
Thiol- and Disulfide-Based Stimulus-Responsive Soft Materials and Self-Assembling Systems


Abstract
1. Introduction
1.1. Thiols and Disulfides in Materials Chemistry
1.2. Some Important Reactions of Thiols and Disulfides
1.3. Biological Relavance of Thiols and Disulfides
1.4. Thiols and Disulfides in Materials Science
1.5. Applications of Thiomers and Disulfide-Containing Polymers
2. Factors Influencing the Reactivity of Thiols and Disulfides
2.1. Factors Influencing the Bond Strengths of Thiols and Disulfides
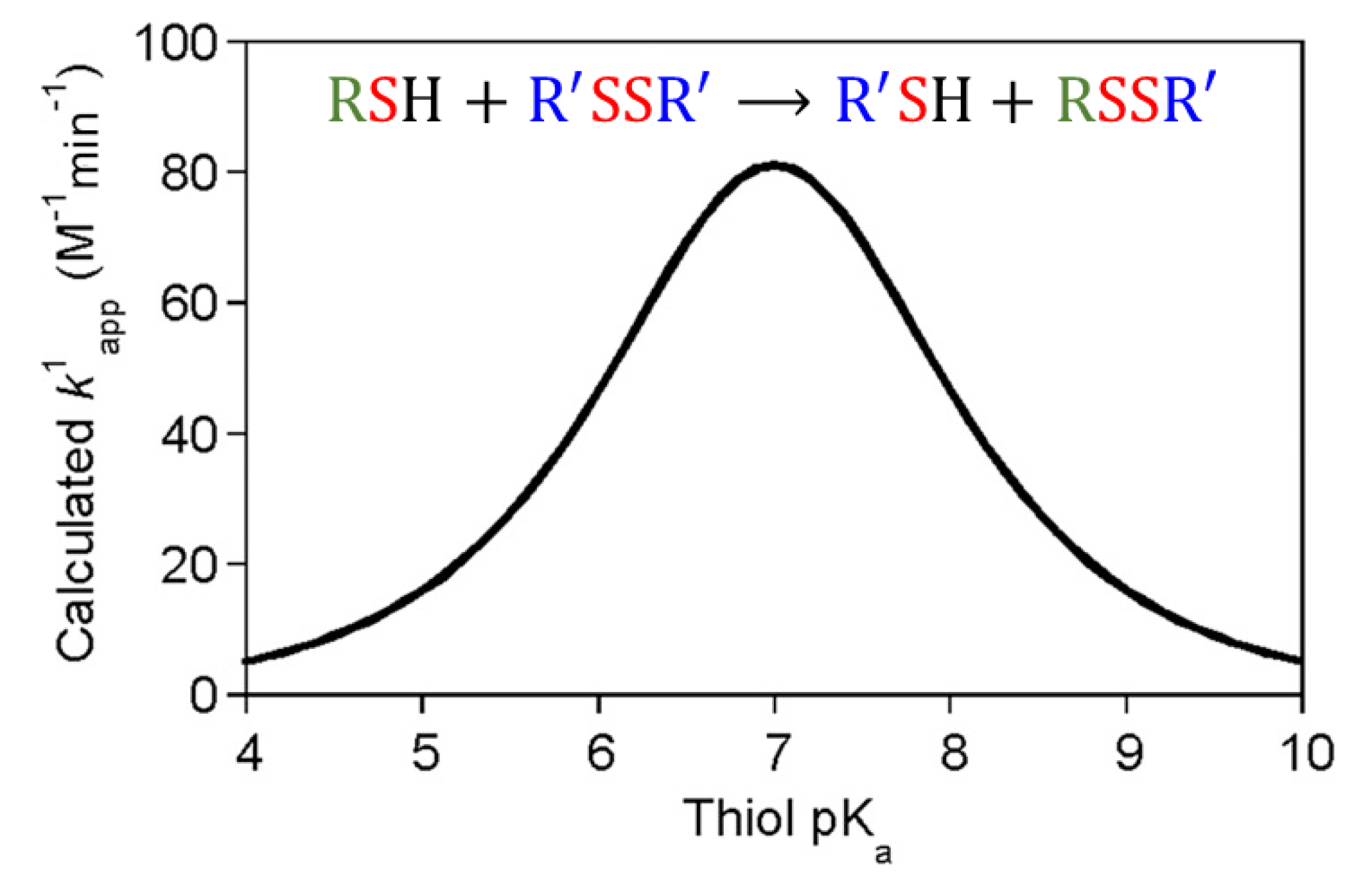
2.2. Reactivity of Thiols and Disulfides in Thiol-Disulfide Exchange Reactions
2.3. Factors Influencing the Redox Chemistry of Thiols and Disulfides
2.3.1. Reduction of Disulfides with Thiols by Exchange Reactions
2.3.2. Direct Reduction of Thiols with Non-Thiol Reducing Agents
2.4. Oxidation of Thiols to Disulfides
2.5. Radical Reactions and Radical Coupling of Thiols and Disulfides
3. Reaction Types and Applications
3.1. Thiol-Disulfide Exchange-Based and Redox Reactions
3.1.1. Natural Keratin Protection and Regenerated Keratin Enhancement
3.1.2. Mucoadhesion
3.1.3. Organic/Inorganic Hybrid Materials and Thiolated Organosilica Nanoparticles
3.1.4. Redox-Reversible Gelation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer Backbone | Thiol or Disulfide Ligand | Gelated Solvents | Oxidizing Agent(s) | Reducing Agent(s) | Number of Reversible Cycles 1 | Reference |
|---|---|---|---|---|---|---|
| Poly(acrylate)-based core-crosslinked star copolymer (polyEGDA-polyBA) (Figure 13) | DSDMA | DMF, CHCl3,THF | FeCl3 and O2 or I2 and O2 | n-Bu3P | One full cycle | [83,84] |
| Branched trithiols: TMMP and TEMPIC Branched tetrathiol: PEMP Branched hexathiol: DPMP (Figure 15) | N/A, disulfides formed during oxidation of branched monomers | DMSO | DMSO or Albright-Goldman oxidation 2 | DTT | One full cycle | [25] |
| Poly (2(dimethylamino) ethyl methacrylate) (Figure 17b) | 1,2,3 triazole-based | Aqueous phosphate buffer | Heating in air | tris(2-carboxyethyl) phosphine hydrochloride | Five cycles | [86] |
| PEG functionalized chitosan (Figure 19) | BDS-functionalized or disulfide-conjugated to thiolated methyl red dye or GSH | Water | n/a, formed as disulfides | DTT | One direction | [87] |
| Poly(styrene-co-4-vinylbenzene chloride) and PEG triblock copolymer | 1,2,3-triazole derivative | [EMI][TFSA] ionic liquid with DCM | Heated in air | DTT | Six redox cycles | [26,88] |
| Poly(benzyl ether)- PEG copolymer (ScIP) (Figure 23) | Pyridine disulfide | DMF | n/a | DTT | Non-reversible | [89] |
3.1.5. Redox-Triggered Drug Release and Disulfide-Diselenide Chemistry
3.1.6. Loading Small Molecule Cargo into Networks
3.1.7. Thiol-Disulfide Exchange and Redox Reactions That Initiate Degradation or Cascade Reactions
Cascade Reactions Triggering Decomposition
Cascade Reactions Resulting in Polymerization or Material Rearrangements
3.2. Disulfide-Disulfide Metathesis-Based Systems
3.2.1. Introduction to Self-Healing Materials and Current Challenges in the Field
3.2.2. Quantification of Self-Healing Behavior
3.2.3. Disulfide-Based Self-Healing
| Polymer Structure | Secondary Self- Healing Interaction | External Stimuli | Self-Healing Efficency 1 | Time to Self-Heal 2 | Tensile Strength 3 (MPa) | Strain at Break 3 | Reference |
|---|---|---|---|---|---|---|---|
 polyEGDA-polyBA star polymers | none | none | 100% | Variable 4 | n/a | n/a | [84] |
 random PDMS-based co-polymer | imine bonding | none 5 | 95% | 4 h | 0.14 | 2200% | [16] |
 = PTMG = PTMG | H-bonding | 60 °C | 100% | 6 h | 5.01 | n/a | [143] |
 IP-SS: co-polymerized with PTMG | none | 25 °C | 77–100% | 2 h | 6.8 | 920% | [12] |
 4,4’-methylenebis(phenyl isocyanate) based copolymer of PTMG (HM-SS) | none | 25 °C | 70–89% | 2 h | 4.5 | 490% | [12] |
 M-SS: co-polymerized with PTMG | none | 25 °C | 0–4% | 2 h | 30.4 | 940% | [12] |
 H-SS: co-polymerized with PTMG | none | 25 °C | 4–30% | 2 h | 9.5 | 750% | [12] |
 co-polymerized with 2-ethylhexyl methacrylate via disulfide linkages | none | none | 100% 7 | 3–30 min. | n/a | n/a | [161] |
 co-polymerized with PTMG | shape memory | 100 °C (microwave) | 74–91% 6 | 10 min. | 22.9–31.9 6 | 850–1160% 6 | [13] |
 disulfide-liquid crystal elastomer | shape memory | 0.015–0.12% | 350% | [32] | |||
 thiuram disulfide crosslinked polytetra(ethylene glycol) | none | table-top lamp | 87–97% | 1 min. | 4.2 | 200% | [163] |
 PU incorporating BiTEMPS | none | 100 °C | 86–93% | 24 h | 0.14 | 440% | [162] |
 poly(hexyl methacrylate) incorporating BiTEMPS | none | 120 °C, 70 kPa | 85–92% | 24 h | 7.6 | 280 | [164] |
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jin, Y.; Yu, C.; Denman, R.J.; Zhang, W. Recent advances in dynamic covalent chemistry. Chem. Soc. Rev. 2013, 42, 6334–6654. [Google Scholar] [CrossRef] [PubMed]
- Leichner, C.; Jelkmann, M.; Bernkop-Schnürch, A. Thiolated polymers: Bioinspired polymers utilizing one of the most important bridging structures in nature. Adv. Drug Deliv. Rev. 2019, 151–152, 191–221. [Google Scholar] [CrossRef]
- Bej, R.; Dey, P.; Ghosh, S. Disulfide chemistry in responsive aggregation of amphiphilic systems. Soft Matter. 2020, 16, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Urban, M.W. Self-healing polymeric materials. Chem. Soc. Rev. 2013, 42, 7446–7467. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Urban, M.W. Self-healing polymers. Nat. Rev. Mater. 2020, 5, 563–583. [Google Scholar] [CrossRef]
- Utrera-Barrios, S.; Verdejo, R.; Lopez-Manchado, M.A.; Hernandez Santana, M. Evolution of self-healing elastomers, from extrinsic to combined intrinsic mechanisms: A review. Mater. Horiz. 2020, 7, 2882–2902. [Google Scholar] [CrossRef]
- Nagy, P. Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation mediated pathways. Antioxid. Redox Signal. 2013, 18, 1623–1641. [Google Scholar] [CrossRef]
- Seidi, F.; Jenjob, R.; Crespy, D. Designing Smart Polymer Conjugates for Controlled Release of Payloads. Chem. Rev. 2018, 118, 3965–4036. [Google Scholar] [CrossRef]
- Zhang, X.; Han, L.; Liu, M.; Wang, K.; Tao, L.; Wan, Q.; Wei, Y. Recent progress and advances in redox-responsive polymers as controlled delivery nanoplatforms. Mater. Chem. Front. 2017, 1, 807–822. [Google Scholar] [CrossRef]
- Dutta, K.; Das, R.; Medeiros, J.; Thayumanavan, S. Disulfide Bridging Strategies in Viral and Nonviral Platforms for Nucleic Acid Delivery. Biochemistry 2021, 60, 966–990. [Google Scholar] [CrossRef]
- Wang, Q.; Guan, J.; Wana, J.; Li, Z. Disulfide based prodrugs for cancer therapy. RSC Adv. 2020, 10, 24397–24409. [Google Scholar] [CrossRef]
- Saitoa, G.; Swanson, J.A.; Lee, K.-D. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: Role and site of cellular reducing activities. Adv. Drug Deliv. Rev. 2003, 55, 199–215. [Google Scholar] [CrossRef]
- Kim, S.-M.; Jeon, H.; Shin, S.-H.; Park, S.-A.; Jegal, J.; Hwang, S.Y.; Oh, D.X.; Park, J. Superior Toughness and Fast Self-Healing and Room Temperature Engineered by Transparent Elastomers. Adv. Mater. 2018, 30, 1705145. [Google Scholar] [CrossRef]
- Ling, L.; Li, J.; Zhang, G.; Sun, R.; Wong, C.-P. Self-Healing and Shape Memory Linear Polyurethane Based on Disulfide Linkages with Excellent Mechanical Property. Macromol. Res. 2018, 26, 365–373. [Google Scholar] [CrossRef]
- Zhuang, J.; Gordon, M.R.; Ventura, J.; Li, L.; Thayumanavan, S. Multi-stimuli responsive macromolecules and their assemblies. Chem. Soc. Rev. 2013, 42, 7421–7435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.P.; Rong, M.Z.; Zhang, M.Q. Polymer engineering based on reversible covalent chemistry: A promising innovative pathway towards new materials and new functionalities. Prog. Polym. Sci. 2018, 80, 39–93. [Google Scholar] [CrossRef]
- Lv, C.; Zhao, K.; Zheng, J.A. Highly Stretchable Self-Healing Poly(dimethylsiloxane) Elastomer with Reprocessability and Degradability. Macromol. Rapid Commun. 2018, 39, 17008686. [Google Scholar] [CrossRef] [PubMed]
- Tomei, M.R.; Cinti, S.; Interino, N.; Manovella, V.; Moscone, D.; Arduini, F. Paper-based electroanalytical strip for user-friendly blood glutathione detection. Sen. Actuators B. Chem. 2019, 294, 291–297. [Google Scholar] [CrossRef]
- Yin, W.; Ke, W.; Lu, N.; Wang, Y.; Japir, A.A.-W.M.M.; Mohammed, F.; Wang, Y.; Pan, Y.; Ge, Z. Glutathione and Reactive Oxygen Species Dual-Responsive Block Copolymer Prodrugs for Boosting Tumor Site-Specific Drug Release and Enhanced Antitumor Efficacy. Biomacromolecules 2020, 21, 921–929. [Google Scholar] [CrossRef]
- Gao, Y.; Dong, C.-M. Reduction and thermo-sensitive core-cross-linked polypeptide hybrid micelles for triggered and intracellular drug release. Polym. Chem. 2017, 8, 1223–1232. [Google Scholar] [CrossRef]
- Subramanian, H.; Moorthy, R.; Sibi, M.P. Thiyl Radicals: From Simple Radical Additions to Asymmetric Catalysis. Angew. Chem. Int. Ed. 2014, 53, 13660–13662. [Google Scholar] [CrossRef]
- Denes, F.; Pichowicz, M.; Poive, G.; Renaud, P. Thiyl Radicals in Organic Synthesis. Chem. Rev. 2014, 114, 2587–2693. [Google Scholar] [CrossRef] [PubMed]
- Winther, J.R.; Thorpe, C. Quantification of thiols and disulfides. Biochim. Biophys. Acta 2014, 1840, 838–846. [Google Scholar] [CrossRef]
- Jocelyn, P.C. Chemical Reduction of Disulfides. In Methods in Enzymology; Jakoby, W.B., Griffith, O., Eds.; Academic Press Inc.: Cambridge, MA, USA, 1987; Volume 143, pp. 246–256. [Google Scholar]
- Naga, N.; Moriyama, K.; Furukawa, H. Synthesis and Properties of Multifunctional Thiol Crosslinked Gels Containing Disulfide Bond in the Network Structure. J. Polym. Sci. Polym. Chem. 2017, 55, 3749–3756. [Google Scholar] [CrossRef]
- Wei, C.; Chen, M.; Liu, D.; Zhou, W.; Khan, M.; Wu, X.; Huang, N.; Li, L. A recyclable disulfide bond chemically cross-linking, high toughness, high conductivity ion gel based on re-shaping and restructuring in the gel state. Polym. Chem. 2015, 6, 4067–4070. [Google Scholar] [CrossRef]
- Patai, S. Oxidation of Thiols. In The Chemistry of the Thiol Group; Patai’s Chemistry of Functional Groups; Wiley: London, UK, 1974; Volume 2, pp. 785–839. [Google Scholar]
- Witt, D. Recent Developments in Disulfide Bond Formation. Synthesis 2008, 16, 2491–2509. [Google Scholar] [CrossRef]
- Mandal, B. Recent advances in S-S bond formation. RSC Adv. 2014, 4, 13854–13881. [Google Scholar] [CrossRef]
- Rowan, S.J.; Cantrill, S.J.; Cousins, R.L.; Sanders, J.K.M.; Stoddart, J.F. Dynamic Covalent Chemistry. Angew. Chem. Int. Ed. 2002, 41, 898–952. [Google Scholar] [CrossRef]
- Zhang, X.; Waymouth, R.M. 1,2-Dithiolane-Derived Dynamic Covalent Materials: Cooperative Self-Assembly and Reversible Cross-Linking. J. Am. Chem. Soc. 2017, 139, 3822–3833. [Google Scholar] [CrossRef]
- Wang, Z.; Tian, H.; He, Q.; Cai, S. Reprogrammable, Reprocessible, and Self-Healable Liquid Crystal Elastomer with Exchangeable Disulfide Bonds. ACS Appl. Mater. Interfaces 2017, 9, 331119. [Google Scholar] [CrossRef]
- Wang, B.; Yang, W.; McKittrick, J.; Meyers, M.A. Keratin: Structure, mechanical properties, occurrence in biological organisms, and efforts at bioinspiration. Prog. Mater. Sci. 2016, 76, 229–318. [Google Scholar] [CrossRef]
- Shavandi, A.; Silva, T.H.; Bekhit, A.A.; Bekhit, A.E.-D.A. Keratin: Dissolution, extraction and biomedical application. Biomater. Sci. 2017, 5, 1699–1735. [Google Scholar] [CrossRef]
- Mi, X.; Xu, H.; Yang, Y. Submicron amino acid particles reinforced 100% keratin biomedical films with enhanced wet properties via interfacial strengthening. Colloids Surf. B Biointerfaces 2019, 177, 33–40. [Google Scholar] [CrossRef]
- Xu, H.; Ma, Z.; Yang, Y. Dissolution and regeneration of wool via controlled disintegration and disentanglement of highly crosslinked keratin. J. Mater. Sci. 2014, 49, 7513–7521. [Google Scholar] [CrossRef]
- Vasconcelos, A.; Freddi, G.; Cavaco-Paulo, A. Biodegradable materials based on silk fibroin and keratin. Biomacromolecules 2008, 9, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Mi, X.; Mu, B.; Li, W.; Xu, H.; Yang, Y. From Poultry Wastes to Quality Protein Products via Restoration of the Secondary Structure with Extended Disulfide Linkages. ACS Sustain. Chem. Eng. 2020, 8, 1396–1405. [Google Scholar] [CrossRef]
- Leichner, C.; Steinbring, C.; Baus, R.A.; Baecker, D.; Gust, R.; Bernkop-Schnurch, A. Reactive keratin derivatives: A promising strategy for covalent binding to hair. J. Colloid Interface Sci. 2019, 534, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.; Cavaco-Paulo, A. Protein disulphide isomerase-mediated grafting of cysteine-containing peptides onto over-bleached hair. Biocatal. Biotransf. 2012, 30, 10–19. [Google Scholar] [CrossRef]
- Roddick-Lanzilotta, A.; Kelly, R.; Scott, S.; Chahal, S. New keratin isolates: Actives for natural hair protection. J. Cosmet. Sci. 2007, 58, 405–511. [Google Scholar]
- Barba, C.; Scott, S.; Roddick-Lanzilotta, A.; Kelly, R.; Manich, A.M.; Parra, J.L.; Coderch, L. Restoring important hair properties with wool keratin proteins and peptides. Fibers Polym. 2010, 11, 1055–1061. [Google Scholar] [CrossRef]
- Griesser, J.; Hetényi, G.; Bernkop-Schnürch, A. Thiolated Hyaluronic Acid as Versatile Mucoadhesive Polymer: From the Chemistry Behind to Product Developments—What are the Capabilities? Polymers 2018, 10, 243. [Google Scholar] [CrossRef]
- Laffleur, F.; Bernkop-Schnürch, A. Thiomers: Promising platform for macromolecular drug delivery. Future Med. Chem. 2012, 4, 2205–2216. [Google Scholar] [CrossRef]
- Shah, K.U.; Shah, S.U.; Dilawar, N.; Khan, G.M.; Gibaud, S. Thiomers and their potential applications in drug delivery. Expert. Opin. Drug Deliv. 2017, 14, 601–610. [Google Scholar] [CrossRef]
- Duggan, S.; Cummins, W.; O’Donovan, O.; Hughes, H.; Owens, E. Thiolated polymers as mucoadhesive drug delivery systems. Eur. J. Pharm. Sci. 2012, 100, 64–78. [Google Scholar] [CrossRef]
- Bonengel, S.; Bernkop-Schnürch, A. Thiomers—From bench to market. J. Control. Release 2014, 195, 120–129. [Google Scholar] [CrossRef]
- Ijaz, M.; Bernkop-Schnürch, A. Preactivated thiomers: Their role in drug delivery. Expert Opin. Drug Deliv. 2015, 12, 1269–1281. [Google Scholar] [CrossRef]
- Smart, J.D. The basics and underlying mechanisms of mucoadhesion. Adv. Drug Deliv. Rev. 2005, 57, 1556–1568. [Google Scholar] [CrossRef]
- Alexander Ajazuddin, A.; Khan, J.; Giri, T.K.; Tripathi, D.K.; Saraf, S. Advancement in stimuli triggered in situ gelling delivery for local and systemic route. Expert Opin. Drug Deliv. 2012, 9, 1573–1593. [Google Scholar] [CrossRef]
- Allen, A.; Hutton, D.; McQueen, S.; Garner, A. Dimensions of gastroduodenal surface pH gradients exceed those of adherent mucus gel layers. Gastroenterology 1983, 85, 463–466. [Google Scholar] [CrossRef]
- Tian, H.; He, Z.; Sun, C.; Yang, C.; Zhao, P.; Liu, L.; Leong, K.W.; Mao, H.Q.; Liu, Z.; Chen, Y. Uniform core-shell nanoparticles with thiolated hyaluronic acid coating to enhance oral delivery of insulin. Adv. Healthc. Mater. 2018, 7, e1800285. [Google Scholar] [CrossRef]
- Denora, N.; Lopedota, A.; Perrone, M.; Laquintana, V.; Iacobazzi, R.M.; Milella, A.; Fanizza, E.; Depalo, N.; Cutrignelli, A.; Lopalco, A.; et al. Spray-dried mucoadhesives for intravesical drug delivery using N-acetylcysteine- and glutathione-glycol chitosan conjugates. Acta Biomater. 2016, 43, 170–184. [Google Scholar] [CrossRef]
- Perrone, M.; Lopalco, A.; Lopedota, A.; Cutrignelli, A.; Laquintana, V.; Franco, M.; Bernkop-Schnürch, A.; Denora, N. S-preactivated thiolated glycol chitosan useful to combine mucoadhesion and drug delivery. Eur. J. Pharm. Biopharm. 2018, 132, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Kassem, A.A.; Farid, R.M.; Issa, D.A.; Khalil, D.S.; Abd-El-Razzak, M.Y.; Saudi, H.I.; Eltokhey, H.M.; El-Zamarany, E.A. Development of mucoadhesive microbeads using thiolated sodium alginate for intrapocket delivery of resveratrol. Int. J. Pharm. 2015, 487, 305–415. [Google Scholar] [CrossRef]
- Mahajan, H.; Shaikh, H.; Gattani, S.; Nerkar, P. In situ gelling system based on thiolated gellan gum as new carrier for nasal administration of dimenhydrinate. Int. J. Pharm. Sci. Nanotech. 2009, 2, 544–550. [Google Scholar] [CrossRef]
- More, M.P.; Bhamare, M.S.; Bhavsar, C.J.; Patil, P.O.; Deshmukh, P.K. Development of novel thiolated carboxymethyl-gellan gum as potential mucoadhesive polymer: Application of DoE. Adv. Mater. Sci. 2017, 2, 1–9. [Google Scholar] [CrossRef]
- Jelkmann, M.; Bonengel, S.; Menzel, C.; Markovic, S.; Bernkop-Schnürch, A. New perspectives of starch: Synthesis and in vitro assessment of novel thiolated mucoadhesive derivatives. Int. J. Pharm. 2018, 546, 70–77. [Google Scholar] [CrossRef]
- Perrone, M.; Lopalco, A.; Lopedota, A.; Cutrignelli, A.; Laquintana, V.; Douglas, J.; Franco, M.; Liberati, E.; Russo, V.; Tongiani, S.; et al. Preactivated thiolated glycogen as mucoadhesive polymer for drug delivery. Eur.J. Pharm. Biopharm. 2017, 119, 161–169. [Google Scholar] [CrossRef]
- Laffleur, F.; Messirek, A. Development of mucoadhesive thio-carboxymethyl cellulose for application in buccal delivery of drugs. Ther. Deliv. 2016, 7, 63–71. [Google Scholar] [CrossRef]
- Leonaviciute, G.; Bonengel, S.; Mahmood, A.; Ahmad Idrees, M.; Bernkop-Schnürch, A. S-protected thiolated hydroxyethyl cellulose (HEC): Novel mucoadhesive excipient with improved stability. Carbohydr. Polym. 2016, 144, 514–521. [Google Scholar] [CrossRef]
- Zaman, M.; Hanif, M.; Sultana, K.; Atta-Ur-Rehman. Synthesis of thiolated arabinoxylan and its application as sustained release mucoadhesive film former. Biomed. Mater. 2018, 13, 025019. [Google Scholar] [CrossRef]
- Bhatia, M.; Ahuja, M.; Mehta, M. Thiol derivatization of Xanthan gum and its evaluation as a mucoadhesive polymer. Carbohydr. Polym. 2015, 131, 119–124. [Google Scholar] [CrossRef]
- Menzel, C.; Jelkmann, M.; Laffleur, F.; Bernkop-Schnürch, A. Nasal drug delivery: Design of a novel mucoadhesive and in situ gelling polymer. Int. J. Pharm. 2017, 517, 196–202. [Google Scholar] [CrossRef]
- Ijaz, M.; Ahmad, M.; Akhtar, N.; Laffleur, F.; Bernkop-Schnürch, A. Thiolated α-Cyclodextrin: The invisible choice to prolong ocular Drug residence time. J. Pharm. Sci. 2016, 105, 2848–2854. [Google Scholar] [CrossRef]
- Moghadam, A.; Ijaz, M.; Asim, H.M.; Mahmood, A.; Jelkmann, M.; Matuszczak, B. Bernkop-Schnürch, A. Non-ionic thiolated cyclodextrins—The next generation. Int. J. Nanomed. 2018, 13, 4003–4013. [Google Scholar] [CrossRef]
- Asim, M.H.; Moghadam, A.; Ijaz, M.; Mahmood, A.; Götz, R.X.; Matuszczak, B.; Bernkop-Schnürch, A. S-protected thiolated cyclodextrins as mucoadhesive oligomers for drug delivery. J. Colloid Interface Sci. 2018, 531, 261–268. [Google Scholar] [CrossRef]
- Bonengel, S.; Haupstein, S.; Perera, G.; Bernkop-Schnürch, A. Thiolated and S-protected hydrophobically modified cross-linked poly (acrylic acid)–a new generation of multifunctional polymers. Eur. J. Pharm. Biopharm. 2014, 88, 390–396. [Google Scholar] [CrossRef]
- Werle, M.; Hironaka, K.; Takeuchi, H.; Hoyer, H. Development and in vitro characterization of liposomes coated with thiolated poly(acrylic acid) for oral drug delivery. Drug Dev. Ind. Pharm. 2009, 35, 209–215. [Google Scholar] [CrossRef]
- Leonaviciute, G.; Adamovic, N.T.; Lam, H.T.; Rohrer, J.; Partenhauser, A.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems (SEDDS): Proof-of-concept how to make them mucoadhesive. Eur. J. Pharm. Biopharm. 2017, 112, 51–57. [Google Scholar] [CrossRef]
- Liang, J.; Struckhoff, J.J.; Du, H.; Hamilton, P.D.; Ravi, N. Synthesis and characterization of in situ forming anionic hydrogel as vitreous substitutes. J. Biomed. Mater. Res. B Appl. Biomater. 2017, 105, 977–988. [Google Scholar] [CrossRef]
- Partenhauser, A.; Laffleur, F.; Roher, J.; Bernkop-Schnürch, A. Thiolated silicone oil: Synthesis, gelling, and mucoadhesive properties. Acta Biomater. 2015, 16, 169–177. [Google Scholar] [CrossRef]
- Fürst, A.L.; Baus, R.; Lupo, N.; Bernkop-Schnürch, A. Entirely S-protected thiolated silicone: A novel solid hydrophobic muco- and skin adhesive. J. Pharm. Sci. 2019, 108, 2887–2894. [Google Scholar] [CrossRef] [PubMed]
- Ways, T.M.M.; Lau, W.M.; Ng, K.W.; Khutoryanskiy, V.V. Synthesis of thiolated, PEGylated and POZylated silica nanoparticles and evaluation of their retention on rat intestinal mucosa in vitro. Eur. J. Pharm. Sci. 2018, 122, 230–238. [Google Scholar] [CrossRef]
- Kalarickal, N.C.; Rimmer, R.; Sarker, P.; Leroux, J.C. Thiol-Functionalized Poly(ethylene glycol)-b-polyesters: Synthesis and characterization. Macromolecules 2007, 40, 1874–1880. [Google Scholar] [CrossRef]
- Budai-Szűcs, M.; Horvát, G.; Szilágyi, B.; Gyarmati, B.; Szilágyi, A.; Berkó, S.; Szabó-Révész, P.; Sandri, G.; Bonferoni, M.C.; Caramella, C.; et al. Cationic thiolated poly(aspartamide) polymer as a potential excipient for artificial tear formulations. J. Ophthalmol. 2016, 2016, 2647264. [Google Scholar] [CrossRef]
- Duggan, S.; Hughes, H.; Owens, E.; Duggan, E.; Cummins, W.; O’Donovan, O. Synthesis and characterisation of mucoadhesive thiolated polyallylamine. Int. J. Pharm. 2016, 499, 368–375. [Google Scholar] [CrossRef]
- Mekaru, H.; Lu, J.; Tamanoi, F. Development of Mesoporous Silica-Based Nanoparticles with Controlled Release Capability for Cancer Therapy. Adv. Drug Deliv. Rev. 2015, 95, 40–49. [Google Scholar] [CrossRef]
- Park, K. Controlled drug delivery systems: Past forward and future back. J. Control. Release 2014, 190, 3–8. [Google Scholar] [CrossRef]
- Zhang, L.; Lui, Y.; Zhang, K.; Chen, Y.; Luo, X. Redox-responsive comparison of diselenide micelles with disulfide micelles. Colloid Polym. Sci. 2019, 297, 225–238. [Google Scholar] [CrossRef]
- Mekaru, H.; Yoshigoe, A.; Nakamura, M.; Doura, T.; Tamanoi, F. Biodegradability of Disulfide-Organosilica Nanoparticles Evaluated by Soft X-ray Photoelectron Spectroscopy: Cancer Therapy Implications. ACS Appl. Nano Mater. 2019, 2, 479–488. [Google Scholar] [CrossRef]
- Doura, T.; Nishio, T.; Tamanoi, F.; Nakamura, M. Relationship between the glutathione-responsive degradability of thiol-organosilica nanoparticles and the chemical structures. J. Mater. Res. 2019, 34, 1266–1278. [Google Scholar] [CrossRef]
- Kamada, J.; Koynov, K.; Corten, C.; Juhari, A.; Yoon, J.A.; Urban, M.W.; Balzars, A.C.; Matyjaszewski, K. Redox Responsive Behavior of Thiol/Disulfide-Functionalized Star Polymers Synthesized via Atom Transfer Radical Polymerization. Macromolecules 2010, 43, 4133–4139. [Google Scholar] [CrossRef]
- Yoon, J.A.; Kamada, J.; Koynov, K.; Mohin, J.; Nicolay, R.; Zhang, Y.; Balazs, A.C.; Kowalewski, T.; Matyjaszewski, K. Self-Healing Polymer Films Based on Thiol—Disulfide Exchange Reactions and Self-Healing Kinetics Using Atomic Force Microscopy. Macromolecules 2012, 45, 142–149. [Google Scholar] [CrossRef]
- Van der Zwaag, S. (Ed.) Self-Healing Materials: An Alternative Approach to 20 Centuries of Materials Science; Springer: Berlin, Germany, 2007. [Google Scholar]
- Mocny, P.; Klock, H.-A. Reversibly Cross-Linking Polymer Brushes Using Interchain Disulfide Bonds. Macromolecules 2020, 53, 731–740. [Google Scholar] [CrossRef]
- Arslan, M. Fabrication and reversible disulfide functionalization of PEGylated chitosan-based hydrogels: Platforms for selective immobilization and release of thiol-containing molecules. Eur. Poly. J. 2020, 126, 109543. [Google Scholar] [CrossRef]
- Wei, C.; Chen, M.; Liu, D.; Zhou, W.; Khan, M.; Wu, X.; Huang, N.; Li, L. Synthesis of recyclable, chemically cross-linked, high toughness, high conductivity ion gels by sequential triblock copolymer self-assembly and disulfide bond cross-linking. RSC Adv. 2015, 5, 22638–22646. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, Y.; Zhang, B.; Li, H.; Cheng, Z.; Shi, J.; Xiong, J.; Bai, Y.; Zhang, K. Functionalizable, Side Chain-Immolative Poly(benzyl ether)s. ACS Macro Lett. 2019, 8, 399–402. [Google Scholar] [CrossRef]
- Wiltshire, J.T.; Qiao, G.G. Recent Advances in Star Polymer Design: Degradability and the Potential for Drug Delivery. Aust. J. Chem. 2007, 60, 699–705. [Google Scholar] [CrossRef]
- Xu, F.J.; Zhang, Z.X.; Ping, Y.; Li, J.; Kang, E.T.; Neoh, K.G. Star-Shaped Cationic Polymers by Atom Transfer Radical Polymerization from β-Cyclodextrin Cores for Nonviral Gene Delivery. Biomacromolecules 2009, 10, 285–293. [Google Scholar] [CrossRef]
- Kim, B.-S.; Gao, H.; Argun, A.A.; Matyjaszewski, K.; Hammond, P.T. All-Star Polymer Multilayers as pH-Responsive Nanofilms. Macromolecules 2009, 42, 368–375. [Google Scholar] [CrossRef]
- Connal, L.A.; Vestberg, R.; Hawker, C.J.; Qiao, G.G. Fabrication of Reversibly Crosslinkable, 3-Dimensionally Conformal Polymeric Microstructures. Adv. Funct. Mater. 2008, 18, 3315–3322. [Google Scholar] [CrossRef]
- Barner-Kowollik, C.; Du Prez, F.E.; Espeel, P.; Hawker, C.J.; Junkers, T.; Schlaad, H.; Van Camp, W. “Clicking” Polymers or Just Efficient Linking: What Is the Difference? Angew. Chem. Int. Ed. 2011, 50, 60–62. [Google Scholar] [CrossRef]
- Imaizumi, S.; Kokubo, H.; Watanabe, M. Polymer Actuators Using Ion-Gel Electrolytes Prepared by Self-Assembly of ABA-Triblock Copolymers. Macromolecules 2012, 45, 401–409. [Google Scholar] [CrossRef]
- Zhang, S.; Lee, K.H.; Sun, J.; Frisbie, C.D.; Lodge, T.P. Viscoelastic Properties, Ionic Conductivity, and Materials Design Considerations for Poly(styrene-b-ethylene oxide-b-styrene)-Based Ion Gel Electrolytes. Macromolecules 2011, 44, 8981–8989. [Google Scholar] [CrossRef]
- Ye, Y.-S.; Rick, J.; Hwang, B.J. Ionic liquid polymer electrolytes. J. Mater. Chem. A 2013, 1, 2719–2743. [Google Scholar] [CrossRef]
- Yue, D.; Cheng, G.; He, Y.; Nie, Y.; Jiang, Q.; Cai, X.; Gu, Z. Influence of reduction-sensitive diselenide bonds and disulfide bonds on oligoethylenimine conjugates for gene delivery. J. Mater. Chem. B 2014, 2, 7210–7221. [Google Scholar] [CrossRef]
- Kim, J.O.; Sahay, G.; Kabanov, A.V.; Bronich, T.K. Polymeric micelles with ionic cores containing biodegradable cross-links for delivery of chemotherapeutic agents. Biomacromolecules 2010, 11, 919–926. [Google Scholar] [CrossRef][Green Version]
- Wei, C.; Zhang, Y.; Song, Z.; Xia, Y.; Xu, H.; Lang, M. Enhanced bioreduction-responsive biodegradable diselenide-containing poly(ester urethane) nanocarriers. Biomater. Sci. 2017, 5, 669–677. [Google Scholar] [CrossRef]
- Perera, M.M.; Chimala, C.; Elhusain-Elnegres, A.; Heaton, P.; Ayres, N. Reversibly Softening and Stiffening Organogels Using a Wavelength-Controlled Disulfide-Diselenide Exchange. ACS Macro Lett. 2020, 9, 1552–1557. [Google Scholar] [CrossRef]
- Quinn, J.F.; Whittaker, M.R.; Davis, T.P. Glutathione responsive polymers and their application in drug delivery systems. Polym. Chem. 2017, 8, 97–126. [Google Scholar] [CrossRef]
- Huo, M.; Yuan, J.; Tao, L.; Wei, Y. Redox-Responsive Polymers for Drug Delivery: From Molecular Design to Applications. Polym. Chem. 2014, 5, 1519–1528. [Google Scholar] [CrossRef]
- Cheng, R.; Feng, F.; Meng, F.H.; Deng, C.; Feijen, J.; Zhong, Z.Y. Glutathione-Responsive Nano-vehicles as a Promising Platform for Targeted Intracellular Drug and Gene Delivery. J. Control. Release 2011, 152, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, Y.F.; Zhong, Z.Y. Reduction-Sensitive Polymeric Nanomedicines: An Emerging Multifunctional Platform for Targeted Cancer Therapy. Adv. Drug Deliv. Rev. 2018, 132, 16–32. [Google Scholar] [CrossRef]
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-Responsive Nanocarriers for Drug Delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef]
- Wierzba, A.; Mojciechowska, M.; Trylska, J.; Gryko, D. Vitamin B12 Suitably Tailored for Disulfide-Based Conjugation. Bioconjugate Chem. 2016, 27, 189–197. [Google Scholar] [CrossRef]
- Banerjee, R. (Ed.) Chemistry and Biochemistry of B12; John Wiley & Sons, Inc.: New York, NY, USA, 1999. [Google Scholar]
- Clardy, S.M.; Allis, D.G.; Fairchild, T.J.; Doyle, R.P. Vitamin B12 in Drug Delivery: Breaking Through the Barriers to a B12 Bioconjugate. Pharmaceutical. Expert Opin. Drug Deliv. 2011, 8, 127–140. [Google Scholar] [CrossRef]
- Fazen, C.H.; Valentin, D.; Fairchild, T.J.; Doyle, R.P. Oral Delivery of the Appetite Suppressing Peptide hPYY(3−36) through the Vitamin B12 Uptake Pathway. J. Med. Chem. 2011, 54, 8707–8711. [Google Scholar] [CrossRef] [PubMed]
- Petrus, A.K.; Fairchild, T.J.; Doyle, R.P. Traveling the Vitamin B12 Pathway: Oral Delivery of Protein and Peptide Drugs. Angew. Chem. Int. Ed. 2011, 48, 1028. [Google Scholar] [CrossRef]
- Song, Q.; Yang, J.; Hall, S.C.L.; Gurnani, P.; Perrier, S. Pyridyl Disulfide Reaction Chemistry: An Efficient Strategy toward Redox-Responsive Cyclic Peptide–Polymer Conjugates. ACS Macro Lett. 2019, 8, 1347–1352. [Google Scholar] [CrossRef]
- Schnabel, W. Polymer Degradation: Principles and Practical Applications; Hanser International: Munich, Germany, 1981. [Google Scholar]
- Hamid, S.H. (Ed.) Handbook of Polymer Degradation; Marcel Dekker: New York, NY, USA, 2000. [Google Scholar]
- Delplace, V.; Nicolas, J. Degradable vinyl polymers for biomedical applications. Nat. Chem. 2015, 7, 771–784. [Google Scholar] [CrossRef]
- Edlund, U.; Albertsson, A.-C. Degradable polymer microspheres for controlled drug delivery. Adv. Polym. Sci. 2002, 157, 67–112. [Google Scholar] [CrossRef]
- Tsarevsky, N.V.; Matyjaszewski, K. Combining Atom Transfer Radical Polymerization and Disulfide/Thiol Redox Chemistry: A Route to Well-Defined (Bio)degradable Polymeric Materials. Macromolecules 2005, 38, 3087–3092. [Google Scholar] [CrossRef]
- Bej, R.; Ghosh, S. Glutathione Triggered Cascade Degradation of Amphiphilic Poly(disulfide)-Drug Conjugate and Targeted Release. Bioconjugate Chem. 2019, 30, 101–110. [Google Scholar] [CrossRef]
- Grek, C.L.; Tew, K.D. Redox Metabolism and Malignancy. Curr. Opin. Pharmacol. 2010, 10, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J.; Hu, X.L.; Shen, Q.; Xing, D. Mitochondria-Specific Drug Release and Reactive Oxygen Species Burst Induced by Polyprodrug Nanoreactors Can Enhance Chemotherapy. Nat. Commun. 2019, 10, 1704. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Liu, S.Y. Functional Block Copolymer Assemblies Responsive to Tumor and Intracellular Microenvironments for Site- Specific Drug Delivery and Enhanced Imaging Performance. Chem. Soc. Rev. 2013, 42, 7289–7325. [Google Scholar] [CrossRef]
- Li, L.; Wang, X.; Yang, J.; Ye, X.; Wu, C. Degradation Kinetics of Model Hyperbranched Chains with Uniform Subchanins and Controlled Locations of Cleavable Disulfide Linkages. Macromolecules 2014, 47, 650–658. [Google Scholar] [CrossRef]
- Deng, R.; Yue, Y.; Jin, F.; Chen, Y.; Kung, H.-F.; Lin, M.C.M.; Wu, C. Revisit the complexation of PEI and DNA—How to make low cytotoxic and highly efficient PEI gene transfection non-viral vectors with a controllable chain length and structure? J. Control. Release 2009, 140, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Tsarevsky, N.V.; Huang, J.; Matyjaszewski, K.J. Synthesis of hyperbranched degradable polymers by atom transfer radical (Co)polymerization of inimers with ester or disulfide groups. Polym. Sci. A Polym. Chem. 2009, 47, 6839–6851. [Google Scholar] [CrossRef]
- Xu, J.; Tao, L.; Liu, J.; Bulmus, V.; Davis, T.P. Synthesis of Functionalized and Biodegradable Hyperbranched Polymers from Novel AB2 Macromonomers Prepared by RAFT Polymerization. Macromolecules 2009, 42, 6893–6901. [Google Scholar] [CrossRef]
- Yang, W.; Pan, C. Synthesis and Fluorescent Properties of Biodegradable Hyperbranched Poly(amido amine)s. Macromol. Rapid Commun. 2009, 30, 2096–2101. [Google Scholar] [CrossRef]
- Yan, J.; Hong, C.; You, Y. An Easy Method To Convert the Topologies of Macromolecules after Polymerization. Macromolecules 2011, 44, 1247–1251. [Google Scholar] [CrossRef]
- Gallogly, M.M.; Starke, D.W.; Mieyal, J.J. Mechanistic and kinetic details of catalysis of thiol-disulfide exchange by glutaredoxins and potential mechanisms of regulation. Antioxid. Redox Signal. 2009, 11, 1059–1081. [Google Scholar] [CrossRef]
- Lukesh, J.C.; Palte, M.J.; Raines, R.T. A potent, versatile disulfide-reducing agent from aspartic acid. J. Am. Chem. Soc. 2012, 134, 4057–4059. [Google Scholar] [CrossRef] [PubMed]
- Wiita, A.P.; Ainavarapu, R.K.; Huang, H.H.; Fernandez, J.M. Force-dependent chemical kinetics of disulfide bond reduction observed with single-molecule techniques. Proc. Natl. Acad. Sci. USA 2006, 103, 7222–7227. [Google Scholar] [CrossRef]
- Singh, R.; Whitesides, G.M. Comparisons of rate constants for thiolate-disulfide interchange in water and in polar aprotic solvents using dynamic proton NMR line shape analysis. J. Am. Chem. Soc. 1990, 112, 1190–1197. [Google Scholar] [CrossRef]
- Zhang, X.; Dai, Y. Amphiphilic graft polymer with reduction breakable main chain prepared via click polymerization and grafting onto. J. Nanopart. Res. 2018, 20, 147. [Google Scholar] [CrossRef]
- Lee, A.L.Z.; Ng, V.W.L.; Gao, S.J.; Hedrick, J.L.; Yang, Y.Y. Injectable Hydrogels from Triblock Copolymers of Vitamin E-Functionalized Polycarbonate and Poly(ethylene glycol) for Subcutaneous Delivery of Antibodies for Cancer Therapy. Adv. Funct. Mater. 2014, 24, 1538–1550. [Google Scholar] [CrossRef]
- Taribagil, R.R.; Hillmyer, M.A.; Lodge, T.P. Hydrogels from ABA and ABC Triblock Polymers. Macromolecules 2010, 43, 5396–5404. [Google Scholar] [CrossRef]
- Tsitsilianis, C. Responsive reversible hydrogels from associative “smart” macromolecules. Soft Matter 2010, 6, 2372–2388. [Google Scholar] [CrossRef]
- Winnik, M.A.; Yekta, A. Associative polymers in aqueous solution. Curr. Opin. Colloid Interface Sci. 1997, 2, 424–436. [Google Scholar] [CrossRef]
- Mortensen, K.; Brown, W.; Jorgensen, E. Phase Behavior of Poly(propylene oxide)-Poly(ethylene oxide)-Poly(propylene oxide) Triblock Copolymer Melt and Aqueous Solution. Macromolecules 1994, 27, 5654–5666. [Google Scholar] [CrossRef]
- Komaromy, D.; Stuart, M.C.A.; Santiago, G.M.; Tezcan, M.; Krasnikov, V.V.; Otto, S. Self-Assembly Can Direct Dynamic Covalent Bond Formation toward Diversity or Specificity. J. Am. Chem. Soc. 2017, 139, 6234–6241. [Google Scholar] [CrossRef]
- Malinskii, Y.M.; Prokopenko, V.V.; Ivanova, N.A.; Kargin, V.A. Investigation of self-healing of cracks in polymers I. Effect of temperature and crosslinks on self-healing of cracks in polyvinyl acetate. Polym. Mech. 1970, 6, 240–244. [Google Scholar] [CrossRef]
- Malinskii, Y.M.; Prokopenko, V.V.; Ivanova, N.A.; Kargin, V.A. Investigation of self-healing of cracks in polymers 2. Effect of molecular weight of a polymer and the environment on self-healing of cracks in polyvinyl acetate. Polym. Mech. 1970, 6, 382–384. [Google Scholar] [CrossRef]
- Malinskii, Y.M.; Prokopenko, V.V.; Ivanova, N.A.; Kargin, V.A. Investigation of the self-healing of cracks in polymers 3. Effect of medium and layer thickness on self-healing in polyvinyl acetate. Polym. Mech. 1970, 6, 969–972. [Google Scholar] [CrossRef]
- Zhai, L.; Narkar, A.; Ahn, K. Self-healing polymers with nanomaterials and nanostructures. Nano Today 2020, 30, 100826. [Google Scholar] [CrossRef]
- Jian, X.; Hu, Y.; Zhou, W.; Xiao, L. Self-healing polyurethane based on disulfide bond and hydrogen bond. Polym. Adv. Technol. 2018, 29, 463–469. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, D. Self-healing polyurethane/attapulgite nanocomposites based on disulfide bonds and shape memory effect. Mater. Chem. Phys. 2017, 195, 40–48. [Google Scholar] [CrossRef]
- Chang, K.; Jia, H.; Gu, S.-Y. A transparent, highly stretchable, self-healing polyurethane based on disulfide bonds. Eur. Polym. J. 2019, 112, 822–831. [Google Scholar] [CrossRef]
- Ha, Y.-M.; Kim, Y.-O.; Ahn, S.; Lee, S.-K.; Lee, J.-S.; Park, M.; Chung, J.W.; Jung, Y.C. Robust and stretchable self-healing polyurethane based on polycarbonate diol with different soft-segment molecular weight for flexible devices. Eur. Polym. J. 2019, 118, 36–44. [Google Scholar] [CrossRef]
- Michal, B.T.; Jaye, C.A.; Spencer, E.J.; Ro, S.J. Inherently Photohealable and Thermal Shape Memory Polydisulfide Networks. ACS Macro Lett. 2013, 2, 694–699. [Google Scholar] [CrossRef]
- Lee, S.-H.; Shin, S.-R.; Lee, D.-S. Self-healing of crosslinked PU via dual-dynamic covalent bonds of a Schiff base from cystine and vanillin. Mater. Des. 2019, 172, 107774. [Google Scholar] [CrossRef]
- Dai, X.; Du, Y.; Wang, Y.; Liu, Y.; Xu, N.; Li, Y.; Shan, D.; Xu, B.B.; Kong, J. Stretchable Self-Healing Polymeric Networks with Recyclability and Dual Responsiveness. ACS Appl. Polym. Mater. 2020, 2, 1065–1072. [Google Scholar] [CrossRef]
- Rekondo, A.; Martin, R.; de Luzuriaga, A.R.; Cabanero, G.; Grande, H.J.; Odriozola, I. Catalyst-free room-temperature self-healing elastomers based on aromatic disulfide metathesis. Mater. Horiz. 2014, 1, 237–240. [Google Scholar] [CrossRef]
- Xu, W.M.; Rong, M.Z.; Zhang, M.Q. Sunlight driven self-healing, reshaping and recycling of a robust, transparent and yellowing-resistant polymer. J. Mater. Chem. A 2016, 4, 10683–10690. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, X.; Wang, W. A tough polyurethane elastomer with self-healing ability. Mater. Des. 2017, 127, 30–36. [Google Scholar] [CrossRef]
- Li, T.; Xie, Z.; Xu, J.; Weng, Y.; Guo, B.-H. Design of a self-healing cross-linked polyurea with dynamic cross-links based on disulfide bonds and hydrogen bonding. Eur. Polym. J. 2018, 107, 249–257. [Google Scholar] [CrossRef]
- Cheng, B.; Lu, X.; Zhou, J.; Qin, R.; Yang, Y. Dual Cross-Linked Self-Healing and Recyclable Epoxidized Natural Rubber Based on Multiple Reversible Effects. ACS Sustain. Chem. Eng. 2019, 7, 4443–4455. [Google Scholar] [CrossRef]
- Hu, J.; Mo, R.; Jiang, X.; Sheng, X.; Zhang, X. Towards mechanical robust yet self-healing polyurethane elastomers via combination of dynamic main chain and dangling quadruple hydrogen bonds. Polymer 2019, 183, 121912. [Google Scholar] [CrossRef]
- Li, X.; Yu, R.; He, Y.; Zhang, Y.; Yang, X.; Zhao, X.; Huang, W. Self-Healing Polyurethane Elastomers Based on a Disulfide Bond by Digital Light Processing 3D Printing. ACS Macro Lett. 2019, 8, 1511–1516. [Google Scholar] [CrossRef]
- Yao, W.; Chen, X.; Tian, Q.; Luo, C.; Zhang, X.; Peng, H.; Wu, W. Directly printing of upconversion fluorescence-responsive elastomers for self-healable optical application. Chem. Eng. J. 2020, 384, 123375. [Google Scholar] [CrossRef]
- Liu, M.; Zhong, J.; Li, Z.; Rong, J.; Yang, K.; Zhou, J.; Shen, L.; Gao, F.; Huang, X.; He, H. A high stiffness and self-healable polyurethane based on disulfide bonds and hydrogen bonding. Eur. Polym. J. 2020, 124, 109475. [Google Scholar] [CrossRef]
- Li, Y.-h.; Guo, W.-j.; Li, W.-j.; Liu, X.; Zhu, H.; Zhang, J.-p.; Liu, X.-j.; Wei, L.-h.; Sun, A.-l. Tuning hard phase towards synergistic improvement of toughness and self-healing ability of poly(urethane urea) by dual chain extenders and coordinative bonds. Chem. Eng. J. 2020, 393, 124583. [Google Scholar] [CrossRef]
- Wool, R.P.; O’Connor, K.M. A theory crack healing in polymers. J. Appl. Phys. 1981, 52, 5953–5963. [Google Scholar] [CrossRef]
- An, S.Y.; Noh, S.M.; Nam, J.H.; Oh, J.K. Dual Sulfide-Disulfide Crosslinked Networks with Rapid and Room Temperature Self-Healability. Macromol. Rapid Commun. 2015, 36, 1255–1260. [Google Scholar] [CrossRef]
- Takahashi, A.; Goseki, R.; Otsuka, H. Thermally Adjustable Dynamic Disulfide Linkages Mediated by Highly Air-Stable 2,2,6,6-Tetramethyl-piperidine-1-sulfanyl (TEMPS) Radicals. Angew. Chem. Int. Ed. 2017, 56, 2016–2021. [Google Scholar] [CrossRef] [PubMed]
- Takahasihi, A.; Goseki, R.; Ito, K.; Otsuka, H. Thermally Healable and Reprocessable Bis(hindered amin8o)disulfide-Cross-Linked Polymethacrylate Networks. ACS Macro Lett. 2017, 6, 1280–1284. [Google Scholar] [CrossRef]
- Amamoto, Y.; Otsuka, H.; Takahara, A.; Matyjaszewski, K. Self-Healing of Covalently Cross-Linked Polymers by Reshuffling Thiuram Disulfide Moieties in Air under Visible Light. Adv. Mater. 2012, 24, 3975–3980. [Google Scholar] [CrossRef] [PubMed]




































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beaupre, D.M.; Weiss, R.G. Thiol- and Disulfide-Based Stimulus-Responsive Soft Materials and Self-Assembling Systems. Molecules 2021, 26, 3332. https://doi.org/10.3390/molecules26113332
Beaupre DM, Weiss RG. Thiol- and Disulfide-Based Stimulus-Responsive Soft Materials and Self-Assembling Systems. Molecules. 2021; 26(11):3332. https://doi.org/10.3390/molecules26113332
Chicago/Turabian StyleBeaupre, Danielle M., and Richard G. Weiss. 2021. "Thiol- and Disulfide-Based Stimulus-Responsive Soft Materials and Self-Assembling Systems" Molecules 26, no. 11: 3332. https://doi.org/10.3390/molecules26113332
APA StyleBeaupre, D. M., & Weiss, R. G. (2021). Thiol- and Disulfide-Based Stimulus-Responsive Soft Materials and Self-Assembling Systems. Molecules, 26(11), 3332. https://doi.org/10.3390/molecules26113332