Determination of d-Cycloserine Impurities in Pharmaceutical Dosage Forms: Comparison of the International Pharmacopoeia HPLC–UV Method and the DOSY NMR Method

 ,
,  , and
, and
Abstract
1. Introduction
2. Results and Discussion
2.1. Verification of The International Pharmacopoeia (Ph. Int.) HPLC Method for Determination of Impurities in Samples Containing d-Cycloserine
2.1.1. Repeatability of d-Cycloserine Chromatographic Peak Areas
2.1.2. Determination of the Limit of Quantification (LOQ) for d-Cycloserine
2.1.3. Linearity of the d-cycloserine Signal
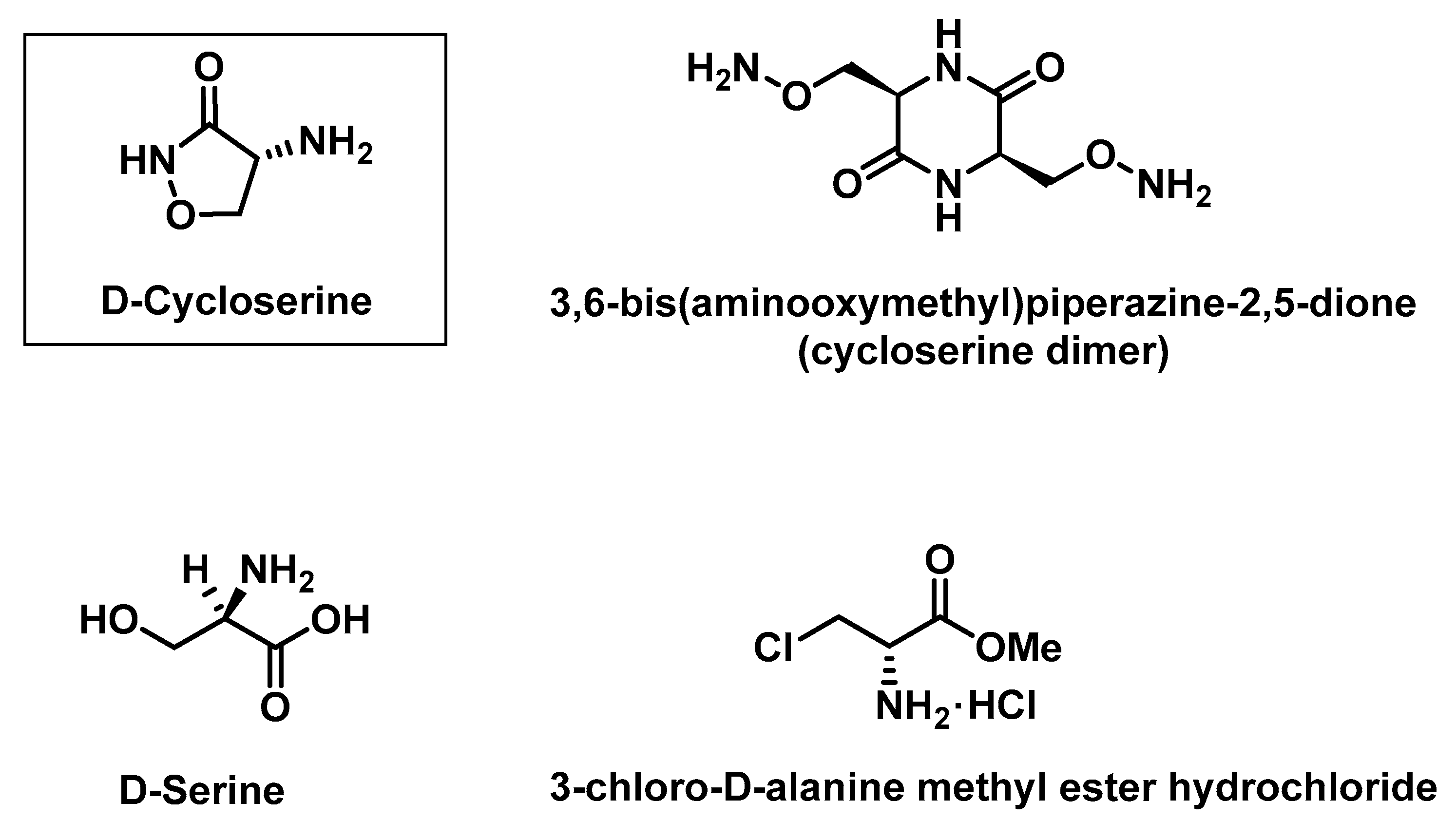
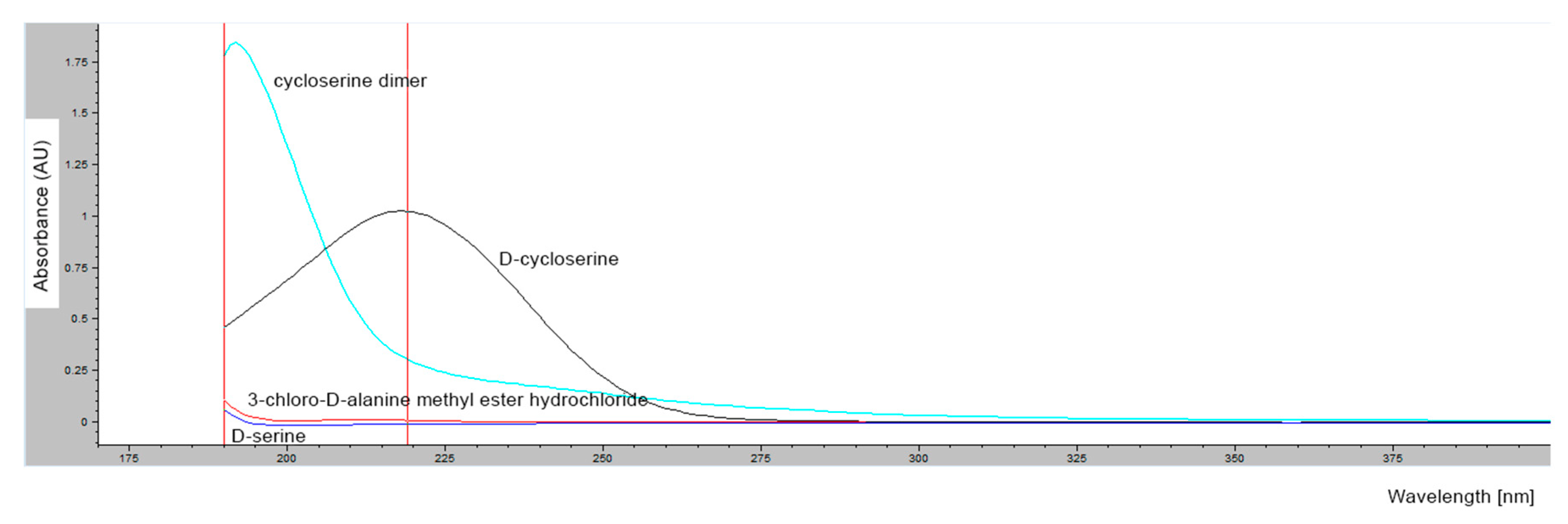
2.1.4. Determination of Retention Times of d-Cycloserine and Its Impurities
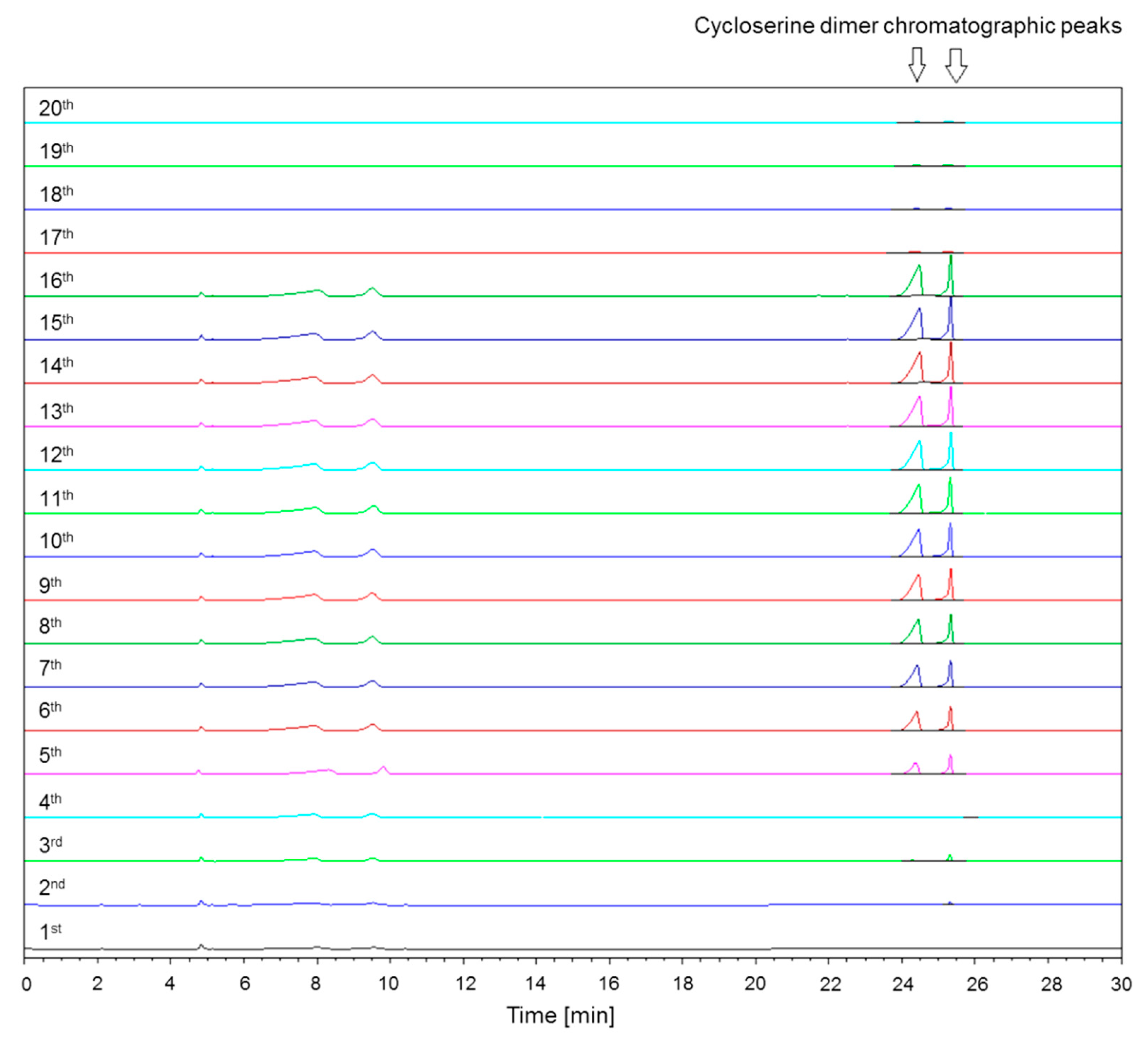
2.1.5. Saturation of Column Free Silanol Groups with Cycloserine Dimer and Repeatability of Cycloserine Dimer Chromatographic Peak Areas
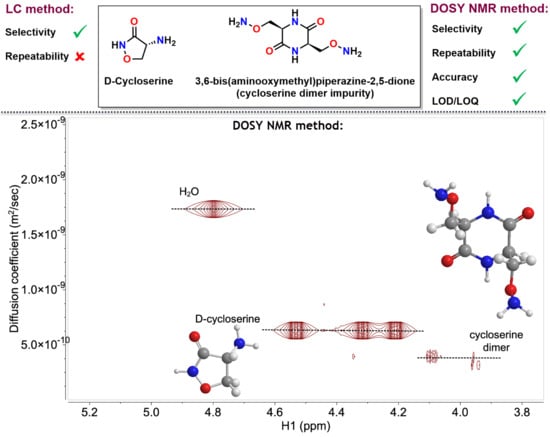
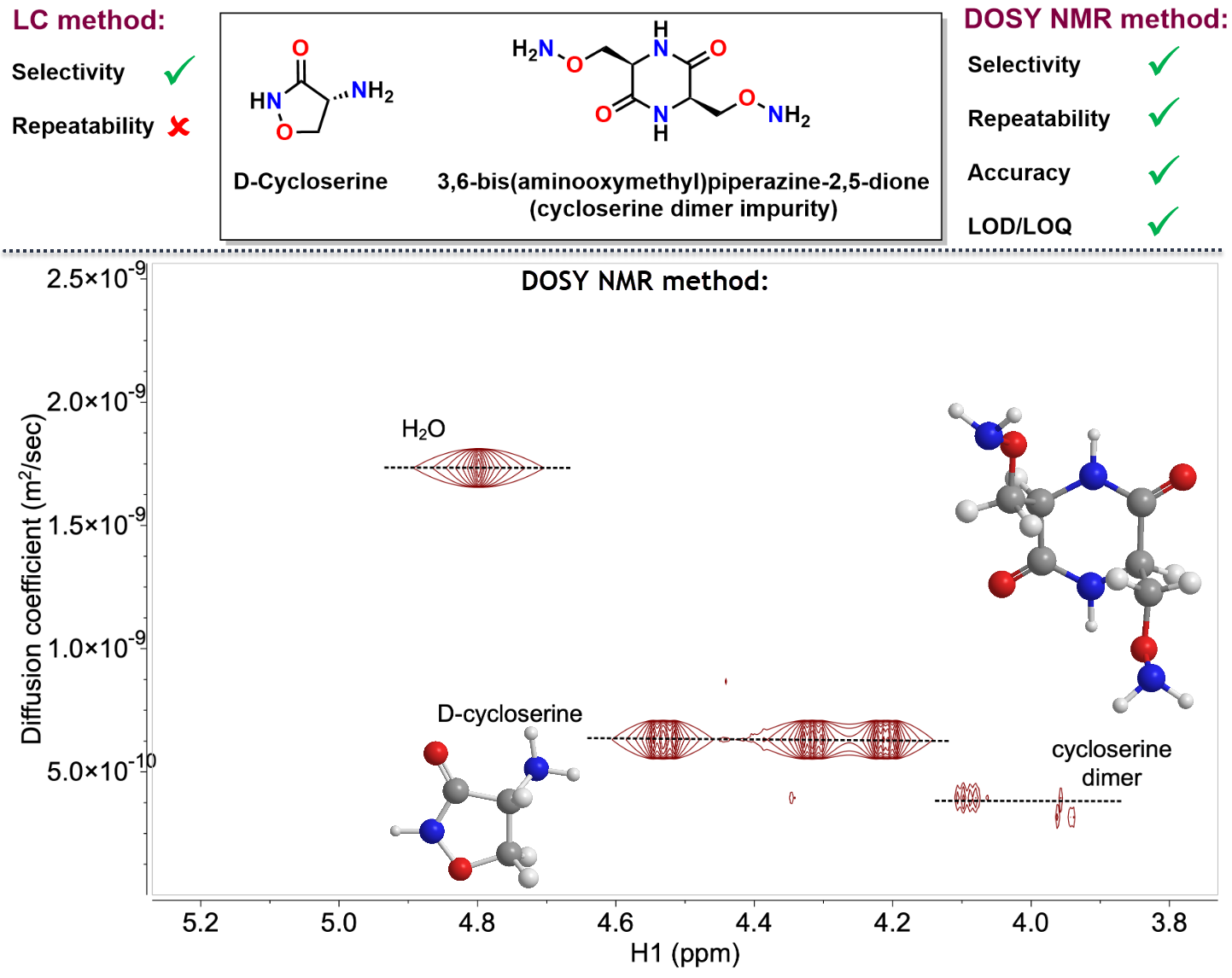
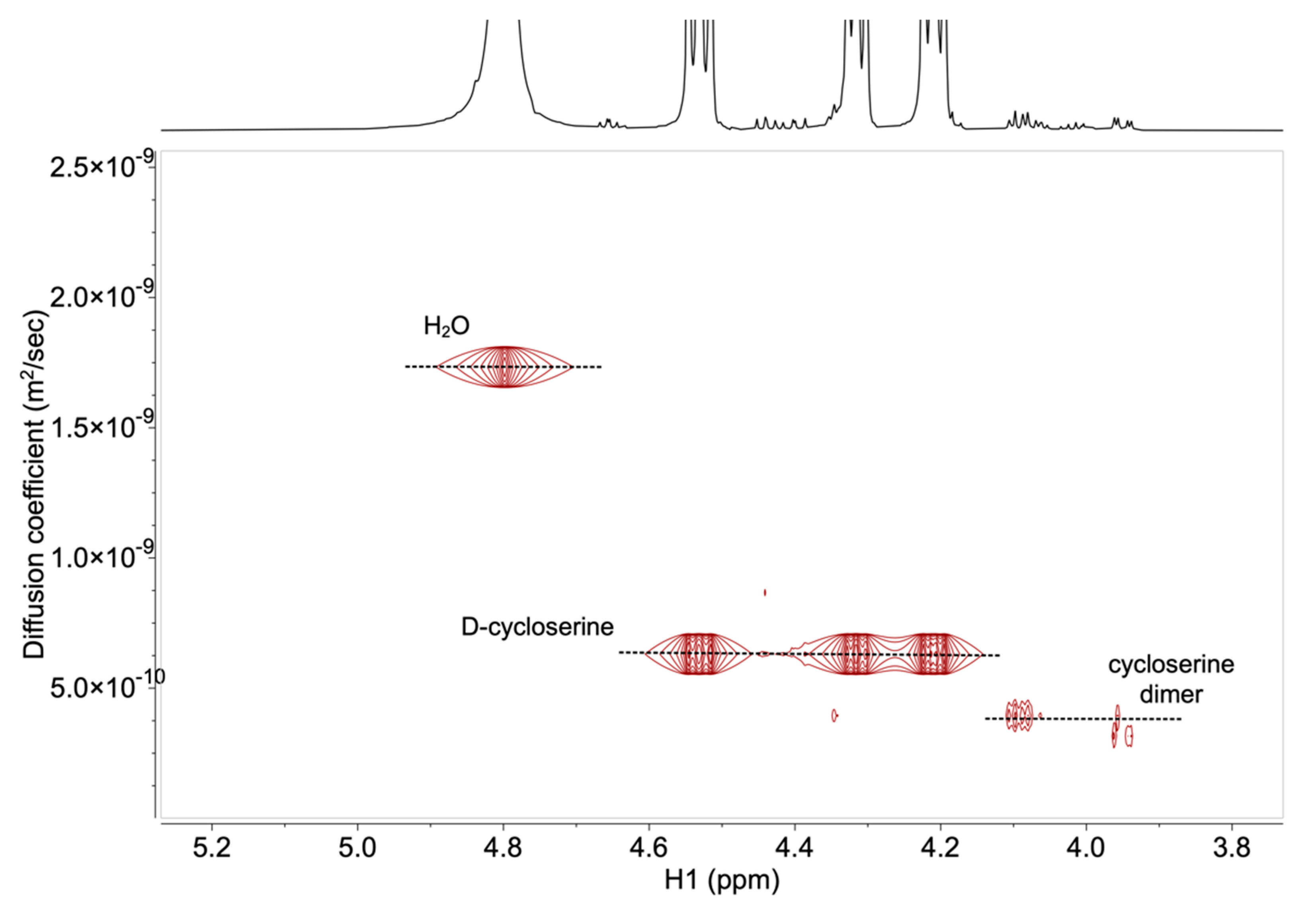
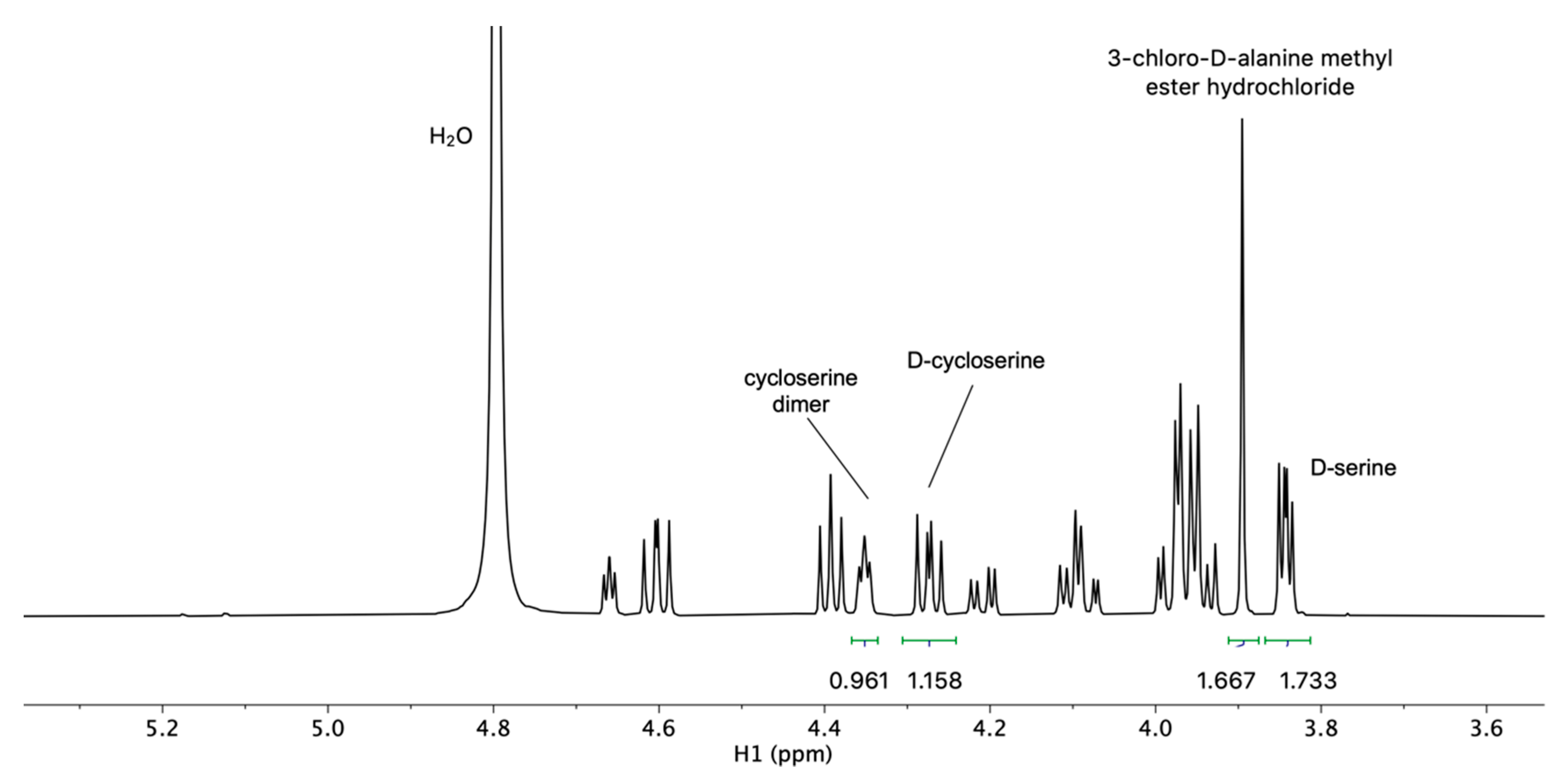
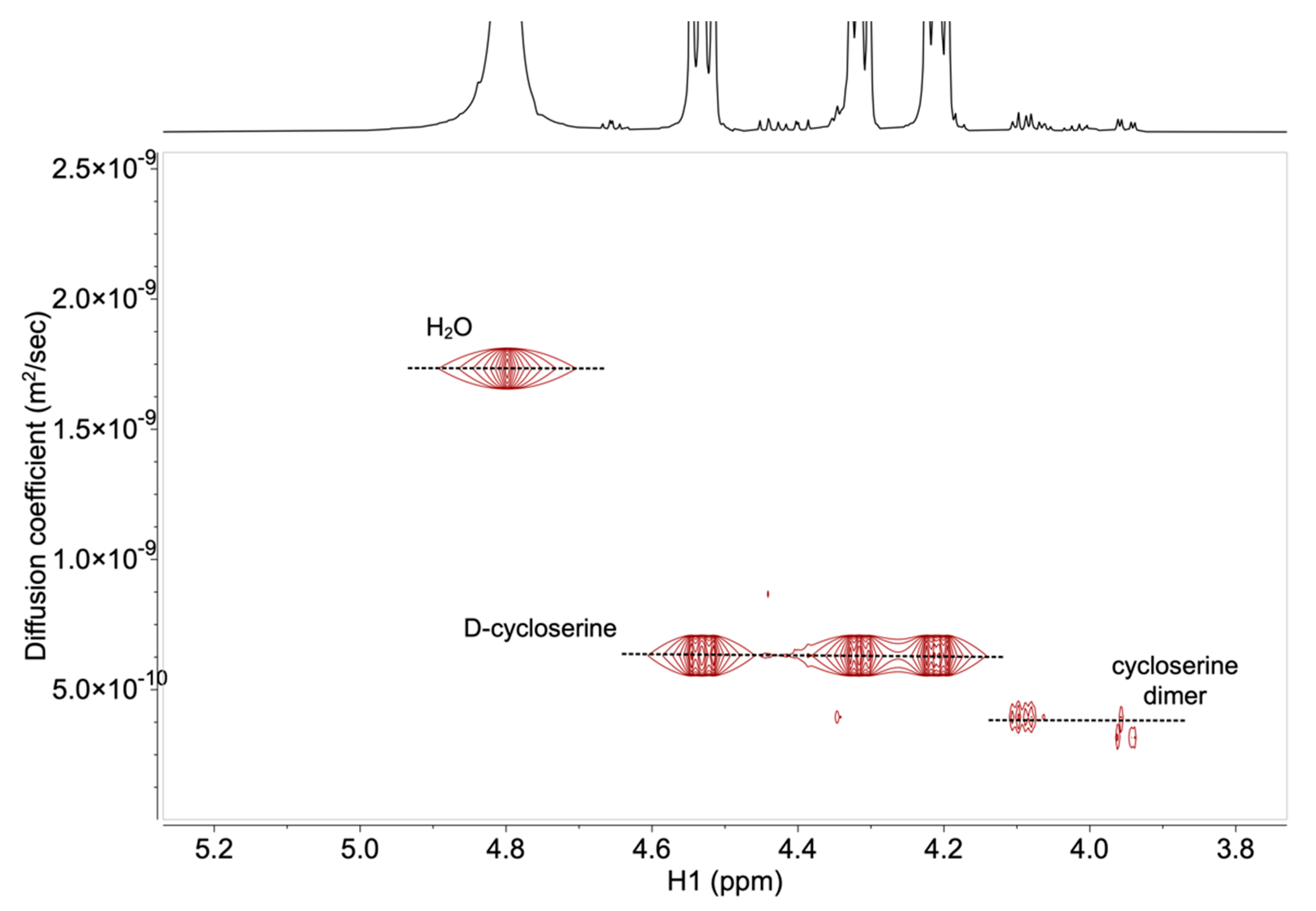
2.2. Development and Verification of the 1H DOSY NMR Method for Determination of Impurities in Samples Containing d-Cycloserine
3. Materials and Methods
3.1. Chemicals
3.2. Instrumentation
3.3. Verification of The International Pharmacopoeia (Ph. Int.) HPLC Method for Determination of Impurities in Samples Containing d-Cycloserine
3.3.1. Chromatographic Conditions
3.3.2. Standard and Sample Preparation
3.4. Development and Verification of the 1H DOSY NMR Method for Determination of Impurities in Samples Containing d-Cycloserine
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hong, W.; Cheng, L.; Xie, J. Molecular basis underlying Mycobacterium tuberculosis d-cycloserine resistance. Is there a role for ubiquinone and menaquinone metabolic pathways? Exp. Opin. Ther. Targets 2014, 18, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Schade, S.; Paulus, W. d-cycloserine in neuropsychiatric diseases: A systematic review. Int. J. Neuropsychopharmacol. 2016, 19, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Pendela, M.; Dragovic, S.; Bockx, L.; Hoogmartens, J.; Van Schepdael, A.; Adams, E. Development of a liquid chromatographic method for the determination of related substances and assay of d-cycloserine. J. Pharm. Biomed. Anal. 2008, 47, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Burge, L.; Sorgen, C.M. Evaluation of polar-embedded and polar-endcapped C12-C18 stationary phase vulnerabilities toward highly nucleophilic compounds: A case study. LCGC N. Am. 2005, 23, 762–765. [Google Scholar]
- Musson, D.G.; Maglietto, S.M.; Hwang, S.S.; Gravellese, D.; Bayne, W.F. Simultaneous quantification of cycloserine and its prodrug acetylacetonylcycloserine in plasma and urine by high-performance liquid chromatography using ultraviolet absorbance and fluorescence after post-column derivatization. J. Chromatogr. 1987, 414, 121–129. [Google Scholar] [CrossRef]
- Pesek, J.J.; Matyska, M.T.; Dang, A. Analysis of cycloserine and related compounds using aqueous normal phase chromatography/mass spectrometry. J. Pharm. Biomed. Anal. 2012, 64–65, 72–76. [Google Scholar] [CrossRef]
- Yaroshenko, D.V.; Grigoriev, A.V.; Sidorova, A.A. Development and validation of a LC-MS/MS method for d-cycloserine determination in human plasma for bioequivalence study. Anal. Bioanal. Chem. 2014, 406, 923–927. [Google Scholar] [CrossRef]
- David, V.; Ionescu, M.; Dumitrescu, V. Determination of cycloserine in human plasma by high-performance liquid chromatography with fluorescence detection, using derivatization with p-benzoquinone. J. Chromatogr. B 2001, 761, 27–33. [Google Scholar] [CrossRef]
- Yoo, G.S.; Choi, K.; Stewart, J.T. Second derivative ultraviolet spectrophotometry and high performance liquid chromatography with fluorometric detection of cycloserine using 9-chloro-10-methyl acridinium triflate as a new UV and fluorescent labeling agent. Anal. Lett. 1990, 23, 1245–1263. [Google Scholar] [CrossRef]
- World Health Organization. Monographs: Pharmaceutical substances: Cycloserine (Cycloserinum). In The International Pharmacopoeia, 9th ed.; 2019; Available online: http://apps.who.int/phint/en/p/docf/ (accessed on 20 March 2020).
- World Health Organization. Monographs: Dosage forms: Specific monographs: Cycloserine capsules (Cycloserini capsulae). In The International Pharmacopoeia, 9th ed.; 2019; Available online: http://apps.who.int/phint/en/p/docf/ (accessed on 20 March 2020).
- Kaushal, G.; Ramirez, R.; Alambo, D.; Taupradist, W.; Choksi, K.; Sirbu, C. Initial characterization of d-cycloserine for future formulation development for anxiety disorders. Drug Discov. Ther. 2011, 5, 253–260. [Google Scholar] [CrossRef][Green Version]
- The United States Pharmacopeia: Cycloserine; United States Pharmacopoeial convention, Inc.: Rockville, MD, USA, 2019; Volume 42, pp. 1187–1188.
- Hemanth Kumar, A.K.; Kumar Polisetty, A.; Sudha, V.; Vijayakumar, A.; Ramachandran, G. A selective and sensitive high performance liquid chromatography assay for the determination of cycloserine in human plasma. Indian J. Tuberc. 2018, 65, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, E.S.; Ovcharov, M.V.; Barsegyan, S.S.; Chistyakov, V.V. Structure of chemical compounds, methods of analysis and process control. Determination of cycloserine in blood plasma by HPLC/MS: Application to bioequivalence studies. Pharm. Chem. J. 2016, 50, 195–199. [Google Scholar] [CrossRef]
- Patel, D.S.; Sharma, N.; Patel, M.C.; Patel, B.N.; Shrivastav, P.S.; Sanyal, M. Development and validation of a selective and sensitive LC–MS/MS method for determination of cycloserine in human plasma: Application to bioequivalence study. J. Chromatogr. B 2011, 879, 2265–2273. [Google Scholar] [CrossRef] [PubMed]
- Polagani, S.R.; Pilli, N.R.; Maddela, R.; Gajula, R.; Gandu, V. A rapid and sensitive liquid chromatography–tandem mass spectrometric assay for cycloserine in 50μL of human plasma: Its pharmacokinetic application. J. Pharm. Biomed. Anal. 2013, 76, 21–27. [Google Scholar] [CrossRef]
- Mao, Z.; Wang, X.; Li, B.; Jin, J.; Xu, M.; Liu, Y.; Di, X. A simplified LC-MS/MS method for rapid determination of cycloserine in small-volume human plasma using protein precipitation coupled with dilution techniques to overcome matrix effects and its application to a pharmacokinetic study. Anal. Bioanal. Chem. 2017, 409, 3025–3032. [Google Scholar] [CrossRef]
- Zhang, W.; Wan, S.; Chen, L.; Wang, X.; Wang, Z.; Huang, Y. Determination of cycloserine in microdialysis samples using liquid chromatography-tandem mass spectrometry with benzoyl chloride derivatization. Biomed. Chromatogr. 2018, 32, e4187. [Google Scholar] [CrossRef]
- Karthikeyan, K.; Arularasu, G.T.; Ramadhas, R.; Chandrasekara Pillai, K. Development and validation of indirect RP-HPLC method for enantiomeric purity determination of d-cycloserine drug substance. J. Pharm. Biomed. Anal. 2011, 54, 850–854. [Google Scholar] [CrossRef]
- Fraschetti, C.; Filippi, A.; Mannina, L.; Sobolev, A.P.; Speranza, M. Role of the solvent on the stability of cycloserine under ESI-MS conditions. J. Mass. Spectrom. 2014, 49, 608–612. [Google Scholar] [CrossRef]
- Lassen, F.O.; Stammer, C.H. Cycloserine dimer hydrolysis and its equilibration with cycloserine. J. Org. Chem. 1971, 36, 2631–2634. [Google Scholar] [CrossRef]
- Lynch Miller, J.; Neuhaus, F.C.; Lassen, F.O.; Stammer, C.H. The reactions of 3,6-bis(aminooxymethyl)-2,5-piperazinedione with acid and alkali. A kinetic study. J. Org. Chem. 1968, 33, 3908–3913. [Google Scholar] [CrossRef]
- Heaton, J.C.; Smith, N.W.; McCalley, D.V. Retention characteristics of some antibiotic and anti-retroviral compounds in hydrophilic interaction chromatography using isocratic elution, and gradient elution with repeatable partial equilibration. Anal. Chim. Acta 2019, 1045, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Trefi, S.; Gilard, V.; Balayssac, S.; Malet-Martino, M.; Martino, R. Quality assessment of fluoxetine and fluvoxamine pharmaceutical formulations purchased in different countries or via the internet by 19F and 2D DOSY 1H NMR. J. Pharm. Biomed. Anal. 2008, 46, 707–722. [Google Scholar] [CrossRef] [PubMed]
- Balayssac, S.; Retailleau, E.; Bertrand, G.; Escot, M.P.; Martino, R.; Malet-Martino, M.; Gilard, V. Characterization of heroin samples by 1H NMR and 2D DOSY 1H NMR. Forensic Sci. Int. 2014, 234, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Balayssac, S.; Trefi, S.; Gilard, V.; Malet-Martino, M.; Martino, R.; Delsuc, M.A. 2D and 3D DOSY 1H NMR, a useful tool for analysis of complex mixtures: Application to herbal drugs or dietary supplements for erectile dysfunction. J. Pharm. Biomed. Anal. 2009, 50, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-K.; Jenny Park, K.-J. Simple and efficient synthetic routes to d-cycloserine. Tetrahedron Lett. 2012, 53, 1668–1670. [Google Scholar] [CrossRef]
- Kim, H.-K.; Jenny Park, K.-J. Novel practical synthesis of d-cycloserine. Tetrahedron Lett. 2012, 53, 4090–4092. [Google Scholar] [CrossRef]
- Fu, D.; Wu, Z.; Yang, Y.; Wen, W. Process of Preparation of 3-halo-d-alanine Methyl Ester or Acid Salt Thereof. Patent Application No. WO 2016155596, 6 October 2016. [Google Scholar]
- Liu, H. Synthesis Method of 3-chloro-d-alanine Methyl Ester Hydrochloride. Chinese Patent Application CN 106518695, 22 March 2017. [Google Scholar]
- ACD/Labs Percepta Predictors. Software Modules to Profile Compounds for Physicochemical, ADME, and Toxicity Properties. Available online: http://www.acdlabs.com/products/percepta/predictors.php (accessed on 20 March 2020).
- Malz, F.; Jancke, H. Validation of quantitative NMR. J. Pharm. Biomed. Anal. 2005, 38, 813–823. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solution Name | Volume WS [mL] | % d-Cycloserine 1 | Concentration [µg mL−1] |
|---|---|---|---|
| LOQ1 | 0.005 | 0.001 | 0.005 |
| LOQ2 | 0.05 | 0.01 | 0.05 |
| LOQ3 | 0.10 | 0.02 | 0.10 |
| LOQ4 | 0.15 | 0.03 | 0.15 |
| LOQ5 = Lin1 | 0.25 | 0.05 | 0.25 |
| Lin2 | 0.30 | 0.06 | 0.30 |
| Lin3 | 0.35 | 0.07 | 0.35 |
| Lin4 | 1.0 | 0.2 | 1.0 |
| Lin5 | 1.2 | 0.24 | 1.2 |
| Lin6 | 1.5 | 0.3 | 1.5 |
| Lin7 | 2.0 | 0.4 | 2.0 |
| Lin8 | 2.4 | 0.48 | 2.4 |
| Lin9 | 2.65 | 0.53 | 2.65 |
| Injection | Area [µV*sec] Cycloserine Dimer 1 | Area [µV*sec] Cycloserine Dimer 2 | Total Area [µV*sec] Cycloserine Dimers 1 & 2 |
|---|---|---|---|
| 1 | 0 | 32794 | 32794 |
| 2 | 0 | 136069 | 136069 |
| 3 | 94450 | 435062 | 529512 |
| 4 | 0 | 0 | 0 |
| 5 | 1571131 | 1261973 | 2833104 |
| 6 | 2813514 | 158198 | 2971712 |
| 7 | 3532956 | 1786999 | 5319955 |
| 8 | 4015227 | 1960635 | 5975862 |
| 9 | 4402109 | 2117970 | 6520079 |
| 10 | 4723430 | 2260472 | 6983902 |
| 11 | 4995383 | 2392454 | 7387837 |
| 12 | 5222891 | 2516870 | 7739761 |
| 13 | 5419184 | 2634801 | 8053985 |
| 14 | 5593956 | 2748004 | 8341960 |
| 15 | 5745123 | 2855549 | 8600672 |
| 16 | 5509693 | 2779019 | 8288712 |
| 17 | 136259 | 151090 | 287349 |
| 18 | 100282 | 132602 | 232884 |
| 19 | 79771 | 124348 | 204119 |
| 20 | 65357 | 119038 | 184395 |
| RSD (%) (1–20): | 90.0 | 87.7 | 88.5 |
| RSD (%) (8–20): | 68.9 | 65.8 | 67.8 |
| RSD (%) (8–16): | 11.6 | 12.7 | 11.9 |
| Concentration [μg mL−1] | S/N at δ 4.4 ppm | S/N at δ 4.1 ppm | S/N at δ 4.0 ppm | |
|---|---|---|---|---|
| 1H | 20 | 39.1 | 42.4 | 63.2 |
| 10 | 17.5 | 26.1 | 30.9 | |
| 4 | 11.3 | 13.1 | 12.3 | |
| 2 | 9.9 | 7.7 | 7.7 | |
| 1H DOSY 1 | 20 | 12.5 | 20.7 | 23.9 |
| 10 | 6.6 | 11.0 | 16.4 |
| Compound | Replicate | mx | Ix | Nx | mRS | Calculated Px (%) | Actual Px (%) |
|---|---|---|---|---|---|---|---|
| d-cycloserine 1 | replicate 1 | 2.908 | 1.365 | 1 | 2.256 | 93.122 | 99.6 |
| replicate 2 | 1.954 | 1.158 | 1 | 1.788 | 93.181 | 99.6 | |
| replicate 3 | 2.084 | 0.909 | 1 | 2.445 | 93.782 | 99.6 | |
| average: | 93.4 | 99.6 | |||||
| Cycloserine dimer 2 | replicate 1 | 1.447 | 0.712 | 2 | 2.256 | 97.627 | 95.0 |
| replicate 2 | 1.581 | 0.961 | 2 | 1.788 | 95.582 | 95.0 | |
| replicate 3 | 1.584 | 0.714 | 2 | 2.445 | 96.926 | 95.0 | |
| average: | 96.7 | 95.0 | |||||
| 3-chloro-d-alanine methyl ester HCl 3 | replicate 1 | 1.491 | 1.255 | 3 | 2.256 | 94.886 | 97.3 |
| replicate 2 | 1.560 | 1.667 | 3 | 1.788 | 95.472 | 97.3 | |
| replicate 3 | 1.988 | 1.610 | 3 | 2.445 | 98.943 | 97.3 | |
| average: | 96.4 | 97.3 | |||||
| d-serine 4 | replicate 1 | 1.557 | 0.810 | 1 | 2.256 | 106.240 | 100.0 |
| replicate 2 | 2.728 | 1.733 | 1 | 1.788 | 102.820 | 100.0 | |
| replicate 3 | 3.531 | 1.631 | 1 | 2.445 | 102.233 | 100.0 | |
| average: | 103.8 | 100.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makuc, D.; Švab, Ž.; Naumoska, K.; Plavec, J.; Časar, Z. Determination of d-Cycloserine Impurities in Pharmaceutical Dosage Forms: Comparison of the International Pharmacopoeia HPLC–UV Method and the DOSY NMR Method. Molecules 2020, 25, 1684. https://doi.org/10.3390/molecules25071684
Makuc D, Švab Ž, Naumoska K, Plavec J, Časar Z. Determination of d-Cycloserine Impurities in Pharmaceutical Dosage Forms: Comparison of the International Pharmacopoeia HPLC–UV Method and the DOSY NMR Method. Molecules. 2020; 25(7):1684. https://doi.org/10.3390/molecules25071684
Chicago/Turabian StyleMakuc, Damjan, Živa Švab, Katerina Naumoska, Janez Plavec, and Zdenko Časar. 2020. "Determination of d-Cycloserine Impurities in Pharmaceutical Dosage Forms: Comparison of the International Pharmacopoeia HPLC–UV Method and the DOSY NMR Method" Molecules 25, no. 7: 1684. https://doi.org/10.3390/molecules25071684
APA StyleMakuc, D., Švab, Ž., Naumoska, K., Plavec, J., & Časar, Z. (2020). Determination of d-Cycloserine Impurities in Pharmaceutical Dosage Forms: Comparison of the International Pharmacopoeia HPLC–UV Method and the DOSY NMR Method. Molecules, 25(7), 1684. https://doi.org/10.3390/molecules25071684