
Synthesis and Evaluation of the Tetracyclic Ring-System of Isocryptolepine and Regioisomers for Antimalarial, Antiproliferative and Antimicrobial Activities

 , , , and
, , , and
Abstract
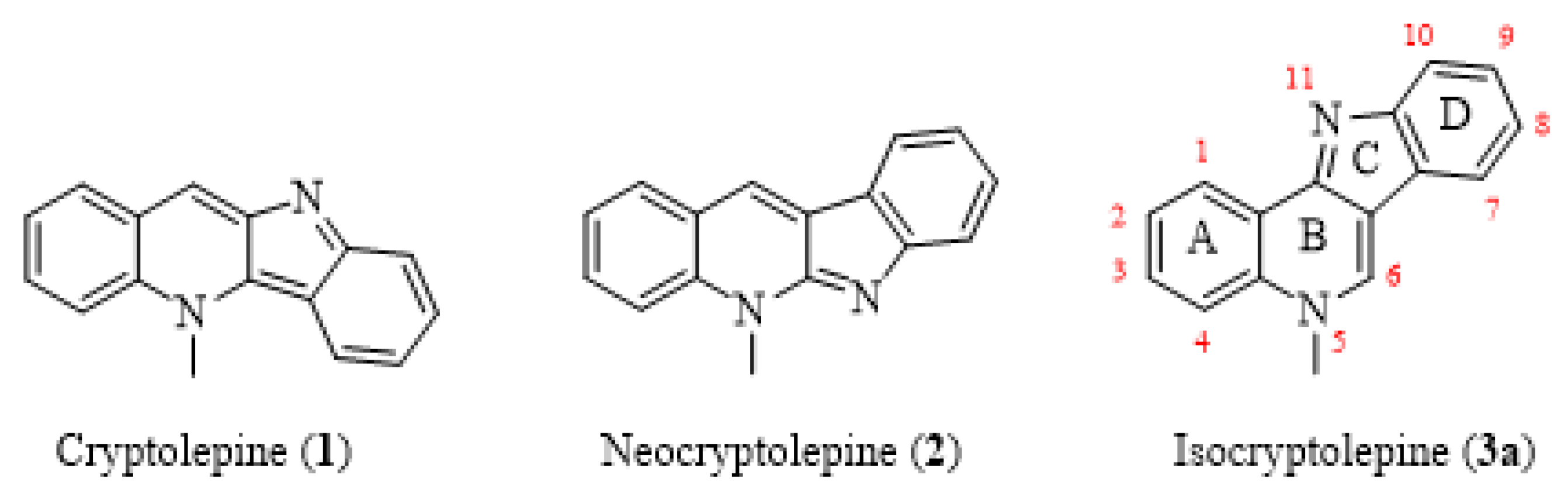
1. Introduction
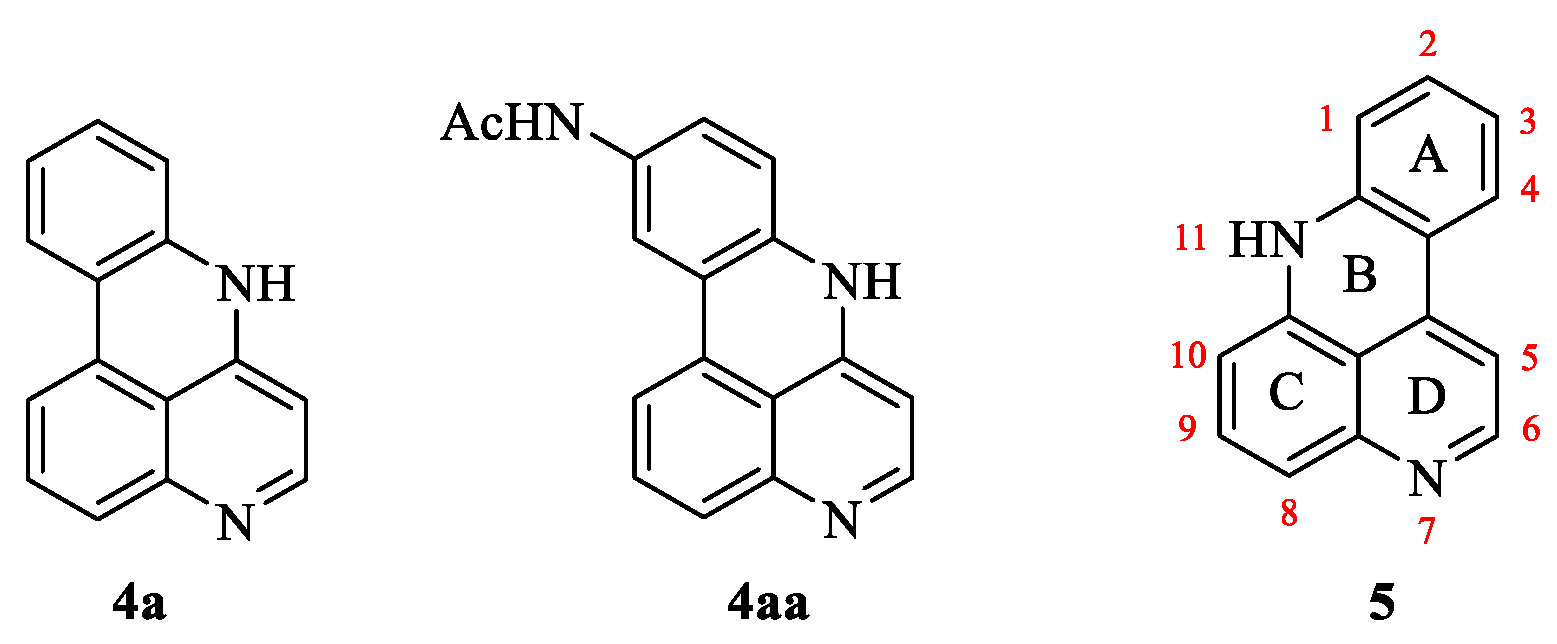
2. Results and Discussion
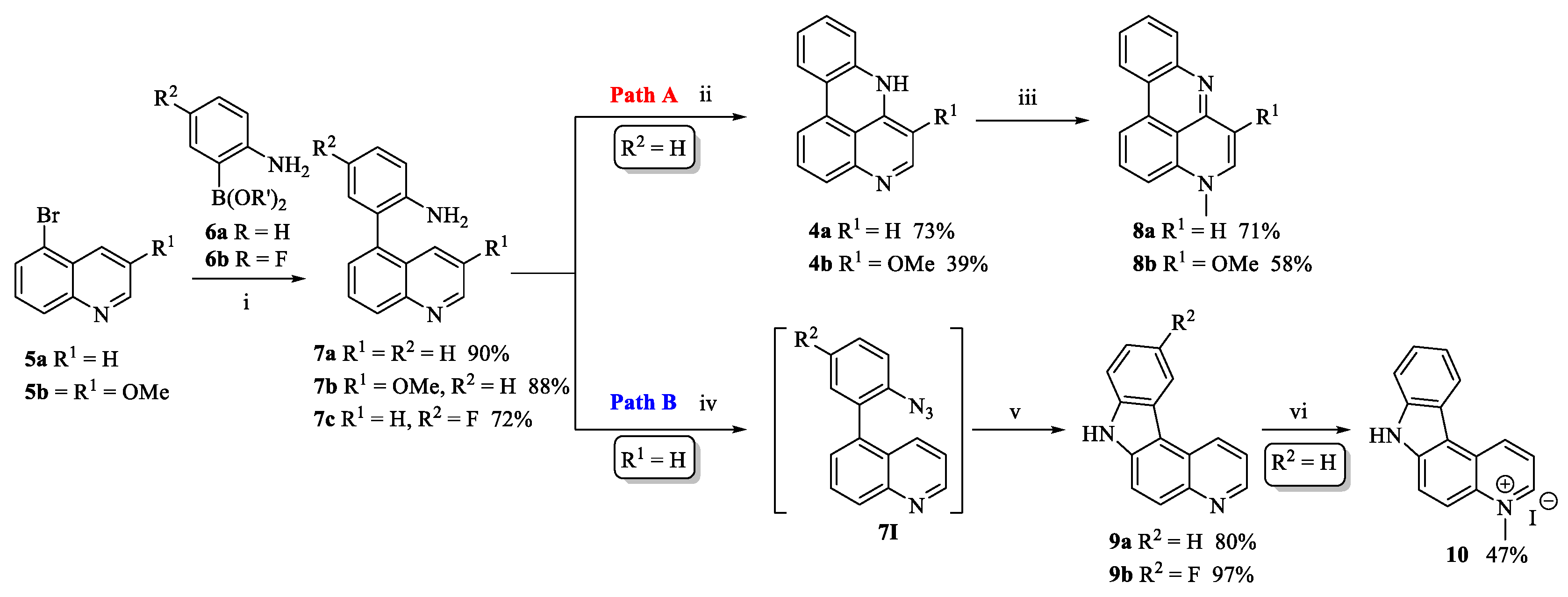
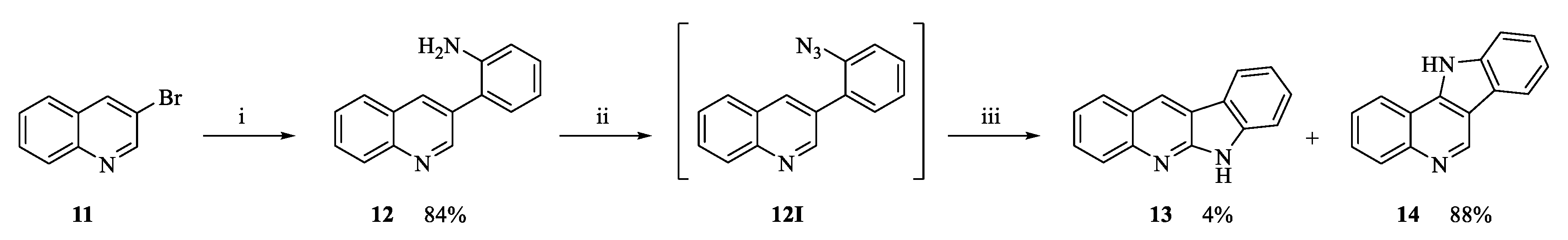
2.1. Chemistry
2.2. Antiplasmodial Assay
2.3. Antiproliferative Assay
2.4. Antimicrobial and Biofilm Iinhibition Assay
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. 4-Fluoro-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline (6b)
3.1.3. 4-Fluoro-2-(quinolin-5-yl)aniline (7c)
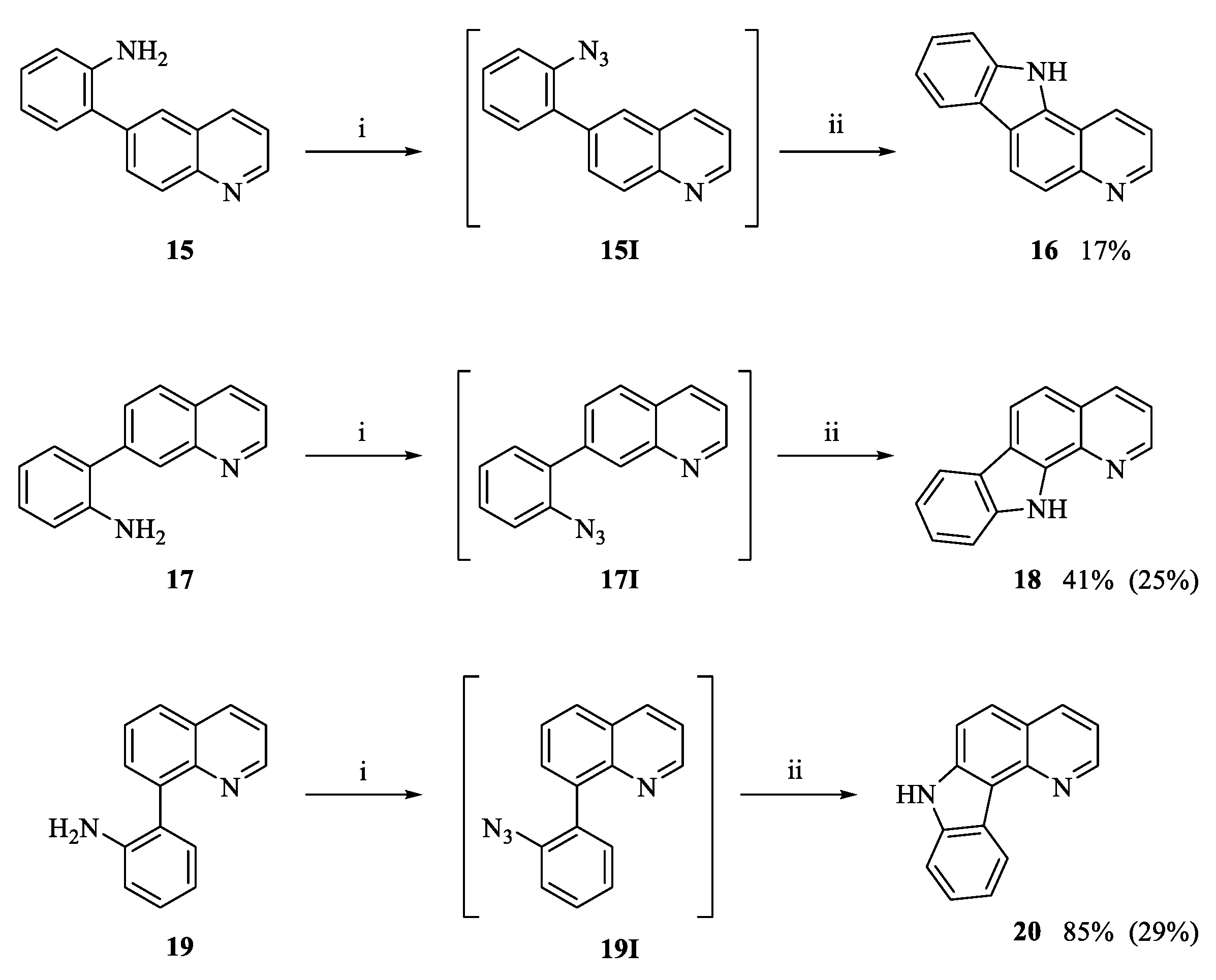
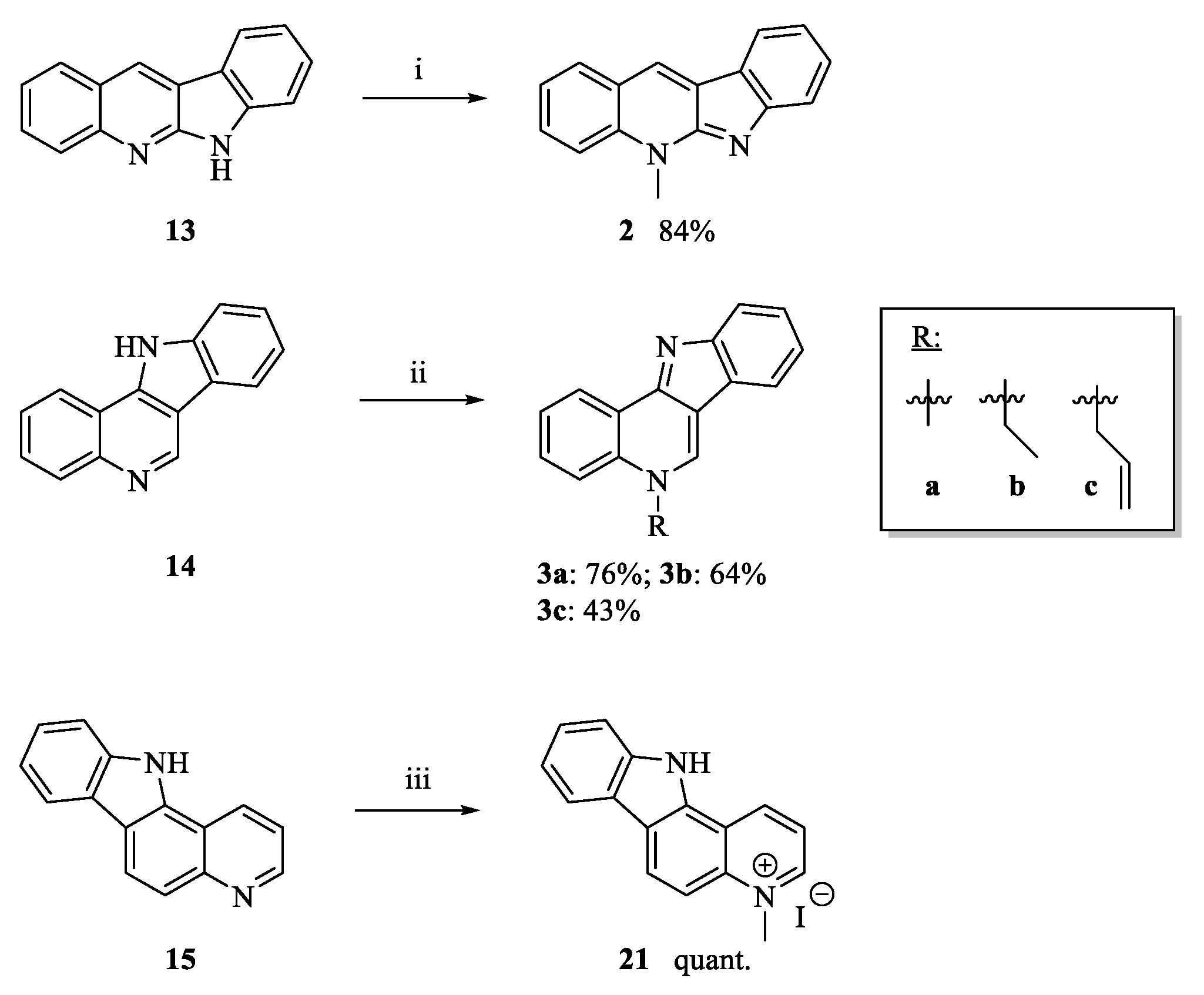
3.1.4. Intramolecular Cyclization to Form Tetracycles 9, 13, 14, 18 and 20
General Procedures
7H-Pyrido[2,3-c]carbazole (9a)
10-Fluoro-7H-pyrido[2,3-c]carbazole (9b)
6H-Indolo[2,3-b]quinoline (13) and 11H-indolo[3,2-c]quinoline (14)
Characterization of Compound 13
Characterization of Compound 14
11H-Pyrido[2,3-a]carbazole (18)
7H-Pyrido[3,2-c]carbazole (20)
3.1.5. Neocryptolepine (2)
3.1.6. 5-Ethyl-5H-indolo[3,2-c]quinoline (3b)
3.1.7. 5-Allyl-5H-indolo[3,2-c]quinoline (3c)
3.1.8. 4-Methyl-4H-pyrido[4,3,2-gh]phenanthridine (8a)
3.1.9. 6-Methoxy-4-methyl-4H-pyrido[4,3,2-gh]phenanthridine (8b)
3.1.10. 4-Methyl-7H-pyrido[2,3-c]carbazolium Iodide (10)
3.1.11. 4-Methyl-11H-pyrido[3,2-a]carbazolium Iodide (21)
3.2. Biological Testing Assay
3.2.1. General
Antiplasmodial Imaging Assay
Cytotoxicity Assay
Antiproliferative Assay
3.2.2. Growth Inhibition Assay
3.2.3. Biofilm Formation Inhibition Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization. World Malaria Report. 2020. Available online: https://www.who.int/publications/i/item/9789240015791 (accessed on 11 January 2021).
- Collins, F.H.; Paskewitz, S.M. Malaria: Current and future prospects for control. Annu. Rev. Entomol. 1995, 40, 195–219. [Google Scholar] [CrossRef]
- Achan, J.; Talisuna, A.O.; Erhart, A.; Yeka, A.; Tibenderana, J.K.; Baliraine, F.N.; Rosenthal, P.J.; D’Alessandro, U. Quinine, an old anti-malarial drug in a modern world: Role in the treatment of malaria. Malar. J. 2011, 10, 1–12. [Google Scholar] [CrossRef]
- Van Baelen, G.; Hostyn, S.; Dhooghe, L.; Tapolcsányi, P.; Mátyus, P.; Lemière, G.; Dommisse, R.; Kaiser, M.; Brun, R.; Cos, P.; et al. Structure-activity relationship of antiparasitic and cytotoxic indoloquinoline alkaloids, and their tricyclic and bicyclic analogues. Bioorg. Med. Chem. 2009, 17, 7209–7217. [Google Scholar] [CrossRef] [PubMed]
- Tse, E.G.; Korsik, M.; Todd, M.H. The past, present and future of anti-malarial medicines. Malar. J. 2019, 18, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Noedl, H.; Se, Y.; Scaecher, K.; Smith, B.L.; Socheat, D.; Fukuda, M.M. Evidence of artemisinin-resistant malaria in western Cambodia. N. Engl. J. Med. 2008, 359, 2619–2620. [Google Scholar] [CrossRef]
- Amato, R.; Pearson, R.D.; Almagro-Garcia, J.; Amaratunga, C.; Lim, P. Origins of the current outbreak of multidrug-resistant malaria in southeast Asia: A retrospective genetic study. Lancet Infect. Dis. 2018, 18, 337–345. [Google Scholar] [CrossRef]
- World Health Organization. World Cancer Report 2020: Cancer Research for Cancer Prevention. Available online: www.iarc.fr/cards_page/world-cancer-report/ (accessed on 24 August 2020).
- Sidoryk, K.; Jaromin, A.; Edward, J.A.; Świtalska, M.; Stefanska, J.; Cmoch, P.; Zagrodzka, J.; Szczepek, W.; Peczynska-Czoch, W.; Wietrzyk, J.; et al. Searching for new derivatives of neocryptolepine: A synthesis, antiproliferative, antimicrobial and antifungal activities. Eur. J. Med. Chem. 2014, 78, 304–313. [Google Scholar] [CrossRef]
- Stamm, A.M.; Long, M.N.; Belcher, B. Higher overall nosocomial infection rate because of increased attack rate of methicillin-resistant Staphylococcus aureus. Am. J. Infect. Control 1993, 21, 70–74. [Google Scholar] [CrossRef]
- Drenkard, E. Antimicrobial resistance of Preudomonas aeruginosa biofilms. Microb. Infect. 2003, 5, 1213–1219. [Google Scholar] [CrossRef]
- Teng, C.P.; Zhou, T.; Ye, E.; Liu, S.; Koh, L.D.; Low, M.; Loh, X.J.; Win, Y.; Zhang, L.; Han, M.-Y. Effective Targeted Photothermal Ablation of Multidrug Resistant Bacteria and Their Biofilms with NIR-Absorbing Gold Nanocrosses. Adv. Healthcare Mater. 2016, 5, 2122–2130. [Google Scholar] [CrossRef]
- Rizzato, C.; Torres, J.; Kasamatsu, E.; Carmorlinga-Ponce, M.; Bravo, M.M.; Canzian, F.; Kato, I. Potential role of biofilm formation in the development of digestive tract cancer with special reference to Heliobacter pylori infection. Front Microbiol. 2019, 10, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.H.; Dejea, C.M.; Edler, D.; Hoang, L.T.; Santidrian, A.F.; Felding, B.H.; Ivanisevic, J.; Cho, K.; Wick, E.C.; Hechenbleikner, E.M.; et al. Metabolism links bacterial biofilms and colon carcinogenesis. Cell Metab. 2015, 21, 891–897. [Google Scholar] [CrossRef]
- Drewes, J.L.; White, J.R.; Dejea, C.M.; Fathi, P.; Iyadorai, T.; Vadivelu, J.; Roslani, A.C.; Wick, E.C.; Mongodin, E.F.; Loke, M.F.; et al. High-resolution bacterial 16S rRNA gene profile meta-analysis and biofilm status reveal common colorectal cancer consortia. NPJ Biofilms Microbiomes 2017, 3, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H. Discovery and development of natural product-derived chemotherapeutic agents based on a medicinal chemistry approach. J. Nat. Prod. 2010, 73, 500–516. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Vuorela, P.; Leinonen, M.; Saikku, P.; Tammela, P.; Rauhad, J.-P.; Wennberg, T.; Vuorela, H. Natural products in the process of finding new drug candidates. Curr. Med. Chem. 2004, 11, 1375–1389. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Bracca, A.B.J.; Heredia, D.A.; Larghi, E.L.; Kaufman, T.S. Neocryptolepine (cryprotackieine), a unique bioactive natural product: Isolation, synthesis, and profile of its biological activity. Eur. J. Med. Chem. 2014, 2014, 7979–8003. [Google Scholar] [CrossRef]
- Madapa, S.; Tusi, Z.; Batra, S. Advances in the syntheses of quinoline and quinoline-annulated ring systems. Curr. Org. Chem. 2008, 12, 1116–1183. [Google Scholar] [CrossRef]
- Sydnes, M.O. Recent progress in the synthesis of antimalarial indoloquinoline natural products and analogues. In Studies in Natural Products Chemistry: Bioactive Natural Products; Rahman, A., Ed.; Elsevier: Karachi, Pakistan, 2020; pp. 59–84. [Google Scholar]
- Sydnes, M.O. Synthetic Strategies for the Synthesis of Indoloquinoline Natural Products. In Targets in Heterocyclic Systems; Attanasi, O.A., Merino, P., Spinelli, D., Eds.; Italian Society of Chemistry: Rome, Italy, 2019; Volume 23, pp. 201–219. [Google Scholar]
- Wang, N.; Wicht, K.J.; Imai, K.; Wang, M.-Q.; Ngoc, T.A.; Kiguchi, R.; Kaiser, M.; Egan, T.J.; Inokuchi, T. Synthesis, β-haematin inhibition, and in vitro antimalarial testing of isocryptolepine analogues: SAR study of indolo[3,2-c]quinolines with various substituents at C2, C6 and N11. Bioorg. Med. Chem. 2014, 22, 2629–2642. [Google Scholar] [CrossRef]
- Aroonkit, P.; Thongsornkleeb, C.; Tummatorn, J.; Krajangsri, S.; Mungthin, M.; Ruchirawat, S. Synthesis of isocryptolepine analogues and their structure-activity relationship studies as antiplasmodial and antiproliferative agents. Eur. J. Med. Chem. 2015, 94, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Pousset, J.L.; Martin, M.T.; Jossang, A.; Bodo, A. Isocryptolepine from Cryptolepis sanguinolenta. Phytochemistry 1995, 39, 735–736. [Google Scholar] [CrossRef]
- Sharaf, M.H.H.; Schiff, P.L.; Tackie, J.A.N.; Phoebe, C.H.; Johnson, J.R.L.; Minick, D.; Andrews, C.W.; Crouch, R.C.; Martin, G.E. The isolation and structure determination of cryptomisrine, a novel indolo[3,2-b]quinoline dimeric alkaloid from cryptolepis sanguinolenta. J. Heterocycl. Chem. 1996, 33, 789–797. [Google Scholar] [CrossRef]
- Lavrado, J.; Moreira, R.; Paulo, A. Indoloquinolines as scaffolds for drug discovery. Curr. Med. Chem. 2010, 17, 2348–2370. [Google Scholar] [CrossRef] [PubMed]
- Cimanga, K.; De Bruyne, T.; Pieters, L.; Vlietnck, A.J. In Vitro and in Vivo Antiplasmodial Activity of Cryptolepine and Related Alkaloids from Cryptolepis sanguinolenta. J. Nat. Prod. 1997, 60, 688–691. [Google Scholar] [CrossRef] [PubMed]
- Sofowora, A. Medicinal Plants and Traditional Medicine in Africa; John Wiley & Sons: Chichester, UK, 1982; pp. 183–256. [Google Scholar]
- Kirby, G.C.; Paine, A.; Warhurst, D.C.; Noamese, B.K.; Phillipson, J.D. In vitro and in vivo antimalarial activity of cryptolepine, a plant-derived indoloquinoline. Phytother. Res. 1995, 9, 359–363. [Google Scholar] [CrossRef]
- Grellier, P.; Ramiaramanana, L.; Millerioux, V.; Deharo, E.; Shrével, J.; Frappier, F.; Trigalo, F.; Bodo, B.; Pousset, J.L. Antimalarial Activity of Cryptolepine and Isocryptolepine, Alkaloids Isolated from Cryptolepis sanguinolenta. Phytother. Res. 1996, 10, 317–321. [Google Scholar] [CrossRef]
- Olajide, O.A.; Heiss, E.H.; Schachner, D.; Wright, C.W.; Vollmar, A.M.; Dirsch, V.M. Synthetic cryptolepine inhibits DNA binding of NF-κB. Bioorg. Med. Chem. 2007, 15, 43–49. [Google Scholar] [CrossRef]
- Bierer, D.E.; Fort, D.M.; Mendez, C.D.; Luo, J.; Imbach, P.A.; Dubenko, L.G.; Jolad, S.D.; Gerber, R.E.; Litvak, J.; Lu, Q.; et al. Ethnobotanical-Directed Discovery of the Antihyperglycemic Properties of Cryptolepine: Its Isolation from Cryptolepis sanguinolenta, Synthesis, and in Vitro and in Vivo Activities. J. Med. Chem. 1998, 41, 894–901. [Google Scholar] [CrossRef]
- Rauwald, H.W.; Kober, M.; Mutschler, E.; Lambrecht, G. Cryptolepis sanguinolenta: Antimuscarinic properties of cryptolepine and the alkaloid fraction at M1, M2 and M3 receptors. Planta Med. 1992, 58, 486–488. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Liu, H.; Zhang, S.-Y.; Li, H.; Ma, K.-Y.; Liu, Y.-Q.; Yin, X.-D.; Zhou, R.; Yan, Y.-F.; Wang, R.-X.; et al. Design, Synthesis, and Antifungal Evaluation of Cryptolepine Derivatives against Phytopathogenic Fungi. J. Agric. Food Chem. 2021, 69, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Ablordeppey, S.Y.; Fan, P.; Li, S.; Clark, A.M.; Hufford, C.D. Substituted indoloquinolines as new antifungal agents. Bioorg. Med. Chem. 2002, 10, 1337–1346. [Google Scholar] [CrossRef]
- Singh, M.; Singh, M.P.; Ablordeppey, S. In vitro studies with liposomal cryptolepine. Drug Dev. Ind. Pharm. 1996, 22, 377–381. [Google Scholar] [CrossRef]
- Paulo, A.; Duarte, A.; Gomes, E.T. In vitro antibacerial screening of Cryptolepis sanguinolenta alkaloids. J. Ethnopharmacol. 1994, 44, 127–130. [Google Scholar] [CrossRef]
- Cimanga, K.; De Bruyne, T.; Lasure, A.; Van Poel, B.; Pieters, L.; Claeys, M.; Berghe, D.V.; Kambu, K.; Tona, L.; Vlietinch, A. In vitro biological activities of alkaloids from Cryptolepis sanguinolenta. Planta Med. 1996, 62, 22–27. [Google Scholar] [CrossRef]
- Zhao, M.; Kamada, T.; Takeuchi, A.; Nishioka, H.; Kuroda, T.; Takeuchi, Y. Structure-activity relationship of indoloquinoline analogs anti-MRSA. Bioorg. Med. Chem. 2015, 25, 5551–5554. [Google Scholar] [CrossRef]
- Karou, D.; Savadogo, A.; Canini, A.; Yameogo, S.; Montesano, C.; Simpore, J.; Colizzi, V.; Traore, A.S. African ethnopharmacology and new drug discovery. Afr. J. Biotechnol. 2007, 5, 195–200. [Google Scholar]
- Lu, C.-M.; Chen, Y.-L.; Chen, H.-L.; Chen, C.-A.; Lu, P.-J.; Yang, C.-N.; Tzeng, C.-C. Synthesis and antiproliferative evaluation of certain indolo[3,2-c]quinoline derivatives. Bioorg. Med. Chem. 2010, 18, 1948–1957. [Google Scholar] [CrossRef]
- Dassonneville, L.; Lansiaux, A.; Wattelet, A.; Watterz, N.; Mahieu, C.; Van Miert, S.; Pieters, L.; Bailly, C. Cytotoxicity and cell cycle effects of the plant alkaloids cryptolepine and neocryptolepine: Relation to drug-induced apoptosis. Eur. J. Pharmacol. 2000, 409, 9–18. [Google Scholar] [CrossRef]
- Zhu, H.; Gooderham, N.J. Mechanisms of induction of cell cycle arrest and cell death by cryptolepine in human lung adenocarcinoma A549 cells. Toxicol. Sci. 2006, 91, 132–139. [Google Scholar] [CrossRef]
- Matsui, T.-A.; Sowa, Y.; Murata, H.; Takagi, K.; Nakanishi, R.; Aoki, S.; Yoshikawa, M.; Kobayashi, M.; Sakabe, T.; Kubo, T.; et al. The plant alkaloid cryptolepine induced p21WAF1/CIP1 and cell cycle arrest in a human osteosarcoma cell line. Int. J. Oncol. 2007, 31, 915–922. [Google Scholar] [PubMed]
- Bonjean, K.; De Pauw-Gillet, M.C.; Defresne, M.P.; Colson, P.; Houssier, C.; Dassonneville, L.; Bailly, C.; Greimers, R.; Wright, C.W.; Quetin-Leclercq, J.; et al. The DNA intercalating alkaloid cryptolepine interferes with topoisomerase II and inhibits primarily DNA synthesis in B16 melanoma cells. Biochemistry 1998, 37, 5136–5146. [Google Scholar] [CrossRef]
- Dassonneville, L.; Bonjean, K.; De Pauw-Gillet, M.C.; Colson, P.; Houssier, C.; Quetin-Leclercq, J.; Angenot, L.; Bailly, C. Stimulation of topoisomerase II-mediated DNA cleavage by three DNA-intercalating plant alkaloids: Cryptolepine, matadine and serpentine. Biochemistry 1999, 38, 7719–7726. [Google Scholar] [CrossRef]
- Whittel, L.R.; Batty, K.T.; Wong, R.P.M.; Bolitho, E.M.; Fox, S.A.; Davis, T.M.E.; Murray, P.E. Synthesis and antimalarial evaluation of novel isocryptolepine derivatives. Bioorg. Med. Chem. 2011, 19, 7519–7525. [Google Scholar] [CrossRef] [PubMed]
- Van Miert, S.; Hostyn, S.; Maes, B.U.W.; Cimanga, K.; Brun, R.; Kaiser, M.; Mátyus, P.; Dommisse, R.A.; Lemière, G.; Vlietinck, A.; et al. Isoneocryptolepine, a synthetic indoloquinoline alkaloid, as an antiplasmodial lead compound. J. Nat. Prod. 2005, 68, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C.; Laine, W.; Baldeyrou, B.; De Pauw-Gillet, M.C.; Colson, P.; Houssier, C.; Cimanga, K.; Van Miert, S.; Vlietinck, A.J.; Pieters, L. DNA intercalation, topoisomerase II inhibition and cytotoxic activity of the plant alkaloid neocryptolepine. Anti-Cancer Drug Des. 2000, 15, 191–201. [Google Scholar]
- Håheim, K.S.; Helgeland, I.T.U.; Lindbäck, E.; Sydnes, M.O. Mapping the reactivity of the quinoline ring-systems—Synthesis of the tetracyclic ring-system of isocryptolepine and regioisomers. Tetrahedron 2019, 75, 2924–2957. [Google Scholar] [CrossRef]
- Schmitz, F.J.; Deguzman, F.S.; Hossain, M.B.; Vanderhelm, D. Cytotoxic aromatic alkaloids from the ascidian Amphicarpa meridiana and Leptoclinides sp.: Meridine and 11-hydroxyascididemin. J. Org. Chem. 1991, 56, 804–808. [Google Scholar] [CrossRef]
- Gunawardana, G.P.; Koehn, F.E.; Lee, A.Y.; Clardy, J.; Hee, H.Y.; Faulkner, D.J. Pyridoacridine alkaloids from deep-water marine sponges of the family Pacchastrellidae: Structure revision of dercitin and related compounds and correlation with the kuanoniamines. J. Org. Chem. 1992, 57, 1523–1526. [Google Scholar] [CrossRef]
- Molinski, T.F. Marine pyridoacridine alkaloids: Structure, synthesis, and biological chemistry. Chem. Rev. 1993, 93, 1825–1838. [Google Scholar] [CrossRef]
- Eder, C.; Schupp, P.; Proksch, P.; Wray, V.; Steube, K.; Müller, C.E.; Frobenius, W.; Herderich, M.; van Soest, R.W.M. Bioactive Pyridoacridine Alkaloids from the Micronesian Sponge Oceanapia sp. J. Nat. Prod. 1998, 61, 301–305. [Google Scholar] [CrossRef]
- Marshall, K.M.; Barrows, L.R. Biological activities of pyridoacridines. Nat. Prod. Rep. 2004, 61, 731–751. [Google Scholar] [CrossRef] [PubMed]
- Delfourne, E.; Bastide, J. Marine pyridoacridine alkaloids and synthetic analogues as antitumor agents. Med. Res. Rev. 2003, 23, 234–252. [Google Scholar] [CrossRef] [PubMed]
- De Guzman, F.S.; Carte, B.; Troupe, N.; Faulkner, D.J.; Harper, M.K.; Concepcion, G.P.; Mangalindan, G.C.; Matsumoto, S.S.; Barrows, L.R.; Ireland, C.M. Neoamphimedine: A new pyridoacridine topoisomerase II inhibitor which catenates DNA. J. Org. Chem. 1999, 64, 1400–1402. [Google Scholar] [CrossRef]
- Feng, Y.R.; Davis, A.; Sykes, M.L.; Avery, V.M.; Carroll, A.R.; Camp, D.; Quinn, R.J. Antitrypanosomal pyridoacridine alkaloids from the Australian ascidian Polysyncraton echinatum. Tetrahedron Lett. 2010, 51, 2477–2479. [Google Scholar] [CrossRef]
- Fuente, J.A.D.L.; Martin, M.J.; Blanco, M.D.M.; Alfonso, E.P.; Avendano, C.; Mendez, J.C. A C-Ring regioisomer of the marine alkaloid meridine exhibits selective in vitro cytotoxicity for solid tumors. Bioorg. Med. Chem. 2001, 9, 1807–1814. [Google Scholar] [CrossRef]
- Helgeland, I.T.U.; Sydnes, M.O. A Concise Synthesis of Isocryptolepine by C-C Cross-Coupling Followed by a Tandem C-H Activation and C-N Bond Formation. SynOpen 2017, 1, 41–44. [Google Scholar] [CrossRef][Green Version]
- Mehra, M.K.; Sharma, S.; Rangan, K.; Kumar, D. Substrate of Solvent-Controlled PdII-Catalyzed Regioselective Arylation of Quinolin-4(1H)-ones Using Diaryliodonium Salts: Facile Access to Benzoxocine and Aaptamine Analogues. Eur. J. Org. Chem. 2020, 2020, 2409–2413. [Google Scholar] [CrossRef]
- Beauchard, A.; Chabane, H.; Sinbandhit, S.; Guenot, P.; Thiéry, V.; Besson, T. Synthesis of original thiazoloindolo[3,2-c]quinoline and novel 8-N-substituted-11H-indolo[3,2-c]quinoline derivatives from benzotriazoles. Part I. Tetrahedron 2006, 62, 1895–1903. [Google Scholar] [CrossRef]
- Timári, G.; Soós, T.; Hajós, G. A convenient synthesis of two new indoloquinoline alkaloids. Synlett 1997, 1997, 1067–1068. [Google Scholar] [CrossRef]
- Hostyn, S.; Maes, B.U.W.; Pieters, L.; Lemière, G.L.F.; Mátyus, P.; Hajós, G.; Dommisse, R.A. Synthesis of the benzo-β-carboline isoneocryptolepine: The missing indoloquinoline isomer in the alkaloid series cryptolepine, neocryptolepine and isocryptolepine. Tetrahedron 2005, 61, 1571–1577. [Google Scholar] [CrossRef]
- Miller, M.; Vogel, J.C.; Tsang, W.; Merrit, A.; Procter, D.J. Formation of N-heterocycles by the reaction of thiols with glyoxamides: Exploring a connective Pummerer-type cyclisation. Org. Biomol. Chem. 2009, 7, 589–597. [Google Scholar] [CrossRef]
- Jonckers, T.H.M.; van Miert, S.; Cimanga, K.; Bailly, C.; Colson, P.; De Pauw-Gillet, M.C.; van den Heuvel, H.; Claeys, M.; Lemière, F.; Esmans, E.L.; et al. Synthesis, cytotoxicity, and antiplasmodial and antitrypanosomal activity of new neocryptolepine derivatives. J. Med. Chem. 2002, 45, 3497–3508. [Google Scholar] [CrossRef]
- Go, M.L.; Koh, H.L.; Ngiam, T.L.; Phillipson, J.D.; Kirby, G.C.; Oneill, M.J.; Warhurst, D.C. Synthesis and in vitro antimalarial activity of some indolo[3,2-c]quinolines. Eur. J. Med. Chem. 1992, 27, 391–394. [Google Scholar] [CrossRef]
- Ansah, C.; Gooderham, N.J. The Popular Herbal Antimalarial, Extract of Cryptolepis sanguinolenta, Is Potently Cytotoxic. Toxicol. Sci. 2002, 70, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.-J.; Switalska, M.; Wang, L.; Yonezawa, M.; El-Sayed, I.E.-T.; Wietrzyk, J.; Inokuchi, T. In vitro antiproliferative activity of 11-aminoalkylamino-substituted 5H-indolo[2,3-b]quinolines; improving activity of neocryptolepines by installation of ester substituent. Med. Chem. Res. 2013, 22, 4492–4504. [Google Scholar] [CrossRef]
- Iorio, F.; Bosotti, R.; Scacheri, E.; Belcastro, V.; Mithbaokar, P.; Ferriro, R.; Murino, L.; Tagliaferri, R.; Brunetti-Pierri, N.; Isacchi, A.; et al. Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc. Natl. Acad. Sci. USA 2012, 107, 14621–14626. [Google Scholar] [CrossRef]
- Peczynska-Czoch, W.; Pognan, F.; Kaczmarek, L.; Boratynski, J. Synthesis and structure-activity relationship of methyl-substituted indolo[2,3-b]quinolines: Novel cytotoxic, DNA topoisomerase II inhibitors. J. Med. Chem. 1994, 37, 3503–3510. [Google Scholar] [CrossRef]
- Cimanga, K.; De Bruyne, T.; Pieters, L.; Totte, J.; Tona, L.; Kambu, K.; Vanden Berghe, D.; Vlietinck, A.J. Antibacterial and antifungal activiies of neocryptolepine, biscryptolepine and cryptoquindoline, alkaloids isolated from Cryptolepis sanguinolenta. Phytomedicine 1998, 5, 209–214. [Google Scholar] [CrossRef]
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzyme Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef]
- Miller, C.M.; McCarthy, F.O. Isolation, biological activity and synthesis of the natural product ellipticine and related pyridocarbazoles. RSC Adv. 2012, 2, 8883–8918. [Google Scholar] [CrossRef]
- Rosenau, C.P.; Jelier, B.J.; Gossert, A.D.; Togni, A. Exposing the origins of irreproducibility in fluorine NMR spectroscopy. Angew. Chem. Int. Ed. 2018, 57, 9528–9533. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Driver, T.G. Efficient Synthesis of 3H-Indoles Enables by the Lead-Mediated α-Arylation of β-Ketoesters or γ-Lactams Using Aryl Azides. Org. Lett. 2014, 16, 2916–2919. [Google Scholar] [CrossRef]
- Alajarin, M.; Molina, P.; Vidal, A. Formal total synthesis of the alkaloid cryptotackieine (neocryptolepine). J. Nat. Prod. 1997, 60, 747–748. [Google Scholar] [CrossRef]
- Duffy, S.A.; Avery, V.M. Development and optimization of a novel 384-well anti-malarial imaging assay validated for high-throughput screening. Am. J. Trop. Med. Hyg. 2012, 86, 84–92. [Google Scholar] [CrossRef]
- Fletcher, S.; Avery, V.M. A novel approach for the discovery of chemically diverse anti-malarial compounds targeting the Plasmodium falciparum Coenzyme A synthesis pathway. Malar. J. 2014, 13, 343. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | 3D7 IC50 (nM) | Cytotoxicity IC50 (nM) | SI a |
|---|---|---|---|---|
| 1 | Neocryptolepine (2) | 7249 ± 6 | >20,000 | 2.8 |
| 2 | Isocryptolepine (3a) | 1211 ± 84 | 2074 ± 70 | 1.7 |
| 3 | 3b | 1318 ± 5 | 3078 ± 49 | 2.3 |
| 4 | 3c | 1198 ± 32 | 3152 ± 40 | 2.6 |
| 5 | 4a | 548 ± 3 | 2834 ± 92 | 5.2 |
| 6 | 4b | 866 ± 2 | 3657 ± 2 | 4.2 |
| 7 | 8a | 1698 ± 5 | 7410 ± 207 | 4.4 |
| 8 | 8b | 1546 ± 27 | 5057 ± 45 | 3.3 |
| 9 | 9a | 6825 ± 61 | >80,000 | 11.7 |
| 10 | 9b | NT b | NT b | - |
| 11 | 10 | 128 ± 2 | NA c | 213.9 |
| 12 | 13 | NA c | NA c | - |
| 13 | 14 | 977 ± 11 | 18460 ± 183 | 18.9 |
| 14 | 16 | NA c | NA c | - |
| 15 | 18 | NA c | NA c | - |
| 16 | 20 | 2414 ± 42 | NA c | 16.6 |
| 17 | 21 | 380 ± 0.5 | NA c | 105.4 |
| 18 | Chloroquine | 24 ± 1 | >4000 | 165 |
| 19 | DHA | 1 ± 0.07 | NA c | 74 |
| 20 | Puromycin | 93 ± 2 | 3 ± 3 | 0.03 |
| Entry | Compound | HCT116 IC50 (nM) | MDA-MB-231 IC50 (nM) | PC-3 IC50 (nM) |
|---|---|---|---|---|
| 13 | Neocryptolepine (2) | 6218 ± 90 | 10,435 ± 375 | 27% at 80 µM |
| 14 | Isocryptolepine (3a) | 667 ± 45 | 695 ± 130 | 1821 ± 7 |
| 15 | 3b | 742 ± 11 | 998 ± 300 | 2440 ± 94 |
| 16 | 3c | 1243 ± 80 | 3064 ± 467 | 1296 ± 51 |
| 1 | 4a | 721 ± 27 | 594 ± 140 | 1630 ± 173 |
| 2 | 4b | 166 ± 16 a | 1002 ± 297 | 24 ± 3 b |
| 3 | 8a | 444 ± 52 | 360 ± 51 | 2571 ± 114 |
| 4 | 8b | 871 ± 172 | 814 ± 162 | 4539 ± 361 |
| 5 | 9a | 20,015 ± 1665 | 21,540 ± 2480 | 17,790 ± 1640 |
| 6 | 9b | NT c | NT c | NT c |
| 7 | 10 | 38% at 40 µM | 24% at 40 µM | 36% at 40 µM |
| 8 | 13 | NA d | NA d | NA d |
| 9 | 14 | 3573 ± 309 | 36% at 80 µM e | 30% at 80 µM f |
| 10 | 16 | 82% at 80 µM | 80% at 80 µM | NA d |
| 11 | 18 | NT c | NT c | NT c |
| 12 | 20 | 17,030 g | 16,415 ± 2305 | 47% at 40 µM |
| 17 | 21 | NA d | NA d | NA d |
| 18 | Puromycin | 85 | 300 | 270 |
| 19 | Doxorubicin | 150 | 590 | 830 |
| Tested Strain | MIC (µM) | |||||||
|---|---|---|---|---|---|---|---|---|
| 2 | 3a | 4a a | 4b a | 8a a | 9a | 9b | Gentamycin | |
| E. faecalis (ATCC 29122) | NA b | 100 | 100 | NA b | 75 | NA b | NA b | 8 |
| E. coli (ATCC 259233) | NA b | 100 | NA b | 50 | NA b | NA b | NA b | 0.13 |
| P. aeruginosa (ATCC 27853) | NA b | NA b | NA b | NA b | NA b | NA b | NA b | 0.25 |
| S. aureus (ATCC 25923) | NA b | 100 | 100 | NA b | 75 | NA b | NA b | 0.06 |
| Streptococcus agalactiae (ATCC 12386) | 100 | 100 | NA b | 75 | NA b | 100 | NA b | 4 |
| MBIC (µM) | ||||||||
| S. epidermis (ATCC 35984) | NA b | 100 | NA b | NA b | NA b | 100 | 100 | NT c |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Håheim, K.S.; Lindbäck, E.; Tan, K.N.; Albrigtsen, M.; Urdal Helgeland, I.T.; Lauga, C.; Matringe, T.; Kennedy, E.K.; Andersen, J.H.; Avery, V.M.; et al. Synthesis and Evaluation of the Tetracyclic Ring-System of Isocryptolepine and Regioisomers for Antimalarial, Antiproliferative and Antimicrobial Activities. Molecules 2021, 26, 3268. https://doi.org/10.3390/molecules26113268
Håheim KS, Lindbäck E, Tan KN, Albrigtsen M, Urdal Helgeland IT, Lauga C, Matringe T, Kennedy EK, Andersen JH, Avery VM, et al. Synthesis and Evaluation of the Tetracyclic Ring-System of Isocryptolepine and Regioisomers for Antimalarial, Antiproliferative and Antimicrobial Activities. Molecules. 2021; 26(11):3268. https://doi.org/10.3390/molecules26113268
Chicago/Turabian StyleHåheim, Katja S., Emil Lindbäck, Kah Ni Tan, Marte Albrigtsen, Ida T. Urdal Helgeland, Clémence Lauga, Théodora Matringe, Emily K. Kennedy, Jeanette H. Andersen, Vicky M. Avery, and et al. 2021. "Synthesis and Evaluation of the Tetracyclic Ring-System of Isocryptolepine and Regioisomers for Antimalarial, Antiproliferative and Antimicrobial Activities" Molecules 26, no. 11: 3268. https://doi.org/10.3390/molecules26113268
APA StyleHåheim, K. S., Lindbäck, E., Tan, K. N., Albrigtsen, M., Urdal Helgeland, I. T., Lauga, C., Matringe, T., Kennedy, E. K., Andersen, J. H., Avery, V. M., & Sydnes, M. O. (2021). Synthesis and Evaluation of the Tetracyclic Ring-System of Isocryptolepine and Regioisomers for Antimalarial, Antiproliferative and Antimicrobial Activities. Molecules, 26(11), 3268. https://doi.org/10.3390/molecules26113268