
Toward Multitasking Pharmacological COX-Targeting Agents: Non-Steroidal Anti-Inflammatory Prodrugs with Antiproliferative Effects

 ,
,  ,
,  ,
, 
 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
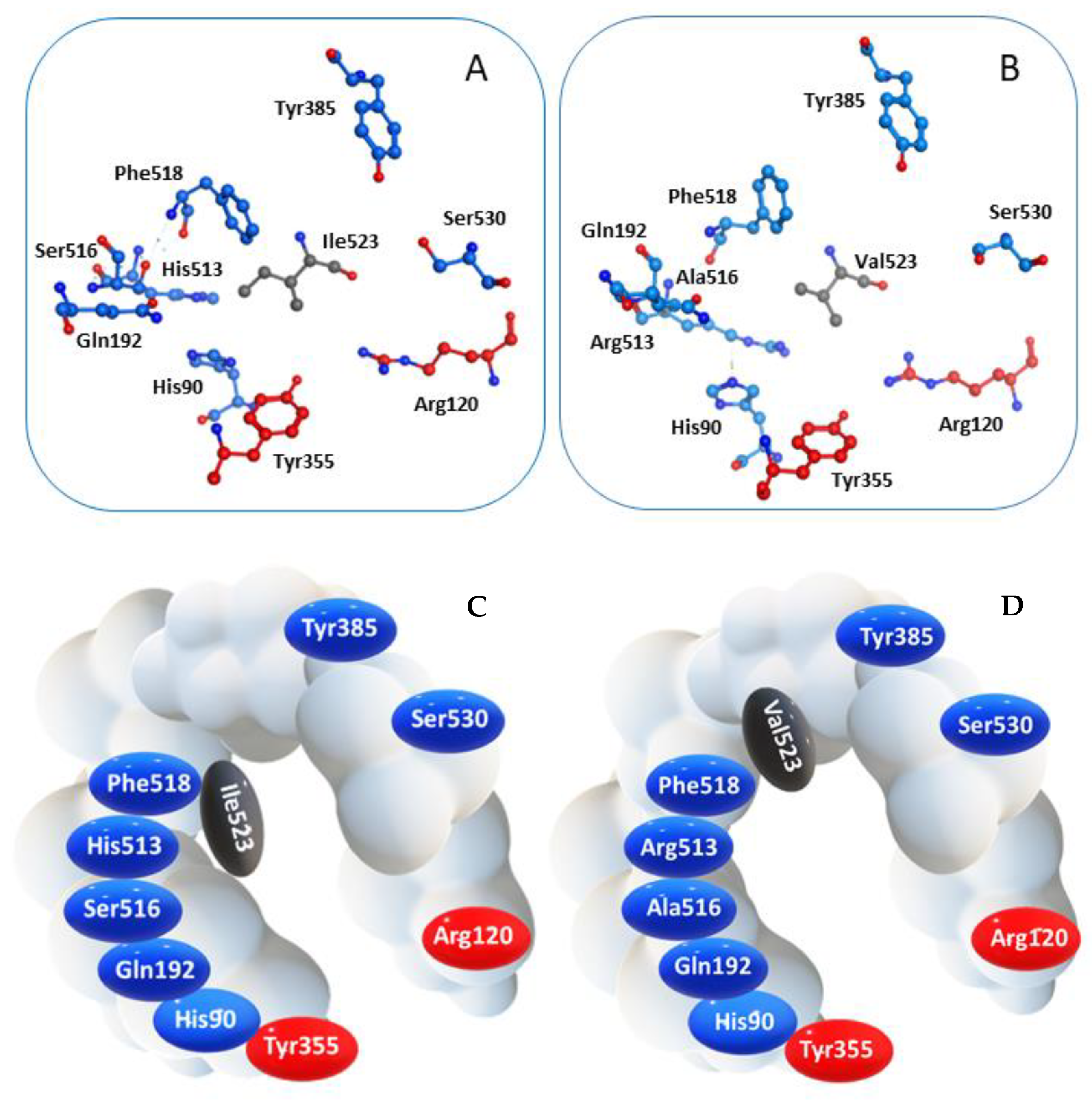
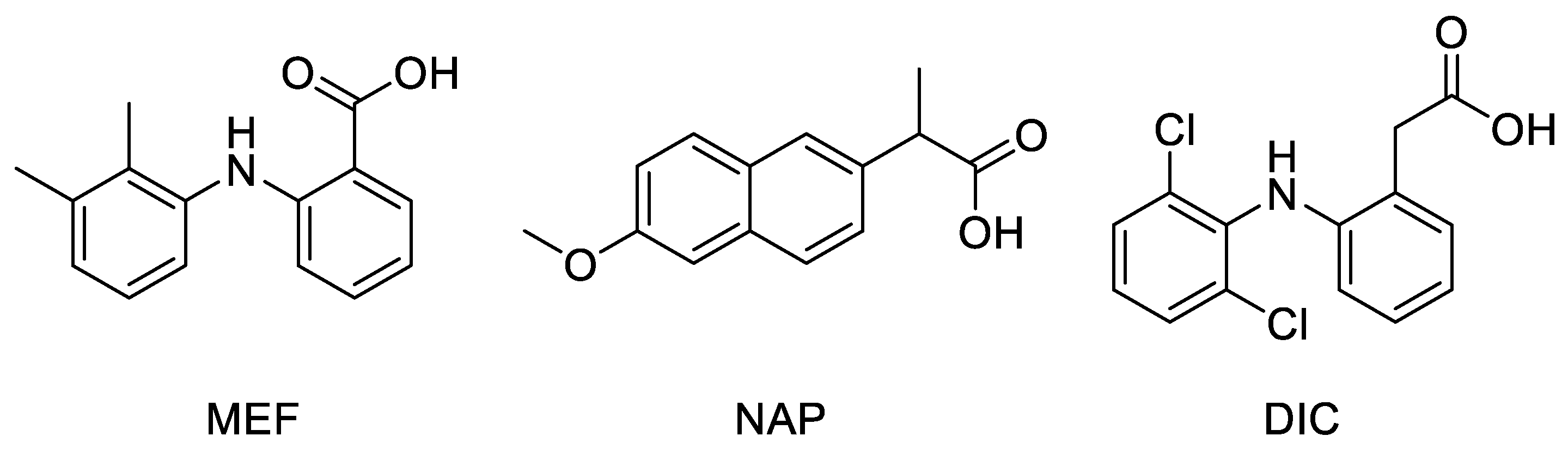
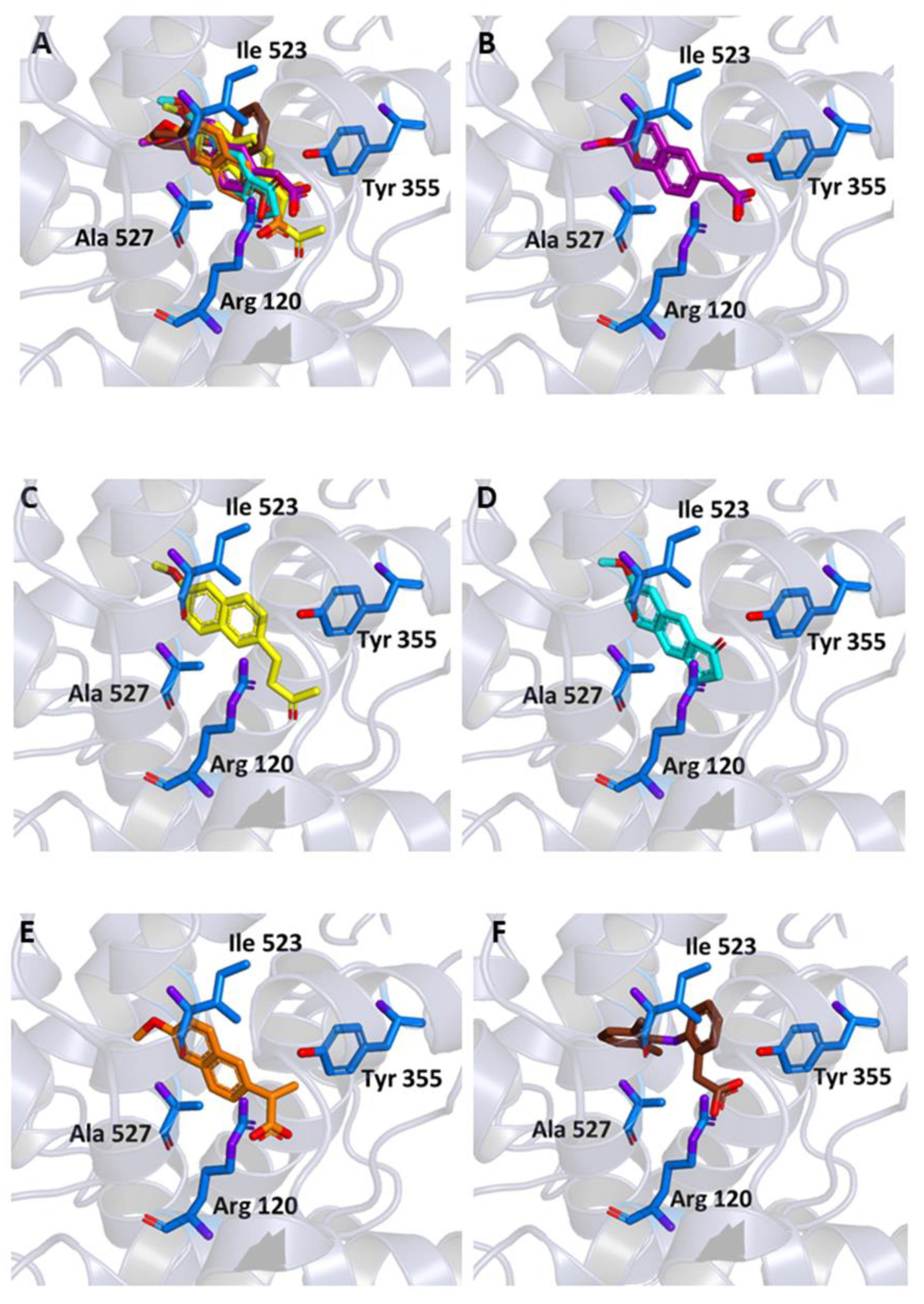
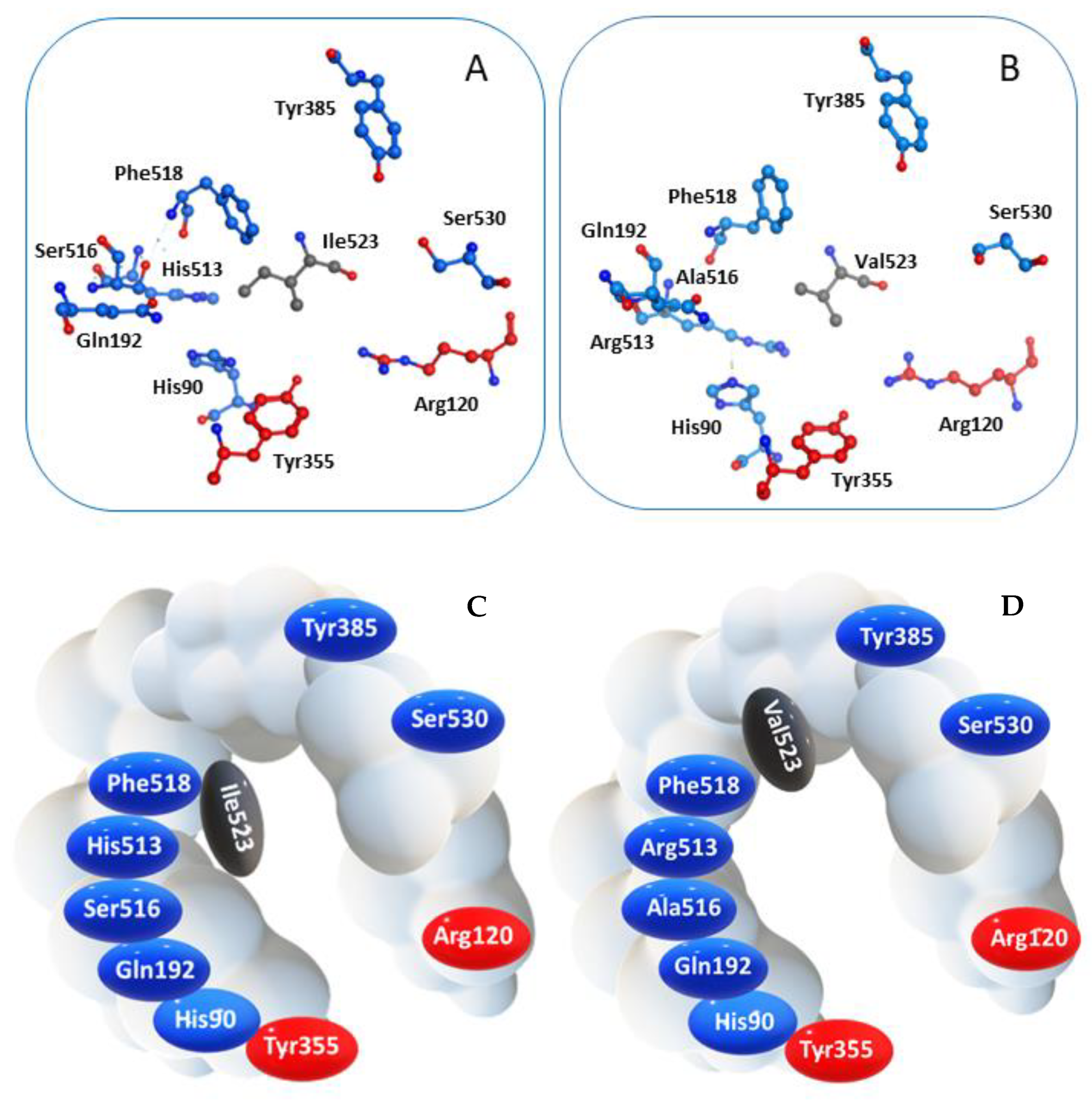
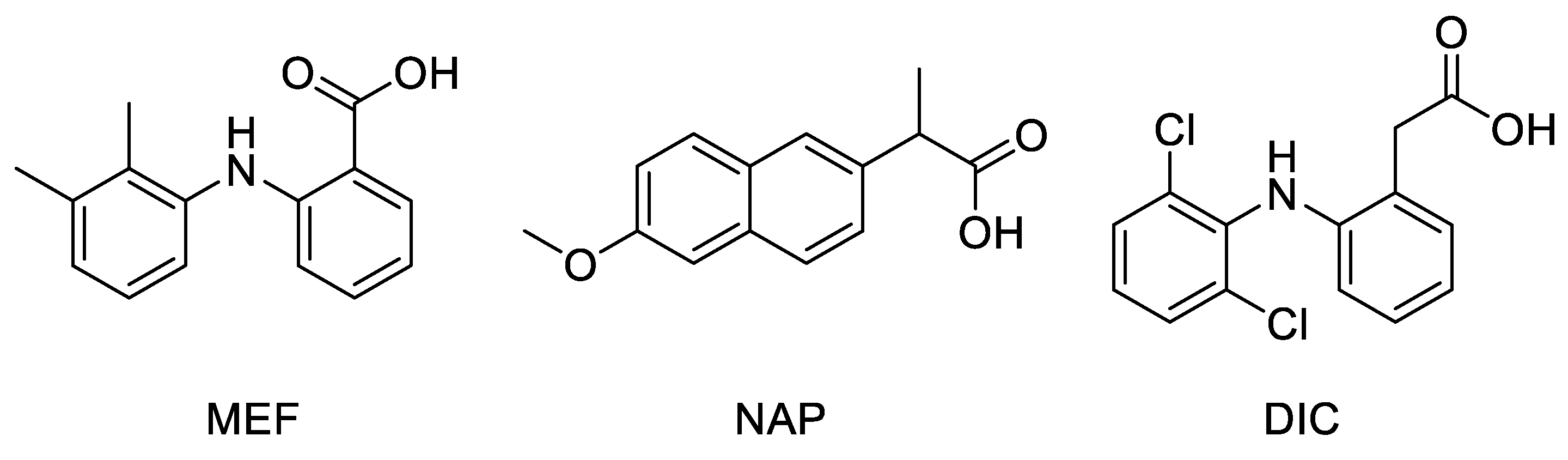
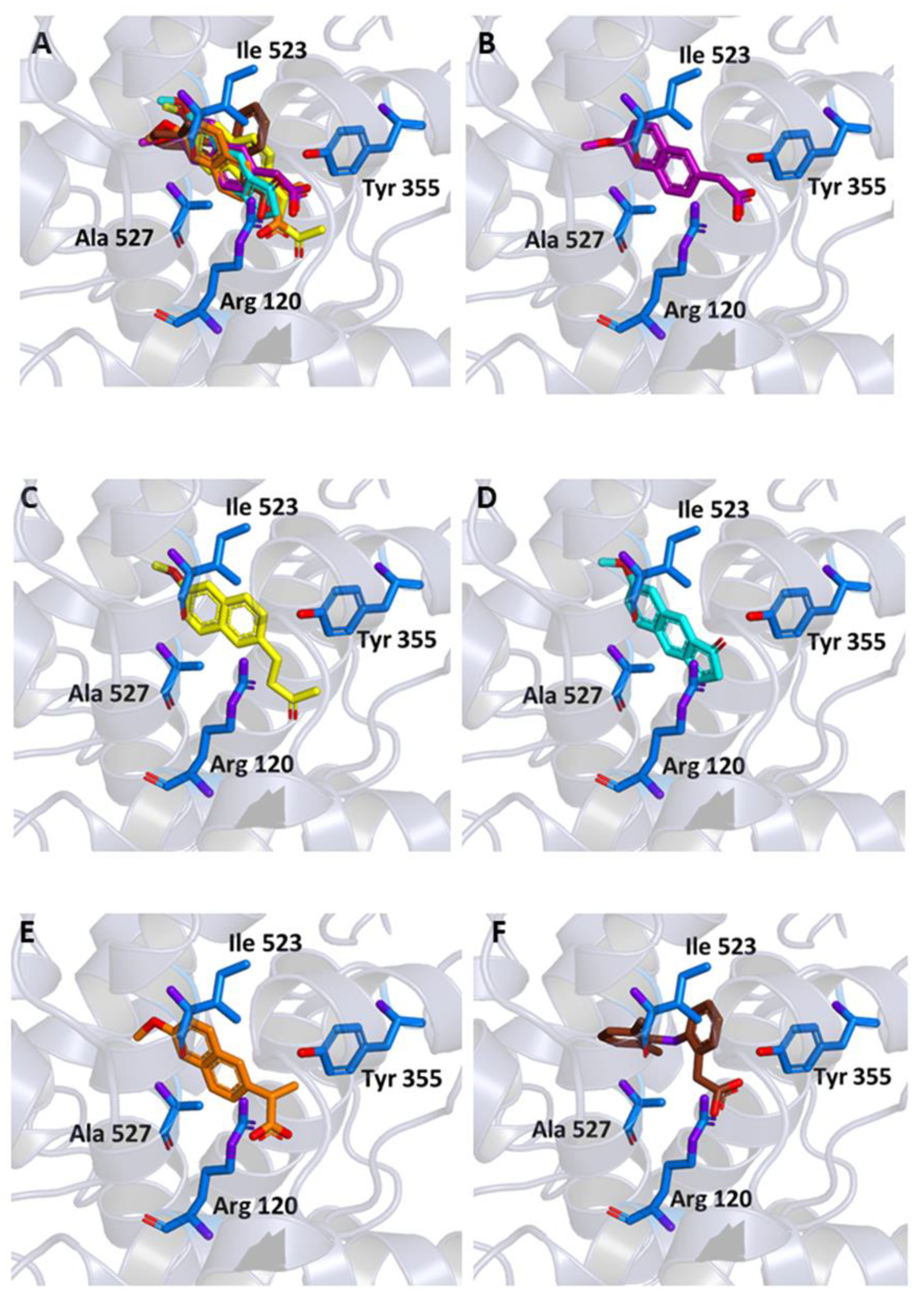
2.1. Molecular Docking
2.2. Biological Assays
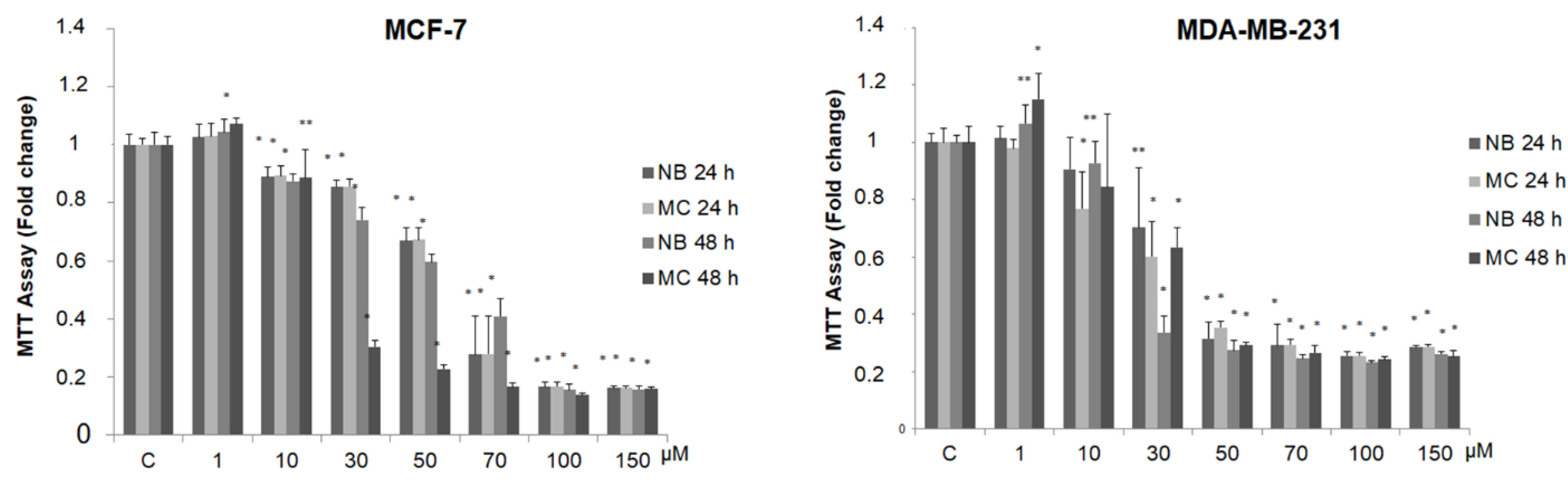
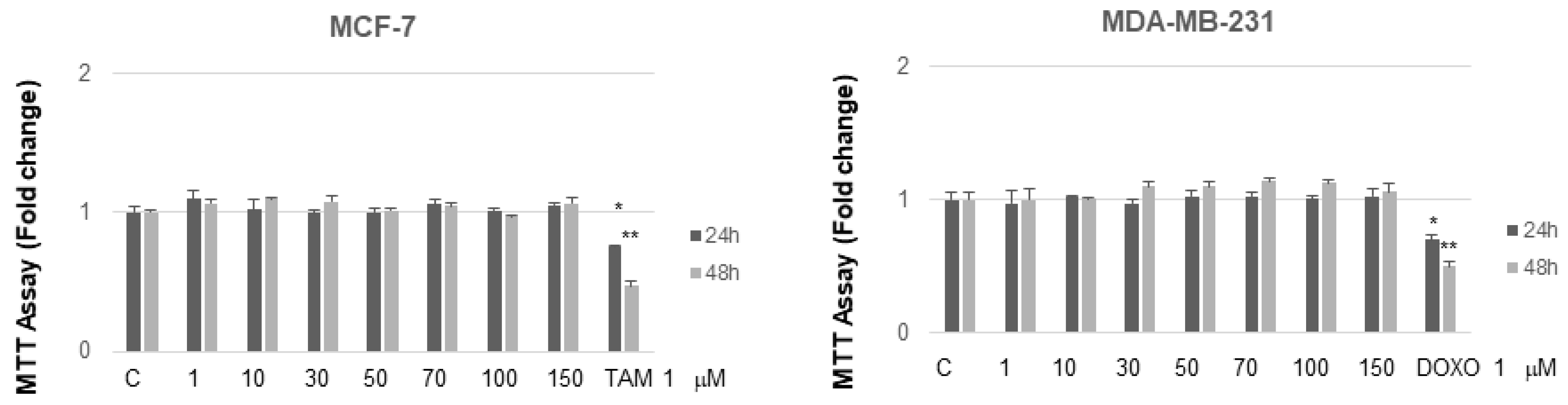
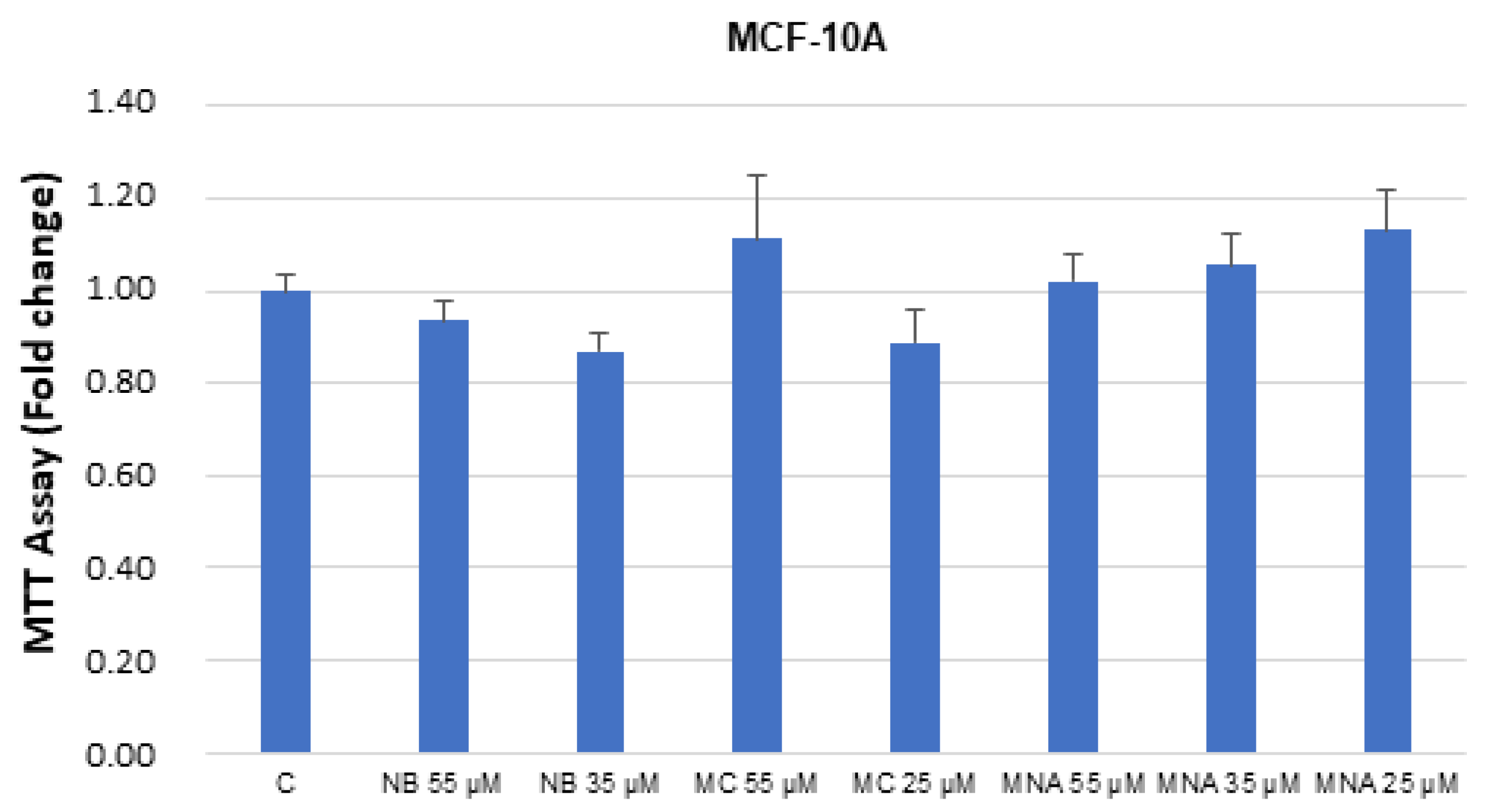
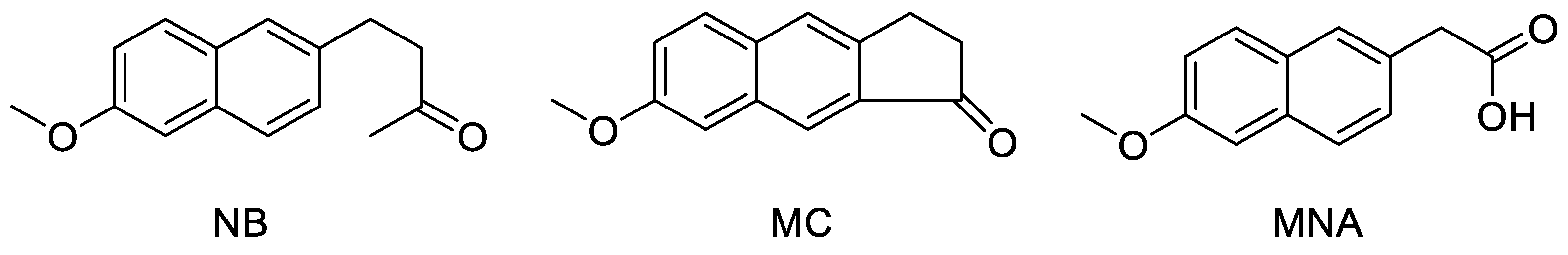
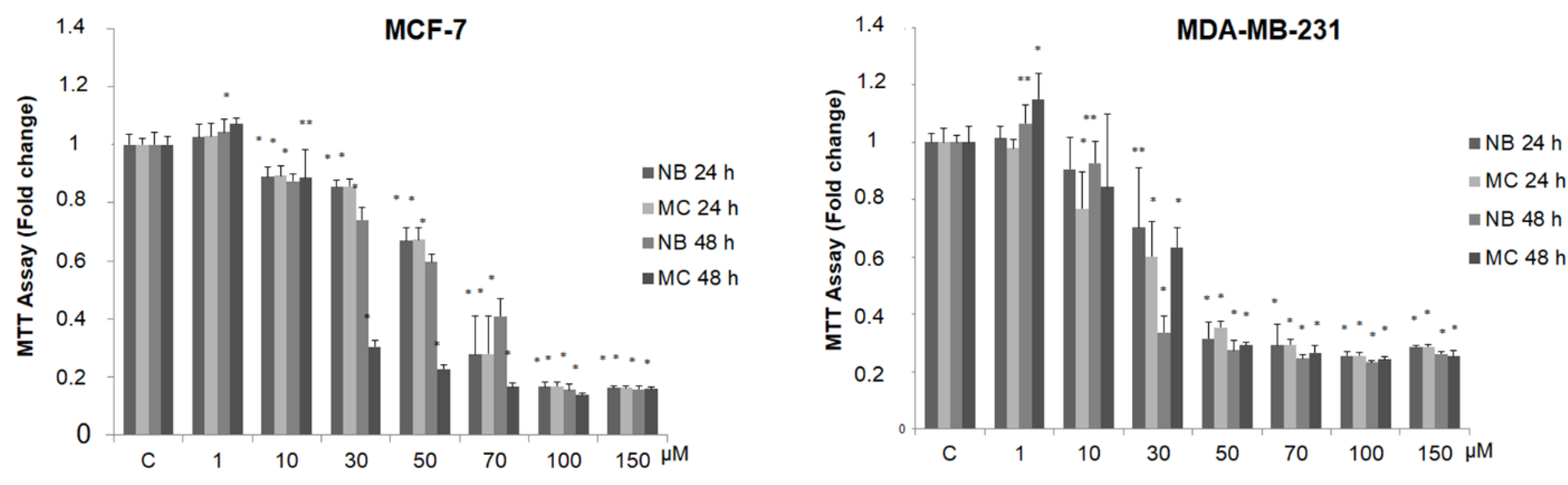
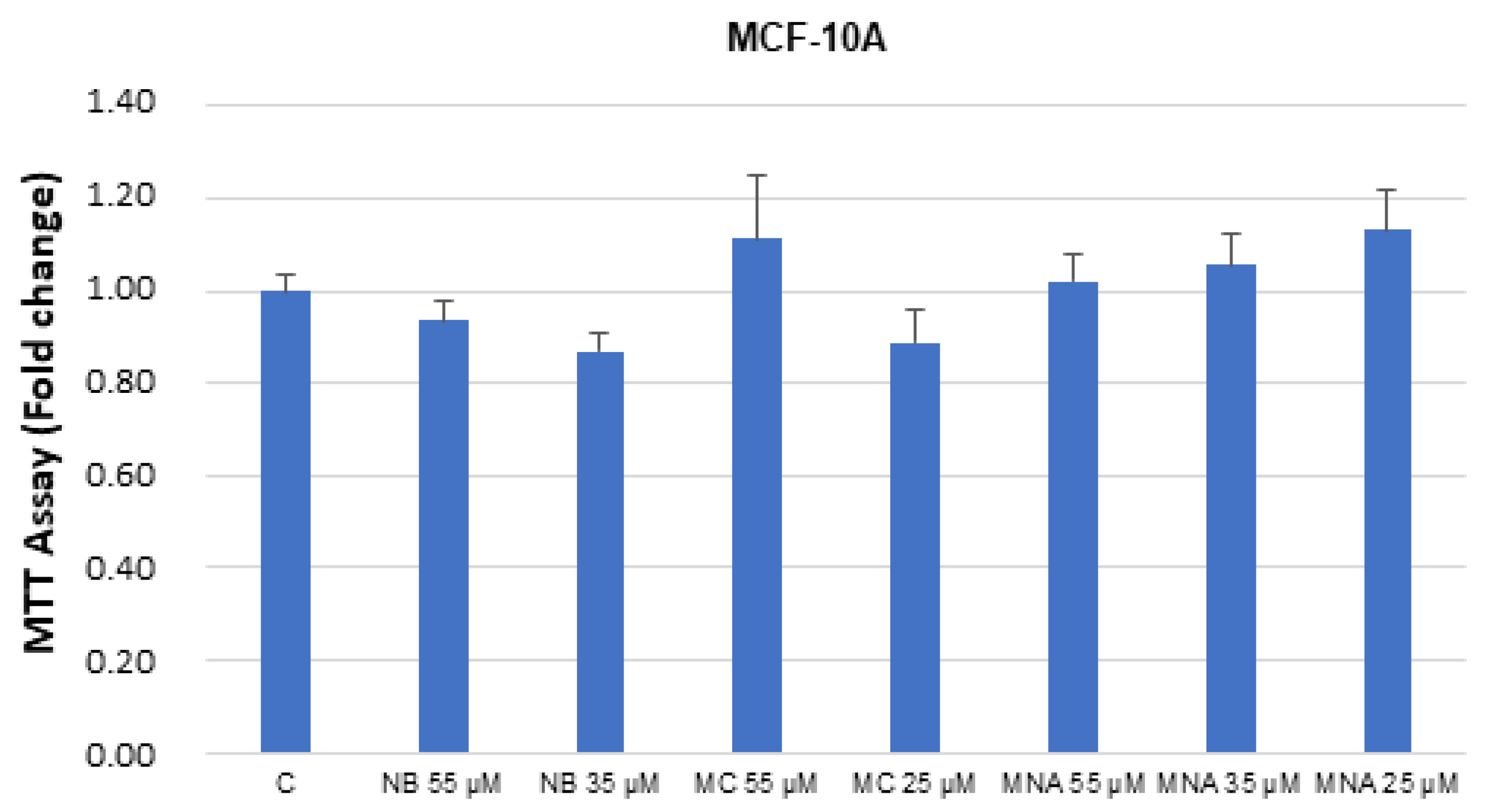
2.3. NB and MC Inhibit Breast Cancer Cell Survival
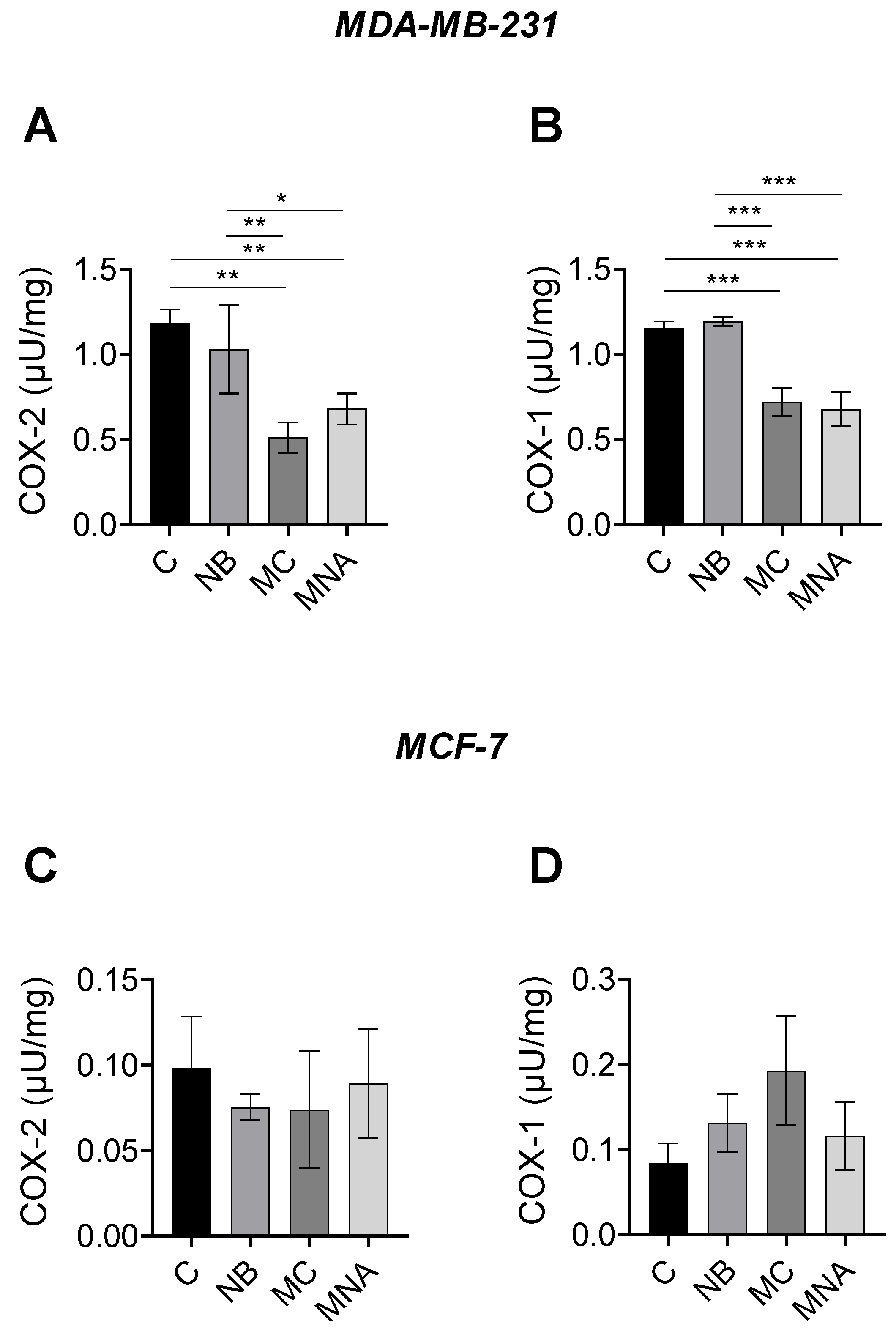
2.4. Effects of NB, MC, and MNA on COX-1 and COX-2 Activity in MCF-7 and MDA-MB-231 Cells
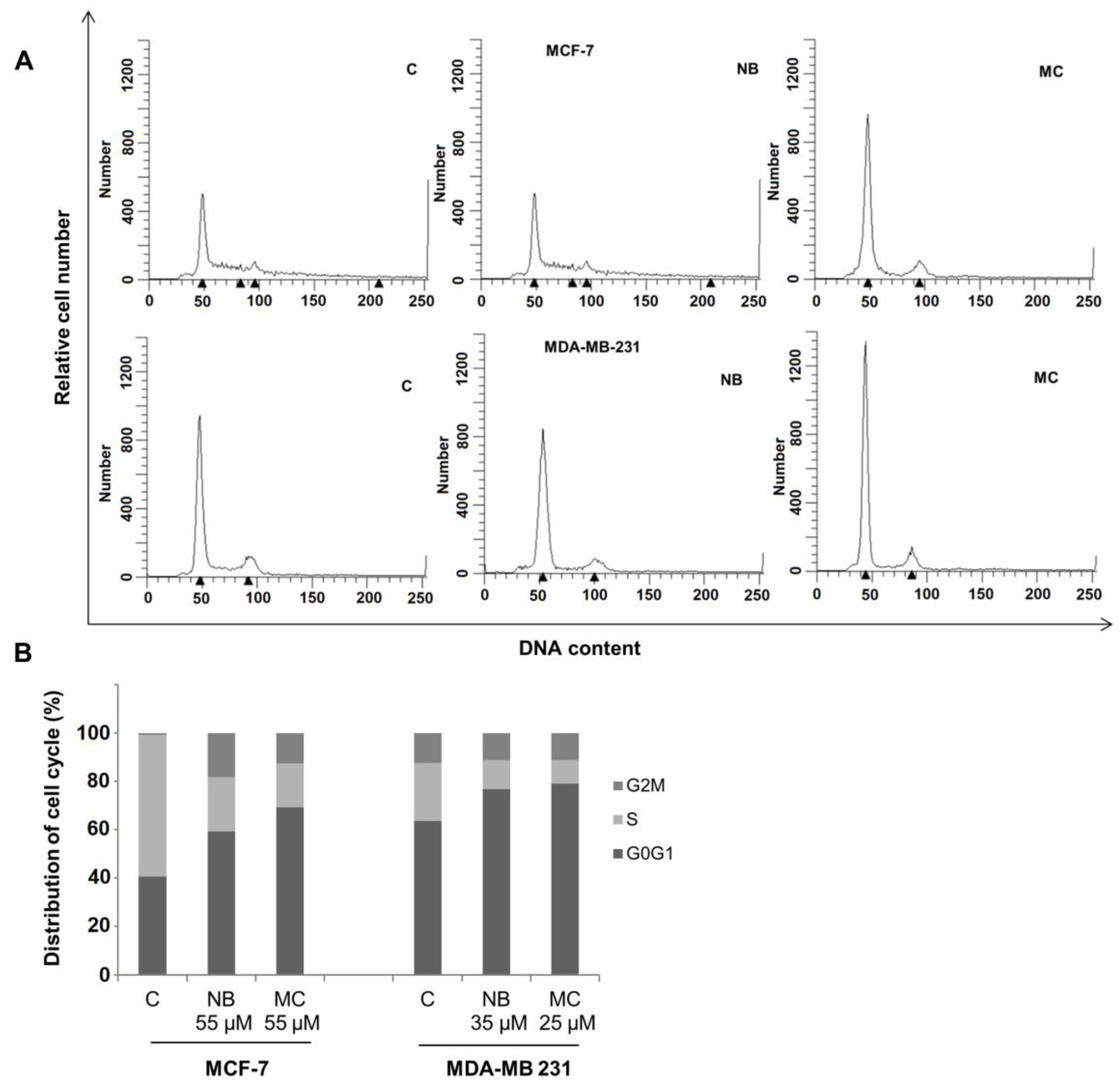
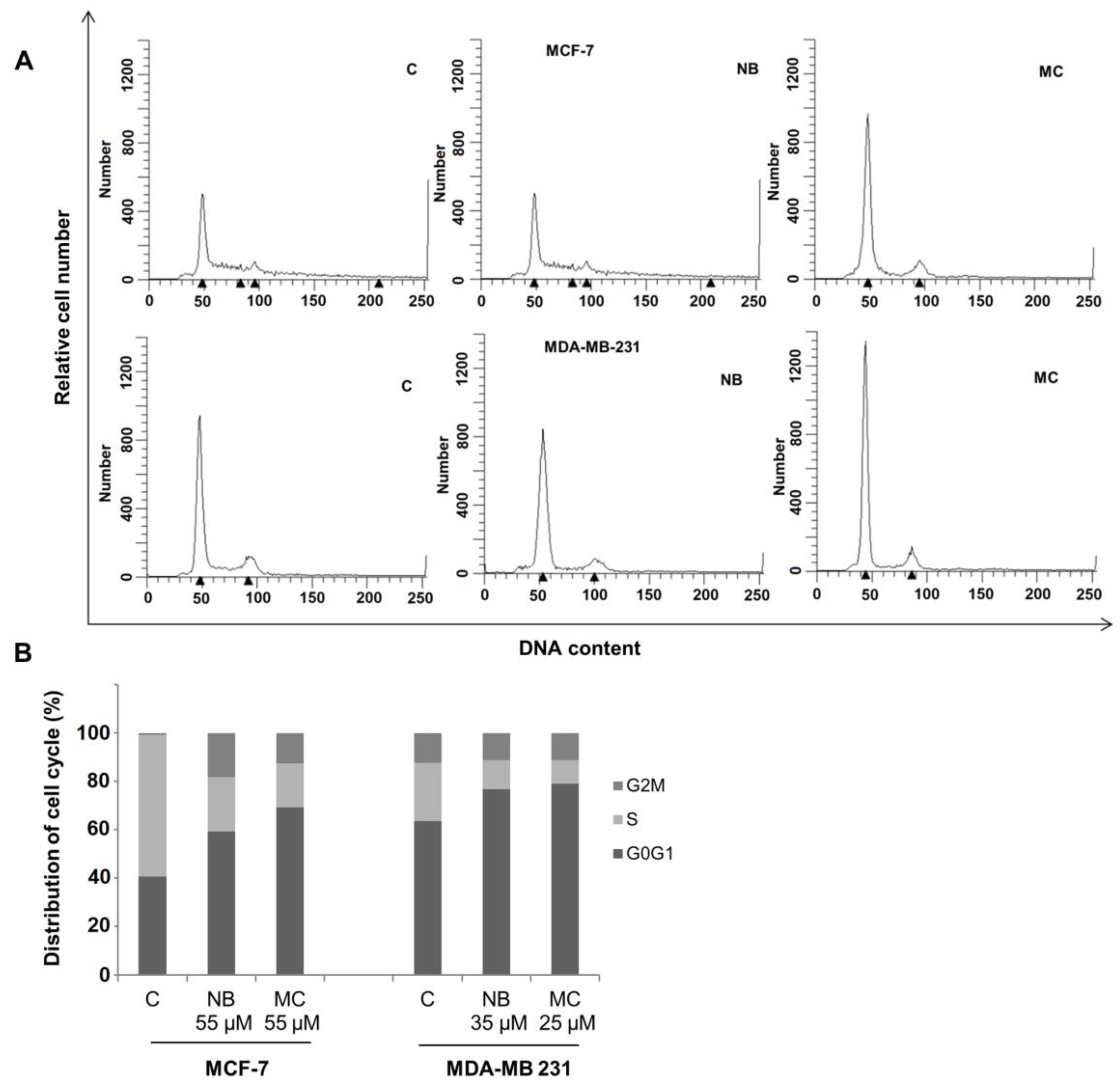
2.5. Effects of NB and MC on Cell Cycle
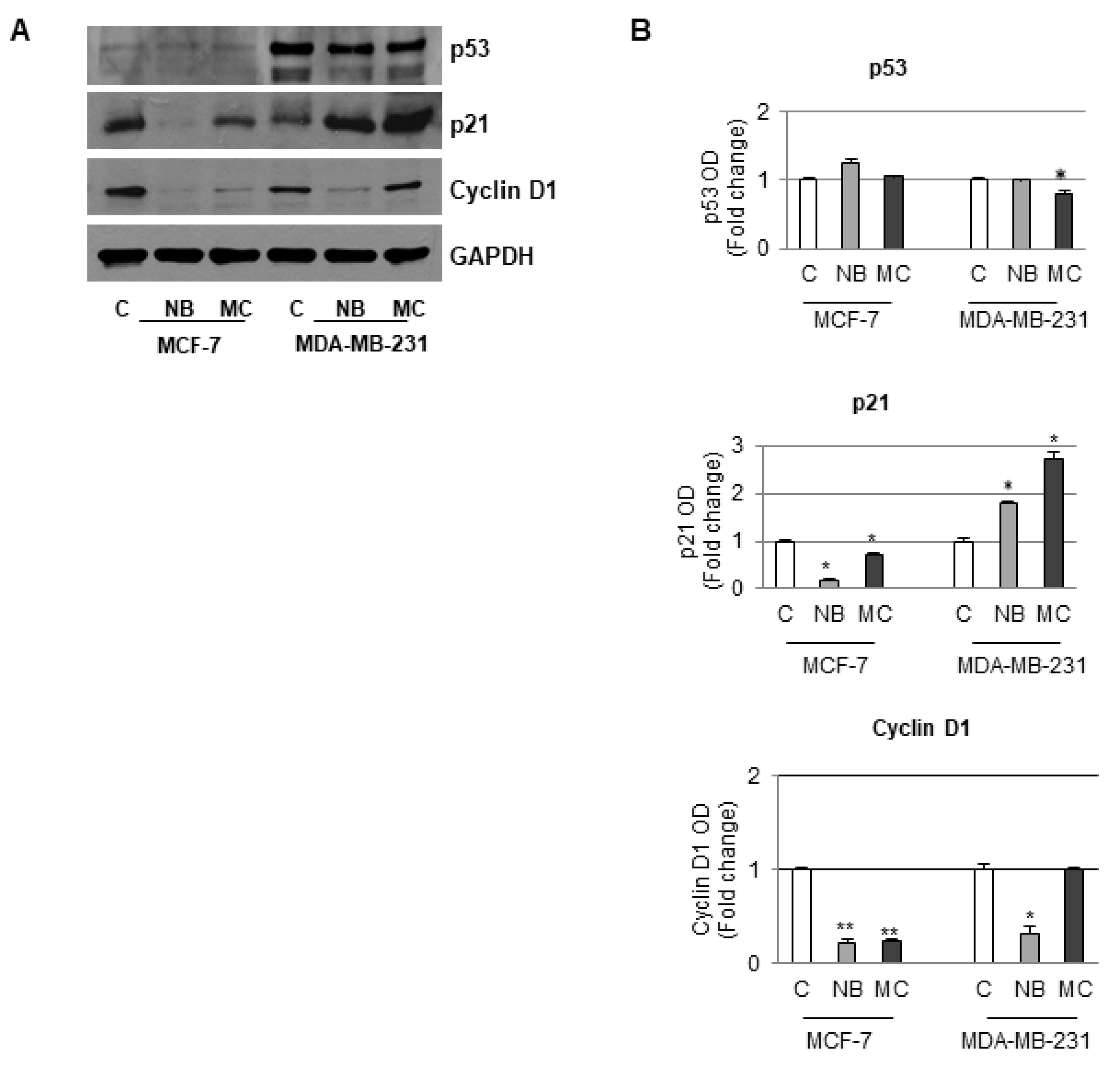
2.6. Effects of NB and MC on the Expression of Cell Cycle Checkpoint Proteins
3. Materials and Methods
3.1. Molecular Docking
3.2. Cell Culture
3.3. Compound Dilutions
3.4. Cell Viability Assays
3.5. COX Activity Assay
3.6. Cell Cycle Analysis
3.7. Immunoblotting Analysis
3.8. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bennett, A.; Mcdonald, A.M.; Stamford, I.F.; Charlier, E.M.; Simpson, J.S.; Zebro, T. Prostaglandins and Breast Cancer. Lancet 1977, 310, 624–626. [Google Scholar] [CrossRef]
- Bennett, A. The production of prostanoids in human cancers, and their implications for tumor progression. Prog. Lipid Res. 1986, 25, 539–542. [Google Scholar] [CrossRef]
- McLemore, T.L.; Hubbard, W.C.; Litterst, C.L.; Liu, M.C.; Miller, S.; McMahon, N.A.; Eggleston, J.; Boyd, M.R. Profiles of prostaglandin biosynthesis in normal lung and tumor tissue from lung cancer patients. Cancer Res. 1988, 48, 3140–3147. [Google Scholar] [PubMed]
- Rigas, B.; Goldman, I.S.; Levine, L. Altered eicosanoid levels in human colon cancer. J. Lab. Clin. Med. 1993, 122, 518–523. [Google Scholar]
- Rocca, C.; Pasqua, T.; Cerra, M.C.; Angelone, T. Cardiac Damage in Anthracyclines Therapy: Focus on Oxidative Stress and Inflammation. Antioxid. Redox Signal. 2020, 32, 1081–1097. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.; Scavello, F.; Colombo, B.; Gasparri, A.M.; Dallatomasina, A.; Granieri, M.C.; Amelio, D.; Pasqua, T.; Cerra, M.C.; Tota, B.; et al. Physiological levels of chromogranin A prevent doxorubicin-induced cardiotoxicity without impairing its anticancer activity. FASEB J. 2019, 33, 7734–7747. [Google Scholar] [CrossRef]
- Crofford, L.J. COX-1 and COX-2 tissue expression: Implications and predictions. J. Reumatol. Suppl. 1997, 49, 15–19. [Google Scholar]
- Łanocha-Arendarczyk, N.; Baranowska-Bosiacka, I.; Kot, K.; Gutowska, I.; Kolasa-Wołosiuk, A.; Chlubek, D.; Kosik-Bogacka, D. Expression and Activity of COX-1 and COX-2 in Acanthamoeba sp.-Infected Lungs According to the Host Immunological Status. Int. J. Mol. Sci. 2018, 19, 121. [Google Scholar] [CrossRef] [Green Version]
- Reddy, B.S.; Rao, C.V.; Seibert, K. Evaluation of cyclooxygenase-2 inhibitor for potential chemopreventive properties in colon carcinogenesis. Cancer Res. 1996, 56, 4566–4569. [Google Scholar]
- Mahesh, G.; Kumar, K.A.; Reddanna, P. Overview on the discovery and development of anti-inflammatory drugs: Should the focus be on synthesis or degradation of pge2? J. Inflamm. Res. 2021, 14, 253–263. [Google Scholar] [CrossRef]
- Miki, Y.; Mukae, S.; Murakami, M.; Ishikawa, Y.; Ishii, T.; Ohki, H.; Matsumoto, M.; Komiyama, K. Butyrate Inhibits Oral Cancer Cell Proliferation and Regulates Expression of Secretory Phospholipase A2-X and COX-2. Anticancer Res. 2007, 27, 1493–1502. [Google Scholar]
- Mahboubi Rabbani, S.M.I.; Zarghi, A. Selective COX-2 inhibitors as anticancer agents: A patent review (2014–2018). Expert Opin. Ther. Pat. 2019, 29, 407–427. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Fukutake, M.; Yokota, S.; Ishida, K.; Wakabayashi, K.; Sugimura, T. Suppression of azoxymethane-induced aberrant crypt foci in rat colon by nimesulide, a selective inhibitor of cyclooxygenase 2. J. Cancer Res. Clin. Oncol. 1996, 122, 219–222. [Google Scholar] [CrossRef]
- Hara, A.; Yoshimi, N.; Niwa, M.; Ino, N.; Mori, H. Apoptosis induced by NS-398, a selective cyclooxygenase-2 inhibitor, in human colorectal cancer cell lines. Jpn. J. Cancer Res. 1997, 88, 600–604. [Google Scholar] [PubMed]
- Eberhart, C.E.; Coffey, R.J.; Radhika, A.; Giardiello, F.M.; Ferrenbach, S.; Dubois, R.N. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994, 107, 1183–1188. [Google Scholar] [CrossRef]
- Ma, Y.; Yu, P.; Lin, S.; Li, Q.; Fang, Z.; Huang, Z. The association between nonsteroidal anti-inflammatory drugs and skin cancer: Different responses in American and European populations. Pharmacol. Res. 2020, 152, 104499. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.R.; Yousef, G.M.; Ni, H. Cancer and platelet crosstalk: Opportunities and challenges of aspirin and other antiplatelet agents. Blood 2018, 131, 1777–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Placke, T.; Salih, H.R.; Kopp, H.-G. GITR Ligand Provided by Thrombopoietic Cells Inhibits NK Cell Antitumor Activity. J. Immunol. 2012, 189, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Lian, L.; Li, W.; Li, Z.Y.; Mao, Y.X.; Zhang, Y.T.; Zhao, Y.M.; Chen, K.; Duan, W.M.; Tao, M. Inhibition of MCF-7 breast cancer cell-induced platelet aggregation using a combination of antiplatelet drugs. Oncol. Lett. 2013, 5, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Prescott, S.M.; Fitzpatrick, F.A. Cyclooxygenase-2 and carcinogenesis. Biochim. Biophys. Acta Rev. Cancer 2000, 1470, M69–M70. [Google Scholar] [CrossRef]
- Frejborg, E.; Salo, T.; Salem, A. Role of cyclooxygenase-2 in head and neck tumorigenesis. Int. J. Mol. Sci. 2020, 21, 9246. [Google Scholar] [CrossRef]
- Toyokuni, S.; Okamoto, K.; Yodoi, J.; Hiai, H. Persistent oxidative stress in cancer. FEBS Lett. 1995, 358, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B. Oxidative stress and cancer: Have we moved forward? Biochem. J. 2007, 401, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.J.; Wilson, L.C.; Lee, C.H.; Eling, T.E. Dual function of nonsteroidal anti-inflammatory drugs (NSAIDs): Inhibition of cyclooxygenase and induction of NSAID-activated gene. J. Pharmacol. Exp. Ther. 2002, 301, 1126–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinsley, H.N.; Grizzle, W.E.; Abadi, A.; Keeton, A.; Zhu, B.; Xi, Y.; Piazza, G.A. New NSAID targets and derivatives for colorectal cancer chemoprevention. Recent Results Cancer Res. 2013, 191, 105–120. [Google Scholar] [PubMed]
- Matsunaga, K.; Yoshimi, N.; Yamada, Y.; Shimizu, M.; Kawabata, K.; Ozawa, Y.; Hara, A.; Mori, H. Inhibitory effects of Nabumetone, a cyclooxygenase-2 inhibitor, and esculetin, a lipoxygenase inhibitor, on N-methyl-N-nitrosourea-induced mammary carcinogenesis in rats. Jpn. J. Cancer Res. 1998, 89, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.J.; Jiang, C.; Lu, J.; Mehta, R.G.; Piazza, G.A.; Paranka, N.S.; Pamukcu, R.; Ahnen, D.J. Sulfone metabolite of sulindac inhibits mammary carcinogenesis. Cancer Res. 1997, 57, 267–271. [Google Scholar]
- Harris, R.E.; Beebe-Donk, J.; Doss, H.; Burr Doss, D. Aspirin, ibuprofen, and other non-steroidal anti-inflammatory drugs in cancer prevention: A critical review of non-selective COX-2 blockade (review). Oncol. Rep. 2005, 13, 559–583. [Google Scholar] [CrossRef]
- Khan, Y.S.; Gutiérrez-De-Terán, H.; Åqvist, J. Molecular Mechanisms in the Selectivity of Nonsteroidal Anti-Inflammatory Drugs. Biochemistry 2018, 57, 1236–1248. [Google Scholar] [CrossRef]
- Wu, A.W.; Gu, J.; Li, Z.F.; Ji, J.F.; Xu, G.W. COX-2 expression and tumor angiogenesis in colorectal cancer. World J. Gastroenterol. 2004, 10, 2323–2326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.-C. Microvessel density is a prognostic marker of human gastric cancer. World J. Gastroenterol. 2006, 12, 7598–7603. [Google Scholar] [CrossRef]
- Prabhakar, C.; Reddy, C.B.; Reddy, C.M.; Nageshwar, D.; Devi, A.S.; Babu, J.M.; Vyas, K.; Sarma, M.R.; Reddy, G. Process research and structural studies on nabumetone. Org. Process Res. Dev. 1999, 3, 121–125. [Google Scholar] [CrossRef]
- Friedel, H.A.; Todd, P.A. Nabumetone: A Preliminary Review of its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Efficacy in Rheumatic Diseases. Drugs 1988, 35, 504–524. [Google Scholar] [CrossRef] [PubMed]
- Grande, F.; Ragno, G.; Muzzalupo, R.; Occhiuzzi, M.A.; Mazzotta, E.; De Luca, M.; Garofalo, A.; Ioele, G. Gel formulation of nabumetone and a newly synthesized analog: Microemulsion as a photoprotective topical delivery system. Pharmaceutics 2020, 12, 423. [Google Scholar] [CrossRef] [PubMed]
- Jagdale, S.C.; Deore, G.K.; Chabukswar, A.R. Development of Microemulsion Based Nabumetone Transdermal Delivery for Treatment of Arthritis. Recent Pat. Drug Deliv. Formul. 2018, 12, 130–149. [Google Scholar] [CrossRef]
- Madhusudhan, B.; Rambhau, D.; Apte, S.S.; Gopinath, D. Improved in Vitro Permeation of Nabumetone across Rat Skin from 1-O-Alkylglycerol/Lecithin Stabilized o/w Nanoemulsions. J. Dispers. Sci. Technol. 2006, 27, 921–926. [Google Scholar] [CrossRef]
- Pannunzio, A.; Coluccia, M. Cyclooxygenase-1 (COX-1) and COX-1 inhibitors in cancer: A review of oncology and medicinal chemistry literature. Pharmaceuticals 2018, 11, 101. [Google Scholar] [CrossRef] [Green Version]
- Yuan, C.; Rieke, C.J.; Rimon, G.; Wingerd, B.A.; Smith, W.L. Partnering between monomers of cyclooxygenase-2 homodimers. Proc. Natl. Acad. Sci. USA 2006, 103, 6142–6147. [Google Scholar] [CrossRef] [Green Version]
- Yousiff, O.A.; Mahdi, M.F.; Raauff, A.M.R. Design, synthesis, preliminary pharmacological evaluation, molecular docking and ADME studies of some new pyrazoline, isoxazoline and pyrimidine derivatives bearing nabumetone moiety targeting cyclooxygenase enzyme New analogues of nabumetone targeting COX-2 enzyme. J. Contemp. Med. Sci. 2019, 5, 41–50. [Google Scholar]
- Smith, F.G.; Wade, A.W.; Lewis, M.L.; Qi, W. Cyclooxygenase (COX) inhibitors and the newborn kidney. Pharmaceuticals 2012, 5, 1160–1176. [Google Scholar] [CrossRef] [Green Version]
- Ermondi, G.; Caron, G.; Lawrence, R.; Longo, D. Docking studies on NSAID/COX-2 isozyme complexes using Contact Statistics analysis. J. Comput. Aided. Mol. Des. 2004, 18, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Luong, C.; Miller, A.; Barnett, J.; Chow, J.; Ramesha, C.; Browner, M.F. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat. Struct. Biol. 1996, 3, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Orlando, B.J.; Malkowski, M.G. Substrate-selective inhibition of cyclooxygeanse-2 by fenamic acid derivatives is dependent on peroxide tone. J. Biol. Chem. 2016, 291, 15069–15081. [Google Scholar] [CrossRef] [Green Version]
- Zarghi, A.; Arfaei, S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran J. Pharm. Res. 2011, 10, 655–683. [Google Scholar] [PubMed]
- Bao, L.; Hazari, S.; Mehra, S.; Kaushal, D.; Moroz, K.; Dash, S. Increased expression of P-glycoprotein and doxorubicin chemoresistance of metastatic breast cancer is regulated by miR-298. Am. J. Pathol. 2012, 180, 2490–2503. [Google Scholar] [CrossRef] [Green Version]
- Parekh, A.; Das, D.; Das, S.; Dhara, S.; Biswas, K.; Mandal, M.; Das, S. Bioimpedimetric analysis in conjunction with growth dynamics to differentiate aggressiveness of cancer cells. Sci. Rep. 2018, 8, 783. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Medema, R.H.; MacŮrek, L. Checkpoint control and cancer. Oncogene 2012, 31, 2601–2613. [Google Scholar] [CrossRef] [Green Version]
- Pucci, B.; Kasten, M.; Giordano, A. Cell cycle and apoptosis. Neoplasia 2000, 2, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.Y.; Zhang, F.; Hong, C.Q.; Giuliano, A.E.; Cui, X.J.; Zhou, G.J.; Zhang, G.J.; Cui, Y.K. Critical protein GAPDH and its regulatory mechanisms in cancer cells. Cancer Biol. Med. 2015, 12, 10–22. [Google Scholar]
- Panno, M.; Giordano, F.; Palma, M.; Bartella, V.; Rago, V.; Maggiolini, M.; Sisci, D.; Lanzino, M.; De Amicis, F.; Ando, S. Evidence that Bergapten, Independently of its Photoactivation, Enhances p53 Gene Expression and Induces Apoptosis in Human Breast Cancer Cells. Curr. Cancer Drug Targets 2009, 9, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Panno, M.L.; Giordano, F.; Rizza, P.; Pellegrino, M.; Zito, D.; Giordano, C.; Mauro, L.; Catalano, S.; Aquila, S.; Sisci, D.; et al. Bergapten induces ER depletion in breast cancer cells through SMAD4-mediated ubiquitination. Breast Cancer Res. Treat. 2012, 136, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Mauro, L.; Pellegrino, M.; Giordano, F.; Ricchio, E.; Rizza, P.; De Amicis, F.; Catalano, S.; Bonofiglio, D.; Panno, M.L.; Andò, S. Estrogen receptor-α drives adiponectin effects on cyclin D1 expression in breast cancer cells. FASEB J. 2015, 29, 2150–2160. [Google Scholar] [CrossRef] [Green Version]
- Giordano, C.; Rovito, D.; Barone, I.; Mancuso, R.; Bonofiglio, D.; Giordano, F.; Catalano, S.; Gabriele, B.; Andò, S. Benzofuran-2-acetic ester derivatives induce apoptosis in breast cancer cells by upregulating p21Cip/WAF1 gene expression in p53-independent manner. DNA Repair 2017, 51, 20–30. [Google Scholar] [CrossRef]
- Gionfriddo, G.; Plastina, P.; Augimeri, G.; Catalano, S.; Giordano, C.; Barone, I.; Morelli, C.; Giordano, F.; Gelsomino, L.; Sisci, D.; et al. Modulating Tumor-Associated Macrophage Polarization by Synthetic and Natural PPARγ Ligands as a Potential Target in Breast Cancer. Cells 2020, 9, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montalto, F.I.; Giordano, F.; Chiodo, C.; Marsico, S.; Mauro, L.; Sisci, D.; Aquila, S.; Lanzino, M.; Panno, M.L.; Andò, S.; et al. Progesterone Receptor B signaling Reduces Breast Cancer Cell Aggressiveness: Role of Cyclin-D1/Cdk4 Mediating Paxillin Phosphorylation. Cancers 2019, 11, 1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Amicis, F.; Aquila, S.; Morelli, C.; Guido, C.; Santoro, M.; Perrotta, I.; Mauro, L.; Giordano, F.; Nigro, A.; Andò, S.; et al. Bergapten drives autophagy through the up-regulation of PTEN expression in breast cancer cells. Mol. Cancer 2015, 14, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Grande, F.; Rizzuti, B.; Occhiuzzi, M.A.; Ioele, G.; Casacchia, T.; Gelmini, F.; Guzzi, R.; Garofalo, A.; Statti, G. Identification by molecular docking of homoisoflavones from leopoldia comosa as ligands of estrogen receptors. Molecules 2018, 23, 894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casacchia, T.; Occhiuzzi, M.A.; Grande, F.; Rizzuti, B.; Granieri, M.C.; Rocca, C.; Gattuso, A.; Garofalo, A.; Angelone, T.; Statti, G. A pilot study on the nutraceutical properties of the Citrus hybrid Tacle® as a dietary source of polyphenols for supplementation in metabolic disorders. J. Funct. Foods 2019, 52, 370–381. [Google Scholar] [CrossRef]
- PyMOL. The Pymol Molecular Graphics System. Available online: https://pymol.org (accessed on 13 April 2021).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Non-Hydrogen Atoms (Å) | Closest Aromatic Ring (Å) | |
|---|---|---|
| MNA | 1.53 | 0.45 |
| MC | 1.80 | 1.15 |
| NB | 2.84 | 0.90 |
| NAP | 1.54 | 0.45 |
| DIC | 0.42 | 0.65 |
| Compound | MCF-7 | MDA-MB-231 | ||
|---|---|---|---|---|
| IC50 (µmol/L) | 95% CI | IC50 (µmol/L) | 95% CI | |
| NB | 55.03 | 51.99–58.25 | 32.55 | 29.36–36.11 |
| MC | 55.03 | 52.03–58.36 | 24.80 | 18.11–33.97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grande, F.; Giordano, F.; Occhiuzzi, M.A.; Rocca, C.; Ioele, G.; De Luca, M.; Ragno, G.; Panno, M.L.; Rizzuti, B.; Garofalo, A. Toward Multitasking Pharmacological COX-Targeting Agents: Non-Steroidal Anti-Inflammatory Prodrugs with Antiproliferative Effects. Molecules 2021, 26, 3940. https://doi.org/10.3390/molecules26133940
Grande F, Giordano F, Occhiuzzi MA, Rocca C, Ioele G, De Luca M, Ragno G, Panno ML, Rizzuti B, Garofalo A. Toward Multitasking Pharmacological COX-Targeting Agents: Non-Steroidal Anti-Inflammatory Prodrugs with Antiproliferative Effects. Molecules. 2021; 26(13):3940. https://doi.org/10.3390/molecules26133940
Chicago/Turabian StyleGrande, Fedora, Francesca Giordano, Maria Antonietta Occhiuzzi, Carmine Rocca, Giuseppina Ioele, Michele De Luca, Gaetano Ragno, Maria Luisa Panno, Bruno Rizzuti, and Antonio Garofalo. 2021. "Toward Multitasking Pharmacological COX-Targeting Agents: Non-Steroidal Anti-Inflammatory Prodrugs with Antiproliferative Effects" Molecules 26, no. 13: 3940. https://doi.org/10.3390/molecules26133940
APA StyleGrande, F., Giordano, F., Occhiuzzi, M. A., Rocca, C., Ioele, G., De Luca, M., Ragno, G., Panno, M. L., Rizzuti, B., & Garofalo, A. (2021). Toward Multitasking Pharmacological COX-Targeting Agents: Non-Steroidal Anti-Inflammatory Prodrugs with Antiproliferative Effects. Molecules, 26(13), 3940. https://doi.org/10.3390/molecules26133940