
Antinociceptive Efficacy of the µ-Opioid/Nociceptin Peptide-Based Hybrid KGNOP1 in Inflammatory Pain without Rewarding Effects in Mice: An Experimental Assessment and Molecular Docking

 ,
,  and
and 
Abstract
1. Introduction
2. Results
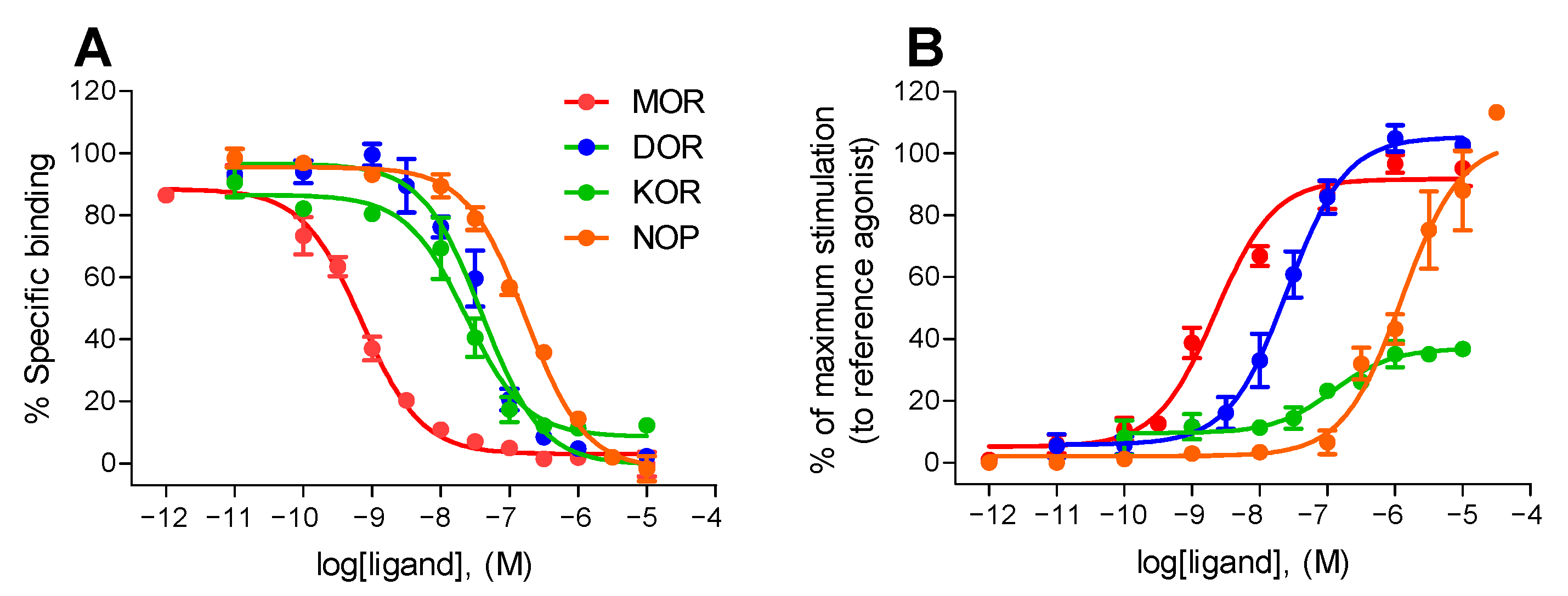
2.1. Pharmacological Properties of KGNOP1 In Vitro
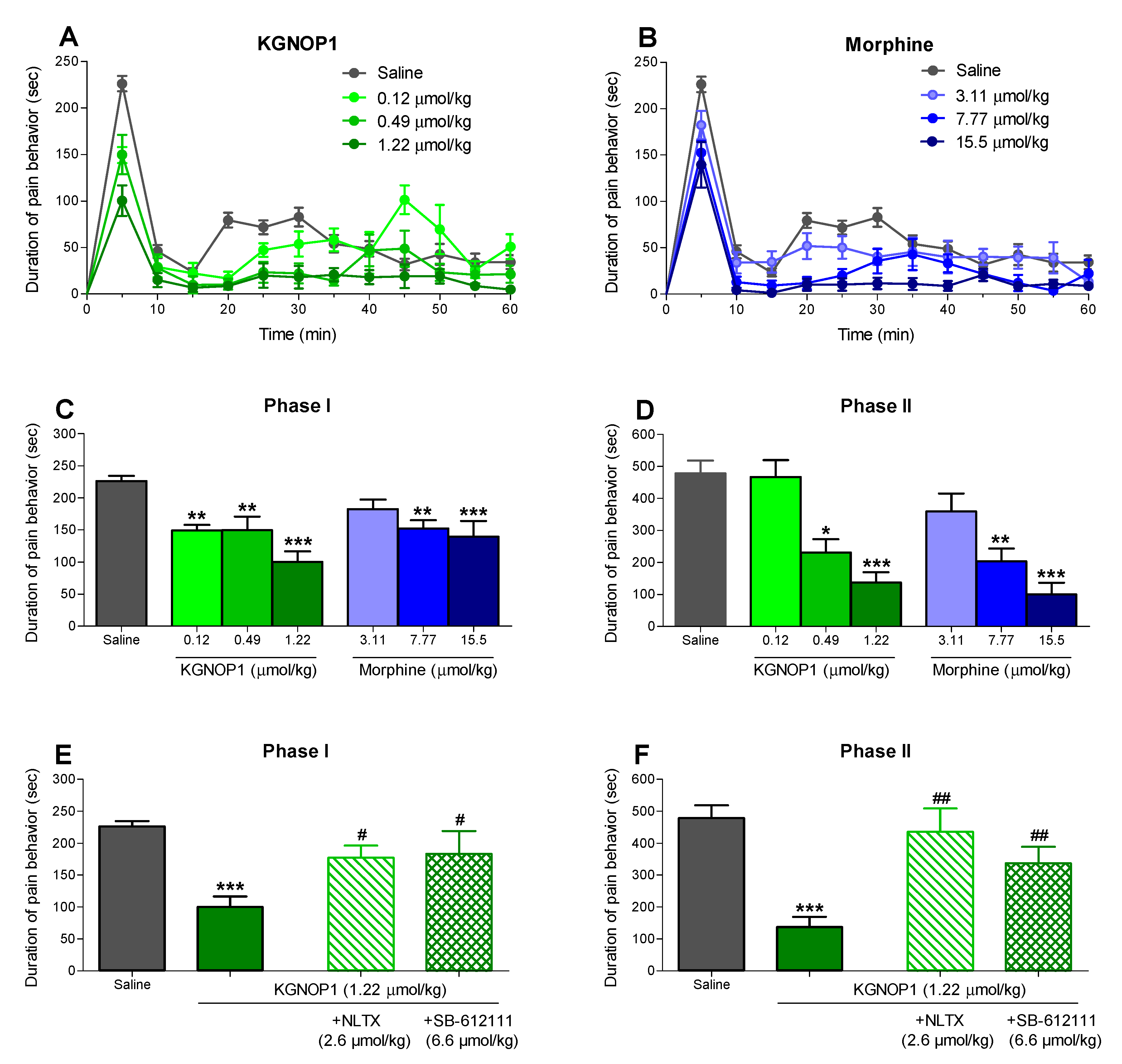
2.2. KGNOP1 Significantly Attenuates Pain Behavior in the Mouse Formalin Test
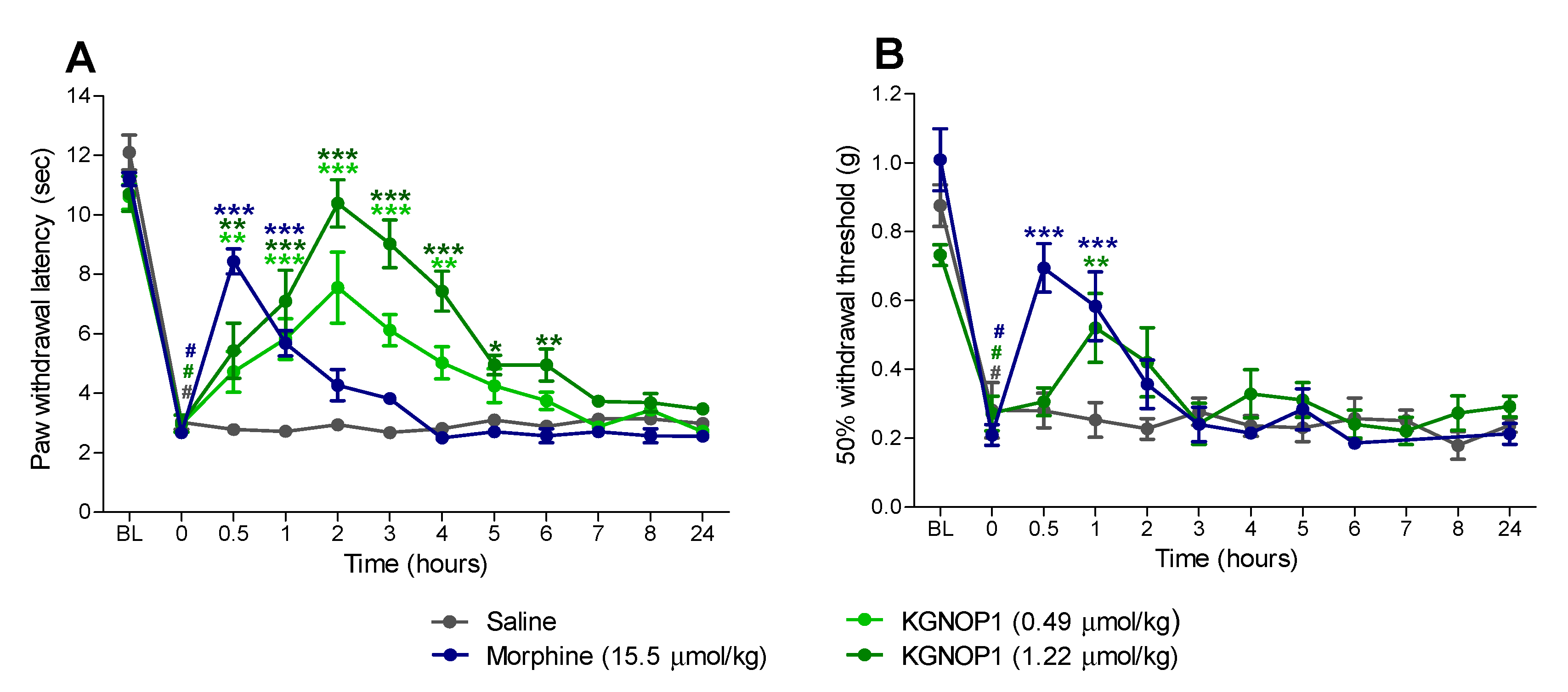
2.3. KGNOP1 Efficiently Reverses Hyperalgesia in Mice with Complete Freund’s Adjuvant-Induced Chronic Inflammatory Pain
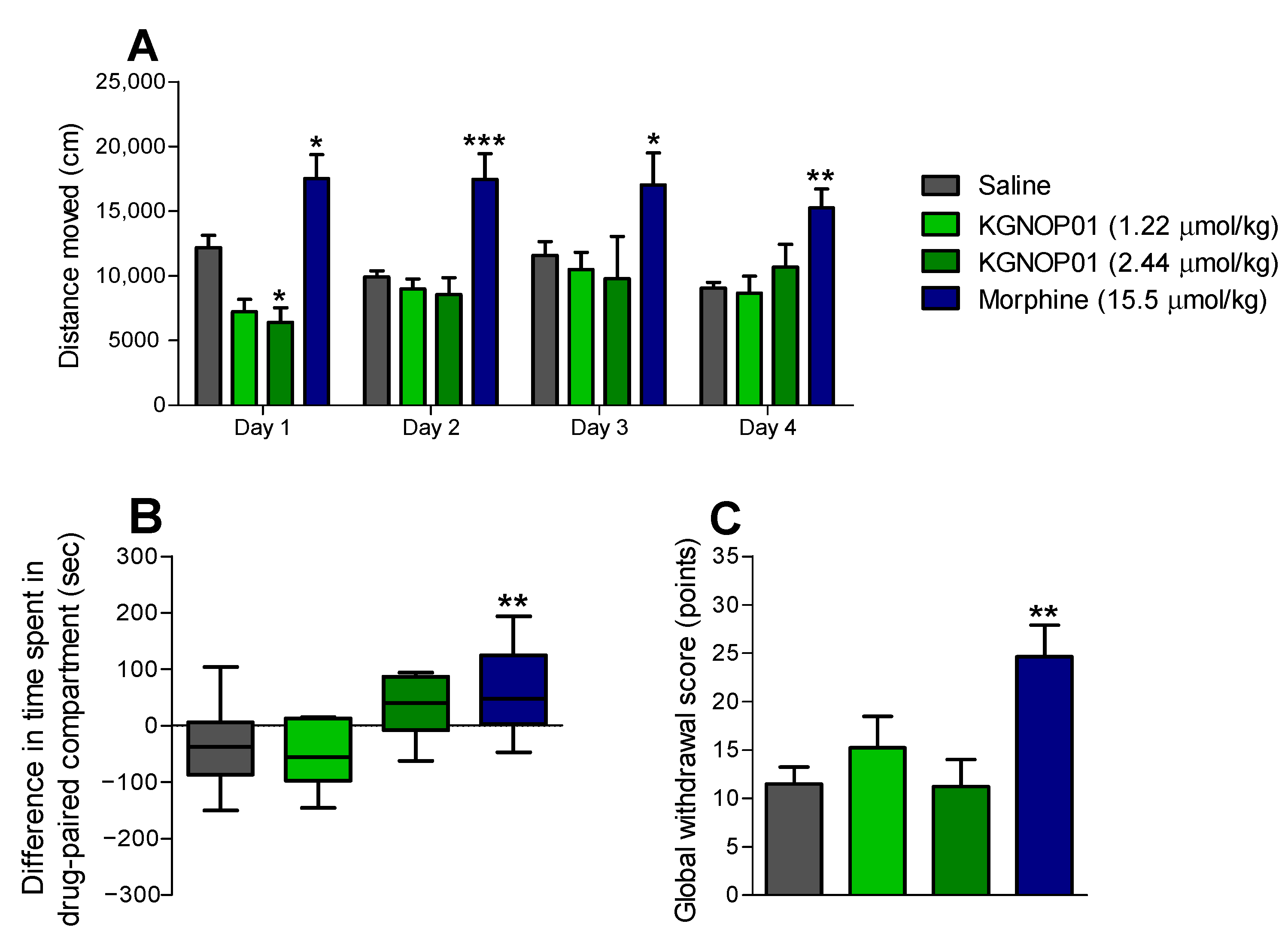
2.4. Chronic Administration of KGNOP1 Does Not Affect Spontaneous Locomotor Activity of Mice
2.5. KGNOP1 Does Not Induce Rewarding Effects in Mice
2.6. Chronic Administration of KGNOP1 Does Not Induce Withdrawal Syndrome in Mice
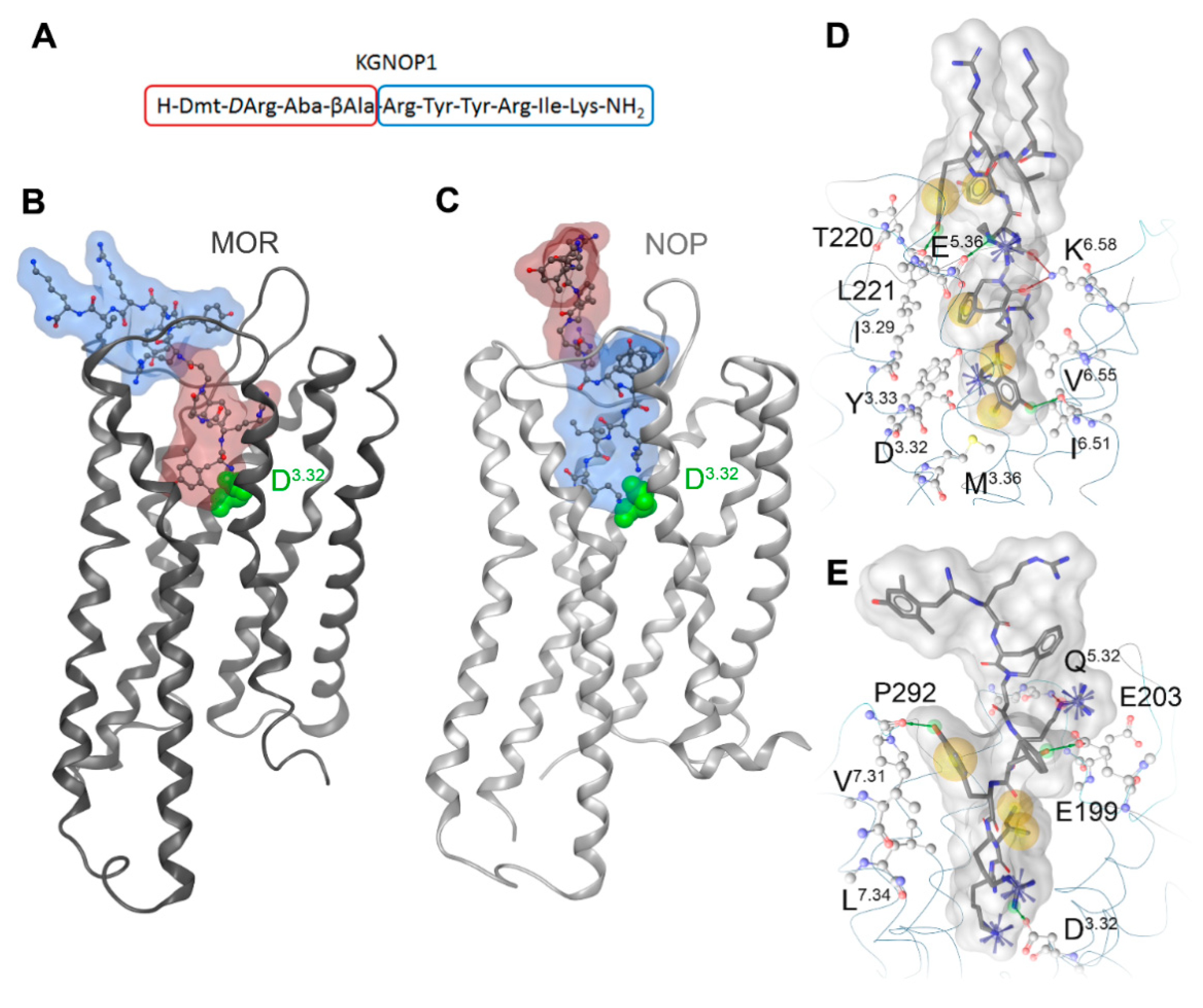
2.7. Molecular Docking of KGNOP1 to the MOR and NOP Receptors
3. Discussion
4. Materials and Methods
4.1. Drugs and Chemicals
4.2. Cell Cultures and Membrane Preparation
4.3. Competitive Radioligand Binding Assays for Opioid Receptors
4.4. [35S]GTPγS Binding Assays for Opioid Receptors
4.5. Animals and Drug Administration
4.6. Formalin-Induced Acute Inflammatory Pain
4.7. Complete Freund’s Adjuvant (CFA)-Induced Chronic Inflammatory Pain
4.8. Open-Field Test
4.9. Conditioned Place Preference
4.10. Naloxone-Precipitated Withdrawal Syndrome
4.11. Molecular Modeling
4.12. Data and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Pasternak, G.W. Mu opioid pharmacology: 40 years to the promised land. Adv. Pharmacol. 2018, 82, 261–291. [Google Scholar] [CrossRef]
- Pasternak, G.W.; Childers, S.R.; Pan, Y.-X. Emerging insights into mu opioid pharmacology. Handb. Exp. Pharmacol. 2019, 258, 89–125. [Google Scholar] [CrossRef]
- Volkow, N.D.; Blanco, C. The changing opioid crisis: Development, challenges and opportunities. Mol. Psychiatry 2021, 26, 218–233. [Google Scholar] [CrossRef]
- Corder, G.; Castro, D.C.; Bruchas, M.R.; Scherrer, G. Endogenous and exogenous opioids in pain. Ann. Rev. Neurosci. 2018, 41, 453–473. [Google Scholar] [CrossRef]
- Darcq, E.; Kieffer, B.L. Opioid receptors: Drivers to addiction? Nat. Rev. Neurosci. 2018, 19, 499–514. [Google Scholar] [CrossRef]
- Azzam, A.A.; McDonald, J.; Lambert, D.G. Hot topics in opioid pharmacology: Mixed and biased opioids. Br. J. Anaesth. 2019, 122, e136–e145. [Google Scholar] [CrossRef]
- Faouzi, A.; Varga, B.R.; Majumdar, S. Biased opioid ligands. Molecules 2020, 25, 4257. [Google Scholar] [CrossRef]
- Kleczkowska, P.; Lipkowski, A.W.; Tourwe, D.; Ballet, S. Hybrid opioid/non-opioid ligands in pain research. Curr. Pharm. Des. 2013, 19, 7435–7450. [Google Scholar] [CrossRef]
- Machelska, H.; Celik, M.Ö. Advances in achieving opioid analgesia without side effects. Front. Pharmacol. 2018, 9, 1388. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.; Kopf, A. Pain therapy—Are there new options on the horizon? Best Pract. Res. Clin. Rheumatol. 2019, 33, 101420. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C. Structural insights into µ-opioid receptor activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H. Structure of the µ-opioid receptor-G i protein complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Ballet, S.; Pietsch, M.; Abell, A.D. Multiple ligands in opioid research. Protein Peptide Lett. 2008, 15, 668–682. [Google Scholar] [CrossRef] [PubMed]
- Günther, T.; Dasgupta, P.; Mann, A.; Miess, E.; Kliewer, A.; Fritzwanker, S.; Steinborn, R.; Schulz, S. Targeting multiple opioid receptors-improved analgesics with reduced side effects? Br. J. Pharmacol. 2018, 175, 2857–2868. [Google Scholar] [CrossRef] [PubMed]
- Hruby, V.J. Multivalent peptide and peptidomimetic ligands for the treatment of pain without toxicities and addiction. Peptides 2019, 116, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Schiller, P.W. Opioid peptide-derived analgesics. In Drug Addiction; Springer: Berlin/Heidelberg, Germany, 2008; pp. 357–366. [Google Scholar]
- Turnaturi, R.; Arico, G.; Ronsisvalle, G.; Parenti, C.; Pasquinucci, L. Multitarget opioid ligands in pain relief: New players in an old game. Eur. J. Med. Chem. 2016, 108, 211–228. [Google Scholar] [CrossRef]
- Anand, J.P.; Montgomery, D. Multifunctional opioid ligands. In Delta Opioid Receptor Pharmacology and Therapeutic Applications; Springer: Berlin/Heidelberg, Germany, 2018; pp. 21–51. [Google Scholar] [CrossRef]
- Cunningham, C.W.; Elballa, W.M.; Vold, S.U. Bifunctional opioid receptor ligands as novel analgesics. Neuropharmacology 2019, 151, 195–207. [Google Scholar] [CrossRef] [PubMed]
- La Rochelle, A.D.; Guillemyn, K.; Dumitrascuta, M.; Martin, C.; Utard, V.; Quillet, R.; Schneider, S.; Daubeuf, F.; Willemse, T.; Mampuys, P.; et al. A bifunctional-biased mu-opioid agonist-neuropeptide FF receptor antagonist as analgesic with improved acute and chronic side effects. Pain 2018, 159, 1705–1718. [Google Scholar] [CrossRef]
- Guillemyn, K.; Starnowska, J.; Lagard, C.; Dyniewicz, J.; Rojewska, E.; Mika, J.; Chung, N.N.; Utard, V.; Kosson, P.; Lipkowski, A.W.; et al. Bifunctional peptide-based opioid agonist-nociceptin antagonist ligands for dual treatment of acute and neuropathic pain. J. Med. Chem. 2016, 59, 3777–3792. [Google Scholar] [CrossRef]
- Lagard, C.; Chevillard, L.; Guillemyn, K.; Risède, P.; Laplanche, J.-L.; Spetea, M.; Ballet, S.; Mégarbane, B. Bifunctional peptide-based opioid agonist/nociceptin antagonist ligand for dual treatment of nociceptive and neuropathic pain. Pain 2017, 158, 505. [Google Scholar] [CrossRef]
- Starnowska, J.; Guillemyn, K.; Makuch, W.; Mika, J.; Ballet, S.; Przewlocka, B. Bifunctional opioid/nociceptin hybrid KGNOP1 effectively attenuates pain-related behaviour in a rat model of neuropathy. Eur. J. Pham. Sci. 2017, 104, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.L.; Thompson, A.A.; Trapella, C.; Guerrini, R.; Malfacini, D.; Patel, N.; Han, G.W.; Cherezov, V.; Caló, G.; Katritch, V. The importance of ligand-receptor conformational pairs in stabilization: Spotlight on the N/OFQ G protein-coupled receptor. Structure 2015, 23, 2291–2299. [Google Scholar] [CrossRef]
- Dumitrascuta, M.; Bermudez, M.; Haddou, T.B.; Guerrieri, E.; Schläfer, L.; Ritsch, A.; Hosztafi, S.; Lantero, A.; Kreutz, C.; Massotte, D.; et al. N-Phenethyl substitution in 14-methoxy-N-methylmorphinan-6-ones turns selective µ opioid receptor ligands into dual µ/δ opioid receptor agonists. Sci. Rep. 2020, 10, 5653. [Google Scholar] [CrossRef]
- Spetea, M.; Eans, S.O.; Ganno, M.L.; Lantero, A.; Mairegger, M.; Toll, L.; Schmidhammer, H.; McLaughlin, J.P. Selective κ receptor partial agonist HS666 produces potent antinociception without inducing aversion after icv administration in mice. Br. J. Pharmacol. 2017, 174, 2444–2456. [Google Scholar] [CrossRef] [PubMed]
- Dubuisson, D.; Dennis, S.G. The formalin test: A quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats. Pain 1977, 4, 161–174. [Google Scholar] [CrossRef]
- Raynor, K.; Kong, H.; Chen, Y.; Yasuda, K.; Yu, L.; Bell, G.I.; Reisine, T. Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol. Pharmacol. 1994, 45, 3330–3334. [Google Scholar]
- Zaratin, P.F.; Petrone, G.; Sbacchi, M.; Garnier, M.; Fossati, C.; Petrillo, P.; Ronzoni, S.; Giardina, G.A.; Scheideler, M.A. Modification of nociception and morphine tolerance by the selective opiate receptor-like orphan receptor antagonist (-)-cis-1-methyl-7-[[4-(2,6-dichlorophenyl)piperidin-1-yl]methyl]-6,7,8,9-tetrahydro-5Hbenzocyclohepten-5-ol (SB-612111). J. Pharmacol. Exp. Ther. 2004, 308, 454–461. [Google Scholar] [CrossRef]
- Le Bars, D.; Gozariu, M.; Cadden, S.W. Animal models of nociception. Pharmacol. Rev. 2001, 53, 597–652. [Google Scholar]
- Dumitrascuta, M.; Bermudez, M.; Ballet, S.; Wolber, G.; Spetea, M. Mechanistic understanding of peptide analogues, DALDA, [Dmt1] DALDA, and KGOP01, binding to the Mu opioid receptor. Molecules 2020, 25, 2087. [Google Scholar] [CrossRef]
- Bannister, K.; Kucharczyk, M.; Dickenson, A.H. Hopes for the future of pain control. Pain Ther. 2017, 6, 117–128. [Google Scholar] [CrossRef]
- Aldrich, J.V.; McLaughlin, J.P. Opioid peptides: Potential for drug development. Drug Discov. Today Technol. 2012, 9, e23–e31. [Google Scholar] [CrossRef] [PubMed]
- Giri, A.K.; Hruby, V.J. Investigational peptide and peptidomimetic μ and δ opioid receptor agonists in the relief of pain. Expert Opin. Invest. Drugs 2014, 23, 227–241. [Google Scholar] [CrossRef]
- Wtorek, K.; Piekielna-Ciesielska, J.; Janecki, T.; Janecka, A. The search for opioid analgesics with limited tolerance liability. Peptides 2020, 130, 170331. [Google Scholar] [CrossRef]
- Funada, M.; Suzuki, T.; Misawa, M. The role of dopamine D1-receptors in morphine-induced hyperlocomotion in mice. Neurosci. Lett. 1994, 169, 1–4. [Google Scholar] [CrossRef]
- Matthes, H.W.; Maldonado, R.; Simonin, F.; Valverde, O.; Slowe, S.; Kitchen, I.; Befort, K.; Dierich, A.; le Meur, M.; Dollé, P.; et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature 1996, 383, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Bohn, L.M.; Gainetdinov, R.R.; Sotnikova, T.D.; Medvedev, I.O.; Lefkowitz, R.J.; Dykstra, L.A.; Caron, M.G. Enhanced rewarding properties of morphine, but not cocaine, in beta(arrestin)-2 knock-out mice. J. Neurosci. 2003, 23, 10265–10273. [Google Scholar] [CrossRef]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hübner, H.; et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef]
- Huang, Y.H.; Wu, Y.W.; Chuang, J.Y.; Chang, Y.C.; Chang, H.F.; Tao, P.L.; Loh, H.H.; Yeh, S.H. Morphine produces potent antinociception, sedation, and hypothermia in humanized mice expressing human mu-opioid receptor splice variants. Pain 2020, 161, 1177–1190. [Google Scholar] [CrossRef]
- Pattinson, K.T. Opioids and the control of respiration. Br. J. Anaesth. 2008, 100, 747–758. [Google Scholar] [CrossRef]
- Li, Y.; van den Pol, A.N. μ-Opioid receptor-mediated depression of the hypothalamic hypocretin/orexin arousal system. J. Neurosci. 2008, 28, 2814–2819. [Google Scholar] [CrossRef] [PubMed]
- Calo, G.; Lambert, D. Nociceptin/orphanin FQ receptor ligands and translational challenges: Focus on cebranopadol as an innovative analgesic. Br. J. Anaesth. 2018, 121, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Toll, L.; Bruchas, M.R.; Cox, B.M.; Zaveri, N.T. Nociceptin/orphanin FQ receptor structure, signaling, ligands, functions, and interactions with opioid systems. Pharmacol. Rev. 2016, 68, 419–457. [Google Scholar] [CrossRef]
- Mansour, A.; Fox, C.A.; Akil, H.; Watson, S.J. Opioid-receptor mRNA expression in the rat CNS: Anatomical and functional implications. Trends Neurosci. 1995, 18, 22–29. [Google Scholar] [CrossRef]
- Neal, C.R., Jr.; Mansour, A.; Reinscheid, R.; Nothacker, H.P.; Civelli, O.; Akil, H.; Watson, S.J., Jr. Opioid receptor-like (ORL1) receptor distribution in the rat central nervous system: Comparison of ORL1 receptor mRNA expression with 125I-[14Tyr]-orphanin FQ binding. J. Comp. Neurol. 1999, 412, 563–605. [Google Scholar] [CrossRef]
- Evans, R.M.; You, H.; Hameed, S.; Altier, C.; Mezghrani, A.; Bourinet, E.; Zamponi, G.W. Heterodimerization of ORL1 and opioid receptors and its consequences for N-type calcium channel regulation. J. Biol. Chem. 2010, 285, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.-X.; Bolan, E.; Pasternak, G.W. Dimerization of morphine and orphanin FQ/nociceptin receptors: Generation of a novel opioid receptor subtype. Biochem. Biophys. Res. Commun. 2002, 297, 659–663. [Google Scholar] [CrossRef]
- Kiguchi, N.; Ding, H.; Ko, M.C. Therapeutic potentials of NOP and MOP receptor coactivation for the treatment of pain and opioid abuse. J. Neurosci. Res. 2020. [Google Scholar] [CrossRef]
- Tzschentke, T.M.; Linz, K.; Koch, T.; Christoph, T. Cebranopadol: A novel first-in-class potent analgesic acting via NOP and opioid receptors. In The Nociceptin/Orphanin FQ Peptide Receptor; Springer: Berlin/Heidelberg, Germany, 2019; pp. 367–398. [Google Scholar] [CrossRef]
- Khroyan, T.V.; Zaveri, N.T.; Polgar, W.E.; Orduna, J.; Olsen, C.; Jiang, F.; Toll, L. SR 16435 [1-(1-(bicyclo [3.3. 1] nonan-9-yl) piperidin-4-yl) indolin-2-one], a novel mixed nociceptin/orphanin FQ/μ-opioid receptor partial agonist: Analgesic and rewarding properties in mice. J. Pharmacol. Exp. Ther. 2007, 320, 934–943. [Google Scholar] [CrossRef]
- Sukhtankar, D.D.; Zaveri, N.T.; Husbands, S.M.; Ko, M.-C. Effects of spinally administered bifunctional nociceptin/orphanin FQ peptide receptor/μ-opioid receptor ligands in mouse models of neuropathic and inflammatory pain. J. Pharmacol. Exp. Ther. 2013, 346, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Kiguchi, N.; Yasuda, D.; Daga, P.R.; Polgar, W.E.; Lu, J.J.; Czoty, P.W.; Kishioka, S.; Zaveri, N.T.; Ko, M.-C. A bifunctional nociceptin and mu opioid receptor agonist is analgesic without opioid side effects in nonhuman primates. Sci. Transl. Med. 2018, 10, eaar3483. [Google Scholar] [CrossRef]
- Kawano, S.; Ito, R.; Nishiyama, M.; Kubo, M.; Matsushima, T.; Minamisawa, M.; Ambo, A.; Sasaki, Y. Receptor binding properties and antinociceptive effects of chimeric peptides consisting of a µ-opioid receptor agonist and an ORL1 receptor antagonist. Biol. Pharm. Bull. 2007, 30, 1260–1264. [Google Scholar] [CrossRef] [PubMed]
- Molinari, S.; Camarda, V.; Rizzi, A.; Marzola, G.; Salvadori, S.; Marzola, E.; Molinari, P.; McDonald, J.; Ko, M.C.; Lambert, D.G.; et al. [Dmt1]N/OFQ(1-13)-NH2: A potent nociceptin/orphanin FQ and opioid receptor universal agonist. Br. J. Pharmacol. 2013, 168, 151–162. [Google Scholar] [CrossRef]
- Bird, M.F.; Cerlesi, M.C.; Brown, M.; Malfacini, D.; Vezzi, V.; Molinari, P.; Micheli, L.; di Cesare, M.L.; Ghelardini, C.; Guerrini, R.; et al. Characterisation of the novel mixed mu-NOP peptide ligand dermorphin-N/OFQ (DeNo). PLoS ONE 2016, 11, e0156897. [Google Scholar] [CrossRef] [PubMed]
- Cerlesi, M.C.; Ding, H.; Bird, M.F.; Kiguchi, N.; Ferrari, F.; Malfacini, D.; Rizzi, A.; Ruzza, C.; Lambert, D.G.; Ko, M.C.; et al. Pharmacological studies on the NOP and opioid receptor agonist PWT2-[Dmt1]N/OFQ(1-13). Eur. J. Pharmacol. 2017, 794, 115–126. [Google Scholar] [CrossRef][Green Version]
- Ribeiro, J.M.L.; Filizola, M. Insights from molecular dynamics simulations of a number of G-Protein coupled receptor targets for the treatment of pain and opioid use disorders. Front. Mol. Neurosci. 2019, 12, 207. [Google Scholar] [CrossRef]
- Manglik, A. Molecular basis of opioid action: From structures to new leads. Biol. Psychiatry 2020, 87, 6–14. [Google Scholar] [CrossRef]
- Kaserer, T.; Steinacher, T.; Kainhofer, R.; Erli, F.; Sturm, S.; Waltenberger, B.; Schuster, D.; Spetea, M. Identification and characterization of plant-derived alkaloids, corydine and corydaline, as novel mu opioid receptor agonists. Sci. Rep. 2020, 10, 13804. [Google Scholar] [CrossRef] [PubMed]
- Kruegel, A.C.; Gassaway, M.M.; Kapoor, A.; Váradi, A.; Majumdar, S.; Filizola, M.; Javitch, J.A.; Sames, D. Synthetic and receptor signaling explorations of the mitragyna alkaloids: Mitragynine as an atypical molecular framework for opioid receptor modulators. J. Am. Chem. Soc. 2016, 138, 6754–6764. [Google Scholar] [CrossRef]
- Noha, S.M.; Schmidhammer, H.; Spetea, M. Molecular docking, molecular dynamics, and structure-activity relationship explorations of 14-oxygenated N-Methylmorphinan-6-ones as potent μ-opioid receptor agonists. ACS Chem. Neurosci. 2017, 8, 1327–1337. [Google Scholar] [CrossRef]
- Obeng, S.; Wang, H.; Jali, A.; Stevens, D.L.; Akbarali, H.I.; Dewey, W.L.; Selley, D.E.; Zhang, Y. Structure-activity relationship studies of 6α-and 6β-indolylacetamidonaltrexamine derivatives as bitopic mu opioid receptor modulators and elaboration of the “message-address concept” to comprehend their functional conversion. ACS Chem. Neurosci. 2018, 10, 1075–1090. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Rizzi, A.; Gavioli, E.C.; Marzola, G.; Spagnolo, B.; Zucchini, S.; Ciccocioppo, R.; Trapella, C.; Regoli, D.; Calò, G. Pharmacological characterization of the nociceptin/orphanin FQ receptor antagonist SB-612111 [(-)-cis-1-methyl-7-[[4-(2,6- dichlorophenyl)piperidin-1-yl]methyl]-6,7,8,9-tetrahydro-5H-benzocyclohepten-5-ol]: In vivo studies. J. Pharmacol. Exp. Therp. 2007, 321, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.; Dubner, R.; Brown, F.; Flores, C.; Joris, J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 1988, 32, 77–88. [Google Scholar] [CrossRef]
- Dixon, W. The up-and-down method for small samples. J. Am. Stat. Assoc. 1965, 60, 967–978. [Google Scholar] [CrossRef]
- Gonzalez-Cano, R.; Boivin, B.; Bullock, D.; Cornelissen, L.; Andrews, N.; Costigan, M. Up-down reader: An open source program for efficiently processing 50% von frey thresholds. Front. Pharmacol. 2018, 9, 433. [Google Scholar] [CrossRef] [PubMed]
- Stanford, S.C. The open field test: Reinventing the wheel. J. Psychopharmacol. 2007, 21, 134–136. [Google Scholar] [CrossRef] [PubMed]
- Bardo, M.T.; Rowlett, J.K.; Harris, M.J. Conditioned place preference using opiate and stimulant drugs: A meta-analysis. Neurosci. Biobehavl. Rev. 1995, 19, 39–51. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Labute, P. Protonate3D: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins 2009, 75, 187–205. [Google Scholar] [CrossRef]
- Schaller, D.; Šribar, D.; Noonan, T.; Deng, L.; Nguyen, T.N.; Pach, S.; Machalz, D.; Bermudez, M.; Wolber, G. Next generation 3D pharmacophore modeling. WIREs Comput. Mol. Sci. 2020, 10, e1468. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Modeling 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, J.T., Jr.; Wilcoxon, F. A simplified method of evaluating dose-effect experiments. J. Pharmacol. Exp. Ther. 1949, 96, 99–113. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Receptor Binding a | [35S]GTPγS Binding b | |
|---|---|---|---|
| Ki (nM) | EC50 (nM) | Emax (%) | |
| MOR | 0.42 ± 0.08 | 2.50 ± 0.81 | 92 ± 4 |
| DOR | 12.2 ± 3.5 | 51.6 ± 17.8 | 106 ± 3 |
| KOR | 13.4 ± 4.3 | 112 ± 32 | 37 ± 3 |
| NOP | 141 ± 23 | 1391 ± 278 | 99 ± 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dumitrascuta, M.; Bermudez, M.; Trovato, O.; De Neve, J.; Ballet, S.; Wolber, G.; Spetea, M. Antinociceptive Efficacy of the µ-Opioid/Nociceptin Peptide-Based Hybrid KGNOP1 in Inflammatory Pain without Rewarding Effects in Mice: An Experimental Assessment and Molecular Docking. Molecules 2021, 26, 3267. https://doi.org/10.3390/molecules26113267
Dumitrascuta M, Bermudez M, Trovato O, De Neve J, Ballet S, Wolber G, Spetea M. Antinociceptive Efficacy of the µ-Opioid/Nociceptin Peptide-Based Hybrid KGNOP1 in Inflammatory Pain without Rewarding Effects in Mice: An Experimental Assessment and Molecular Docking. Molecules. 2021; 26(11):3267. https://doi.org/10.3390/molecules26113267
Chicago/Turabian StyleDumitrascuta, Maria, Marcel Bermudez, Olga Trovato, Jolien De Neve, Steven Ballet, Gerhard Wolber, and Mariana Spetea. 2021. "Antinociceptive Efficacy of the µ-Opioid/Nociceptin Peptide-Based Hybrid KGNOP1 in Inflammatory Pain without Rewarding Effects in Mice: An Experimental Assessment and Molecular Docking" Molecules 26, no. 11: 3267. https://doi.org/10.3390/molecules26113267
APA StyleDumitrascuta, M., Bermudez, M., Trovato, O., De Neve, J., Ballet, S., Wolber, G., & Spetea, M. (2021). Antinociceptive Efficacy of the µ-Opioid/Nociceptin Peptide-Based Hybrid KGNOP1 in Inflammatory Pain without Rewarding Effects in Mice: An Experimental Assessment and Molecular Docking. Molecules, 26(11), 3267. https://doi.org/10.3390/molecules26113267