
Development of Halogenated Pyrazolines as Selective Monoamine Oxidase-B Inhibitors: Deciphering via Molecular Dynamics Approach

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Inhibitory Activities against MAO Enzymes
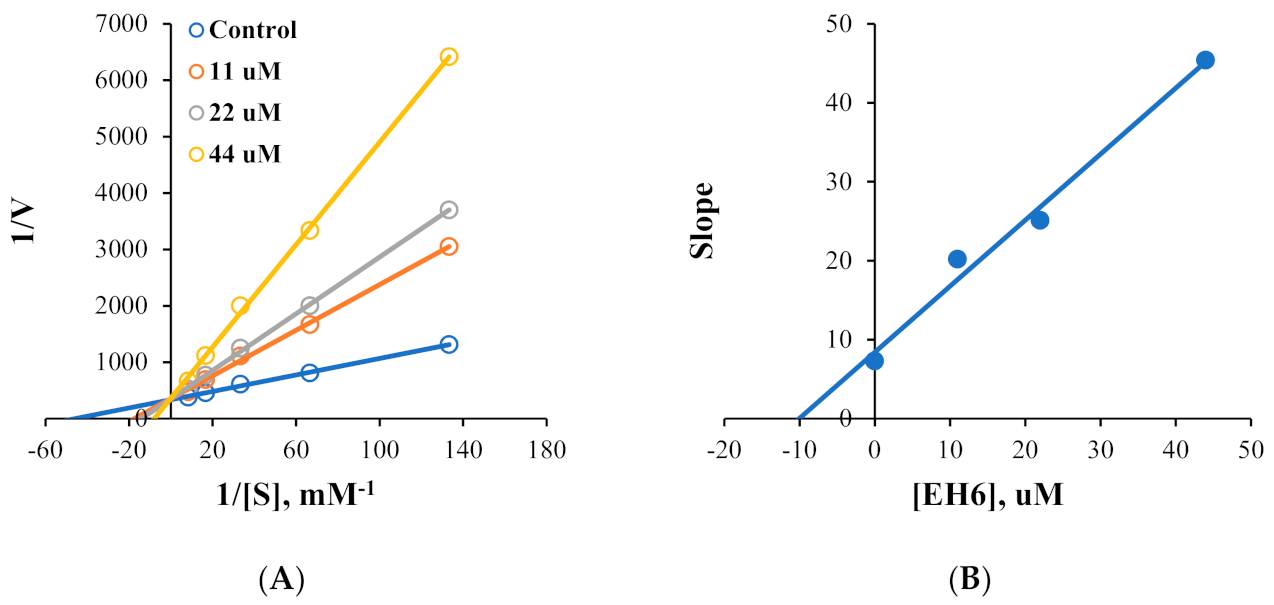
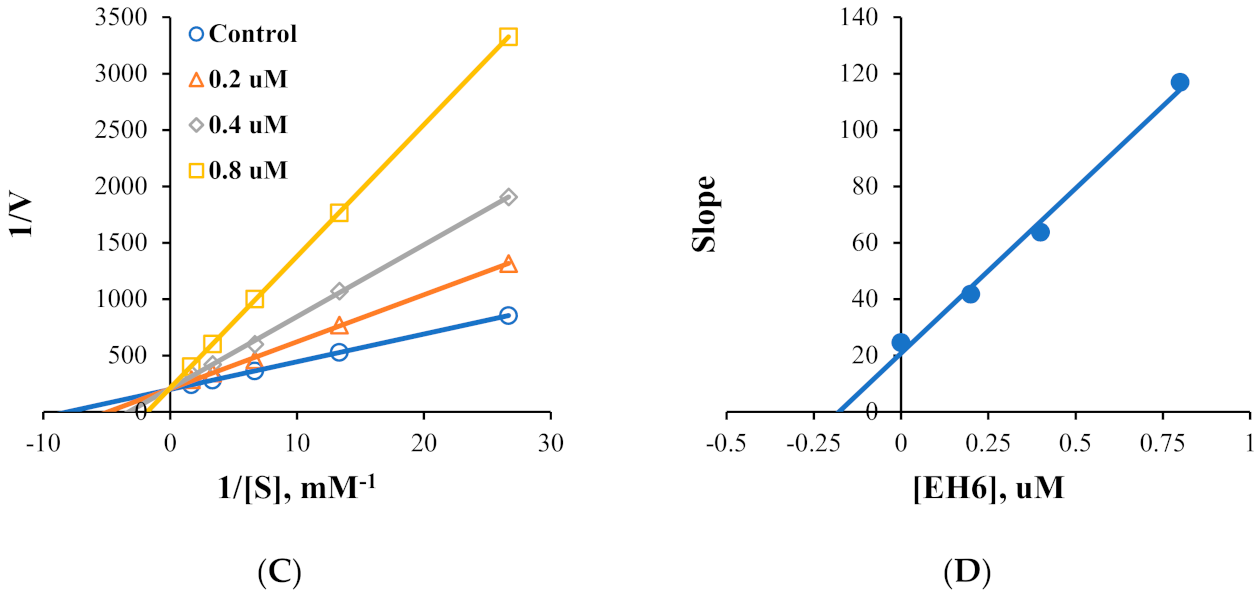
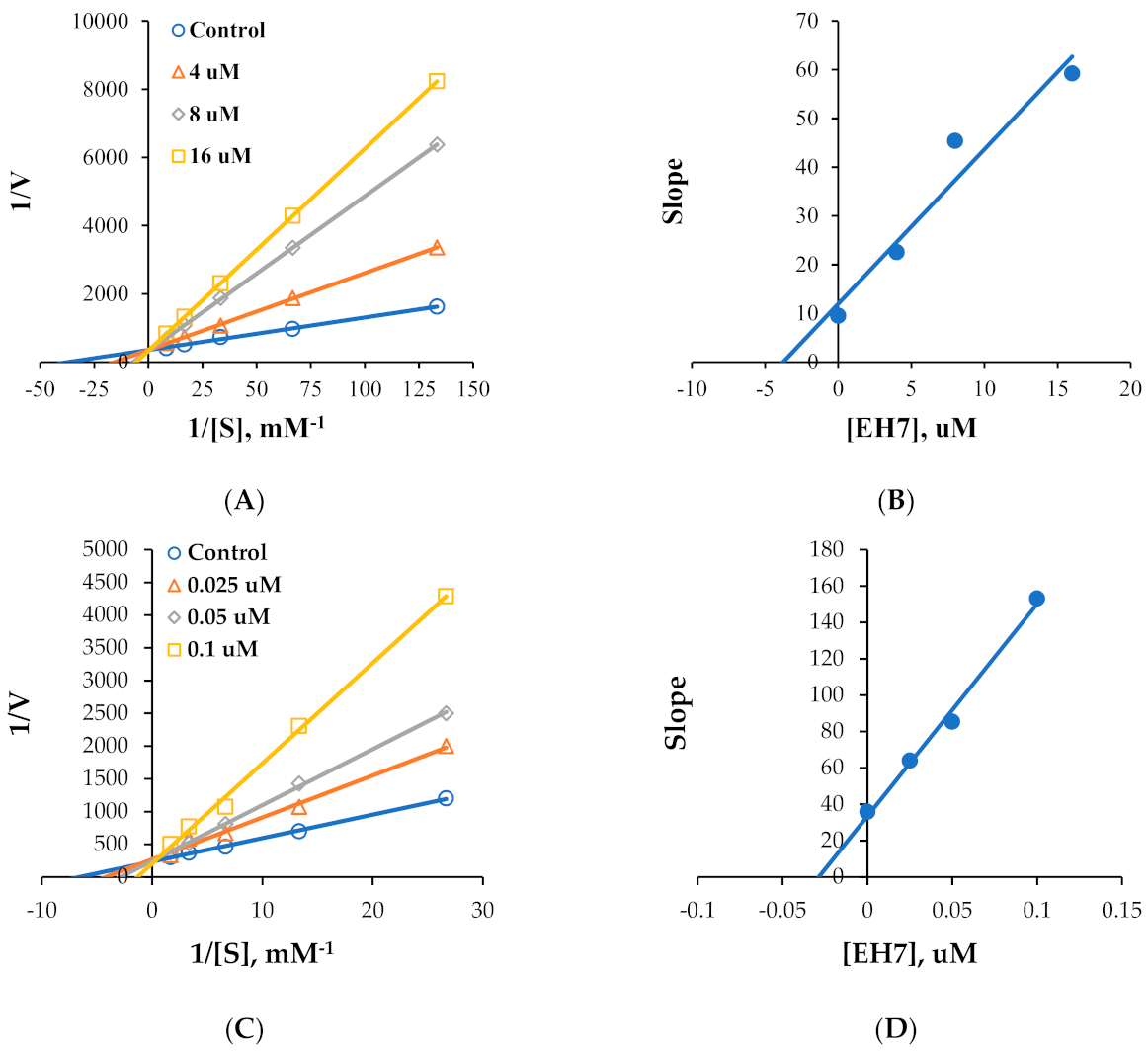
2.2. Kinetics of MAO Inhibition
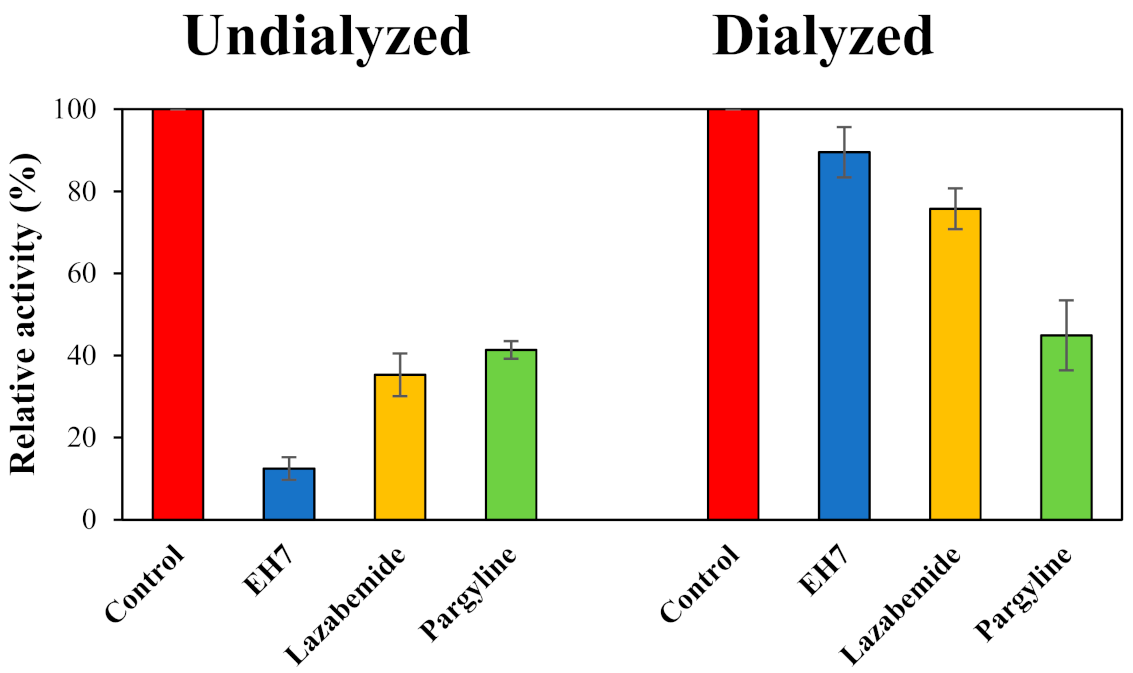
2.3. Reversibility Study
2.4. Blood–Brain Barrier (BBB) Permeation Studies
2.5. Molecular Dynamics Studies
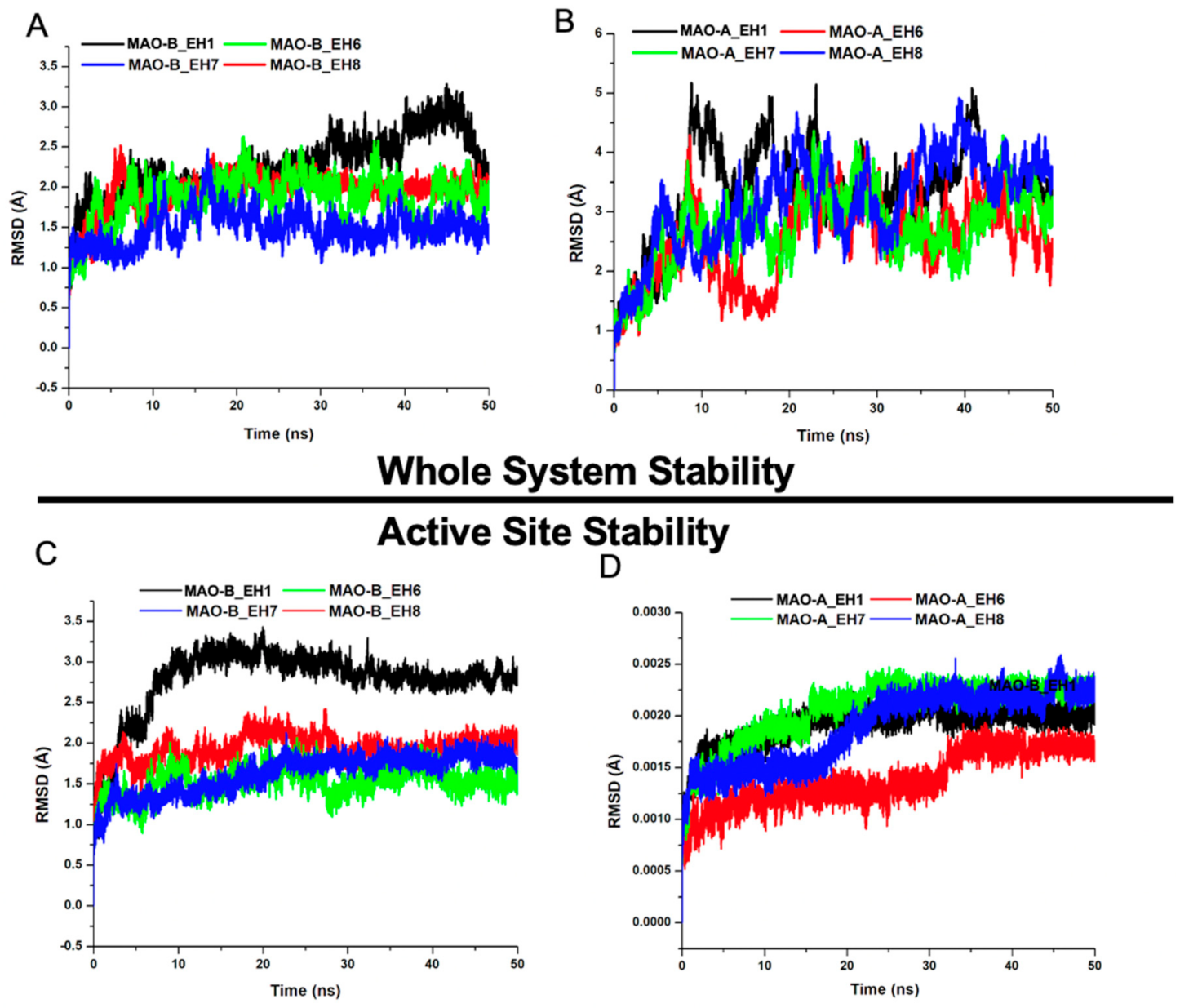
2.5.1. EH1, EH6, EH7, and EH8 Exert Differential Impacts on the Stability of MAO-A and MAO-B
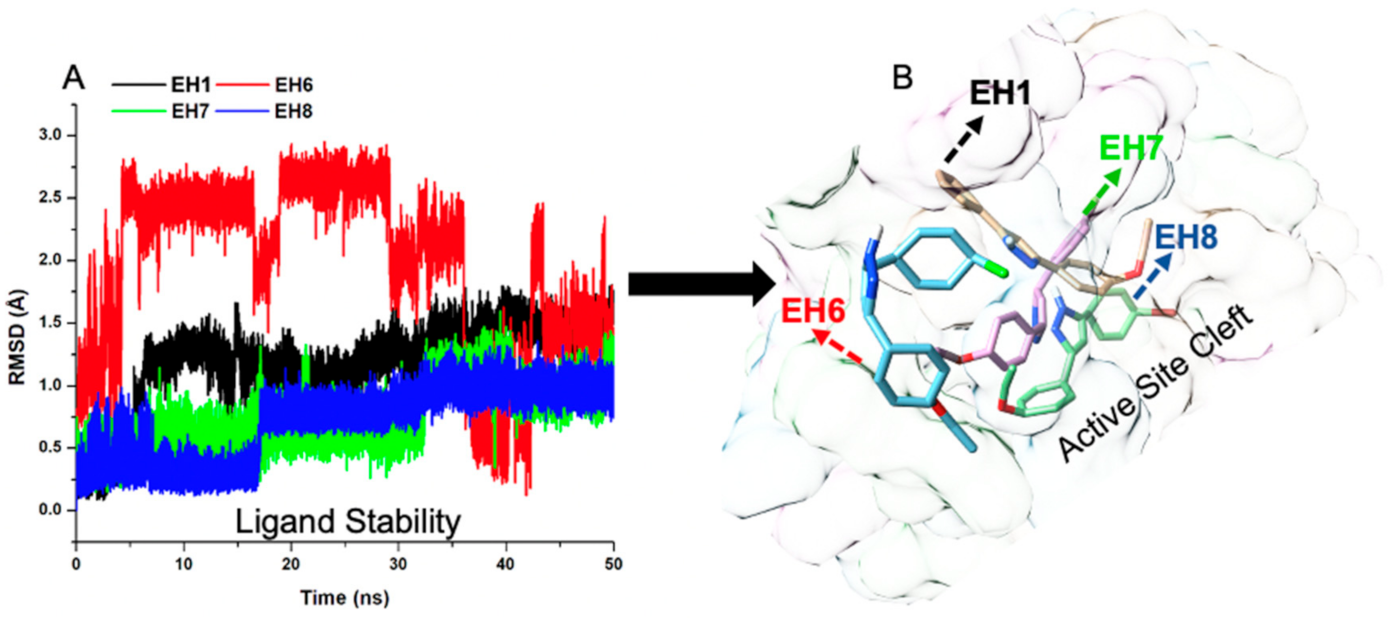
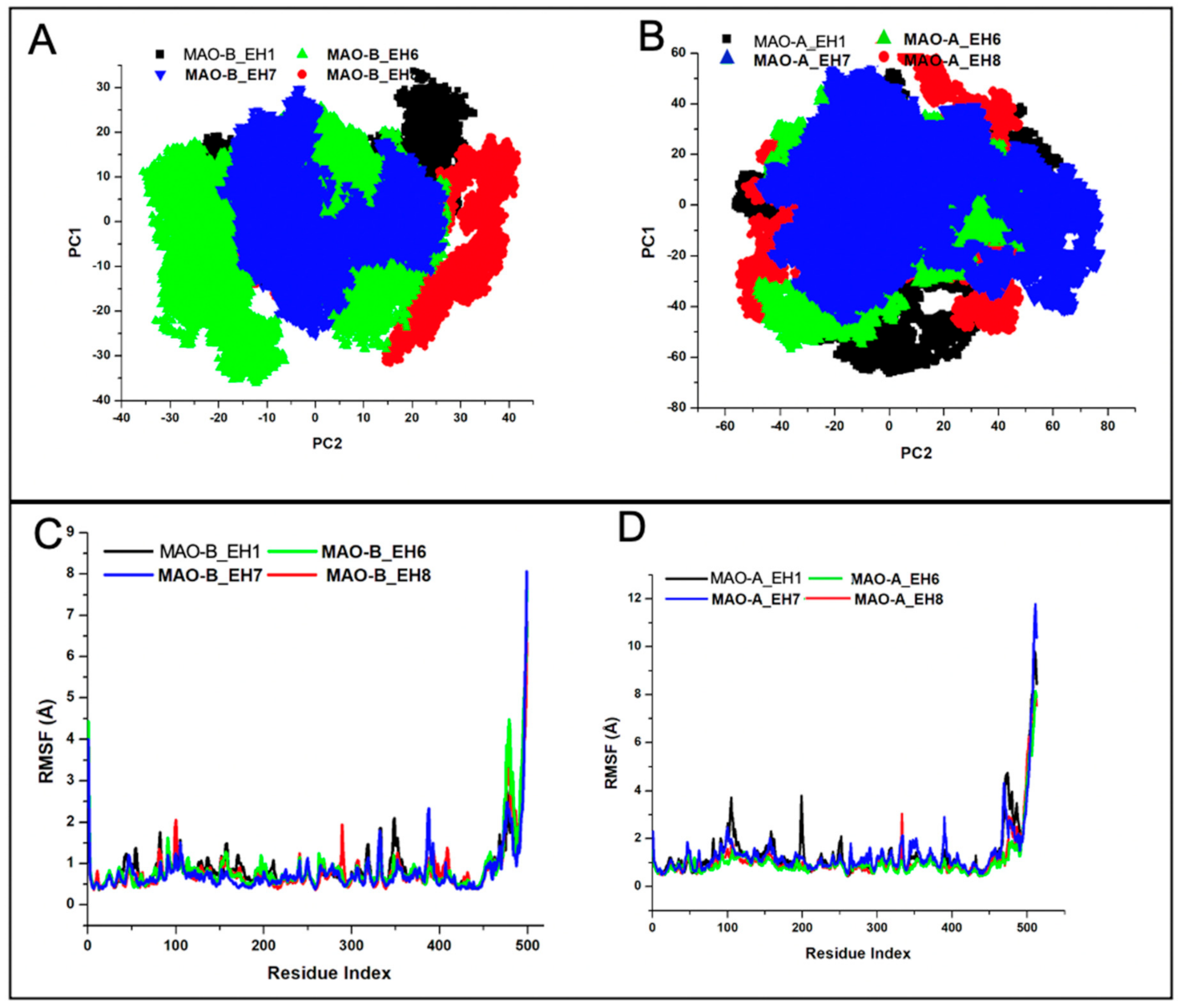
2.5.2. EH7 Exhibited Low Residual Motion and Trajectory Movement upon Binding to MAO-A and MAO-B
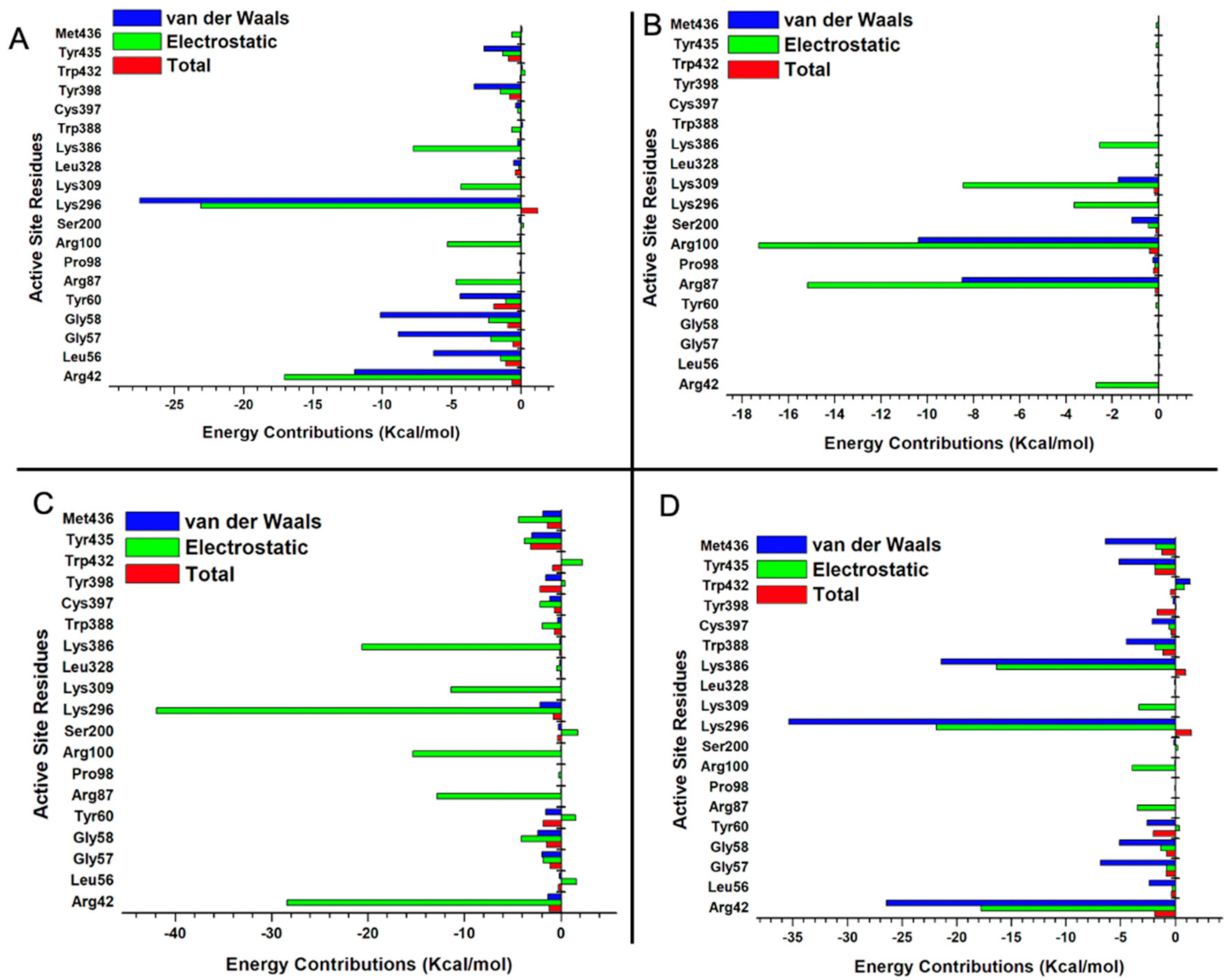
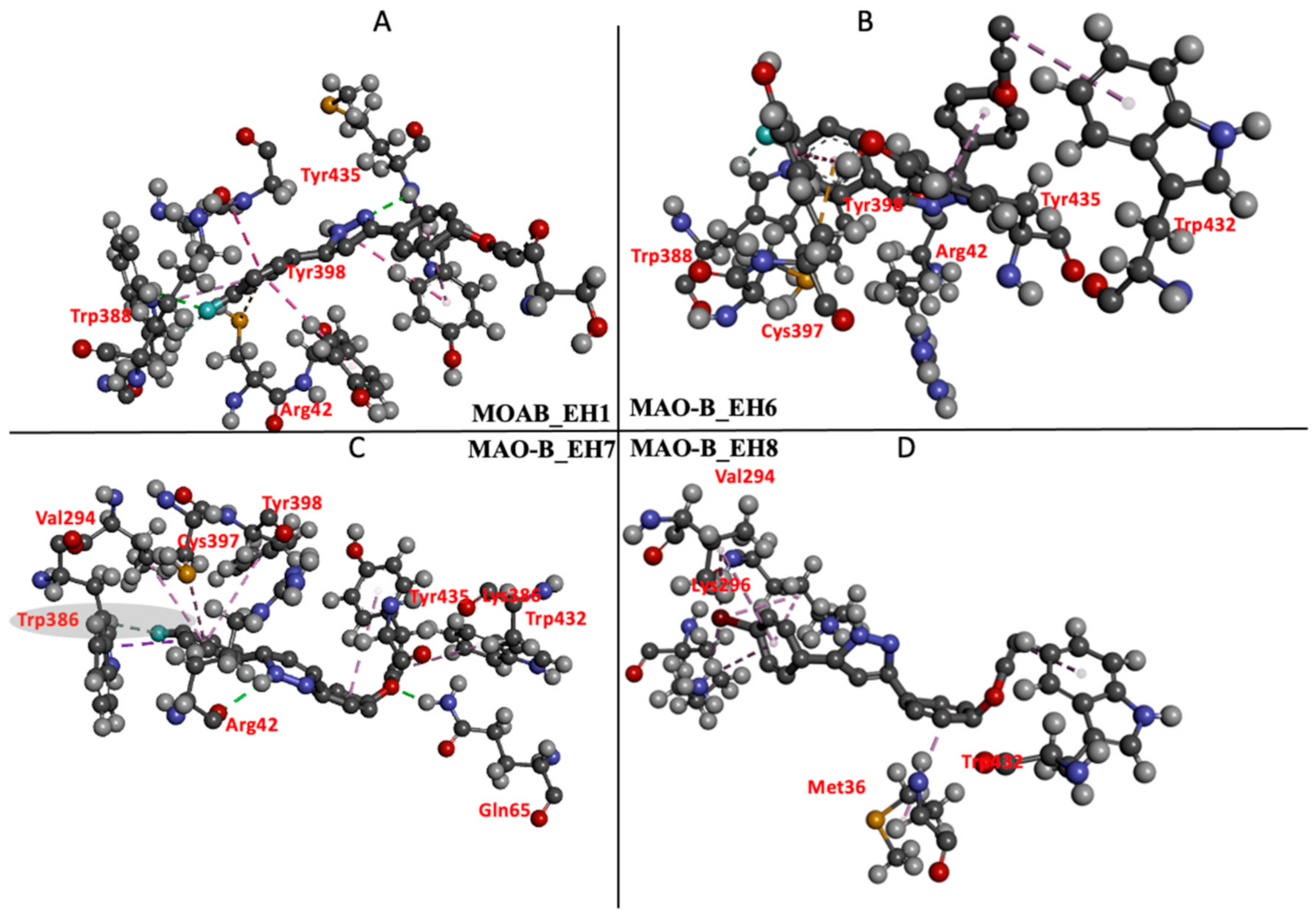
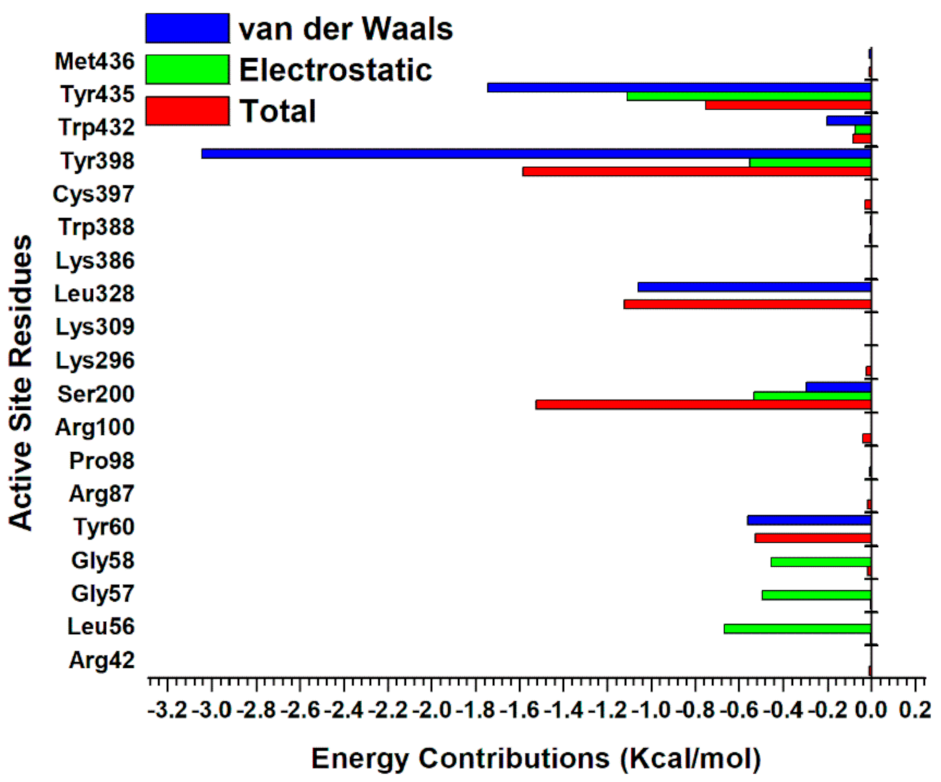
2.5.3. Estimation of Free Binding Energies of EH1, EH6, EH7, and EH8 on MAO-A and MAO-B
2.5.4. Estimation of Free Binding Energies of EH1, EH6, EH7, and EH8 on MAO-A and MAO-B
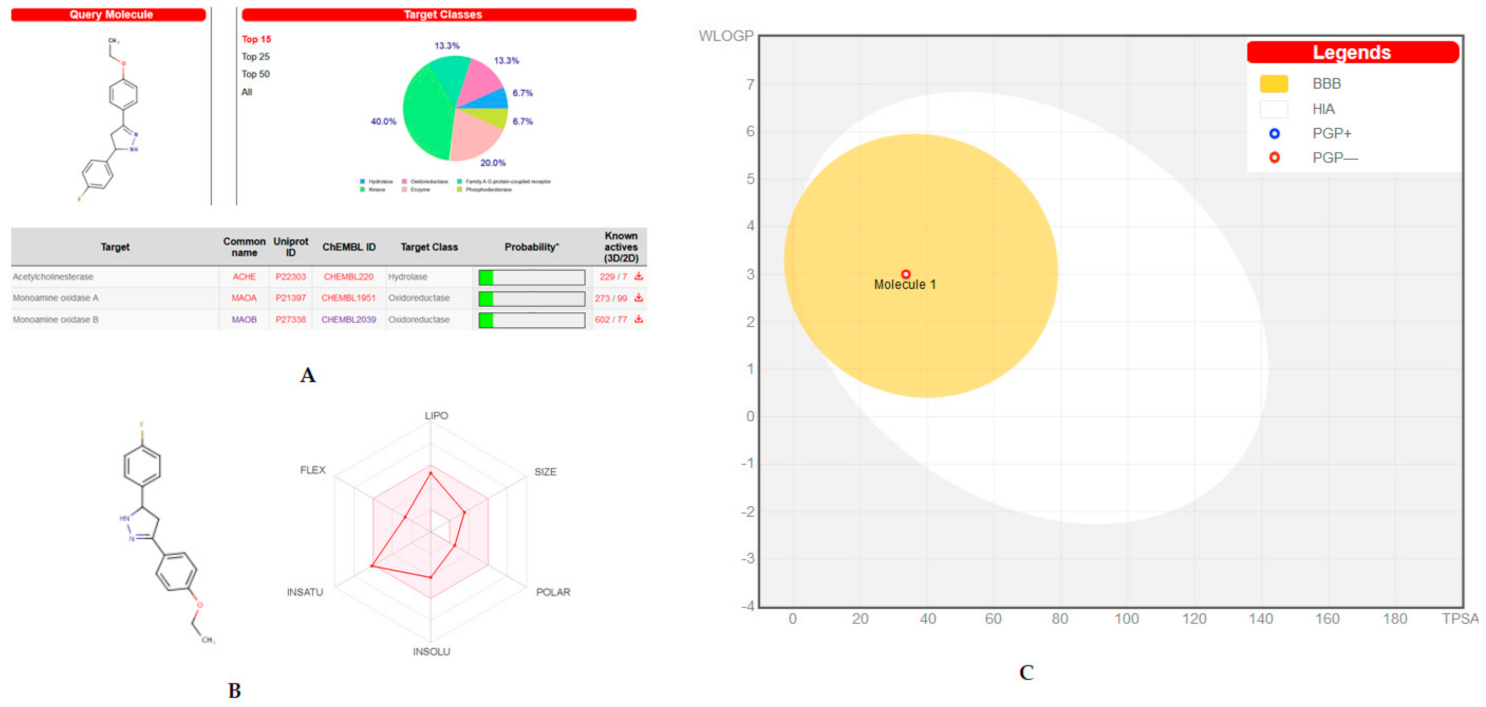
2.6. Target Predicition and ADME Analysis
3. Materials and Methods
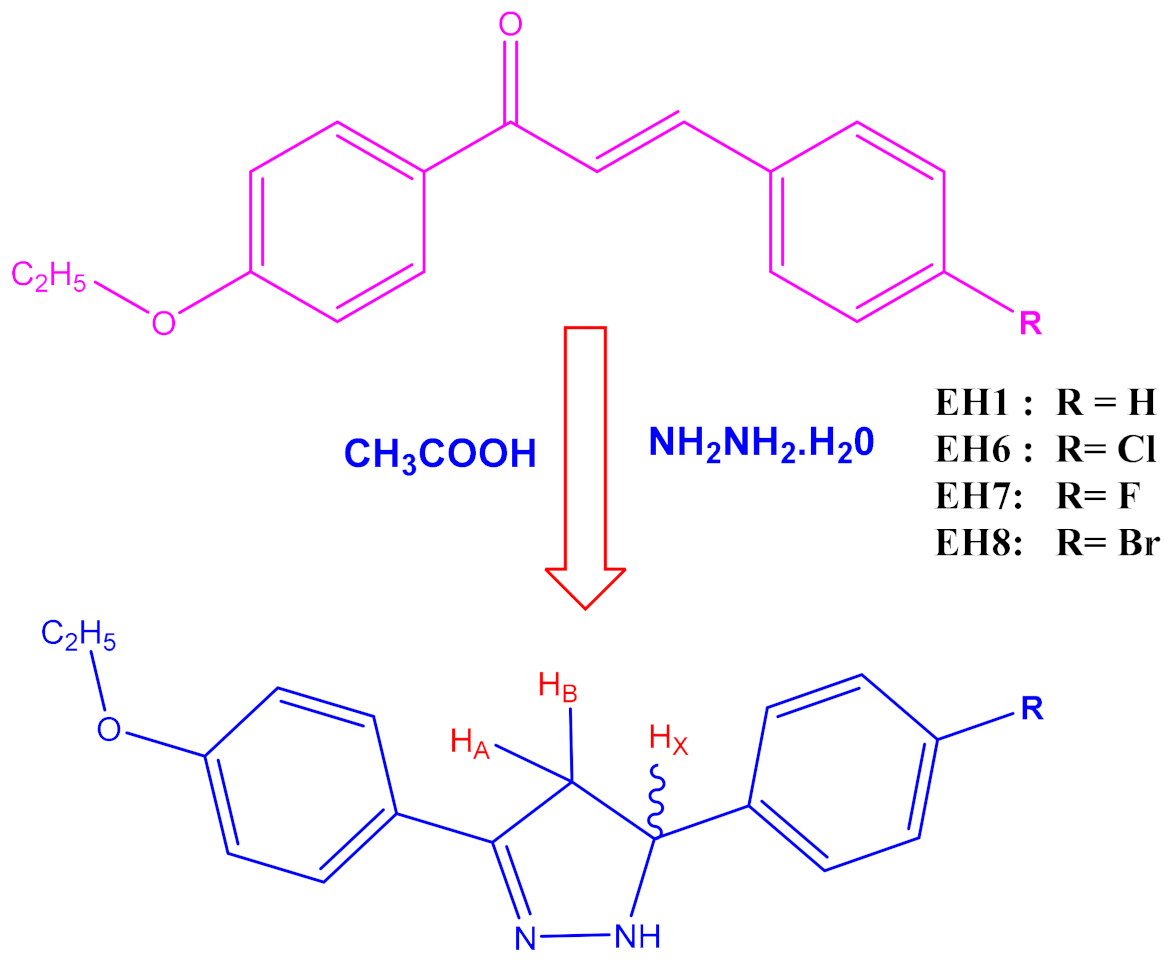
3.1. Synthesis
- 3-(4-ethoxyphenyl)-5-phenyl-4,5-dihydro-1H-pyrazole (EH1): 1H NMR (400 MHz, CDCl3) δ: 1.47–1.45 (t, 3H, J = 8.0 Hz, CH3), 4.15–4.13 (d, 2H, J = 8.0 Hz, CH2), 3.76 (d, 1H, HA), 4.02 (d, 1H, HB), 5.51 (d, 1H, HX), 7.89–7.10 (d, 9H, ArH), 9.21(S, NH). MS (ESI) m/z calcd. for C17H18N2O: 266.3376, found 266.3345.
- 5-(4-chlorophenyl)-3-(4-ethoxyphenyl)-4,5-dihydro-1H-pyrazole (EH6): 1H NMR (400 MHz, CDCl3) δ: 1.47–1.45 (t, 3H, J = 8.0 Hz, CH3), 4.14–4.12 (D, 2H, J = 8.0 Hz, CH2), 3.75 (d, 1H, HA), 4.01 (d, 1H, HB), 5.51 (d, 1H, HX), 7.99–7.08 (d, 8H, ArH) 9.24 (S, NH). MS (ESI) m/z calcd. for C17H17N2OCl: 284.3280, found 284.3276.
- 3-(4-ethoxyphenyl)-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazole (EH7): 1H NMR (400 MHz, CDCl3) δ: 1.47–1.45 (t, 3H, J = 8.0 Hz, CH3), 4.15–4.13 (d, 2H, J = 8.0 Hz, CH2), 3.72 (d, 1H, HA), 4.04 (d, 1H, HB), 5.51 (d, 1H, HX), 7.96–7.03 (d, 8H, ArH) 9.23 (S, NH). MS (ESI) m/z calcd. for C17H17N2OF: 266.3376, found 266.3345.
- 5-(4-bromophenyl)-3-(4-ethoxyphenyl)-4,5-dihydro-1H-pyrazole (EH8):1H NMR (400 MHz, CDCl3) δ: 1.47–1.45 (t, 3H, J = 8.0 Hz, CH3), 4.15–4.13 (q, 2H, J = 8.0 Hz, CH2), 3.76 (d, 1H, HA), 4.02 (d, 1H, HB), 5.51 (d, 1H, HX), 7.89–7.70 (d, 8H, ArH), 9.24(S, NH). C17H17N2OBr: 345.2336, found 345.2340.
3.2. Enzyme Assays
3.3. Analysis of Enzyme Inhibitions and Kinetics
3.4. Analysis of Inhibitor Reversibilities
3.5. Molecular Dynamics Study
3.6. Target Predicition and ADME Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Carradori, S.; Petzer, J.P. Novel monoamine oxidase inhibitors: A patent review (2012−2014). Expert Opin. Ther. Pat. 2015, 25, 91–110. [Google Scholar] [CrossRef]
- Youdim, M.B.; Bakhle, Y.S. Monoamine oxidase: Isoforms and inhibitors in Parkinson´s disease and depressive illness. Br. J. Pharm. 2006, 147, S287–S296. [Google Scholar] [CrossRef]
- Ramsay, R.R. Inhibitor design for monoamine oxidases. Curr. Pharm. Des. 2013, 19, 2529–2539. [Google Scholar] [CrossRef]
- Tripathi, R.K.P.; Ayyannan, S.R. Monoamine oxidase-B inhibitors as potential neurotherapeutic agents: An overview and update. Med. Res. Rev. 2019, 39, 1603–1706. [Google Scholar] [CrossRef] [PubMed]
- Guglielmi, P.; Carradori, S.; Ammazzalorso, A.; Secci, D. Novel approaches to the discovery of selective human monoamine oxidase-B inhibitors: Is there room for improvement? Expert Opin. Drug Discov. 2019, 14, 995–1035. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Sheetal, S.; Mantha, A.K.; Kumar, V. Recent developments on the structure-activity relationship studies of MAO inhibitors and their role in different neurological disorders. RSC Adv. 2016, 6, 42660–42683. [Google Scholar] [CrossRef]
- Kumar, B.; Gupta, V.P.; Kumar, V. A perspective on monoamine oxidase enzyme as drug target. Challenges and opportunities. Curr. Drug Targets 2017, 18, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Tipton, K. 90 years of monoamine oxidase: Some progress and some confusion. J. Neural Transm. 2018, 125, 1519–1551. [Google Scholar] [CrossRef]
- Leuratti, C.; Sardina, M.; Ventura, P.; Assandri, A.; Muller, M.; Brunner, M. Disposition and metabolism of safinamide, a novel drug for Parkinson’s disease, in healthy male volunteers. Pharmacology 2013, 92, 7–16. [Google Scholar] [CrossRef]
- Mao, Q.; Qin, W.; Zhang, A.; Ye, N. Recent advances in dopaminergic strategies for the treatment of Parkinson’s disease. Acta Pharmacol. Sin. 2020, 41, 471–482. [Google Scholar] [CrossRef]
- Hagenow, J.; Hagenow, S.; Grau, K.; Khanfar, M.; Hefke, L.; Proschak, E.; Stark, H. Reversible small molecule inhibitors of MAO A and MAO B with anilide motifs. Drug Des. Dev. Ther. 2020, 14, 371–393. [Google Scholar] [CrossRef] [PubMed]
- Mathew, B.; Suresh, J.; Anbazhagan, S.; Mathew, G.E. Pyrazoline. A promising scaffold for the inhibition of monoamine oxidase. Cent. Nerv. Syst. Agents Med. Chem. 2013, 13, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.J.; Vina, D.; Vazquez-Rodriquez, S.; Uriarte, E.; Santana, L. Focusing on new monoamine oxidase inhibitors. Differently substituted coumarins as an interesting scaffold. Curr. Top. Med. Chem. 2012, 12, 2210–2233. [Google Scholar] [CrossRef] [PubMed]
- Kavully, F.S.; Oh, J.M.; Dev, S.; Kaipakasseri, S.; Palakkathondi, A.; Vengamthodi, A.; Azeez, R.F.A.; Tondo, A.R.; Nicolotti, O.; Kim, H.; et al. Design of enamides as new selective monoamine oxidase-B inhibitors. J. Pharm. Pharmacol. 2020, 72, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, F.; Secci, D.; Bolasco, A.; Chimenti, P.; Bizzarri, B.; Granese, A.; Carradori, S.; Yáñez, M.; Orallo, F.; Ortuso, F.; et al. Synthesis, molecular modeling, and selective inhibitory activity against human monoamine oxidases of 3-carboxamido-7-substituted coumarins. J. Med. Chem. 2009, 52, 1935–1942. [Google Scholar] [CrossRef]
- Gaspar, A.; Texeira, F.; Uriarte, E.; Milhazes, N.; Melo, A.; Cordeiro, M.N.D.S.; Ortuso, F.; Alcaro, S.; Borges, F. Towards the discovery of novel class of monoamine oxidase inhibitors: Structure property activity and docking studies on chromones amides. Chem. Med. Chem. 2011, 6, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Parambi, D.G.T.; Oh, J.M.; Baek, S.C.; Lee, J.P.; Tondo, A.R.; Nicolotti, O.; Hoon, K.; Mathew, B. Design, synthesis and biological evaluation of oxygenated chalcones as potent and selective MAO-B inhibitors. Bioorg. Chem. 2019, 93, 103335. [Google Scholar] [CrossRef]
- Reeta; Baek, S.C.; Lee, J.P.; Rangarajan, T.M.; Ayushee; Singh, R.P.; Singh, M.; Mangiatordi, G.F.; Nicolotti, O.; Kim, H.; et al. Ethyl acetohydroxamate incorporated chalcones: Unveiling a novel class of chalcones for multitarget monoamine oxidase-B inhibitors against Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2019, 8, 643–654. [Google Scholar] [CrossRef]
- Oh, J.M.; Rangarajan, T.M.; Chaudhary, R.; Singh, R.P.; Singh, M.; Singh, R.P.; Tondo, A.R.; Gambacorta, N.; Nicolotti, O.; Mathew, B.; et al. Novel class of chalcone oxime ethers as potent monoamine oxidase-B and acetylcholinesterase inhibitors. Molecules 2020, 25, 2356. [Google Scholar] [CrossRef]
- Mathew, B.; Baek, S.C.; Parambi, D.G.T.; Lee, J.P.; Mathew, G.E.; Jayanthi, S.; Devaraji, D.; Raphael, C.; Vinod, D.; Kondarath, S.S.; et al. Potent and highly selective dual-targeting monoamine oxidase-B inhibitors: Fluorinated chalcones of morpholine versus imidazole. Arch. Pharm. 2019, 352, e1800309. [Google Scholar] [CrossRef]
- Guglielmi, P.; Mathew, B.; Secci, D.; Carradori, S. Chalcones: Unearthing their therapeutic possibility as monoamine oxidase B inhibitors. Eur. J. Med. Chem. 2020, 205, 112650. [Google Scholar] [CrossRef] [PubMed]
- Mathew, B.; Parambi, D.G.T.; Mathew, G.E.; Uddin, M.S.; Inasu, S.T.; Kim, H.; Marathakam, A.; Unnikrishnan, M.K.; Carradori, S. Emerging therapeutic potentials of dual-acting MAO and AChE inhibitors in Alzheimer’s and Parkinson’s diseases. Arch. Pharm. 2019, 352, e1900177. [Google Scholar] [CrossRef] [PubMed]
- Finberg, J.P.M.; Rabey, J.M. Inhibitors of MAO-A and MAO-B in Psychiatry and Neurology. Front. Pharmacol. 2016, 7, 340. [Google Scholar] [CrossRef] [PubMed]
- Carradori, S.; Silvestri, R. New frontiers in selective human MAO-B inhibitors. J. Med. Chem. 2015, 58, 6717–6732. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, A.; Silva, T.; Yáñez, M.; Viña, D.; Orallo, F.; Ortuso, F.; Uriarte, E.; Alcaro, S.; Borges, F. Chromone, a privileged scaffold for the development of monoamine oxidase inhibition. J. Med. Chem. 2011, 54, 5165–5173. [Google Scholar] [CrossRef]
- Reis, J.; Cagide, F.; Chavarria, D.; Silva, T.B.; Fernandes, C.; Gaspar, A.; Uriarte, E.; Remiao, F.; Alcaro, S.; Ortuso, F.; et al. Discovery of new chemical entities of old targets. Insight on the lead optimization of chromones based monoamine oxidase B inhibitors. J. Med. Chem. 2016, 59, 5879–5893. [Google Scholar] [CrossRef]
- Shaaban, M.R.; Mayhoub, A.S.; Farag, A.M. Recent advances in the therapeutic applications of pyrazolines. Expert Opin. Ther. Patents 2012, 22, 253–291. [Google Scholar] [CrossRef]
- Bansal, R.; Singh, R. Steroidal pyrazolines as a promising scaffold in drug discovery. Future Med. Chem. 2020, 12, 949–959. [Google Scholar] [CrossRef]
- Badavath, V.N.; Baysal, I.; Ucar, G.; Sinha, B.N.; Jayaprakash, V. Monoamine oxidase inhibitory activity of novel pyrazoline analogues: Curcumin based design and synthesis. ACS Med. Chem. Lett. 2016, 7, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Goksen, U.S.; Sarigul, S.; Bultinck, P.; Herrebout, W.; Dogan, I.; Yelekci, K.; Ucar, G.; Gokhan Kelekci, N. Absolute configuration and biological profile of pyrazoline enantiomers as MAO inhibitory activity. Chirality 2019, 31, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Vishnu Nayak, B.; Ciftci-Yabanoglu, S.; Jadav, S.S.; Jagrat, M.; Sinha, B.N.; Ucar, G.; Jayaprakash, V. Monoamine oxidase inhibitory activity of 3,5-biaryl-4,5-dihydro-1H-pyrazole-1-carboxylate derivatives. Eur. J. Med. Chem. 2013, 69, 762–767. [Google Scholar] [CrossRef]
- Tong, X.; Chen, R.; Zhang, T.T.; Han, Y.; Tang, W.J.; Liu, X.H. Design and synthesis of novel 2-pyrazoline-1-ethanone derivatives as selective MAO inhibitors. Bioorg. Med. Chem. 2015, 23, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Evranos-Aksoz, B.; Ucar, G.; Tas, S.T.; Aksoz, E.; Yelekci, K.; Erikci, A.; Sara, Y.; Iskit, A.B. New Human Monoamine Oxidase A Inhibitors with Potential Anti- Depressant Activity: Design, Synthesis, Biological Screening and Evaluation of Pharmacological Activity. Comb. Chem. High Throughput Screen 2017, 20, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Guglielmi, P.; Carradori, S.; Poli, G.; Secci, D.; Cirilli, R.; Rotondi, G.; Chimenti, P.; Petzer, A.; Petzer, J.P. Design, Synthesis, Docking Studies and Monoamine Oxidase Inhibition of a Small Library of 1-acetyl- and 1-thiocarbamoyl-3,5-diphenyl-4,5-dihydro-(1H)-pyrazoles. Molecules 2019, 24, 484. [Google Scholar] [CrossRef] [PubMed]
- Hitge, R.; Smit, S.; Petzer, A.; Petzer, J.P. Evaluation of nitrocatechol chalcone and pyrazoline derivatives as inhibitors of catechol-O-methyltransferase and monoamine oxidase. Bioorg. Med. Chem. Lett. 2020, 30, 127188. [Google Scholar] [CrossRef]
- Bolasco, A.; Carradori, S.; Fioravanti, R. Focusing on new monoamine oxidase inhibitors. Expert Opin. Ther. Patents 2010, 20, 909–939. [Google Scholar] [CrossRef]
- Fang, W.Y.; Ravindar, L.; Rakesh, K.P.; Manukumar, H.M.; Shantharam, C.S.; Alharbi, N.S.; Qin, H.L. Synthetic approaches and pharmaceutical application of chlorine containing molecules for drug discovery: A critical review. Eur. J. Med. Chem. 2019, 173, 117–153. [Google Scholar] [CrossRef]
- Mathew, B.; Carradori, S.; Guglielmi, P.; Uddin, M.S.; Kim, H. New aspects of monoamine oxidase B inhibitors. The key role of halogens to open the golden door. Curr. Med. Chem. 2021, 28, 266–283. [Google Scholar] [CrossRef]
- Lakshminarayanan, B.; Baek, S.C.; Lee, J.P.; Kannappan, N.; Mangiatordi, G.F.; Nicolotti, O.; Subburaju, T.; Kim, H.; Mathew, B. Ethoxylated head of chalcones as a new class of multi-targeted MAO inhibitors. ChemistrySelect 2019, 4, 6614–6619. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Pitera, J.W. Expected distributions of root-mean-square positional deviations in proteins. J. Phys. Chem. B. 2014, 118, 6526–6530. [Google Scholar] [CrossRef]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar]
- Martínez, L. Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis. PLoS ONE 2015, 10, e0119264. [Google Scholar] [CrossRef]
- Soremekun, O.S.; Olotu, F.A.; Agoni, C.; Soliman, M.E.S. Drug promiscuity: Exploring the polypharmacology potential of 1, 3, 6-trisubstituted 1, 4-diazepane-7-ones as an inhibitor of the ‘god father’ of immune checkpoint. Comput. Biol. Chem. 2019, 80, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Tubert-Brohman, I.; Sherman, W.; Repasky, M.; Beuming, T. Improved docking of polypeptides with glide. J. Chem. Inf. Model. 2013, 53, 1689–1699. [Google Scholar] [CrossRef]
- Son, S.Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2-Å resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Weedbrook, C.; Pirandola, S.; García-Patrón, R.; Cerf, N.J.; Ralph, T.C.; Shapiro, J.H.; Lloyd, S. Gaussian Quantum Information. Rev. Mod. Phys. 2012, 84, 621. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Pique, M.E.; Lindstrom, L.; Heuy, R.; Forli, S.; Hart, W.E.; Halliday, S.; Belew, R.; Olson, A.J. AutoDock Version 4.2. User Guide. 2009, pp. 1–49. Available online: http://autodock.scripps.edu/faqs-help/manual/autodock-4-2-user-guide/AutoDock4.2_UserGuide.pdf (accessed on 27 May 2021).
- Lee, T.S.; Cerutti, D.S.; Mermelstein, D.; Lin, C.; LeGrand, S.; Giese, T.J.; Roitberg, A.; Case, D.A.; Walker, R.C.; York, D.M. GPU-Accelerated Molecular Dynamics and Free Energy Methods ion Amber18: Performance Enhancements and New Features. J. Chem. Inf. Model. 2018, 58, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Soremekun, O.S.; Olotu, F.A.; Agoni, C.; Soliman, M.E.S. Recruiting monomer for dimer formation: Resolving the antagonistic mechanisms of novel immune check point inhibitors against Programmed Death Ligand-1 in cancer immunotherapy. Mol. Simul. 2019, 45, 777–789. [Google Scholar] [CrossRef]
- Soremekun, O.S.; Soliman, M.E.S. From genomic variation to protein aberration: Mutational analysis of single nucleotide polymorphism present in ULBP6 gene and implication in immune response. Comput. Biol. Med. 2019, 111, 103354. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A boiled-egg too predict gastrointestinal absorption and brain penetration of small molecules. Chem. Med. Chem. 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Residual Activity at 10 µM (%) | IC50 (µM) | SI b | ||||
|---|---|---|---|---|---|---|---|
| MAO-A | MAO-B | AChE | MAO-A | MAO-B | AChE | ||
| EH1 | 99.1 ± 0.27 | 33.7 ± 0.96 | 81.0 ± 6.21 | >40 | 5.38 ± 0.16 | >40 | >7.43 |
| EH6 | 68.1 ± 0.18 | 19.4 ± 1.01 | 63.5 ± 4.99 | 22.3 ± 0.12 | 0.40 ± 0.051 | >40 | 55.8 |
| EH7 | 46.9 ± 1.25 | 4.03 ± 0.53 | 74.0 ± 0.71 | 8.38 ± 0.88 | 0.063 ± 0.0042 | >40 | 133.0 |
| EH8 | 33.5 ± 0.65 | 11.3 ± 0.13 | 53.7 ± 2.15 | 4.31 ± 0.013 | 0.69 ± 0.017 | 10.8 ± 0.15 | 6.3 |
| Toloxatone | 1.08 ± 0.0025 | - | - | ||||
| Lazabemide | - | 0.11 ± 0.016 | - | ||||
| Clorgyline | 0.0070 ± 0.00070 | - | - | ||||
| Pargyline | - | 0.14 ± 0.0059 | - | ||||
| Tacrine | - | - | 0.27 ± 0.019 | ||||
| Compounds | Bibliography Pe (×10−6 cm/s) a | Experimental Pe (×10−6 cm/s) | Prediction |
|---|---|---|---|
| Progesterone | 9.3 | 9.02 ± 0.11 | CNS+ |
| Verapamil | 16.0 | 15.53 ± 0.24 | CNS+ |
| Piroxicam | 2.5 | 2.43 ± 0.30 | CNS+/− |
| Lomefloxacin | 1.1 | 1.12 ± 0.01 | CNS− |
| Dopamine | 0.2 | 0.22 ± 0.01 | CNS− |
| EH1 | 10.34 ± 0.33 | CNS+ | |
| EH6 | 14.26 ± 0.80 | CNS+ | |
| EH7 | 14.13 ± 0.71 | CNS+ | |
| EH8 | 14.56 ± 0.26 | CNS+ |
| Complexes | RMSD | RMSD_AS | RMSF | IC50 (µM) |
|---|---|---|---|---|
| MAO-A_EH1 | 3.35 (0.73) | 1.93 (0.18) | 1.47 (1.28) | >40 |
| MAO-A_EH6 | 2.63 (0.72) | 1.44 (0.27) | 1.15 (1.04) | 22.3 |
| MAO-A_EH7 | 2.74 (0.59) | 2.09 (0.29) | 1.07 (0.96) | 8.38 |
| MAO-A_EH8 | 3.26 (0.74) | 1.96 (0.34) | 1.40 (1.28) | 4.31 |
| MAO-B_EH1 | 2.29 (0.34) | 2.85 (0.37) | 0.88 (0.59) | 5.38 |
| MAO-B_EH6 | 1.61 (0.22) | 1.54 (0.14) | 0.83 (0.60) | 0.40 |
| MAO-B_EH7 | 1.49 (0.26) | 1.60 (0.16) | 0.68 (0.72) | 0.063 |
| MAO-B_EH8 | 1.79 (0.24) | 1.70 (0.25) | 0.75 (0.61) | 0.69 |
| Complexes | Energy (kcal/mol) | ||||||
|---|---|---|---|---|---|---|---|
| ∆EvdW | ∆Eele | ∆Ggas | ∆GGB | ∆GSA | ∆Gsol | ∆Gbind | |
| MAO-B_EH1 | −6.68 | −34.66 | −35.32 | 35.91 | −1.68 | 35.74 | −4.11 |
| (±0.19) | (±4.28) | (±4.20) | (±3.99) | (±0.01) | (±3.98) | (±0.36) | |
| MAO-B_EH6 | −25.46 | −94.22 | −96.77 | 108.35 | −4.28 | 107.92 | −11.15 |
| (±0.15) | (±2.01) | (±2.06) | (±2.64) | (±0.01) | (±2.63) | (±0.88) | |
| MAO-B_EH7 | −36.79 | −32.46 | −85.96 | 55.09 | −5.12 | 49.67 | −36.29 |
| (±0.15) | (±0.54) | (±0.53) | (±0.50) | (±0.01) | (±0.50) | (±0.13) | |
| MAO-B_EH8 | −36.79 | −101.78 | −105.46 | 106.54 | − 5.35 | 106 | −5.48 |
| (±0.15) | (±1.73) | (±1.74) | (±1.56) | (±0.01) | (±1.56) | (±0.37) | |
| MAO-A_EH1 | −33.17 | −12.98 | −13.31 | −12.85 | −4.65 | 12.81 | −5.06 |
| (±0.22) | (±4.07) | (±4.20) | (±2.86) | (±0.01) | (±2.85) | (±1.49) | |
| MAO-A_EH6 | −36.75 | −14.50 | −14.88 | 13.78 | −4.86 | 13.73 | −11.33 |
| (±0.18) | (±2.36) | (±2.39) | (±1.61) | (±0.01) | (±1.61) | (±0.97) | |
| MAO-A_EH7 | −32.75 | −13.21 | −13.54 | −36.79 | −4.85 | 13.21 | −32.55 |
| (±0.15) | (±2.06) | (±2.06) | (±0.15) | (±0.01) | (±1.36) | (±1.07) | |
| MAO-A_EH8 | −7.52 | −12.07 | −12.14 | 12.92 | −2.61 | 12.89 | −7.48 |
| (±0.15) | (±2.49) | (±2.56) | (±2.30) | (±0.01) | (±0.15) | (±0.15) | |
| Energy (kcal/mol) | |||||||
|---|---|---|---|---|---|---|---|
| Complexes | ∆EvdW | ∆Eele | ∆Ggas | ∆GGB | ∆GSA | ∆Gsol | ∆Gbind |
| MAO-B_EH7R | −36.79 | −32.46 | −85.96 | 55.09 | −5.12 | 49.67 | −36.2 |
| (±0.15) | (±0.54) | (±0.53) | (±0.50) | (±0.01) | (±0.50) | (±0.13) | |
| MAO-B_EH7S | −53.42 | −17.59 | −22.93 | 19.88 | −5.62 | 19.31 | −36.17 |
| (±0.08) | (±1.56) | (±0.50) | (±1.49) | (±0.01) | (±0.47) | (±0.12) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nair, A.S.; Oh, J.-M.; Koyiparambath, V.P.; Kumar, S.; Sudevan, S.T.; Soremekun, O.; Soliman, M.E.; Khames, A.; Abdelgawad, M.A.; Pappachen, L.K.; et al. Development of Halogenated Pyrazolines as Selective Monoamine Oxidase-B Inhibitors: Deciphering via Molecular Dynamics Approach. Molecules 2021, 26, 3264. https://doi.org/10.3390/molecules26113264
Nair AS, Oh J-M, Koyiparambath VP, Kumar S, Sudevan ST, Soremekun O, Soliman ME, Khames A, Abdelgawad MA, Pappachen LK, et al. Development of Halogenated Pyrazolines as Selective Monoamine Oxidase-B Inhibitors: Deciphering via Molecular Dynamics Approach. Molecules. 2021; 26(11):3264. https://doi.org/10.3390/molecules26113264
Chicago/Turabian StyleNair, Aathira Sujathan, Jong-Min Oh, Vishal Payyalot Koyiparambath, Sunil Kumar, Sachithra Thazhathuveedu Sudevan, Opeyemi Soremekun, Mahmoud E. Soliman, Ahmed Khames, Mohamed A. Abdelgawad, Leena K. Pappachen, and et al. 2021. "Development of Halogenated Pyrazolines as Selective Monoamine Oxidase-B Inhibitors: Deciphering via Molecular Dynamics Approach" Molecules 26, no. 11: 3264. https://doi.org/10.3390/molecules26113264
APA StyleNair, A. S., Oh, J.-M., Koyiparambath, V. P., Kumar, S., Sudevan, S. T., Soremekun, O., Soliman, M. E., Khames, A., Abdelgawad, M. A., Pappachen, L. K., Mathew, B., & Kim, H. (2021). Development of Halogenated Pyrazolines as Selective Monoamine Oxidase-B Inhibitors: Deciphering via Molecular Dynamics Approach. Molecules, 26(11), 3264. https://doi.org/10.3390/molecules26113264