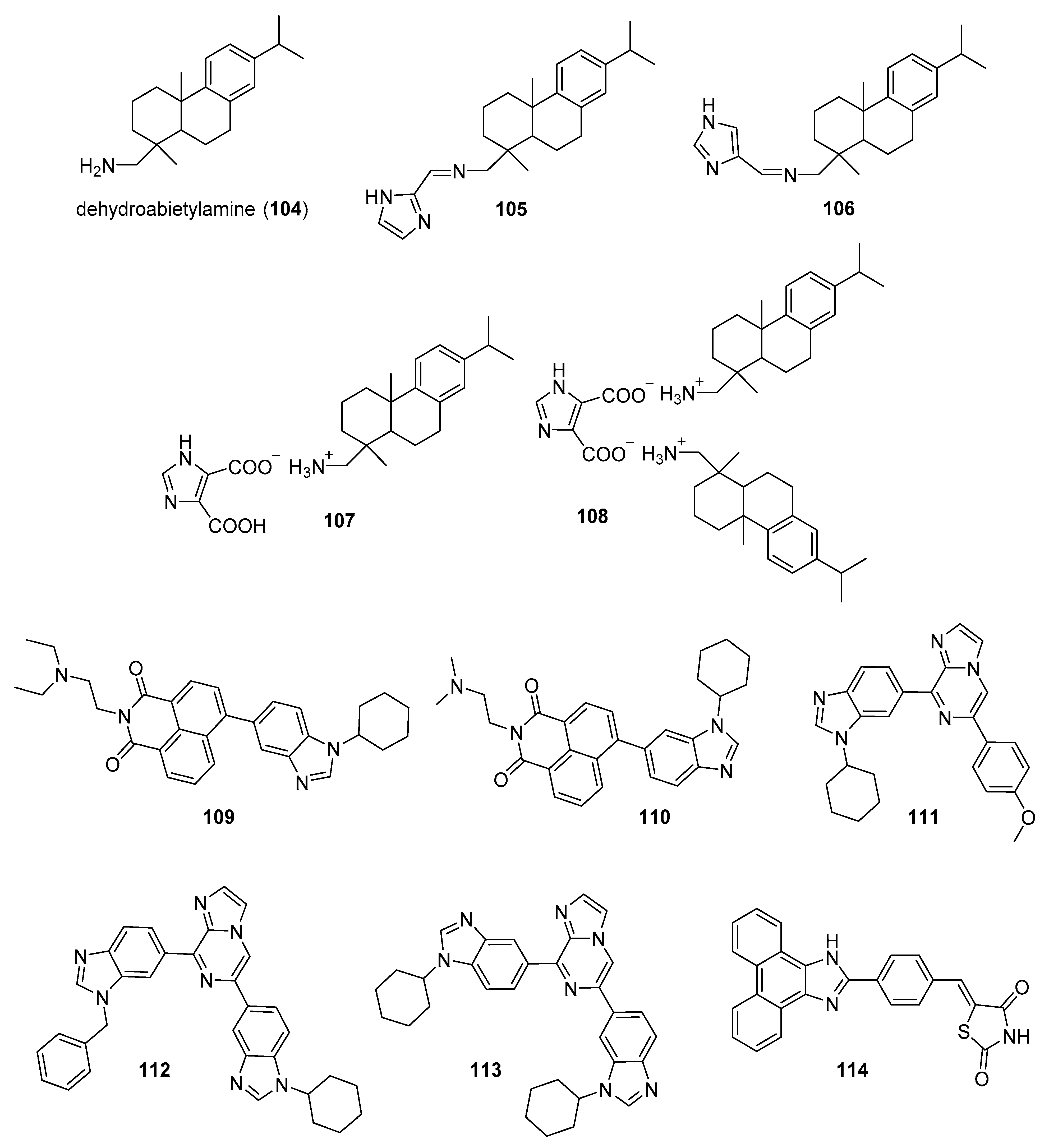
4.1. DNA Intercalators
4.2. G-Quadruplex Stabilizers
4.3. Topoisomerase Inhibitors
4.4. Inhibitors of Minichromosomal Maintenance Proteins (MCMs)
4.5. Poly(ADP-Ribose)polymerase (PARP) Inhibitors
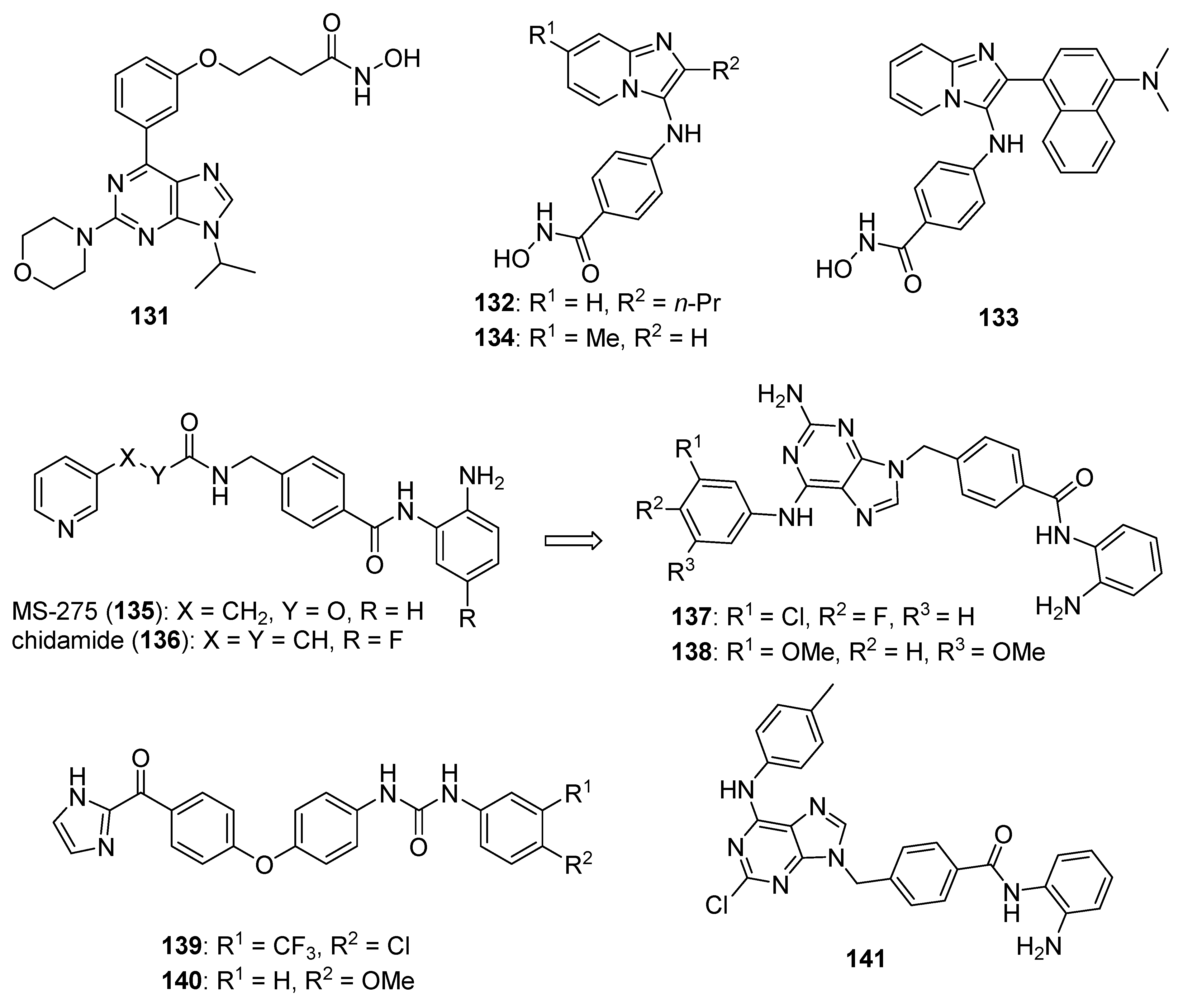
4.6. Histone Deacetylase (HDAC) Inhibitors
4.7. Lysine-Specific Demethylase 1 (KDM1A) Inhibitors
4.8. p53-Murine Double Minute 2 (MDM2) Inhibitors
4.9. Bromodomain and Extraterminal (BET) Protein Inhibitors
4.10. WD Repeat Domain 5 (WDR5) Inhibitors
4.11. Signal Transducer and Activator of Transcription 3 (STAT3) Inhibitors
4.12. Indoleamine-2,3-Dioxygenase (IDO)/Tryptophan 2,3-Dioxygenase (TDO) Signaling Inhibitors
4.13. Aromatase Inhibitors
4.14. Inhibition of Aldehyde Dehydrogenase
4.15. Heme Oxygenase-1 (HO-1) Inhibitors
4.16. Galectin-1 Inhibitors
4.17. Glutathione S-Transferase Inhibitors
4.18. Lipoxygenase Inhibitors
4.19. Estrogen Receptor-α (ER-α) Inhibitors
4.20. ABCB1 Inhibitors
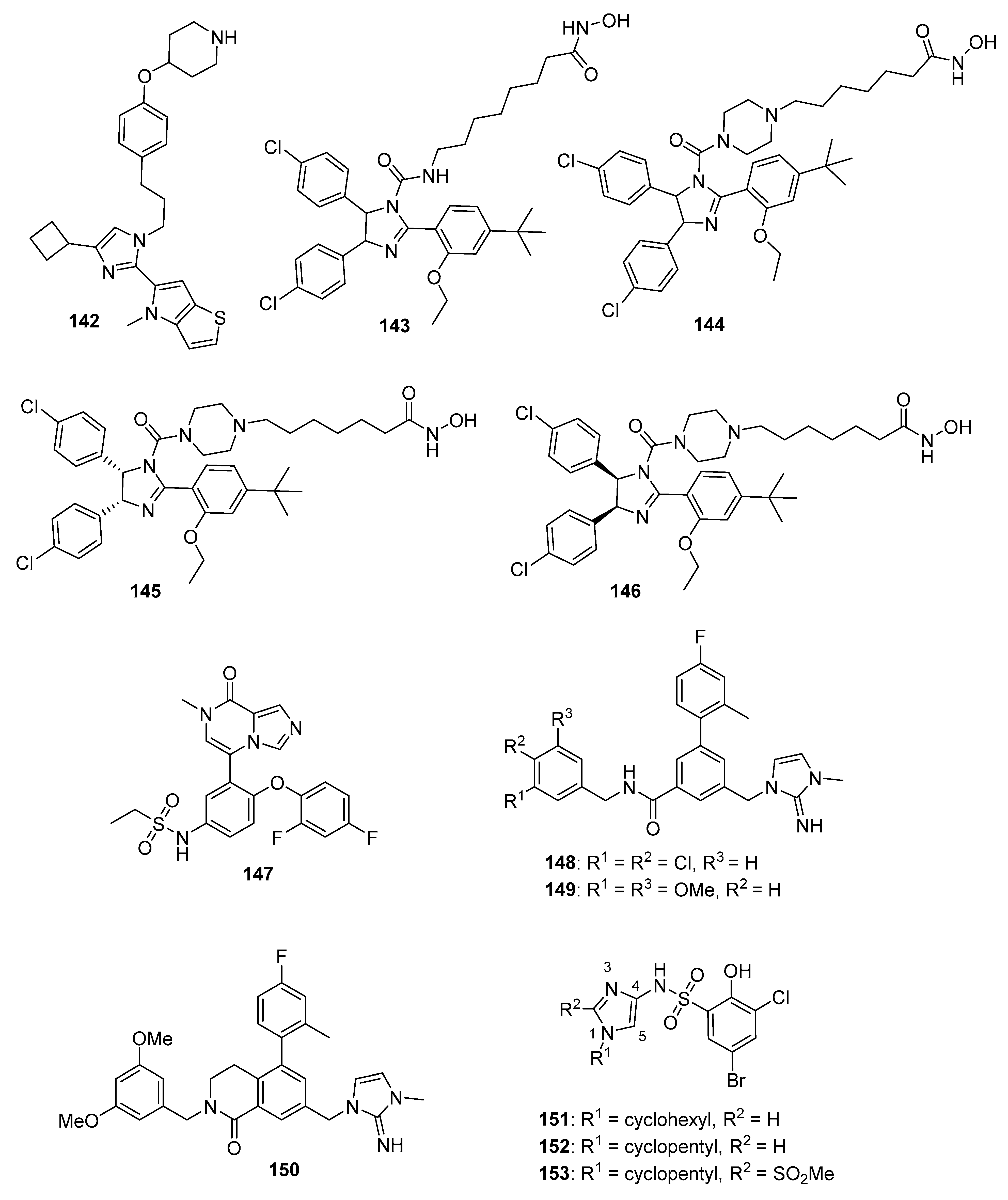
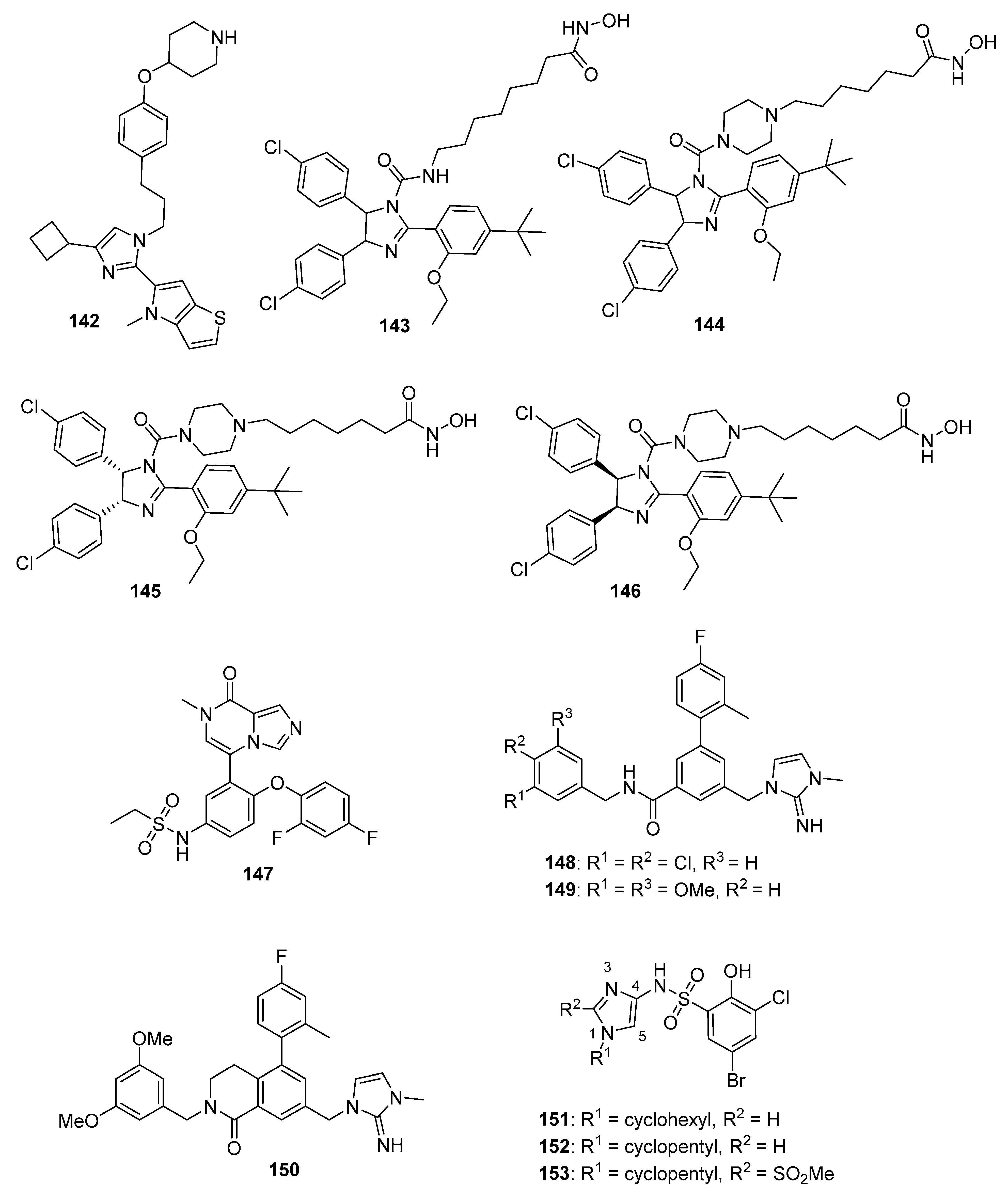
4.21. Heat Shock Protein (HSP) Inhibitors
5. Anticancer Activities Shown by Imidazole Derivatives through Undefined Mechanisms
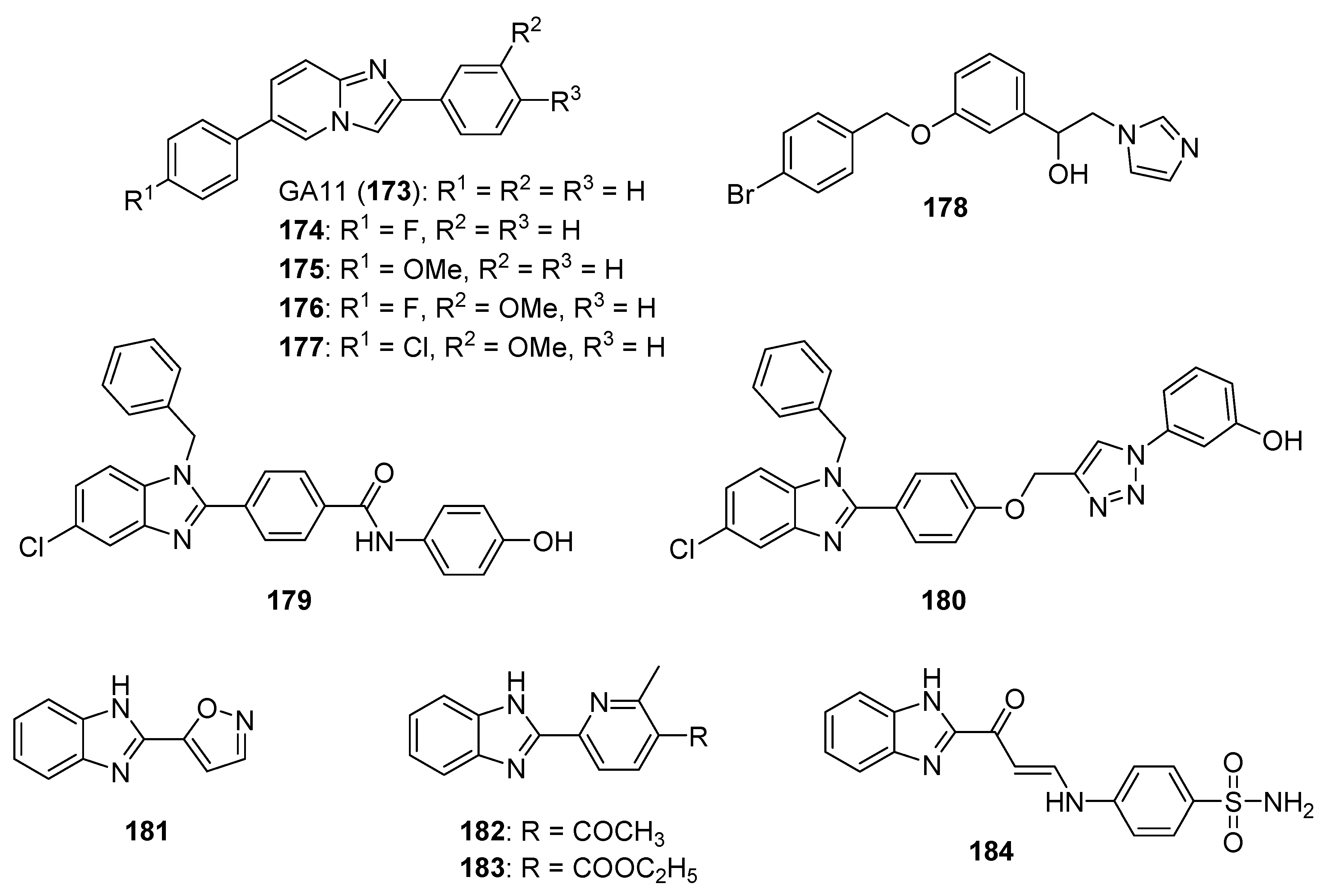
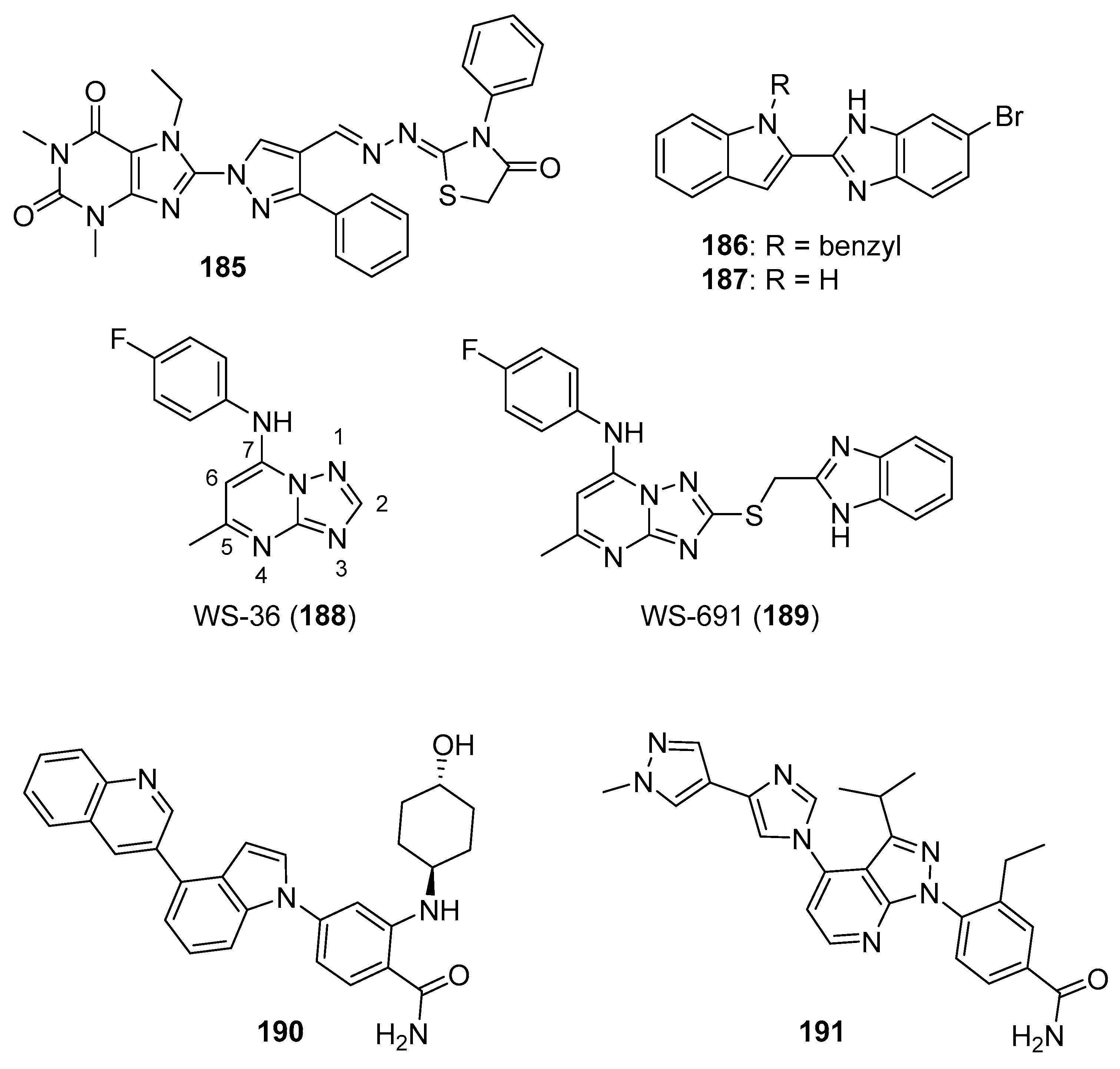
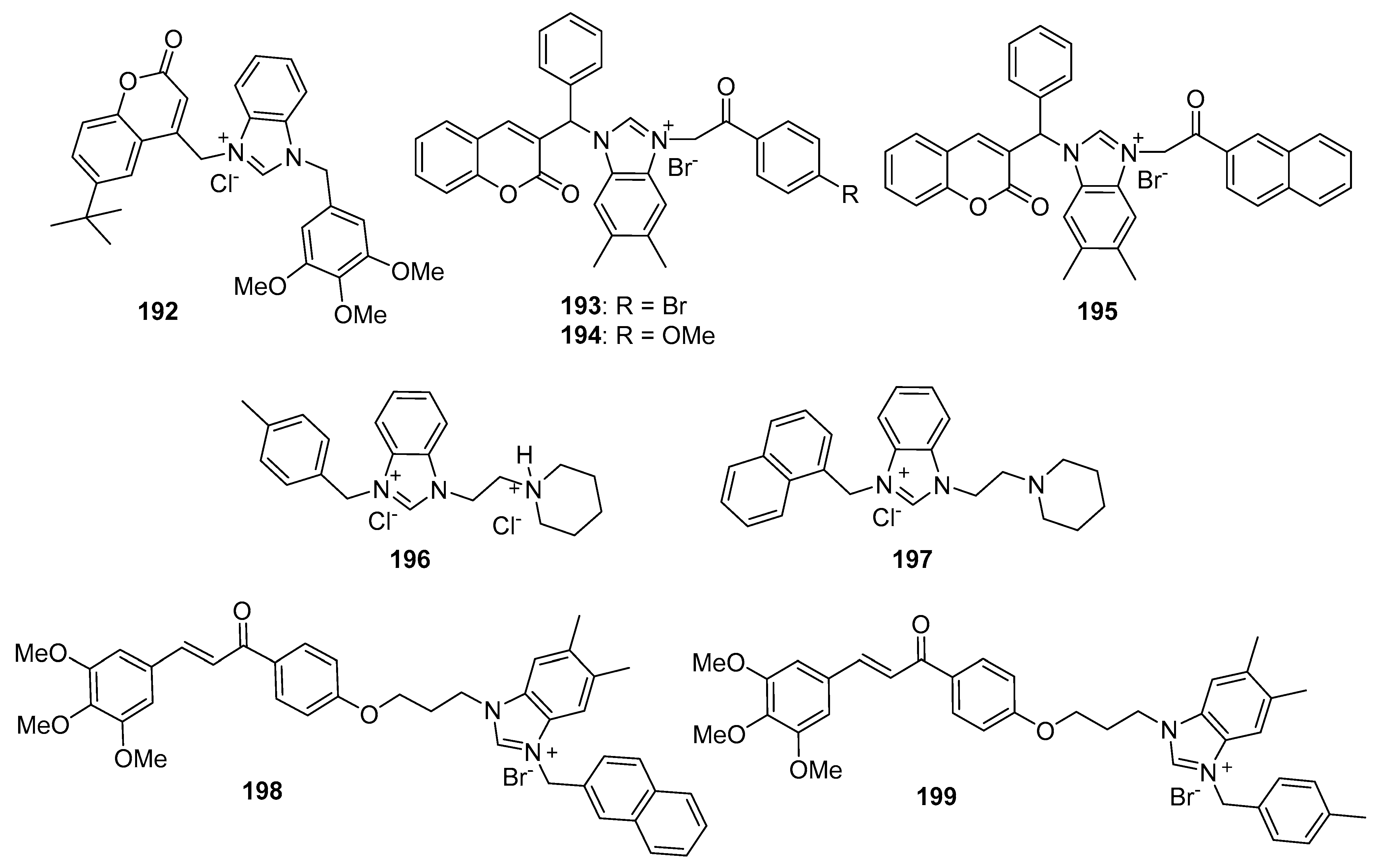
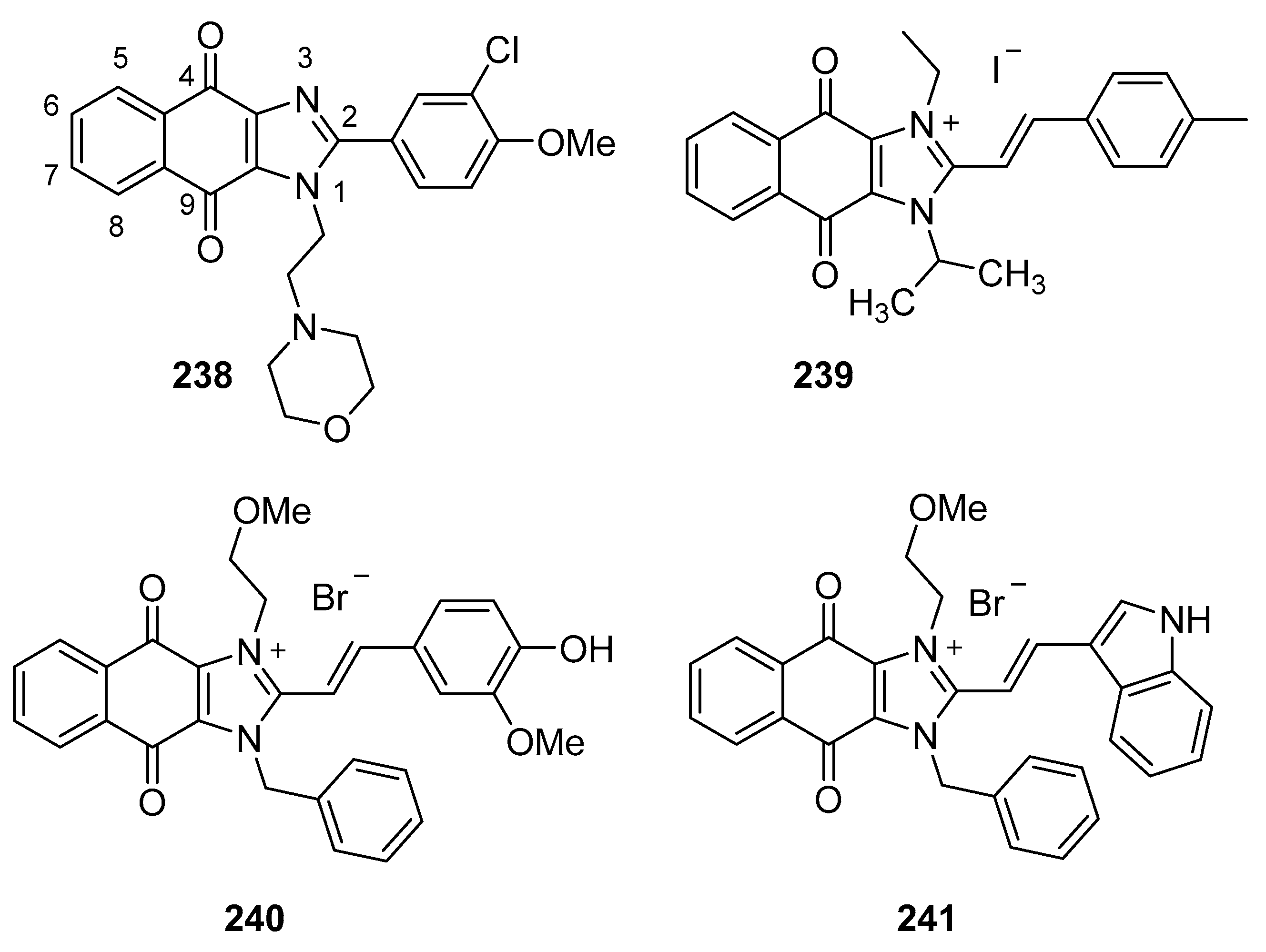
5.1. Benzimidazolium Salts
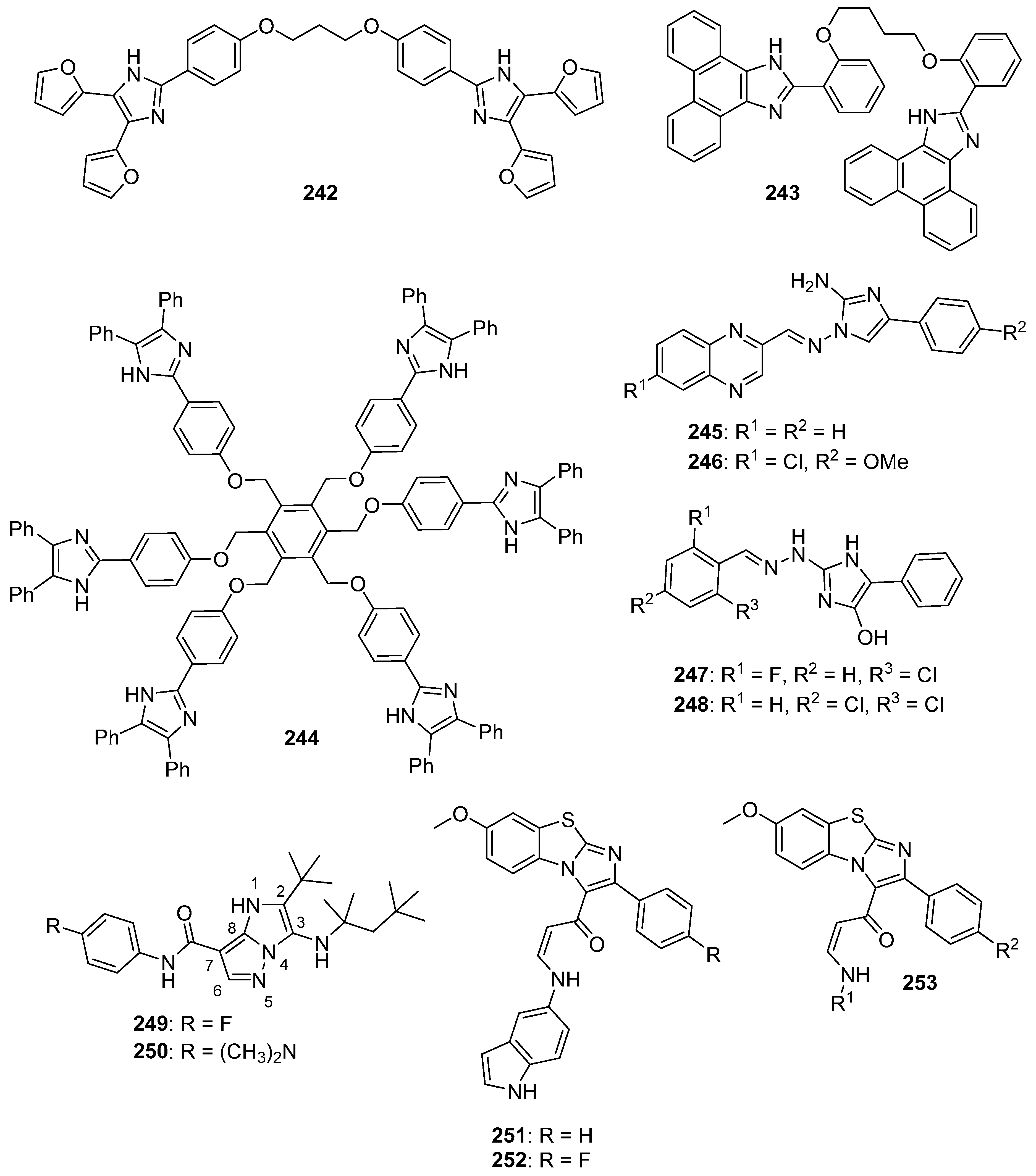
5.2. Benzimidazole Containing and Related Molecules
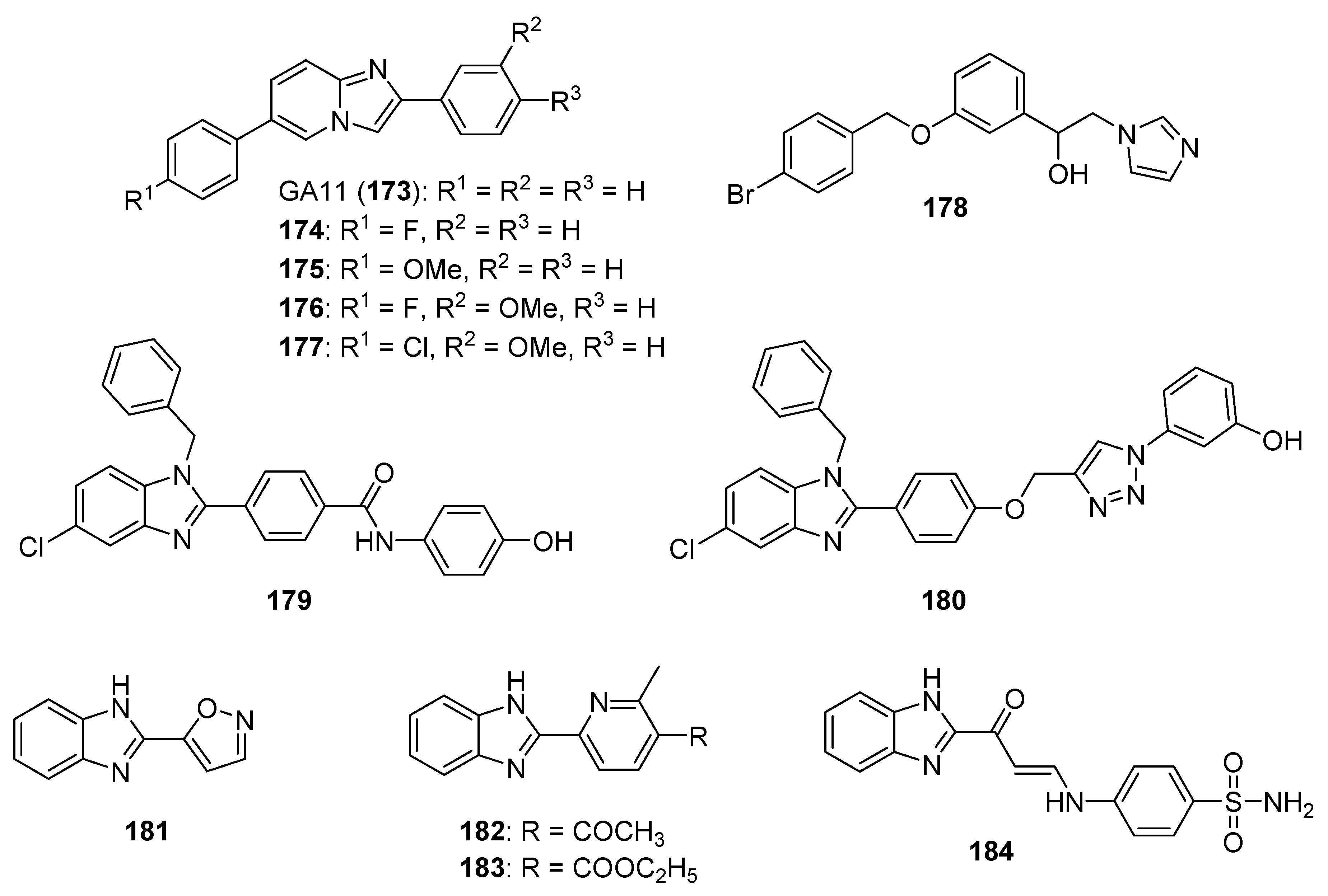
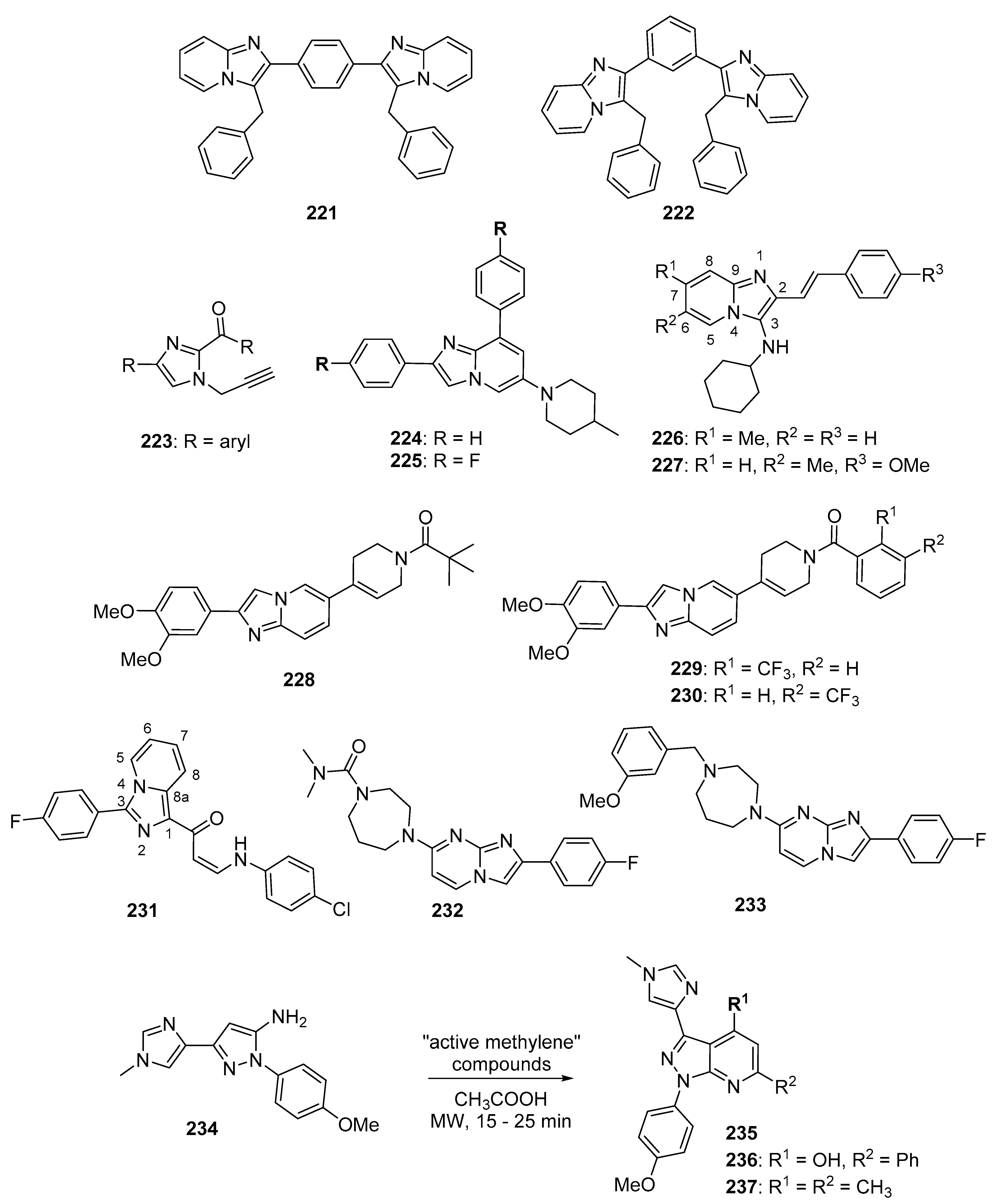
5.3. Imidazopyridines, Imidazopyrimidines, and Related Compounds
5.4. Naphthoquinone Imidazoles
5.5. Polysubstituted Imidazole Derivatives
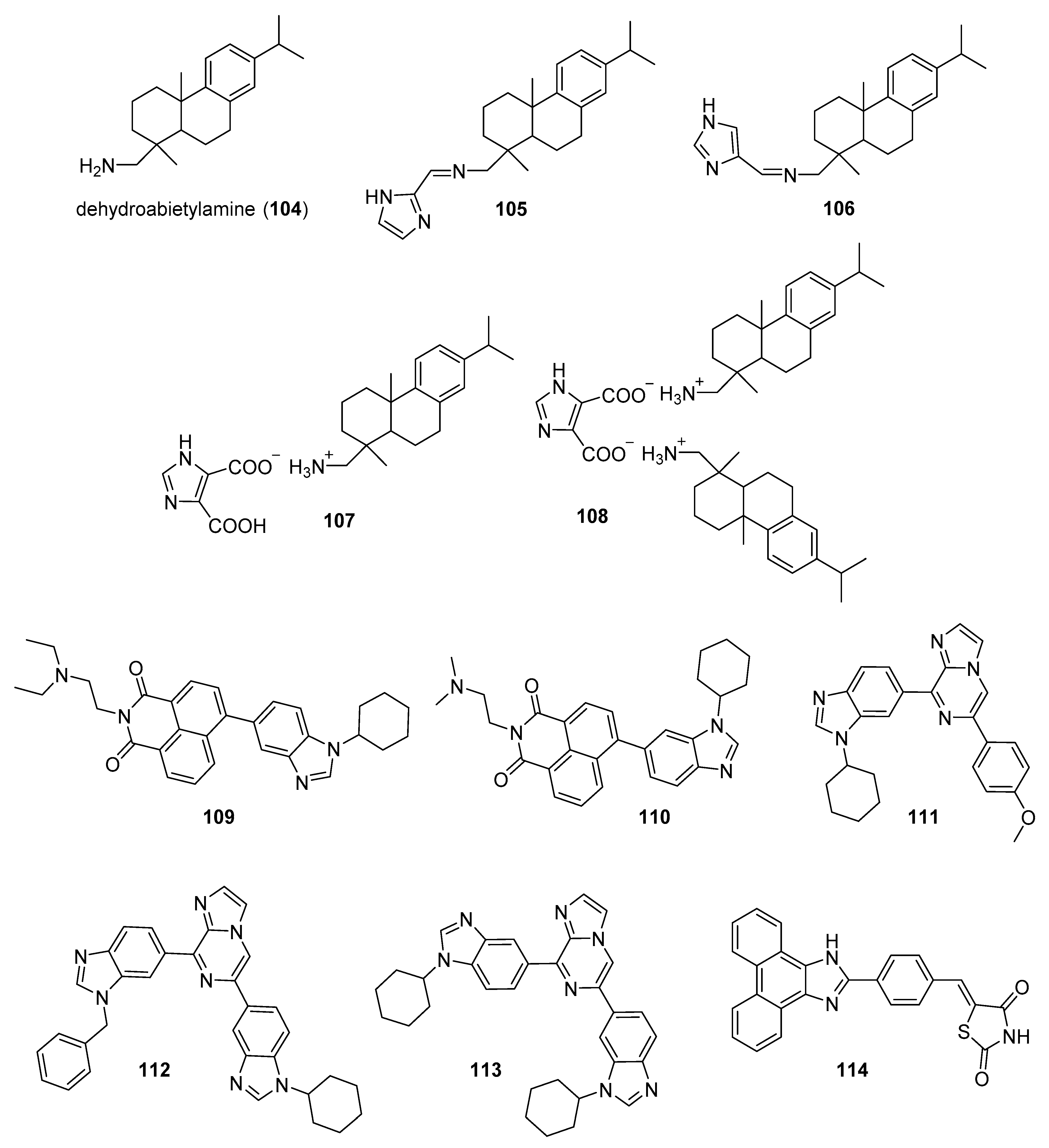
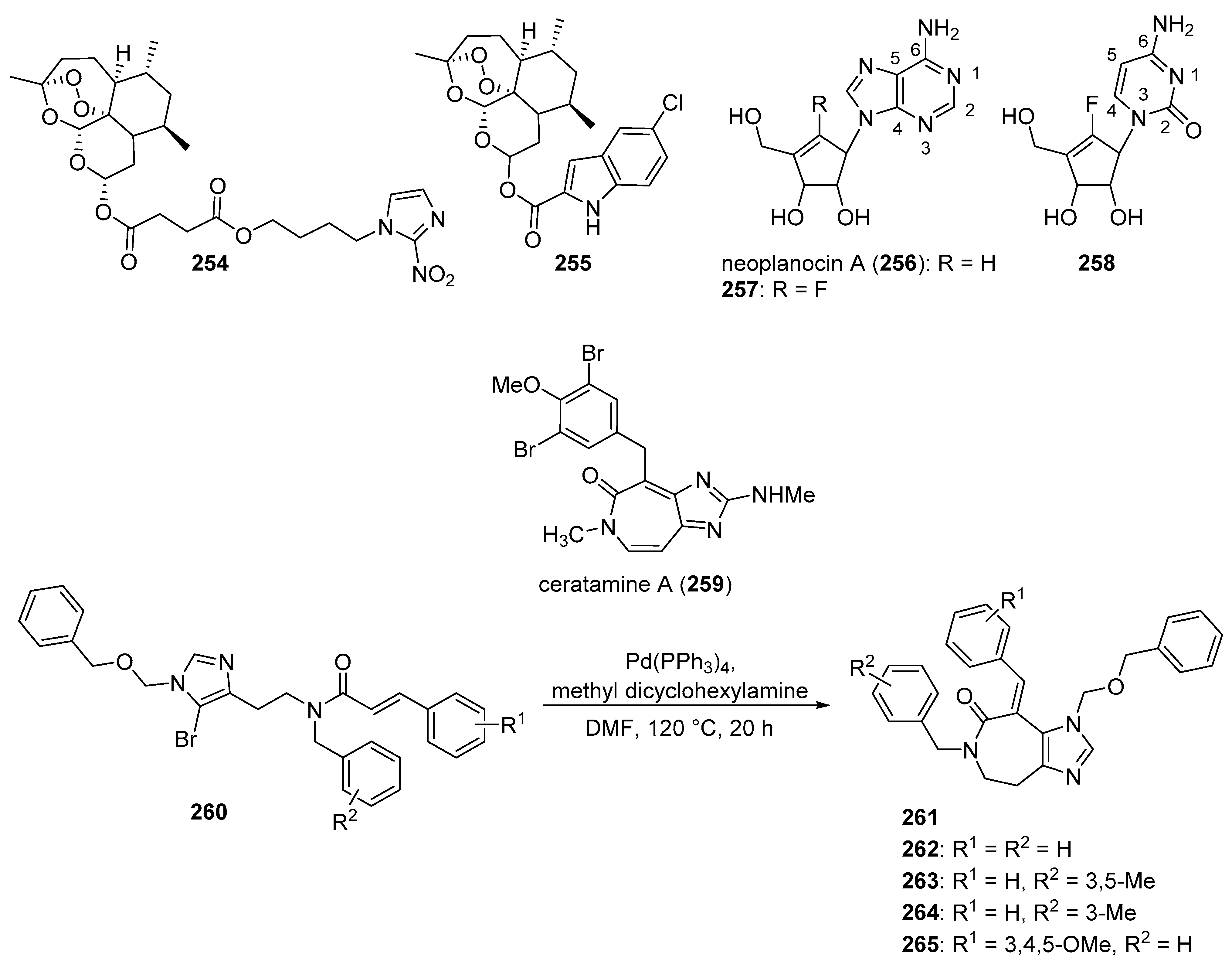
5.6. Natural Product Imidazole Derivatives
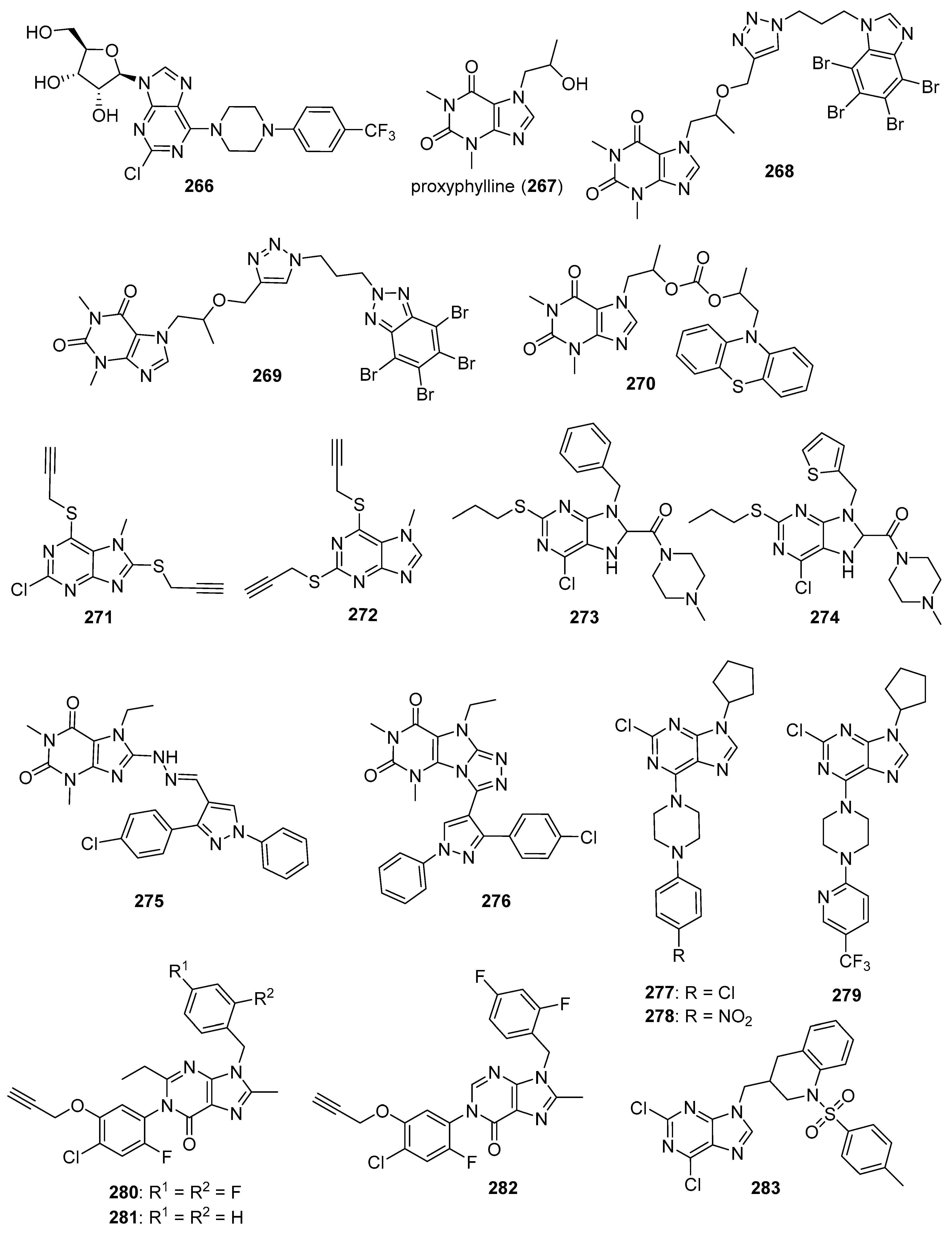
5.7. Purine Derivatives
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
List of Abbreviations and Cell Lines
References
- Wild, C.P.; Weiderpass, E.; Stewart, B.W. World Cancer Report: Cancer Research for Cancer Prevention; International Agency for Research on Cancer: Lyon, France, 2021; Available online: http://publications.iarc.fr/586 (accessed on 8 February 2021).
- Tsimberidou, A.M. Targeted therapy in cancer. Cancer Chemother. Pharmacol. 2015, 76, 1113–1132. [Google Scholar] [CrossRef] [Green Version]
- Seebacher, N.A.; Stacy, A.E.; Porter, G.M.; Merlot, A.M. Clinical development of targeted and immune based anti-cancer therapies. J. Exp. Clin. Cancer Res. 2019, 38, 156. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Alex, J.; Chauhan, M.; Joshi, G.; Kumar, R. A review on pharmacophoric designs of antiproliferative agents. Med. Chem. Res. 2015, 24, 903–920. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Ali, I.; Lone, M.N.; Aboul-Enein, H.Y. Imidazoles as potential anticancer agents. Med. Chem. Commun. 2017, 8, 1742–1773. [Google Scholar] [CrossRef] [PubMed]
- Rani, N.; Sharma, A.; Singh, R. Imidazoles as promising scaffolds for antibacterial activity: A review. Mini Rev. Med. Chem. 2013, 13, 1812–1835. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Liu, X.; Zhu, J.; Fang, Z.; Li, Z.; Pannecouque, C.; de Clercq, E. Synthesis and biological evaluation of imidazole thioacetanilides as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. Bioorg. Med. Chem. 2009, 17, 5775–5781. [Google Scholar] [CrossRef]
- Mishra, R.; Ganguly, S. Imidazole as an anti-epileptic: An overview. Med. Chem. Res. 2012, 21, 3929–3939. [Google Scholar] [CrossRef]
- Fan, Y.L.; Jin, X.H.; Huang, Z.P.; Yu, H.F.; Zeng, Z.G.; Gao, T.; Feng, L.S. Recent advances of imidazole-containing derivatives as anti-tubercular agents. Eur. J. Med. Chem. 2018, 150, 347–365. [Google Scholar] [CrossRef]
- Rani, N.; Sharma, A.; Gupta, G.K.; Singh, R. Imidazoles as potential antifungal agents: A review. Mini Rev. Med. Chem. 2013, 13, 1626–1655. [Google Scholar] [CrossRef] [PubMed]
- Wattanasin, P.; Saetear, P.; Wilairat, P.; Nacapricha, D.; Teerasong, S. Zone fluidics for measurement of octanol–water partition coefficient of drugs. Anal. Chim. Acta 2015, 860, 1–7. [Google Scholar] [CrossRef]
- Molina, P.; Tárraga, A.; Otón, F. Imidazole derivatives: A comprehensive survey of their recognition properties. Org. Biomol. Chem. 2012, 10, 1711–1724. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-S. The roles of imidazole ligands in coordination supramolecular systems. Cryst. Eng. Comm 2016, 18, 6543–6565. [Google Scholar] [CrossRef]
- Zhang, L.; Peng, X.-M.; Damu, G.L.V.; Geng, R.-X.; Zhou, C.-H. Comprehensive review in current developments of imidazole-based medicinal chemistry. Med. Res. Rev. 2014, 34, 340–437. [Google Scholar] [CrossRef] [PubMed]
- Debus, H. Ueber die einwirkung des ammoniaks auf glyoxal. Justus Liebigs Ann. Chem. 1858, 107, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Radziszewski, B. Ueber die constitution des lophins und verwandter verbindungen. Chem. Ber. 1882, 15, 1493–1496. [Google Scholar] [CrossRef] [Green Version]
- Benincori, T.; Brenna, E.; Sannicolo, F. Studies on Wallach’s imidazole synthesis. J. Chem. Soc. Perkin Trans. 1993, 1, 675–679. [Google Scholar] [CrossRef]
- Marckwald, W. Ein beitrag zur kenntniss der imidazole und der constitution des glyoxalins. Chem. Ber. 1892, 25, 2354–2362. [Google Scholar] [CrossRef]
- Sharma, A.; Kumar, V.; Kharb, R.; Kumar, S.; Sharma, P.C.; Pathak, D.P. Imidazole derivatives as potential therapeutic agents. Curr. Pharm. Des. 2016, 22, 3265–3301. [Google Scholar] [CrossRef]
- Vessaly, E.; Soleimani-Amiri, S.; Hosseinian, A.; Edjlali, L.; Bekhradnia, A. New protocols to access imidazoles and their ring fused analogues: Synthesis from N-propargylamines. RSC Adv. 2017, 7, 7079–7091. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.; Nanda, A.K. A review on heterocyclic: Synthesis and their application in medicinal chemistry of imidazole moiety. Sci. J. Chem. 2018, 6, 83–94. [Google Scholar] [CrossRef]
- Soni, J.; Sethiya, A.; Sahiba, N.; Agarwal, D.K.; Agarwal, S. Contemporary progress in the synthetic strategies of imidazole and its biological activities. Curr. Org. Synth. 2019, 16, 1078–1104. [Google Scholar] [CrossRef] [PubMed]
- Shabalin, D.A.; Camp, J.E. Recent advances in the synthesis of imidazoles. Org. Biomol. Chem. 2020, 18, 3950–3964. [Google Scholar] [CrossRef]
- Alaqeel, S.I. Synthetic approaches to benzimidazoles from o-phenylenediamine: A literature review. J. Saudi Chem. Soc. 2017, 21, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Traube, W. Ueber eine neue synthese des guanins und xanthins. Chem. Ber. 1900, 33, 1371–1383. [Google Scholar] [CrossRef] [Green Version]
- Zelli, R.; Zeinyeh, W.; Haudecoeur, R.; Alliot, J.; Boucherle, B.; Callebaut, I.; Décout, J.L. A one-pot synthesis of highly functionalized purines. Org. Lett. 2017, 19, 6360–6363. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, W.; Khan, M.F.; Verma, G.; Shaquiquzzaman, M.; Rizvi, M.A.; Mehdi, S.H.; Akhter, M.; Alam, M.M. Therapeutic evolution of benzimidazole derivatives in the last quinquennial period. Eur. J. Med. Chem. 2017, 126, 705–753. [Google Scholar] [CrossRef] [PubMed]
- Siwach, A.; Verma, P.K. Synthesis and therapeutic potential of imidazole containing compounds. BMC Chem. 2021, 15, 12. [Google Scholar] [CrossRef]
- Florian, S.; Mitchison, T.J. Anti-microtubule drugs. Methods Mol. Biol. 2016, 1413, 403–421. [Google Scholar] [PubMed]
- de Weger, V.A.; Beijnen, J.H.; Schellens, J.H. Cellular and clinical pharmacology of the taxanes docetaxel and paclitaxel—A review. Anticancer Drugs 2014, 25, 488–494. [Google Scholar] [CrossRef]
- Martino, E.; Casamassima, G.; Castiglione, S.; Cellupica, E.; Pantalone, S.; Papagni, F.; Rui, M.; Siciliano, A.M.; Collina, S. Vinca alkaloids and analogues as anti-cancer agents: Looking back, peering ahead. Bioorg. Med. Chem. Lett. 2018, 28, 2816–2826. [Google Scholar] [CrossRef]
- Li, J.; Ren, J.; Sun, W. Systematic review of ixabepilone for treating metastatic breast cancer. Breast Cancer 2017, 24, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Quan, D.; Chen, J.; Ding, J.; Zhao, J.; Lv, L.; Chen, J. Design, synthesis, and biological evaluation of 1-substituted -2-aryl imidazoles targeting tubulin polymerization as potential anticancer agents. Eur. J. Med. Chem. 2019, 184, 111732. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Arnst, K.E.; Wang, Y.; Kumar, G.; Ma, D.; Chen, H.; Wu, Z.; Yang, J.; White, S.W.; Miller, D.D.; et al. Structural modification of the 3,4,5-trimethoxyphenyl moiety in the tubulin inhibitor VERU-111 leads to improved antiproliferative activities. J. Med. Chem. 2018, 61, 7877–7891. [Google Scholar] [CrossRef]
- Wang, Q.; Arnst, K.E.; Wang, Y.; Kumar, G.; Ma, D.; White, S.W.; Miller, D.D.; Li, W.; Li, W. Structure-guided design, synthesis, and biological evaluation of (2-(1H-indol-3-yl)-1H-imidazol-4-yl)(3,4,5-trimethoxyphenyl) methanone (ABI-231) analogues targeting the colchicine binding site in tubulin. J. Med. Chem. 2019, 62, 6734–6750. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Liu, X.; Guan, Q.; Ding, N.; Wei, Q.; Tong, B.; Zhao, M.; Zhang, W.; Ma, L. 5-(3,4,5-trimethoxybenzoyl)-4-methyl-2-(p-tolyl) imidazol (BZML) targets tubulin and DNA to induce anticancer activity and overcome multidrug resistance in colorectal cancer cells. Chem. Biol. Interact. 2020, 315, 108886. [Google Scholar] [CrossRef]
- Sayeed, I.B.; Vishnuvardhan, M.V.P.S.; Nagarajan, A.; Kantevari, S.; Kamal, A. Imidazopyridine linked triazoles as tubulin inhibitors, effectively triggering apoptosis in lung cancer cell line. Bioorg. Chem. 2018, 80, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Narasimha Rao, M.P.; Nagaraju, B.; Kovvuri, J.; Polepalli, S.; Alavala, S.; Vishnuvardhan, M.V.P.S.; Swapna, P.; Nimbarte, V.D.; Lakshmi, J.K.; Jain, N.; et al. Synthesis of imidazo-thiadiazole linked indolinone conjugates and evaluated their microtubule network disrupting and apoptosis inducing ability. Bioorg. Chem. 2018, 76, 420–436. [Google Scholar] [CrossRef] [PubMed]
- Baig, M.F.; Nayak, V.L.; Budaganaboyina, P.; Mullagiri, K.; Sunkari, S.; Gour, J.; Kamal, A. Synthesis and biological evaluation of imidazo[2,1-b]thiazole-benzimidazole conjugates as microtubule-targeting agents. Bioorg. Chem. 2018, 77, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Donthiboina, K.; Anchi, P.; Gurram, S.; Sai Mani, G.; Lakshmi Uppu, J.; Godugu, C.; Shankaraiah, N.; Kamal, A. Synthesis and biological evaluation of substituted N-(2-(1H-benzo[d]imidazol-2-yl)phenyl)cinnamides as tubulin polymerization inhibitors. Bioorg. Chem. 2020, 103, 104191. [Google Scholar] [CrossRef]
- Wang, Y.T.; Shi, T.Q.; Zhu, H.L.; Liu, C.H. Synthesis, biological evaluation and molecular docking of benzimidazole grafted benzsulfamide-containing pyrazole ring derivatives as novel tubulin polymerization inhibitors. Bioorg. Med. Chem. 2019, 27, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hu, X.; Wan, G.; Wang, J.; Li, L.; Wu, X.; Liu, Z.; Yu, L. Discovery of 3-(((9H-purin-6-yl)amino)methyl)-4,6-dimethylpyridin-2(1H)-one derivatives as novel tubulin polymerization inhibitors for treatment of cancer. Eur. J. Med. Chem. 2019, 184, 111728. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, L.; Zhang, Q.; Wang, Q.; Feng, Z.; Xu, Y.; Xia, Y.; Yu, L. Design, synthesis and biological evaluation of a novel tubulin inhibitor SKLB0565 targeting the colchicine binding site. Bioorg. Chem. 2020, 97, 103695. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Le, X.; Huang, F.; Yang, J.; Yang, H.; Ma, J.; Hu, G.; Li, Q.; Chen, Z. Discovery of a dual tubulin polymerization and cell division cycle 20 homologue inhibitor via structural modification on apcin. J. Med. Chem. 2020, 63, 4685–4700. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, J.; Wan, L.; Zhou, X.; Wang, Z.; Wei, W. Targeting Cdc20 as a novel cancer therapeutic strategy. Pharmacol. Ther. 2015, 151, 141–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, P.C.; Wilke, D.V.; Branco, P.C.; Bauermeister, A.; Rezende-Teixeira, P.; Gaudêncio, S.P.; Costa-Lotufo, L.V. Enriching cancer pharmacology with drugs of marine origin. Br. J. Pharmacol. 2020, 177, 3–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Z.; Ma, M.; Zhong, C.; Wang, S.; Fu, Z.; Hou, Y.; Liu, Y.; Zhong, L.; Chu, Y.; Li, F.; et al. Development of novel phenoxy-diketopiperazine-type plinabulin derivatives as potent antimicrotubule agents based on the co-crystal structure. Bioorg. Med. Chem. 2020, 28, 115186. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol. Res. 2020, 152, 104609. [Google Scholar] [CrossRef]
- Karaman, S.; Leppänen, V.M.; Alitalo, K. Vascular endothelial growth factor signaling in development and disease. Development 2018, 145, dev151019. [Google Scholar] [CrossRef] [Green Version]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in signaling and disease: Beyond discovery and development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [Green Version]
- Randrup Hansen, C.; Grimm, D.; Bauer, J.; Wehland, M.; Magnusson, N.E. Effects and side effects of using sorafenib and sunitinib in the treatment of metastatic renal cell carcinoma. Int. J. Mol. Sci. 2017, 18, 461. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Yang, Q.; Liu, T.; Li, K.; Liu, Y.; Zhu, C.; Zhang, Z.; Li, L.; Zhang, C.; Xie, M.; et al. Design, synthesis and in vitro evaluation of 6-amide-2-aryl benzoxazole/benzimidazole derivatives against tumor cells by inhibiting VEGFR-2 kinase. Eur. J. Med. Chem. 2019, 179, 147–165. [Google Scholar] [CrossRef]
- Mostafa, A.S.; Gomaa, R.M.; Elmorsy, M.A. Design and synthesis of 2-phenyl benzimidazole derivatives as VEGFR-2 inhibitors with anti-breast cancer activity. Chem. Biol. Drug Des. 2019, 93, 454–463. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Akhtar, M.J.; Khan, A.A.; Ali, Z.; Dewangan, R.P.; Rafi, M.; Hassan, M.Q.; Akhtar, M.S.; Siddiqui, A.A.; Partap, S.; Pasha, S.; et al. Synthesis of stable benzimidazole derivatives bearing pyrazole as anticancer and EGFR receptor inhibitors. Bioorg. Chem. 2018, 78, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Hei, Y.Y.; Shen, Y.; Wang, J.; Zhang, H.; Zhao, H.Y.; Xin, M.; Cao, Y.X.; Li, Y.; Zhang, S.Q. Synthesis and evaluation of 2,9-disubstituted 8-phenylthio/phenylsulfinyl-9H-purine as new EGFR inhibitors. Bioorg. Med. Chem. 2018, 26, 2173–2185. [Google Scholar] [CrossRef]
- Lei, H.; Fan, S.; Zhang, H.; Liu, Y.J.; Hei, Y.Y.; Zhang, J.J.; Zheng, A.Q.; Xin, M.; Zhang, S.Q. Discovery of novel 9-heterocyclyl substituted 9H-purines as L858R/T790M/C797S mutant EGFR tyrosine kinase inhibitors. Eur. J. Med. Chem. 2020, 186, 111888. [Google Scholar] [CrossRef] [PubMed]
- Abou-Zied, H.A.; Youssif, B.G.M.; Mohamed, M.F.A.; Hayallah, A.M.; Abdel-Aziz, M. EGFR inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg. Chem. 2019, 89, 102997. [Google Scholar] [CrossRef] [PubMed]
- Hisham, M.; Youssif, B.G.M.; Osman, E.E.A.; Hayallah, A.M.; Abdel-Aziz, M. Synthesis and biological evaluation of novel xanthine derivatives as potential apoptotic antitumor agents. Eur. J. Med. Chem. 2019, 176, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Srour, A.M.; Ahmed, N.S.; Abd El-Karim, S.S.; Anwar, M.M.; El-Hallouty, S.M. Design, synthesis, biological evaluation, QSAR analysis and molecular modelling of new thiazol-benzimidazoles as EGFR inhibitors. Bioorg. Med. Chem. 2020, 28, 115657. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Joshi, G.; Kumar, M.; Arora, S.; Kaur, H.; Singh, S.; Munshi, A.; Kumar, R. Anticancer potential of some imidazole and fused imidazole derivatives: Exploring the mechanism via epidermal growth factor receptor (EGFR) inhibition. RSC Med. Chem. 2020, 11, 923–939. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Francini, C.M.; Musumeci, F.; Fallacara, A.L.; Botta, L.; Molinari, A.; Artusi, R.; Mennuni, L.; Angelucci, A.; Schenone, S. Optimization of aminoimidazole derivatives as Src family kinase inhibitors. Molecules 2018, 23, 2369. [Google Scholar] [CrossRef] [Green Version]
- Rezaei, Z.; Mahdi Didehvar, M.; Mahdavi, M.; Azizian, H.; Hamedifar, H.; Mohammed, E.H.M.; Ostad, S.; Amini, M. Anticancer properties of N-alkyl-2, 4-diphenylimidazo [1, 2-a] quinoxalin-1-amine derivatives; kinase inhibitors. Bioorg. Chem. 2019, 90, 103055. [Google Scholar] [CrossRef] [PubMed]
- Rossari, F.; Minutolo, F.; Orciuolo, E. Past, present, and future of Bcr-Abl inhibitors: From chemical development to clinical efficacy. J. Hematol. Oncol. 2018, 11, 84. [Google Scholar] [CrossRef] [Green Version]
- Wen, T.; Wang, J.; Shi, Y.; Qian, H.; Liu, P. Inhibitors targeting Bruton’s tyrosine kinase in cancers: Drug development advances. Leukemia 2021, 35, 312–332. [Google Scholar] [CrossRef]
- Bertrand, J.; Dostálová, H.; Krystof, V.; Jorda, R.; Castro, A.; Mella, J.; Espinosa-Bustos, C.; María Zarate, A.; Salas, C.O. New 2,6,9-trisubstituted purine derivatives as Bcr-Abl and Btk inhibitors and as promising agents against leukemia. Bioorg. Chem. 2020, 94, 103361. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Morris, J.C. Transforming growth factor-β: A therapeutic target for cancer. Hum. Vaccin. Immunother. 2017, 13, 1741–1750. [Google Scholar] [CrossRef]
- Guo, Z.; Song, X.; Zhao, L.M.; Piao, M.G.; Quan, J.; Piao, H.R.; Jin, C.H. Synthesis and biological evaluation of novel benzo[c][1,2,5]thiadiazol-5-yl and thieno[3,2-c]-pyridin-2-yl imidazole derivatives as ALK5 inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Manic, G.; Obrist, F.; Sistigu, A.; Vitale, I. Trial watch: Targeting ATM-CHK2 and ATR-CHK1 pathways for anticancer therapy. Mol. Cell Oncol. 2015, 2, e1012976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meuth, M. Chk1 suppressed cell death. Cell Div. 2010, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Ronco, C.; Martin, A.R.; Demange, L.; Benhida, R. ATM, ATR, CHK1, CHK2 and WEE1 inhibitors in cancer and cancer stem cells. Med. Chem. Commun. 2017, 8, 295–319. [Google Scholar] [CrossRef]
- Galal, S.A.; Khairat, S.H.M.; Ali, H.I.; Shouman, S.A.; Attia, Y.M.; Ali, M.M.; Mahmoud, A.E.; Abdel-Halim, A.H.; Fyiad, A.A.; Tabll, A.; et al. Part II: New candidates of pyrazole-benzimidazole conjugates as checkpoint kinase 2 (Chk2) inhibitors. Eur. J. Med. Chem. 2018, 144, 859–873. [Google Scholar] [CrossRef]
- Galal, S.A.; Khattab, M.; Shouman, S.A.; Ramadan, R.; Kandil, O.M.; Kandil, O.M.; Tabll, A.; El Abd, Y.S.; El-Shenawy, R.; Attia, Y.M.; et al. Part III: Novel checkpoint kinase 2 (Chk2) inhibitors; design, synthesis and biological evaluation of pyrimidine-benzimidazole conjugates. Eur. J. Med. Chem. 2018, 146, 687–708. [Google Scholar] [CrossRef]
- Karoulia, Z.; Gavathiotis, E.; Poulikakos, P.I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 2017, 17, 676–691. [Google Scholar] [CrossRef]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2016, 109, 314–341. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Maksoud, M.S.; Ammar, U.M.; Oh, C.H. Anticancer profile of newly synthesized BRAF inhibitors possess 5-(pyrimidin-4-yl)imidazo[2,1-b]thiazole scaffold. Bioorg. Med. Chem. 2019, 27, 2041–2051. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, G.I. The evolving role of cyclin-dependent kinase inhibitors in cancer management. Clin. Adv. Hematol. Oncol. 2017, 15, 174–177. [Google Scholar] [PubMed]
- Sánchez-Martínez, C.; Lallena, M.J.; Sanfeliciano, S.G.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs: Recent advances (2015-2019). Bioorg. Med. Chem. Lett. 2019, 29, 126637. [Google Scholar] [CrossRef]
- Al-Warhi, T.; Said, M.A.; El Hassab, M.A.; Aljaeed, N.; Ghabour, H.A.; Almahli, H.; Eldehna, W.M.; Abdel-Aziz, H.A. Unexpected synthesis, single-crystal x-ray structure, anticancer activity, and molecular docking studies of certain 2–((imidazole/benzimidazol–2–yl)thio)–1–arylethanones. Crystals 2020, 10, 446. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, W.J.; Yin, L.; Li, H.; Chen, Z.H.; Zhu, D.X.; Song, X.Q.; Cheng, Z.Z.; Song, P.; Wang, Z.; et al. Design and synthesis of 4-(2,3-dihydro-1H-benzo[d]pyrrolo[1,2-a]imidazol-7-yl)-N-(5-(piperazin-1-ylmethyl)pyridine-2-yl)pyrimidin-2-amine as a highly potent and selective cyclin-dependent kinases 4 and 6 inhibitors and the discovery of structure-activity relationships. Bioorg. Med. Chem. Lett. 2018, 28, 974–978. [Google Scholar]
- Ghanem, N.M.; Farouk, F.; George, R.F.; Abbas, S.E.S.; El-Badry, O.M. Design and synthesis of novel imidazo[4,5-b]pyridine based compounds as potent anticancer agents with CDK9 inhibitory activity. Bioorg. Chem. 2018, 80, 565–576. [Google Scholar] [CrossRef]
- Bavetsias, V.; Linardopoulos, S. Aurora kinase inhibitors: Current status and outlook. Front. Oncol. 2015, 5, 278. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.; Zhong, T.; Yang, H.; Yang, Y.; Wang, D.; Yang, X.; Xu, Y.; Fan, Y. Design, synthesis, biological evaluation of 6-(2-amino-1H-benzo[d]imidazole-6-yl)quinazolin-4(3H)-one derivatives as novel anticancer agents with Aurora kinase inhibition. Eur. J. Med. Chem. 2020, 190, 112108. [Google Scholar] [CrossRef]
- Fang, Y.; Zhang, X. Targeting NEK2 as a promising therapeutic approach for cancer treatment. Cell Cycle 2016, 15, 895–907. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chen, Y.; Gu, X.; Xi, J.; Ren, Z.; Wang, S.; Duan, Y.; Li, H.; Zhu, T.; Du, Y.; et al. Design, synthesis, and structure activity relationship (SAR) studies of novel imidazo[1,2-a] pyridine derivatives as Nek2 inhibitors. Bioorg. Med. Chem. 2020, 28, 115775. [Google Scholar] [CrossRef]
- Matheson, C.J.; Coxon, C.R.; Bayliss, R.; Boxall, K.; Carbain, B.; Fry, A.M.; Hardcastle, I.R.; Harnor, S.J.; Mas-Droux, C.; Newell, D.R.; et al. 2-Arylamino-6-ethynylpurines are cysteine-targeting irreversible inhibitors of Nek2 kinase. RSC Med. Chem. 2020, 11, 707–731. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curigliano, G.; Shah, R. Safety and tolerability of phosphatidylinositol-3-kinase (PI3K) inhibitors in oncology. Drug Saf. 2019, 42, 247–262. [Google Scholar] [CrossRef]
- Markham, A. Alpelisib: First global approval. Drugs 2019, 79, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.W.; Yu, L.; Bai, M.X.; Qin, X.C.; Song, M.T.; Zhao, Q.C. Design, synthesis and evaluation of some 1,6-disubstituted-1H-benzo[d]imidazoles derivatives targeted PI3K as anticancer agents. Bioorg. Chem. 2019, 93, 103283. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Lei, F.; Chen, X.; Wang, X.; Cao, L.; Ye, K.; Zhu, W.; Xu, S. Design, synthesis, and antitumor evaluation of quinoline-imidazole derivatives. Arch. Pharm. 2018, 351, e1700407. [Google Scholar] [CrossRef]
- Wu, T.T.; Guo, Q.Q.; Chen, Z.L.; Wang, L.L.; Du, Y.; Chen, R.; Mao, Y.H.; Yang, S.G.; Huang, J.; Wang, J.T.; et al. Design, synthesis and bioevaluation of novel substituted triazines as potential dual PI3K/mTOR inhibitors. Eur. J. Med. Chem. 2020, 204, 112637. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, H.; Liu, Q.; Sun, Y.; Gu, W. Synthesis and anticancer evaluation of novel 1H-benzo[d]imidazole derivatives of dehydroabietic acid as PI3Kα inhibitors. Bioorg. Chem. 2020, 100, 103845. [Google Scholar] [CrossRef]
- Zuo, W.Q.; Hu, R.; Wang, W.L.; Zhu, Y.X.; Xu, Y.; Yu, L.T.; Liu, Z.H.; Wang, N.Y. Identification of a potent and selective phosphatidylinositol 3-kinase δ inhibitor for the treatment of non-Hodgkin’s lymphoma. Bioorg. Chem. 2020, 105, 104344. [Google Scholar] [CrossRef] [PubMed]
- Gaonkar, S.; Savanur, M.A.; Sunagar, M.G.; Puthusseri, B.; Deshapande, N.; Nadaf, A.A.; Khazi, I.A.M. Exploring the potential of newly synthesized 4-methyl-6-morpholino-pyrimidine derivatives as antiproliferative agents. New J. Chem. 2018, 42, 2790–2803. [Google Scholar] [CrossRef]
- Sutherlin, D.P.; Bao, L.; Berry, M.; Castanedo, G.; Chuckowree, I.; Dotson, J.; Folks, A.; Friedman, L.; Goldsmith, R.; Gunzner, J.; et al. Discovery of a potent, selective, and orally available class I phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) kinase inhibitor (GDC-0980) for the treatment of cancer. J. Med. Chem. 2011, 54, 7579–7587. [Google Scholar] [CrossRef] [PubMed]
- Rescifina, A.; Zagni, C.; Varrica, M.G.; Pistarà, V.; Corsaro, A. Recent advances in small organic molecules as DNA intercalating agents: Synthesis, activity, and modeling. Eur. J. Med. Chem. 2014, 74, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Gupta, M.; Kumar, P.; Sharma, A. A comprehensive review on fused heterocyclic as DNA intercalators: Promising anticancer agents. Curr. Pharm. Des. 2021, 27, 15–42. [Google Scholar] [CrossRef]
- Zhao, F.; Wang, W.; Lu, W.; Xu, L.; Yang, S.; Cai, X.M.; Zhou, M.; Lei, M.; Ma, M.; Xu, H.J.; et al. High anticancer potency on tumor cells of dehydroabietylamine Schiff-base derivatives and a copper(II) complex. Eur. J. Med. Chem. 2018, 146, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Lu, W.; Su, F.; Xu, L.; Jiang, D.; Sun, X.; Shi, J.; Zhou, M.; Lin, F.; Cao, F. Synthesis and potential antineoplastic activity of dehydroabietylamine imidazole derivatives. Med. Chem. Commun. 2018, 9, 2091–2099. [Google Scholar] [CrossRef]
- Singh, I.; Luxami, V.; Paul, K. Synthesis and in vitro evaluation of naphthalimide-benzimidazole conjugates as potential antitumor agents. Org. Biomol. Chem. 2019, 17, 5349–5366. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Luxami, V.; Paul, K. Effective synthesis of benzimidazoles-imidazo[1,2-a]pyrazine conjugates: A comparative study of mono-and bis-benzimidazoles for antitumor activity. Eur. J. Med. Chem. 2019, 180, 546–561. [Google Scholar] [CrossRef]
- Singh, I.; Luxami, V.; Paul, K. Synthesis, cytotoxicity, pharmacokinetic profile, binding with DNA and BSA of new imidazo[1,2-a]pyrazine-benzo[d]imidazol-5-yl hybrids. Sci. Rep. 2020, 10, 6534. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Rani, R.; Luxami, V.; Paul, K. Synthesis of 5-(4-(1H-phenanthro[9,10-d]imidazol-2-yl)benzylidene)thiazolidine-2,4-dione as promising DNA and serum albumin-binding agents and evaluation of antitumor activity. Eur. J. Med. Chem. 2019, 166, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, C.; Seimiya, H. G-quadruplex in cancer biology and drug discovery. Biochem. Biophys. Res. Commun. 2020, 531, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Simonsson, T.; Pecinka, P.; Kubista, M. DNA tetraplex formation in the control region of c-myc. Nucleic Acids Res. 1998, 26, 1167–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.Y.; Chen, A.C.; Yin, Q.K.; Li, Z.; Huang, S.M.; Du, G.; He, J.H.; Zan, L.P.; Wang, S.K.; Xu, Y.H.; et al. New disubstituted quindoline derivatives inhibiting Burkitt’s lymphoma cell proliferation by impeding c-MYC transcription. J. Med. Chem. 2017, 60, 5438–5454. [Google Scholar] [CrossRef]
- Wu, Q.; Song, Y.; Liu, R.; Wang, R.; Mei, W.; Chen, W.; Yang, H.; Wang, X. Synthesis, docking studies and antitumor activity of phenanthroimidazole derivatives as promising c-myc G-quadruplex DNA stabilizers. Bioorg. Chem. 2020, 102, 104074. [Google Scholar] [CrossRef]
- Pelliccia, S.; Amato, J.; Capasso, D.; Di Gaetano, S.; Massarotti, A.; Piccolo, M.; Irace, C.; Tron, G.C.; Pagano, B.; Randazzo, A.; et al. Bio-inspired dual-selective BCL-2/c-MYC G-quadruplex binders: Design, synthesis, and anticancer activity of drug-like imidazo[2,1-i]purine derivatives. J. Med. Chem. 2020, 63, 2035–2050. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef]
- Hu, M.H.; Lin, X.T.; Liu, B.; Tan, J.H. Dimeric aryl-substituted imidazoles may inhibit ALT cancer by targeting the multimeric G-quadruplex in telomere. Eur. J. Med. Chem. 2020, 186, 111891. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef]
- Liang, X.; Wu, Q.; Luan, S.; Yin, Z.; He, C.; Yin, L.; Zou, Y.; Yuan, Z.; Li, L.; Song, X.; et al. A comprehensive review of topoisomerase inhibitors as anticancer agents in the past decade. Eur. J. Med. Chem. 2019, 171, 129–168. [Google Scholar] [CrossRef] [PubMed]
- Kundu, B.; Das, S.K.; Paul Chowdhuri, S.; Pal, S.; Sarkar, D.; Ghosh, A.; Mukherjee, A.; Bhattacharya, D.; Das, B.B.; Talukdar, A. Discovery and mechanistic study of tailor-made quinoline derivatives as topoisomerase 1 poison with potent anticancer activity. J. Med. Chem. 2019, 62, 3428–3446. [Google Scholar] [CrossRef]
- Yu, S.; Wang, G.; Shi, Y.; Xu, H.; Zheng, Y.; Chen, Y. MCMs in cancer: Prognostic potential and mechanisms. Anal. Cell Pathol. 2020, 2020, 3750294. [Google Scholar] [CrossRef] [Green Version]
- Gou, K.; Liu, J.; Feng, X.; Li, H.; Yuan, Y.; Xing, C. Expression of minichromosome maintenance proteins (MCM) and cancer prognosis: A meta-analysis. J. Cancer 2018, 9, 1518–1526. [Google Scholar] [CrossRef]
- Lin, C.; Wu, H.; Hsu, Y.; Cheng, T.; Liu, J.; Huang, R.; Hsiao, T.; Wang, C.; Hung, P.; Lan, A.; et al. Suppression of drug-resistant non-small-cell lung cancer with inhibitors targeting minichromosomal maintenance protein. J. Med. Chem. 2020, 63, 3172–3187. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Jain, P.G.; Patel, B.D. Medicinal chemistry approaches of poly ADP-ribose polymerase 1 (PARP1) inhibitors as anticancer agents —A recent update. Eur. J. Med. Chem. 2019, 165, 198–215. [Google Scholar] [CrossRef] [PubMed]
- Min, R.; Wu, W.; Wang, M.; Tang, L.; Chen, D.; Zhao, H.; Zhang, C.; Jiang, Y. Discovery of 2-(1-(3-(4-chloroxyphenyl)-3-oxo-propyl)pyrrolidine-3-yl)-1H-benzo[d]imidazole-4-carboxamide: A potent poly(ADP-ribose) polymerase (PARP) inhibitor for treatment of cancer. Molecules 2019, 24, 1901. [Google Scholar] [CrossRef] [Green Version]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [Green Version]
- Ai, T.; Cui, H.; Chen, L. Multi-targeted histone deacetylase inhibitors in cancer therapy. Curr. Med. Chem. 2012, 19, 475–487. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClure, J.J.; Li, X.; Chou, C.J. Advances and challenges of HDAC inhibitors in cancer therapeutics. Adv. Cancer Res. 2018, 138, 183–211. [Google Scholar] [PubMed]
- Chen, D.; Soh, C.K.; Goh, W.H.; Wang, Z.; Wang, H. Synthesis and biological evaluation of 6-phenylpurine linked hydroxamates as novel histone deacetylase inhibitors. Bioorg. Chem. 2020, 98, 103724. [Google Scholar] [CrossRef]
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Mackwitz, M.K.W.; Hamacher, A.; Osko, J.D.; Held, J.; Schöler, A.; Christianson, D.W.; Kassack, M.U.; Hansen, F.K. Multicomponent synthesis and binding mode of imidazo[1,2-a]pyridine-capped selective HDAC6 inhibitors. Org. Lett. 2018, 20, 3255–3258. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Chang, T.Y.; Lai, M.J.; Hsu, K.C.; Yen, Y.; Lin, T.E.; Lee, S.B.; Liou, J.P. Purine/purine isoster based scaffolds as new derivatives of benzamide class of HDAC inhibitors. Eur. J. Med. Chem. 2020, 196, 112291. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Gong, G.; Chen, X.; Song, R.; Duan, M.; Qiao, R.; Jiao, Y.; Qi, J.; Chen, Y.; Zhu, Y. Design, synthesis and biological evaluation of novel benzoylimidazole derivatives as Raf and histone deacetylases dual inhibitors. Chem. Pharm. Bull. 2019, 67, 1116–1122. [Google Scholar] [CrossRef] [Green Version]
- Yun, F.; Cheng, C.; Ullah, S.; Yuan, Q. Design, synthesis and biological evaluation of novel histone deacetylase1/2 (HDAC1/2) and cyclin-dependent kinase2 (CDK2) dual inhibitors against malignant cancer. Eur. J. Med. Chem. 2020, 198, 112322. [Google Scholar] [CrossRef] [PubMed]
- Majello, B.; Gorini, F.; Saccà, C.D.; Amente, S. Expanding the role of the histone lysine-specific demethylase LSD1 in cancer. Cancers 2019, 11, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romussi, A.; Cappa, A.; Vianello, P.; Brambillasca, S.; Cera, M.R.; Dal Zuffo, R.; Fagà, G.; Fattori, R.; Moretti, L.; Trifirò, P.; et al. Discovery of reversible inhibitors of KDM1A efficacious in acute myeloid leukemia models. ACS Med. Chem. Lett. 2020, 11, 754–759. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, Y.; Aguilar, A.; Bernard, D.; Yang, C.Y. Targeting the MDM2-p53 protein-protein interaction for new cancer therapy: Progress and challenges. Cold Spring Harb. Perspect. Med. 2017, 7, a026245. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 inhibition: An important step forward in cancer therapy. Leukemia 2020, 34, 2858–2874. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Dong, G.; Wu, S.; Fang, K.; Miao, Z.; Wang, W.; Sheng, C. Small molecules simultaneously inhibiting p53-murine double minute 2 (MDM2) interaction and histone deacetylases (HDACs): Discovery of novel multitargeting antitumor agents. J. Med. Chem. 2018, 61, 7245–7260. [Google Scholar] [CrossRef]
- Cochran, A.G.; Conery, A.R.; Sims, R.J., III. Bromodomains: A new target class for drug development. Nat. Rev. Drug Discov. 2019, 18, 609–628. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, P.; Zhao, L.; Zhang, F.; Zhang, B.; Xu, C.; Zhang, H.; Zhou, J. Exploiting the 7-methylimidazo[1,5-a]pyrazin-8(7H)-one scaffold for the development of novel chemical inhibitors for Bromodomain and Extraterminal Domain (BET) family. Bioorg. Chem. 2019, 90, 103044. [Google Scholar] [CrossRef]
- Schapira, M.; Tyers, M.; Torrent, M.; Arrowsmith, C. WD40 repeat domain proteins: A novel target class? Nat. Rev. Drug Discov. 2017, 16, 773–786. [Google Scholar] [CrossRef]
- Lu, K.; Tao, H.; Si, X.; Chen, Q. The histone H3 lysine 4 presenter WDR5 as an oncogenic protein and novel epigenetic target in cancer. Front. Oncol. 2018, 8, 502. [Google Scholar] [CrossRef]
- Thomas, L.R.; Wang, Q.; Grieb, B.C.; Phan, J.; Foshage, A.M.; Sun, Q.; Olejniczak, E.T.; Clark, T.; Dey, S.; Lorey, S.; et al. Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol. Cell 2015, 58, 440–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Teuscher, K.; Aho, E.; Alvarado, J.; Mills, J.; Meyers, K.; Gogliotti, R.; Han, C.; Macdonald, J.; Sai, J.; et al. Discovery and structure-based optimization of potent and selective WD repeat domain 5 (WDR5) inhibitors containing a dihydroisoquinolinone bicyclic core. J. Med. Chem. 2020, 63, 656–675. [Google Scholar] [CrossRef]
- Chacón Simon, S.; Wang, F.; Thomas, L.; Phan, J.; Zhao, B.; Olejniczak, E.; Macdonald, J.; Shaw, J.; Schlund, C.; Payne, W.; et al. Discovery of WD repeat-containing protein 5 (WDR5)-MYC inhibitors using fragment-based methods and structure-based design. J. Med. Chem. 2020, 63, 4315–4333. [Google Scholar] [CrossRef]
- Yang, L.; Lin, S.; Xu, L.; Lin, J.; Zhao, C.; Huang, X. Novel activators and small-molecule inhibitors of STAT3 in cancer. Cytokine Growth Factor Rev. 2019, 49, 10–22. [Google Scholar] [CrossRef]
- Su, J.C.; Chang, C.H.; Wu, S.H.; Shiau, C.W. Novel imidazopyridine suppresses STAT3 activation by targeting SHP-1. J. Enzyme Inhib. Med. Chem. 2018, 33, 1248–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; He, Q.; Wu, K.; Guo, T.; Du, X.; Zhang, H.; Fang, L.; Zheng, N.; Zhang, Q.; Ye, F. Design, synthesis and activity of novel 2,6-disubstituted purine derivatives, potential small molecule inhibitors of signal transducer and activator of transcription 3. Eur. J. Med. Chem. 2019, 179, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Hornyák, L.; Dobos, N.; Koncz, G.; Karányi, Z.; Páll, D.; Szabó, Z.; Halmos, G.; Székvölgyi, L. The role of indoleamine-2,3-dioxygenase in cancer development, diagnostics, and therapy. Front. Immunol. 2018, 9, 151. [Google Scholar] [CrossRef]
- Tu, W.; Yang, F.; Xu, G.; Chi, J.; Liu, Z.; Peng, W.; Hu, B.; Zhang, L.; Wan, H.; Yu, N.; et al. Discovery of imidazoisoindole derivatives as highly potent and orally active indoleamine-2,3-dioxygenase inhibitors. ACS Med. Chem. Lett. 2019, 10, 949–953. [Google Scholar] [CrossRef]
- Tojo, S.; Kohno, T.; Tanaka, T.; Kamioka, S.; Ota, Y.; Ishii, T.; Kamimoto, K.; Asano, S.; Isobe, Y. Crystal structures and structure-activity relationships of imidazothiazole derivatives as IDO1 inhibitors. ACS Med. Chem. Lett. 2014, 5, 1119–1123. [Google Scholar] [CrossRef] [Green Version]
- Griglio, A.; Torre, E.; Serafini, M.; Bianchi, A.; Schmid, R.; Coda Zabetta, G.; Massarotti, A.; Sorba, G.; Pirali, T.; Fallarini, S. A multicomponent approach in the discovery of indoleamine 2,3-dioxygenase 1 inhibitors: Synthesis, biological investigation and docking studies. Bioorg. Med. Chem. Lett. 2018, 28, 651–657. [Google Scholar] [CrossRef]
- Serafini, M.; Torre, E.; Aprile, S.; Massarotti, A.; Fallarini, S.; Pirali, T. Synthesis, docking and biological evaluation of a novel class of imidazothiazoles as IDO1 inhibitors. Molecules 2019, 24, 1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafini, M.; Torre, E.; Aprile, S.; Grosso, E.; Gesù, A.; Griglio, A.; Colombo, G.; Travelli, C.; Paiella, S.; Adamo, A.; et al. Discovery of highly potent benzimidazole derivatives as indoleamine 2,3-dioxygenase-1 (IDO1) inhibitors: From structure-based virtual screening to in vivo pharmacodynamic activity. J. Med. Chem. 2020, 63, 3047–3065. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I. Shagufta Recent developments in steroidal and nonsteroidal aromatase inhibitors for the chemoprevention of estrogen-dependent breast cancer. Eur. J. Med. Chem. 2015, 102, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Kalalinia, F.; Jouya, M.; Komachali, A.K.; Aboutourabzadeh, S.M.; Karimi, G.; Behravan, J.; Abnous, K.; Etemad, L.; Kamali, H.; Hadizadeh, F. Design, synthesis, and biological evaluation of new azole derivatives as potent aromatase inhibitors with potential effects against breast cancer. Anticancer Agents Med. Chem. 2018, 18, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, Y.; Hu, H.; Huang, H.; Bao, Z.; Yang, P.; Wang, Y.; You, G.; Yan, W.; Jiang, T.; et al. ALDH1A3: A marker of mesenchymal phenotype in gliomas associated with cell invasion. PLoS ONE 2015, 10, e0142856. [Google Scholar] [CrossRef]
- Quattrini, L.; Gelardi, E.; Coviello, V.; Sartini, S.; Ferraris, D.M.; Mori, M.; Nakano, I.; Garavaglia, S.; La Motta, C. Imidazo[1,2-a]pyridine derivatives as aldehyde dehydrogenase inhibitors: Novel chemotypes to target glioblastoma stem cells. J. Med. Chem. 2020, 63, 4603–4616. [Google Scholar] [CrossRef] [PubMed]
- Chau, L.Y. Heme oxygenase-1: Emerging target of cancer therapy. J. Biomed. Sci. 2015, 22, 22. [Google Scholar] [CrossRef] [Green Version]
- Ciaffaglione, V.; Intagliata, S.; Pittalà, V.; Marrazzo, A.; Sorrenti, V.; Vanella, L.; Rescifina, A.; Floresta, G.; Sultan, A.; Greish, K.; et al. New arylethanolimidazole derivatives as HO-1 inhibitors with cytotoxicity against MCF-7 breast cancer cells. Int. J. Mol. Sci. 2020, 21, 1923. [Google Scholar] [CrossRef] [Green Version]
- Astorgues-Xerri, L.; Riveiro, M.; Tijeras-Raballand, A.; Serova, M.; Neuzillet, C.; Albert, S.; Raymond, E.; Faivre, S. Unraveling galectin-1 as a novel therapeutic target for cancer. Cancer Treat. Rev. 2014, 40, 307–319. [Google Scholar] [CrossRef]
- Goud, N.S.; Ghouse, S.M.; Vishnu, J.; Komal, D.; Talla, V.; Alvala, R.; Pranay, J.; Kumar, J.; Qureshi, I.A.; Alvala, M. Synthesis of 1-benzyl-1H-benzimidazoles as galectin-1 mediated anticancer agents. Bioorg. Chem. 2019, 89, 103016. [Google Scholar] [CrossRef] [PubMed]
- Sridhar Goud, N.; Pooladanda, V.; Muni Chandra, K.; Lakshmi Soukya, P.S.; Alvala, R.; Kumar, P.; Nagaraj, C.; Dawn Bharath, R.; Qureshi, I.; Godugu, C.; et al. Novel benzimidazole-triazole hybrids as apoptosis inducing agents in lung cancer: Design, synthesis, 18F-radiolabeling & galectin-1 inhibition studies. Bioorg. Chem. 2020, 102, 104125. [Google Scholar]
- Chatterjee, A.; Gupta, S. The multifaceted role of glutathione S-transferases in cancer. Cancer Lett. 2018, 433, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Karim, S.S.; Anwar, M.M.; Zaki, E.R.; Elseginy, S.A.; Nofal, Z.M. Synthesis and molecular modeling of new benzimidazoles as glutathione S-transferase inhibitors and anticancer agents. Future Med. Chem. 2018, 10, 157–181. [Google Scholar] [CrossRef] [PubMed]
- Orafaie, A.; Matin, M.M.; Sadeghian, H. The importance of 15-lipoxygenase inhibitors in cancer treatment. Cancer Metastasis Rev. 2018, 37, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Afifi, O.S.; Shaaban, O.G.; Abd El Razik, H.A.; Shams El-Dine, S.E.A.; Ashour, F.A.; El-Tombary, A.A.; Abu-Serie, M.M. Synthesis and biological evaluation of purine-pyrazole hybrids incorporating thiazole, thiazolidinone or rhodanine moiety as 15-LOX inhibitors endowed with anticancer and antioxidant potential. Bioorg. Chem. 2019, 87, 821–837. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Kumar, S.; Narasimhan, B. Estrogen alpha receptor antagonists for the treatment of breast cancer: A review. Chem. Cent. J. 2018, 12, 107. [Google Scholar] [CrossRef] [Green Version]
- Singla, R.; Gupta, K.B.; Upadhyay, S.; Dhiman, M.; Jaitak, V. Design, synthesis and biological evaluation of novel indole-benzimidazole hybrids targeting estrogen receptor alpha (ER-α). Eur. J. Med. Chem. 2018, 146, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Wang, S.; Wang, S.Q.; Teng, Q.X.; Yang, L.; Lei, Z.N.; Yuan, X.; Huo, J.F.; Chen, X.; Wang, M.; Yu, B.; et al. Structure-based design, synthesis, and biological evaluation of new triazolo[1,5-a]pyrimidine derivatives as highly potent and orally active ABCB1 modulators. J. Med. Chem. 2020, 63, 15979–15996. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, L.; Bolaender, A.; Patel, H.J.; Taldone, T. Heat shock protein (HSP) drug discovery and development: Targeting heat shock proteins in disease. Curr. Top. Med. Chem. 2016, 16, 2753–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, J.; Carter, T.R.; Cohen, M.S.; Blagg, B.S.J. Old and new approaches to target the Hsp90 chaperone. Curr. Cancer Drug Targets 2020, 20, 253–270. [Google Scholar] [CrossRef]
- Uno, T.; Kawai, Y.; Yamashita, S.; Oshiumi, H.; Yoshimura, C.; Mizutani, T.; Suzuki, T.; Chong, K.T.; Shigeno, K.; Ohkubo, M.; et al. Discovery of 3-ethyl-4-(3-isopropyl-4-(4-(1-methyl-1H-pyrazol-4-yl)-1H-imidazol-1-yl)-1H-pyrazolo[3,4-b]pyridin-1-yl)benzamide (TAS-116) as a potent, selective, and orally available HSP90 inhibitor. J. Med. Chem. 2019, 62, 531–551. [Google Scholar] [CrossRef] [PubMed]
- Karataş, M.O.; Tekin, S.; Alici, B.; Sandal, S. Cytotoxic effects of coumarin substituted benzimidazolium salts against human prostate and ovarian cancer cells. J. Chem. Sci. 2019, 131, 69. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.Q.; Chen, X.B.; Ye, P.T.; Yang, Z.X.; Bai, M.J.; Duan, S.Y.; Li, Y.; Yang, X.D. Synthesis and biological evaluation of novel 3-benzylcoumarin-imidazolium salts. Bioorg. Med. Chem. Lett. 2020, 30, 126896. [Google Scholar] [CrossRef]
- Akkoç, S. Derivatives of 1-(2-(piperidin-1-yl)ethyl)-1H-benzo[d]imidazole: Synthesis, characterization, determining of electronic properties and cytotoxicity studies. ChemistrySelect 2019, 4, 4938–4943. [Google Scholar] [CrossRef]
- Yang, J.L.; Ma, Y.H.; Li, Y.H.; Zhang, Y.P.; Tian, H.C.; Huang, Y.C.; Li, Y.; Chen, W.; Yang, L.J. Design, synthesis, and anticancer activity of novel trimethoxyphenyl-derived chalcone-benzimidazolium salts. ACS Omega 2019, 4, 20381–20393. [Google Scholar] [CrossRef]
- Kuang, W.B.; Huang, R.Z.; Qin, J.L.; Lu, X.; Qin, Q.P.; Zou, B.Q.; Chen, Z.F.; Liang, H.; Zhang, Y. Design, synthesis and pharmacological evaluation of new 3-(1H-benzimidazol-2-yl)quinolin-2(1H)-one derivatives as potential antitumor agents. Eur. J. Med. Chem. 2018, 157, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Mohareb, R.M.; Abdallah, A.E.M.; Mohamed, A.A. Synthesis of novel thiophene, thiazole and coumarin derivatives based on benzimidazole nucleus and their cytotoxicity and toxicity evaluations. Chem. Pharm. Bull. 2018, 66, 309–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouiter, M.I.; Boulebd, H.; Pereira, D.M.; Valentão, P.; Andrade, P.B.; Belfaitah, A.; Silva, A.M. New chalcone-type compounds and 2-pyrazoline derivatives: Synthesis and caspase-dependent anticancer activity. Future Med. Chem. 2020, 12, 493–509. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.Y.; Ko, P.W.; Chang, Y.J.; Kapoor, M.; Liang, Y.C.; Chu, H.L.; Lin, H.H.; Horng, J.C.; Hsu, M.H. Design and synthesis of benzimidazole-chalcone derivatives as potential anticancer agents. Molecules 2019, 24, 3259. [Google Scholar] [CrossRef] [Green Version]
- Suk, F.M.; Liu, C.L.; Hsu, M.H.; Chuang, Y.T.; Wang, J.P.; Liao, Y.J. Treatment with a new benzimidazole derivative bearing a pyrrolidine side chain overcomes sorafenib resistance in hepatocellular carcinoma. Sci. Rep. 2019, 9, 17259. [Google Scholar] [CrossRef] [Green Version]
- Rasal, N.; Sonawane, R.; Jagtap, S. Potential 2,4-dimethyl-1H-pyrrole-3-carboxamide bearing benzimidazole template: Design, synthesis, in vitro anticancer and in silico ADME study. Bioorg. Chem. 2020, 97, 103660. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Yang, Y.; Wang, W.; Liu, Q.; Sun, Y.; Gu, W. Synthesis, cytotoxicity and apoptosis-inducing activity of novel 1H-benzo[d]imidazole derivatives of dehydroabietic acid. J. Chin. Chem. Soc. 2020, 67, 1668–1678. [Google Scholar] [CrossRef]
- Bistrović, A.; Krstulović, L.; Harej, A.; Grbčić, P.; Sedić, M.; Koštrun, S.; Pavelić, S.K.; Bajić, M.; Raić-Malić, S. Design, synthesis and biological evaluation of novel benzimidazole amidines as potent multi-target inhibitors for the treatment of non-small cell lung cancer. Eur. J. Med. Chem. 2018, 143, 1616–1634. [Google Scholar] [CrossRef] [PubMed]
- Ashok, D.; Ram Reddy, M.; Nagaraju, N.; Dharavath, R.; Ramakrishna, K.; Gundu, S.; Shravani, P.; Sarasija, M. Microwave-assisted synthesis and in vitro antiproliferative activity of some novel 1,2,3-triazole-based pyrazole aldehydes and their benzimidazole derivatives. Med. Chem. Res. 2020, 29, 699–706. [Google Scholar] [CrossRef]
- Meenakshisundaram, S.; Manickam, M.; Pillaiyar, T. Exploration of imidazole and imidazopyridine dimers as anticancer agents: Design, synthesis, and structure-activity relationship study. Arch. Pharm. (Weinheim) 2019, 352, e1900011. [Google Scholar] [CrossRef]
- Güçlü, D.; Kuzu, B.; Tozlu, İ.; Taşpınar, F.; Canpınar, H.; Taşpınar, M.; Menges, N. Synthesis of novel imidazopyridines and their biological evaluation as potent anticancer agents: A promising candidate for glioblastoma. Bioorg. Med. Chem. Lett. 2018, 28, 2647–2651. [Google Scholar] [CrossRef]
- Khalili, F.; Akrami, S.; Safavi, M.; Mohammadi-Khanaposhtani, M.; Saeedi, M.; Ardestani, S.K.; Larijani, B.; Zonouzi, A.; Tehrani, M.B.; Mahdavi, M. Design, synthesis, in vitro cytotoxic activity evaluation, and study of apoptosis inducing effect of new styrylimidazo[1,2-a]pyridines as potent anti-breast cancer agents. Anticancer Agents Med. Chem. 2019, 19, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Chitti, S.; Singireddi, S.; Santosh Kumar Reddy, P.; Trivedi, P.; Bobde, Y.; Kumar, C.; Rangan, K.; Ghosh, B.; Sekhar, K. Design, synthesis and biological evaluation of 2-(3,4-dimethoxyphenyl)-6 (1,2,3,6-tetrahydropyridin-4-yl)imidazo[1,2-a]pyridine analogues as antiproliferative agents. Bioorg. Med. Chem. Lett. 2019, 29, 2551–2558. [Google Scholar] [CrossRef] [PubMed]
- Mani, G.; Anchi, P.; Sunkari, S.; Donthiboina, K.; Godugu, C.; Shankaraiah, N.; Kamal, A. Synthesis of (Z)-3-(arylamino)-1-(3-phenylimidazo[1,5-a]pyridin-1-yl)prop-2-en-1-ones as potential cytotoxic agents. Bioorg. Med. Chem. Lett. 2020, 30, 127432. [Google Scholar] [CrossRef] [PubMed]
- Mantipally, M.; Gangireddy, M.R.; Gundla, R.; Badavath, V.N.; Mandha, S.R.; Maddipati, V.C. Rational design, molecular docking and synthesis of novel homopiperazine linked imidazo[1,2-a]pyrimidine derivatives as potent cytotoxic and antimicrobial agents. Bioorg. Med. Chem. Lett. 2019, 29, 2248–2253. [Google Scholar] [CrossRef] [PubMed]
- El-Borai, M.A.; Awad, M.K.; Rizk, H.F.; Atlam, F.M. Design, synthesis and docking study of novel imidazolyl pyrazolopyridine derivatives as antitumor agents targeting MCF7 cell line. Curr. Org. Synth. 2018, 15, 275–285. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, Z.; Zhang, W.; Yan, D. 2-Substituted-1-(2-morpholinoethyl)-1H-naphtho[2,3-d]imidazole-4,9-diones: Design, synthesis and antiproliferative activity. Bioorg. Med. Chem. Lett. 2018, 28, 2454–2458. [Google Scholar] [CrossRef]
- Wei, Q.; Li, J.; Tang, F.; Yin, Y.; Zhao, Y.; Yao, Q. Synthesis and biological evaluation of novel 2-arylvinyl-substituted naphtho[2,3-d]imidazolium halide derivatives as potent antitumor agents. Eur. J. Med. Chem. 2018, 144, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Hebishy, A.M.S.; Abdelfattah, M.S.; Elmorsy, A.; Elwahy, A.H.M. ZnO nanoparticles catalyzed synthesis of bis- and poly(imidazoles) as potential anticancer agents. Synth. Commun. 2020, 50, 980–996. [Google Scholar] [CrossRef]
- Ghanbarimasir, Z.; Bekhradnia, A.; Morteza-Semnani, K.; Rafiei, A.; Razzaghi-Asl, N.; Kardan, M. Design, synthesis, biological assessment and molecular docking studies of new 2-aminoimidazole-quinoxaline hybrids as potential anticancer agents. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018, 194, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Yavuz, S.Ç.; Akkoç, S.; Sarıpınar, E. The cytotoxic activities of imidazole derivatives prepared from various guanylhydrazone and phenylglyoxal monohydrate. Synth. Commun. 2019, 49, 3198–3209. [Google Scholar] [CrossRef]
- Demjén, A.; Alföldi, R.; Angyal, A.; Gyuris, M.; Hackler, L., Jr.; Szebeni, G.J.; Wölfling, J.; Puskás, L.G.; Kanizsai, I. Synthesis, cytotoxic characterization, and SAR study of imidazo[1,2-b]pyrazole-7-carboxamides. Arch. Pharm. 2018, 351, e1800062. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.P.; Reddy, T.S.; Sunkari, S.; Rao, A.V.S.; Babu, K.S.; Bhargava, S.K.; Kamal, A. Synthesis of benzo[d]imidazo[2,1-b]thiazole-propenone conjugates as cytotoxic and apoptotic inducing agents. Anticancer Agents Med. Chem. 2019, 19, 347–355. [Google Scholar] [CrossRef]
- Hu, Y.; Li, N.; Zhang, J.; Wang, Y.; Chen, L.; Sun, J. Artemisinin-indole and artemisinin-imidazole hybrids: Synthesis, cytotoxic evaluation and reversal effects on multidrug resistance in MCF-7/ADR cells. Bioorg. Med. Chem. Lett. 2019, 29, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.S.; Jarhad, D.B.; Kim, G.; Nayak, A.; Zhao, L.X.; Yu, J.; Kim, H.R.; Lee, J.Y.; Mulamoottil, V.A.; Chandra, G.; et al. Design, synthesis and anticancer activity of fluorocyclopentenyl-purines and -pyrimidines. Eur. J. Med. Chem. 2018, 155, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Tao, L.; Ji, M.; Chen, X.; Liu, Z. Synthesis and cytotoxicity of novel imidazo[4,5-d]azepine compounds derived from marine natural product ceratamine A. Bioorg. Med. Chem. Lett. 2018, 28, 866–868. [Google Scholar] [CrossRef]
- Tuncbilek, M.; Kucukdumlu, A.; Guven, E.B.; Altiparmak, D.; Cetin-Atalay, R. Synthesis of novel 6-substituted amino-9-(β-D-ribofuranosyl)purine analogs and their bioactivities on human epithelial cancer cells. Bioorg. Med. Chem. Lett. 2018, 28, 235–239. [Google Scholar] [CrossRef]
- Borowiecki, P.; Wińska, P.; Bretner, M.; Gizińska, M.; Koronkiewicz, M.; Staniszewska, M. Synthesis of novel proxyphylline derivatives with dual anti-Candida albicans and anticancer activity. Eur. J. Med. Chem. 2018, 150, 307–333. [Google Scholar] [CrossRef]
- Kowalska, A.; Pluta, K.; Latocha, M. Synthesis and anticancer activity of multisubstituted purines and xanthines with one or two propynylthio and aminobutynylthio groups. Med. Chem. Res. 2018, 27, 1384–1395. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.Q.; Zhao, Y.D.; Liu, X.Y.; Wang, B.; Li, Z.H.; He, Z.X.; Zhang, X.H.; Liang, J.J.; Ma, L.Y.; Liu, H.M. Discovery of 6-chloro-2-(propylthio)-8,9-dihydro-7H-purines containing a carboxamide moiety as potential selective anti-lung cancer agents. Eur. J. Med. Chem. 2018, 151, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, O.G.; Abd El Razik, H.A.; SE, A.S.E.-D.; Ashour, F.A.; El-Tombary, A.A.; Afifi, O.S.; Abu-Serie, M.M. Purines and triazolo[4,3-e]purines containing pyrazole moiety as potential anticancer and antioxidant agents. Future Med. Chem. 2018, 10, 1449–1464. [Google Scholar] [CrossRef]
- Salas, C.; Zarate, A.M.; Kryštof, V.; Mella, J.; Faundez, M.; Brea, J.; Loza, M.I.; Brito, I.; Hendrychová, D.; Jorda, R.; et al. Promising 2,6,9-trisubstituted purine derivatives for anticancer compounds: Synthesis, 3D-QSAR, and preliminary biological assays. Int. J. Mol. Sci. 2020, 21, 161. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Wang, Z.; Xu, F.; Li, Q.; Wang, H.; Bian, Q.; Hu, F. Synthesis and activity investigation of novel 1H-purin-6(9H)-one and 3H-imidazo[4,5-d][1,2,3]triazin-4(7H)-one derivatives. ACS Omega 2019, 4, 15742–15753. [Google Scholar] [CrossRef]
- Fernández-Sáez, N.; Rubio-Ruiz, B.; Campos, J.M.; Unciti-Broceta, A.; Carrión, M.D.; Camacho, M.E. Purine derivatives with heterocyclic moieties and related analogs as new antitumor agents. Future Med. Chem. 2019, 11, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Sutanto, F.; Konstantinidou, M.; Dömling, A. Covalent inhibitors: A rational approach to drug discovery. RSC Med. Chem. 2020, 11, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Antolin, A.A.; Workman, P.; Mestres, J.; Al-Lazikani, B. Polypharmacology in precision oncology: Current applications and future prospects. Curr. Pharm. Des. 2016, 22, 6935–6945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, L.R.; Adams, C.M.; Fesik, S.W.; Eischen, C.M.; Tansey, W.P. Targeting MYC through WDR5. Mol. Cell. Oncol. 2020, 7, 1709388. [Google Scholar] [CrossRef] [Green Version]
- Doi, T.; Kurokawa, Y.; Sawaki, A.; Komatsu, Y.; Ozaka, M.; Takahashi, T.; Naito, Y.; Ohkubo, S.; Nishida, T. Efficacy and safety of TAS-116, an oral inhibitor of heat shock protein 90, in patients with metastatic or unresectable gastrointestinal stromal tumour refractory to imatinib, sunitinib and regorafenib: A phase II, single-arm trial. Eur. J. Cancer 2019, 121, 29–39. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, P.; LaRosa, C.; Antwi, J.; Govindarajan, R.; Werbovetz, K.A. Imidazoles as Potential Anticancer Agents: An Update on Recent Studies. Molecules 2021, 26, 4213. https://doi.org/10.3390/molecules26144213
Sharma P, LaRosa C, Antwi J, Govindarajan R, Werbovetz KA. Imidazoles as Potential Anticancer Agents: An Update on Recent Studies. Molecules. 2021; 26(14):4213. https://doi.org/10.3390/molecules26144213
Chicago/Turabian StyleSharma, Pankaj, Chris LaRosa, Janet Antwi, Rajgopal Govindarajan, and Karl A. Werbovetz. 2021. "Imidazoles as Potential Anticancer Agents: An Update on Recent Studies" Molecules 26, no. 14: 4213. https://doi.org/10.3390/molecules26144213
APA StyleSharma, P., LaRosa, C., Antwi, J., Govindarajan, R., & Werbovetz, K. A. (2021). Imidazoles as Potential Anticancer Agents: An Update on Recent Studies. Molecules, 26(14), 4213. https://doi.org/10.3390/molecules26144213