
Theoretical and Computational Insight into Solvent and Specific Ion Effects for Polyelectrolytes: The Importance of Local Molecular Interactions


Abstract
{kind=link}
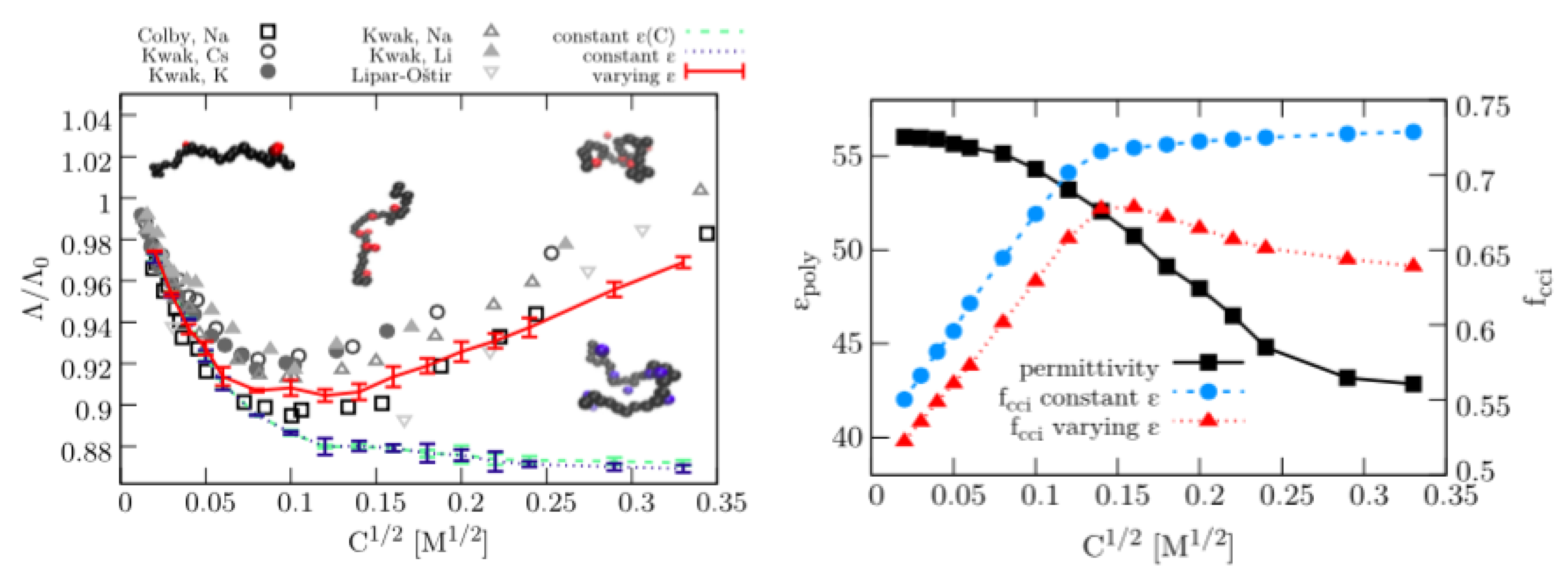{kind=link}
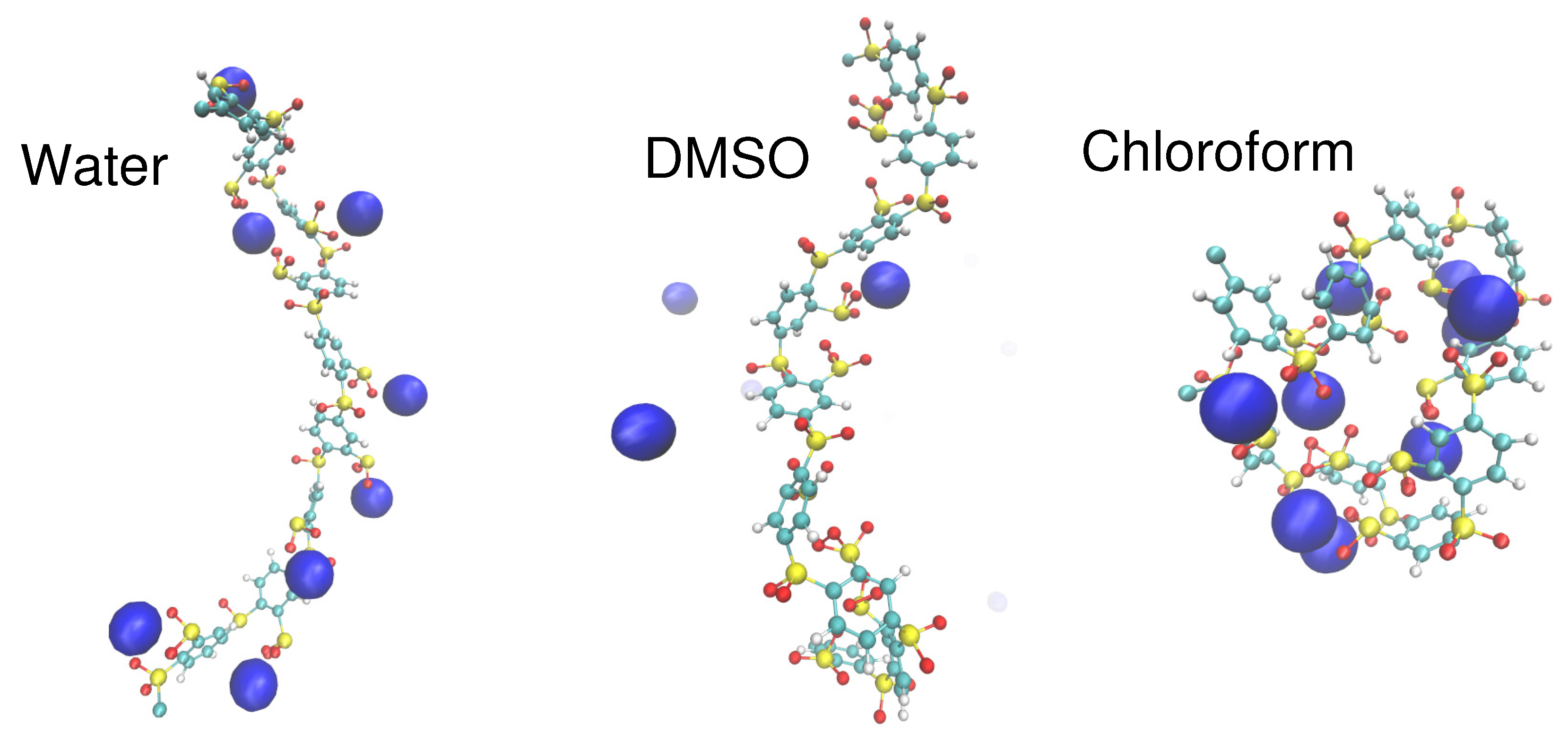{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Theoretical Background: Polyelectrolytes and Ions in Solution
2.1. Electrostatic Screening Effects
2.2. Counterion Condensation Theory
3. Solvent Effects
3.1. Dielectric Decrement Effects
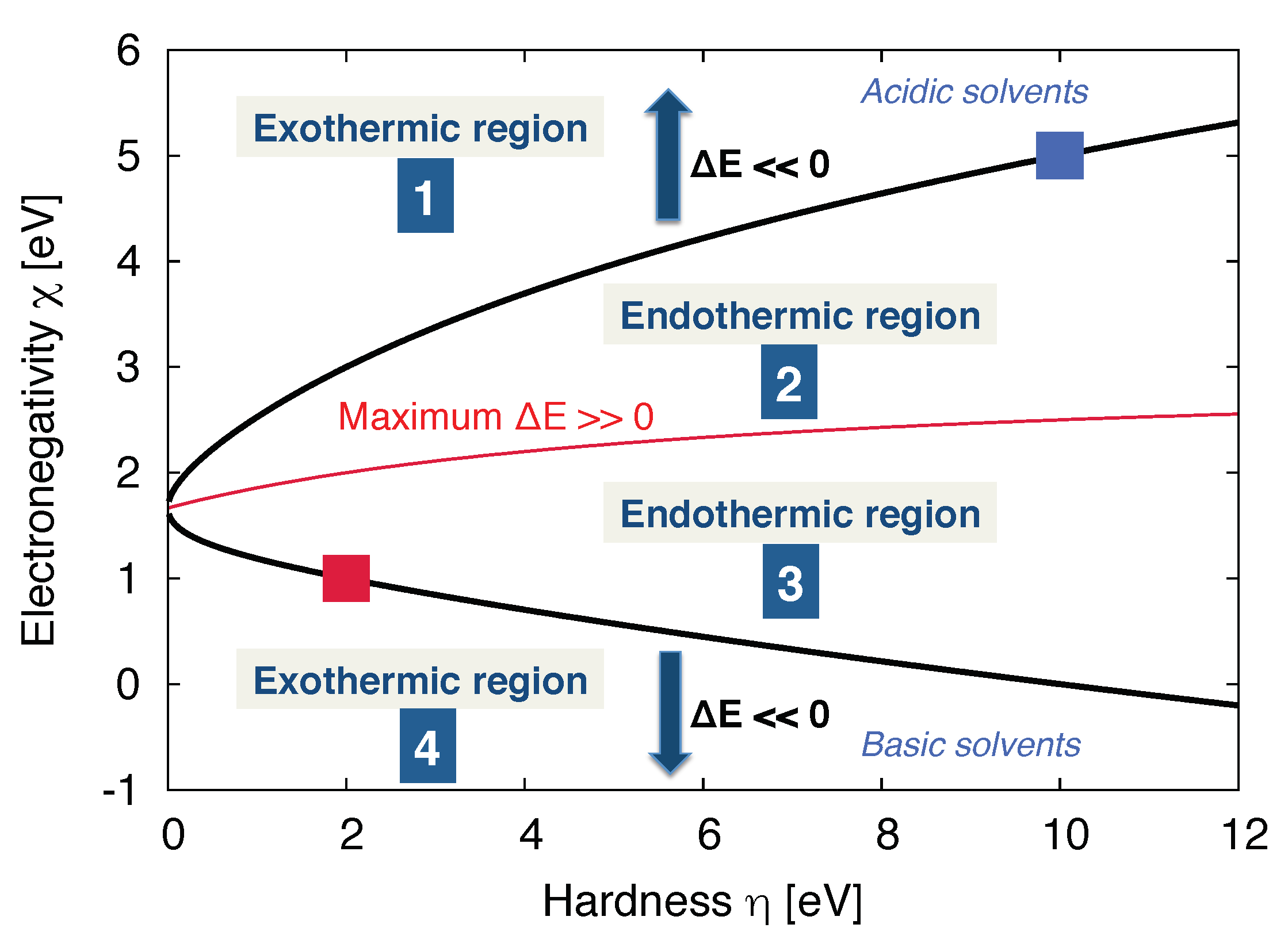
3.2. Molecular Properties of the Solvent: Donor/Acceptor Numbers and Chemical Hardnesses
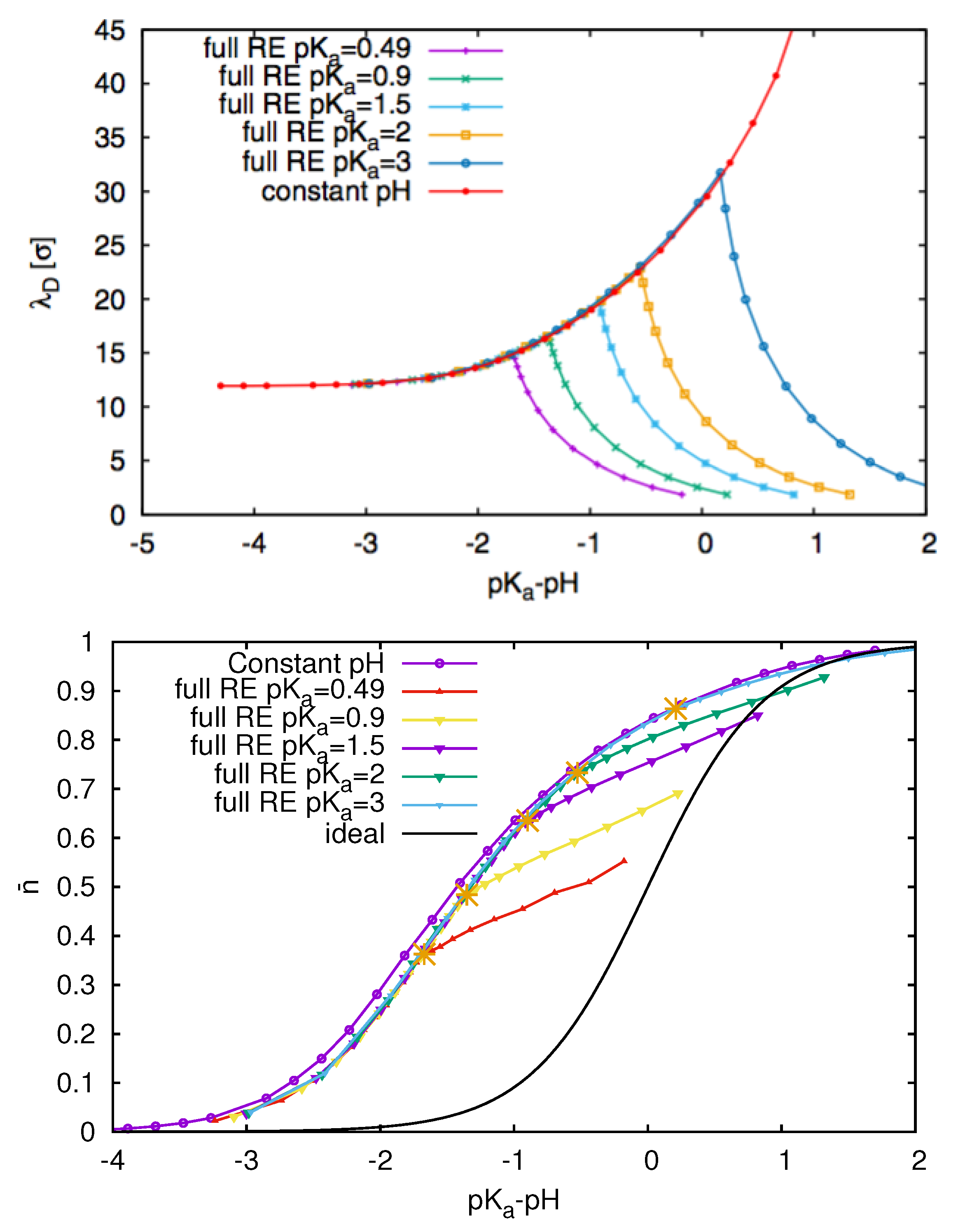
3.3. Weak Polyelectrolytes: pH Value Effects
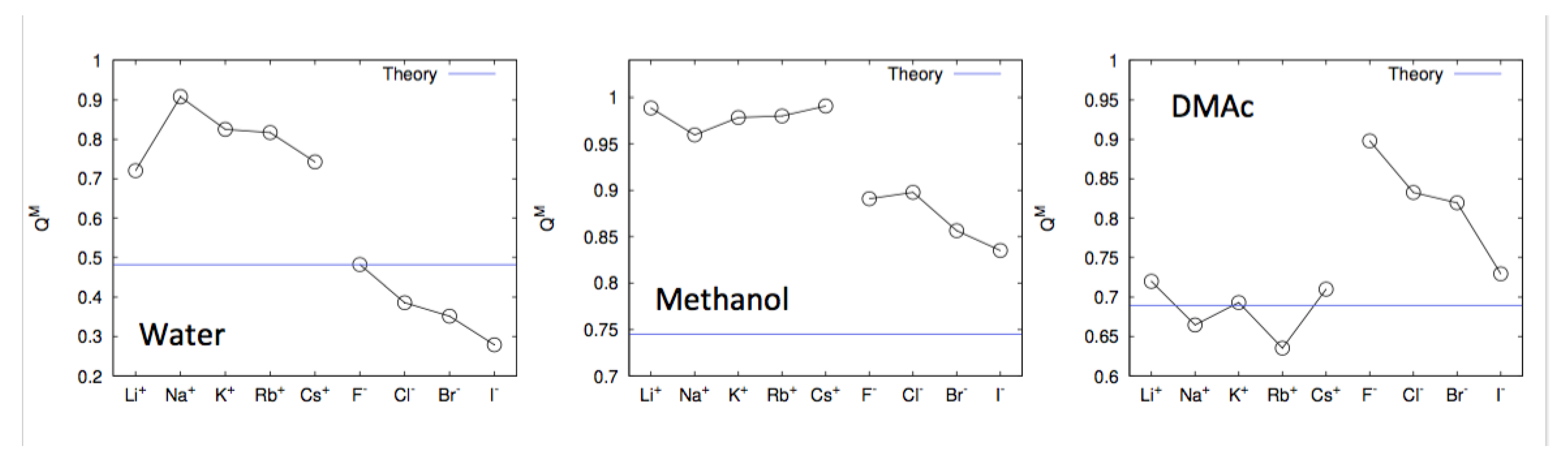
4. Specific Ion Effects
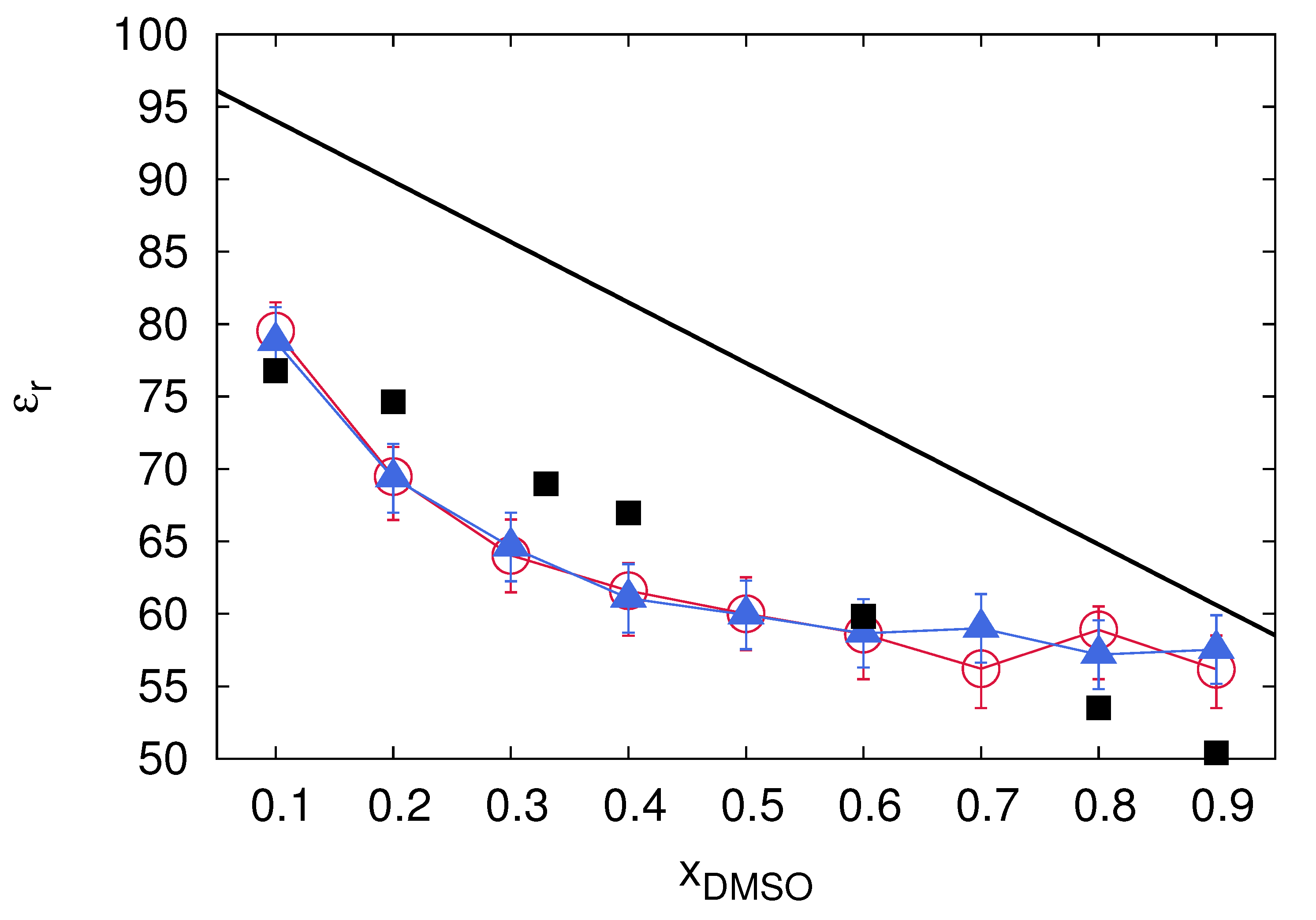
5. Co-Solute and Co-Solvent Effects
6. Summary and Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- McNaught, A.D.; McNaught, A.D. Compendium of Chemical Terminology; Blackwell Science Oxford: Oxford, UK, 1997; Volume 1669. [Google Scholar]
- Dobrynin, A.V.; Colby, R.H.; Rubinstein, M. Scaling theory of polyelectrolyte solutions. Macromolecules 1995, 28, 1859–1871. [Google Scholar] [CrossRef]
- Dobrynin, A.V.; Rubinstein, M. Theory of polyelectrolytes in solutions and at surfaces. Prog. Polym. Sci. 2005, 30, 1049–1118. [Google Scholar] [CrossRef]
- De Gennes, P.G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Oxford University Press: Oxford, UK, 1988. [Google Scholar]
- Boroudjerdi, H.; Kim, Y.W.; Naji, A.; Netz, R.R.; Schlagberger, X.; Serr, A. Statics and dynamics of strongly charged soft matter. Phys. Rep. 2005, 416, 129–199. [Google Scholar] [CrossRef]
- Slater, G.W.; Holm, C.; Chubynsky, M.V.; de Haan, H.W.; Dube, A.; Grass, K.; Hickey, O.A.; Kingsburry, C.; Sean, D.; Shendruk, T.N.; et al. Modeling the separation of macromolecules: A review of current computer simulation methods. Electrophoresis 2009, 30, 792–818. [Google Scholar] [CrossRef]
- Pagonabarraga, I.; Rotenberg, B.; Frenkel, D. Recent advances in the modelling and simulation of electrokinetic effects: Bridging the gap between atomistic and macroscopic descriptions. Phys. Chem. Chem. Phys. 2010, 12, 9566–9580. [Google Scholar] [CrossRef]
- Streek, M.; Schmid, F.; Duong, T.T.; Ros, A. Mechanisms of DNA separation in entropic trap arrays: A Brownian dynamics simulation. J. Biotechnol. 2004, 112, 79–89. [Google Scholar] [CrossRef]
- Frank, S.; Winkler, R.G. Mesoscale hydrodynamic simulation of short polyelectrolytes in electric fields. J. Chem. Phys. 2009, 131, 234905. [Google Scholar] [CrossRef]
- Grass, K.; Böhme, U.; Scheler, U.; Cottet, H.; Holm, C. Importance of hydrodynamic shielding for the dynamic behavior of short polyelectrolyte chains. Phys. Rev. Lett. 2008, 100, 096104. [Google Scholar] [CrossRef]
- Smiatek, J.; Schmid, F. Mesoscopic simulations of electroosmotic flow and electrophoresis in nanochannels. Comput. Phys. Commun. 2011, 182, 1941–1944. [Google Scholar] [CrossRef]
- Smiatek, J.; Schmid, F. Polyelectrolyte electrophoresis in nanochannels: A dissipative particle dynamics simulation. J. Phys. Chem. B 2010, 114, 6266–6272. [Google Scholar] [CrossRef][Green Version]
- Muthukumar, M. Dynamics of polyelectrolyte solutions. J. Chem. Phys. 1997, 107, 2619–2635. [Google Scholar] [CrossRef]
- Krishnamoorthy, A.N.; Holm, C.; Smiatek, J. Specific ion effects for polyelectrolytes in aqueous and non-aqueous media: The importance of the ion solvation behavior. Soft Matter 2018, 14, 6243–6255. [Google Scholar] [CrossRef] [PubMed]
- Smiatek, J.; Holm, C. From the atomistic to the macromolecular scale: Distinct simulation approaches for polyelectrolyte solutions. In Handbook of Materials Modeling; Springer International Publishing: Heidelberg, Germany, 2018. [Google Scholar]
- Smiatek, J.; Harishchandra, R.K.; Rubner, O.; Galla, H.J.; Heuer, A. Properties of compatible solutes in aqueous solution. Biophys. Chem. 2012, 160, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Smiatek, J.; Wohlfarth, A.; Holm, C. The solvation and ion condensation properties for sulfonated polyelectrolytes in different solvents—A computational study. New J. Phys. 2014, 16, 025001. [Google Scholar] [CrossRef]
- Krishnamoorthy, A.N.; Zeman, J.; Holm, C.; Smiatek, J. Preferential solvation and ion association properties in aqueous dimethyl sulfoxide solutions. Phys. Chem. Chem. Phys. 2016, 18, 31312–31322. [Google Scholar] [CrossRef]
- Krishnamoorthy, A.N.; Holm, C.; Smiatek, J. The influence of co-solutes on the chemical equilibrium—A Kirkwood-Buff theory for ion pair association-dissociation processes in ternary electrolyte solutions. J. Phys. Chem. C 2018, 122, 10293–10392. [Google Scholar] [CrossRef]
- Oprzeska-Zingrebe, E.A.; Smiatek, J. Some Notes on the Thermodynamic Accuracy of Coarse-Grained Models. Front. Mol. Biosci. 2019, 6, 87. [Google Scholar] [CrossRef]
- Guenza, M.; Dinpajooh, M.; McCarty, J.; Lyubimov, I. Accuracy, transferability, and efficiency of coarse-grained models of molecular liquids. J. Phys. Chem. B 2018, 122, 10257–10278. [Google Scholar] [CrossRef]
- Onufriev, A.V.; Case, D.A. Generalized Born Implicit Solvent Models for Biomolecules. Annu. Rev. Biophys. 2019, 48, 275–296. [Google Scholar] [CrossRef]
- Landsgesell, J.; Holm, C.; Smiatek, J. Simulation of weak polyelectrolytes: A comparison between the constant pH and the reaction ensemble method. Eur. Phys. J. Spec. Top. 2017, 226, 725–736. [Google Scholar] [CrossRef]
- Landsgesell, J.; Holm, C.; Smiatek, J. Wang–Landau Reaction Ensemble Method: Simulation of Weak Polyelectrolytes and General Acid–Base Reactions. J. Chem. Theory Comput. 2017, 13, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Fahrenberger, F.; Hickey, O.A.; Smiatek, J.; Holm, C. The influence of charged-induced variations in the local permittivity on the static and dynamic properties of polyelectrolyte solutions. J. Chem. Phys. 2015, 143, 243140. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, A.; Fenley, A.T.; Tolokh, I.S.; Onufriev, A.V. Charge hydration asymmetry: The basic principle and how to use it to test and improve water models. J. Phys. Chem. B 2012, 116, 9776–9783. [Google Scholar] [CrossRef] [PubMed]
- Weyman, A.; Bier, M.; Holm, C.; Smiatek, J. Microphase separation and the formation of ion conductivity channels in poly (ionic liquid) s: A coarse-grained molecular dynamics study. J. Chem. Phys. 2018, 148, 193824. [Google Scholar] [CrossRef]
- Holm, C.; Limbach, H.; Kremer, K. Poor-solvent polyelectrolytes. J. Phys. Condens. Matter 2002, 15, S205. [Google Scholar] [CrossRef]
- Limbach, H.; Sayar, M.; Holm, C. Polyelectrolyte bundles. J. Phys. Condens. Matter 2004, 16, S2135. [Google Scholar] [CrossRef]
- Dormidontova, E.E.; Erukhimovich, I.Y.; Khokhlov, A.R. Microphase separation in poor-solvent polyelectrolyte solutions: Phase diagram. Macromol. Theory Simul. 1994, 3, 661–675. [Google Scholar] [CrossRef]
- Cerda, J.J.; Qiao, B.; Holm, C. Understanding polyelectrolyte multilayers: An open challenge for simulations. Soft Matter 2009, 5, 4412–4425. [Google Scholar] [CrossRef]
- Vögele, M.; Holm, C.; Smiatek, J. Coarse-grained simulations of polyelectrolyte complexes: MARTINI models for poly(styrene sulfonate) and poly(diallyldimethylammonium). J. Chem. Phys. 2015, 143, 243151. [Google Scholar] [CrossRef]
- Smiatek, J.; Heuer, A.; Winter, M. Properties of Ion Complexes and their Impact on Charge Transport in Organic Solvent–based Electrolyte Solutions for Lithium Batteries: Insights from a Theoretical Perspective. Batteries 2018, 4, 62. [Google Scholar] [CrossRef]
- Andreev, M.; Prabhu, V.M.; Douglas, J.F.; Tirrell, M.; de Pablo, J.J. Complex coacervation in polyelectrolytes from a coarse-grained model. Macromolecules 2018, 51, 6717–6723. [Google Scholar] [CrossRef]
- Andelman, D. Electrostatic properties of membranes: The Poisson-Boltzmann theory. In Handbook of Biological Physics; Elsevier: Amsterdam, The Netherland, 1995; Volume 1, pp. 603–642. [Google Scholar]
- Grochowski, P.; Trylska, J. Continuum molecular electrostatics, salt effects, and counterion binding—A review of the Poisson–Boltzmann theory and its modifications. Biopolymers 2008, 89, 93–113. [Google Scholar] [CrossRef] [PubMed]
- Israelachvili, J.N. Intermolecular and Surface Forces; Academic Press: Cambridge, MA, USA, 2011. [Google Scholar]
- Hickey, O.A.; Shendruk, T.N.; Harden, J.L.; Slater, G.W. Simulations of free-solution electrophoresis of polyelectrolytes with a finite Debye length using the Debye-Hückel approximation. Phys. Rev. Lett. 2012, 109, 098302. [Google Scholar] [CrossRef] [PubMed]
- Hickey, O.A.; Holm, C.; Smiatek, J. Lattice-Boltzmann simulations of the electrophoretic stretching of polyelectrolytes: The importance of hydrodynamic interactions. J. Chem. Phys. 2014, 140, 164904. [Google Scholar] [CrossRef]
- Roy, T.; Szuttor, K.; Smiatek, J.; Holm, C.; Hardt, S. Stretching of surface-tethered polymers in pressure-driven flow under confinement. Soft Matter 2017, 13, 6189–6196. [Google Scholar] [CrossRef]
- Szuttor, K.; Roy, T.; Hardt, S.; Holm, C.; Smiatek, J. The stretching force on a tethered polymer in pressure-driven flow. J. Chem. Phys. 2017, 147, 034902. [Google Scholar] [CrossRef]
- Manning, G. Limiting Laws and Counterion Condensation in Polyelectrolyte Solutions I. Colligative Properties. J. Chem. Phys. 1969, 51, 924–933. [Google Scholar] [CrossRef]
- Manning, G.S.; Ray, J. Counterion condensation revisited. J. Biomol. Struct. Dyn. 1998, 16, 461–476. [Google Scholar] [CrossRef]
- Oosawa, F. Polyelectrolytes; Marcel Dekker: New York, NY, USA, 1971. [Google Scholar]
- Muthukumar, M. Theory of counter-ion condensation on flexible polyelectrolytes: Adsorption mechanism. J. Chem. Phys. 2004, 120, 9343–9350. [Google Scholar] [CrossRef]
- Dobrynin, A.V.; Rubinstein, M. Counterion condensation and phase separation in solutions of hydrophobic polyelectrolytes. Macromolecules 2001, 34, 1964–1972. [Google Scholar] [CrossRef]
- Dobrynin, A.V. Effect of counterion condensation on rigidity of semiflexible polyelectrolytes. Macromolecules 2006, 39, 9519–9527. [Google Scholar] [CrossRef]
- Manning, G.S. Counterion condensation on charged spheres, cylinders, and planes. J. Phys. Chem. B 2007, 111, 8554–8559. [Google Scholar] [CrossRef] [PubMed]
- Marcus, Y. Ions in Solution and Their Solvation; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Manning, G.S. Counterion condensation theory constructed from different models. Phys. A 1996, 231, 236–253. [Google Scholar] [CrossRef]
- Deserno, M.; Holm, C.; May, S. Fraction of condensed counterions around a charged rod: Comparison of Poisson-Boltzmann theory and computer simulations. Macromolecules 2000, 33, 199–206. [Google Scholar] [CrossRef]
- Deserno, M.; Holm, C. Cell-model and Poisson-Boltzmann-theory: A brief introduction. In Electrostatic Effects in Soft Matter and Biophysics; Holm, C., Kékicheff, P., Podgornik, R., Eds.; NATO Science Series II—Mathematics, Physics and Chemistry; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2001; Volume 46, pp. 27–50. [Google Scholar]
- Heyda, J.; Dzubiella, J. Ion-specific counterion condensation on charged peptides: Poisson–Boltzmann vs. atomistic simulations. Soft Matter 2012, 8, 9338–9344. [Google Scholar] [CrossRef]
- Batys, P.; Luukkonen, S.; Sammalkorpi, M. Ability of Poisson–Boltzmann Equation to Capture Molecular Dynamics Predicted Ion Distribution around Polyelectrolytes. Phys. Chem. Chem. Phys. 2017, 19, 24583–24593. [Google Scholar] [CrossRef]
- Zeman, J.; Holm, C.; Smiatek, J. The Effect of Small Organic Cosolutes on Water Structure and Dynamics. J. Chem. Eng. Data 2019. [Google Scholar] [CrossRef]
- Smiatek, J. Osmolyte effects: Impact on the aqueous solution around charged and neutral spheres. J. Phys. Chem. B 2014, 118, 771–782. [Google Scholar] [CrossRef]
- Schröder, C.; Haberler, M.; Steinhauser, O. On the computation and contribution of conductivity in molecular ionic liquids. J. Chem. Phys. 2008, 128, 134501. [Google Scholar] [CrossRef]
- Michalowsky, J.; Zeman, J.; Holm, C.; Smiatek, J. A polarizable MARTINI model for monovalent ions in aqueous solution. J. Chem. Phys. 2018, 149, 163319. [Google Scholar] [CrossRef]
- Neumann, M. Dipole moment fluctuation formulas in computer simulations of polar systems. Mol. Phys. 1983, 50, 841–858. [Google Scholar] [CrossRef]
- Caillol, J.; Levesque, D.; Weis, J. Theoretical calculation of ionic solution properties. J. Chem. Phys. 1986, 85, 6645–6657. [Google Scholar] [CrossRef]
- Caillol, J.M.; Levesque, D.; Weis, J.J. Electrical properties of polarizable ionic solutions. I. Theoretical aspects. J. Chem. Phys. 1989, 91, 5544–5554. [Google Scholar] [CrossRef]
- Bonthuis, D.J.; Gekle, S.; Netz, R.R. Dielectric profile of interfacial water and its effect on double-layer capacitance. Phys. Rev. Lett. 2011, 107, 166102. [Google Scholar] [CrossRef]
- Gekle, S.; Netz, R.R. Anisotropy in the dielectric spectrum of hydration water and its relation to water dynamics. J. Chem. Phys. 2012, 137, 104704. [Google Scholar] [CrossRef]
- Fahrenberger, F.; Hickey, O.A.; Smiatek, J.; Holm, C. Importance of varying permittivity on the conductivity of polyelectrolyte solutions. Phys. Rev. Lett. 2015, 115, 118301. [Google Scholar] [CrossRef]
- Vögele, M.; Holm, C.; Smiatek, J. Properties of the polarizable MARTINI water model: A comparative study for aqueous electrolyte solutions. J. Mol. Liquids 2015, 212, 103. [Google Scholar] [CrossRef]
- Michalowsky, J.; Schäfer, L.V.; Holm, C.; Smiatek, J. A refined polarizable water model for the coarse-grained MARTINI force field with long-range electrostatic interactions. J. Chem. Phys. 2017, 146, 054501. [Google Scholar] [CrossRef]
- Hahn, M.B.; Solomun, T.; Wellhausen, R.; Hermann, S.; Seitz, H.; Meyer, S.; Kunte, H.J.; Zeman, J.; Uhlig, F.; Smiatek, J.; et al. Influence of the Compatible Solute Ectoine on the Local Water Structure: Implications for the Binding of the Protein G5P to DNA. J. Phys. Chem. B 2015, 119, 15212–15220. [Google Scholar] [CrossRef]
- Hess, B.; van der Vegt, N.F. Cation specific binding with protein surface charges. Proc. Natl. Acad. Sci. USA 2009, 106, 13296–13300. [Google Scholar] [CrossRef]
- Chremos, A.; Douglas, J.F. Communication: Counter-ion solvation and anomalous low-angle scattering in salt-free polyelectrolyte solutions. J. Chem. Phys. 2017, 147, 241103. [Google Scholar] [CrossRef] [PubMed]
- Chremos, A.; Douglas, J.F. Polyelectrolyte association and solvation. J. Chem. Phys. 2018, 149, 163305. [Google Scholar] [CrossRef] [PubMed]
- Chremos, A.; Douglas, J.F. The influence of polymer and ion solvation on the conformational properties of flexible polyelectrolytes. Gels 2018, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Wohlfarth, A.; Smiatek, J.; Kreuer, K.D.; Takamuku, S.; Jannasch, P.; Meier, J. Proton dissociation of sulfonated polysulfones: Influence of molecular structure and conformation. Macromolecules 2015, 48, 1134–1143. [Google Scholar] [CrossRef]
- Krishnamoorthy, A.N.; Oldiges, K.; Heuer, A.; Winter, M.; Cekic-Laskovic, I.; Holm, C.; Smiatek, J. Electrolyte solvents for high voltage lithium ion batteries: Ion pairing mechanisms, ionic conductivity, and specific anion effects in adiponitrile. Phys. Chem. Chem. Phys. 2018. [Google Scholar] [CrossRef]
- Borodin, O.; Smith, G.D. LiTFSI structure and transport in ethylene carbonate from molecular dynamics simulations. J. Phys. Chem. B 2006, 110, 4971–4977. [Google Scholar] [CrossRef]
- Lesch, V.; Heuer, A.; Holm, C.; Smiatek, J. Properties of Apolar Solutes in Alkyl Imidazolium-Based Ionic Liquids: The Importance of Local Interactions. ChemPhysChem 2016, 17, 387–394. [Google Scholar] [CrossRef]
- Nandy, A.; Smiatek, J. Mixtures of LiTFSI and urea: Ideal thermodynamic behavior as key to the formation of deep eutectic solvents? Phys. Chem. Chem. Phys. 2019, 21, 12279–12287. [Google Scholar] [CrossRef]
- Gutmann, V. Empirical parameters for donor and acceptor properties of solvents. Electrochim. Acta 1976, 21, 661–670. [Google Scholar] [CrossRef]
- Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Smiatek, J. Enthalpic contributions to solvent–solute and solvent–ion interactions: Electronic perturbation as key to the understanding of molecular attraction. J. Chem. Phys. 2019, 150, 174112. [Google Scholar] [CrossRef]
- Smiatek, J. Specific Ion Effects and the Law of Matching Solvent Affinities: A Conceptual Density Functional Theory Approach. J. Phys. Chem. B 2020, 124, 2191–2197. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Giri, S. Electrophilicity index within a conceptual DFT framework. Ann. Rep. Phys. Chem. C 2009, 105, 13–39. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef]
- Chen, W.; Morrow, B.H.; Shi, C.; Shen, J.K. Recent development and application of constant pH molecular dynamics. Mol. Simul. 2014, 40, 830–838. [Google Scholar] [CrossRef]
- Reed, C.E.; Reed, W.F. Monte Carlo study of titration of linear polyelectrolytes. J. Chem. Phys. 1992, 96, 1609–1620. [Google Scholar] [CrossRef]
- Mongan, J.; Case, D.A.; McCammon, J.A. Constant pH molecular dynamics in generalized Born implicit solvent. J. Comput. Chem. 2004, 25, 2038–2048. [Google Scholar] [CrossRef]
- Heath Turner, C.; Brennan, J.K.; Lisal, M.; Smith, W.R.; Karl Johnson, J.; Gubbins, K.E. Simulation of chemical reaction equilibria by the reaction ensemble Monte Carlo method: A review. Mol. Simul. 2008, 34, 119–146. [Google Scholar] [CrossRef]
- Smith, P.E. Cosolvent interactions with biomolecules:Relating computer simulation data to experimental thermodynamic data. J. Phys. Chem. B 2004, 108, 18716–18724. [Google Scholar] [CrossRef]
- Mazzini, V.; Craig, V.S. Specific-ion effects in non-aqueous systems. Curr. Opin. Colloid Int. Sci. 2016, 23, 82–93. [Google Scholar] [CrossRef]
- Mazzini, V.; Craig, V.S. What is the fundamental ion-specific series for anions and cations? Ion specificity in standard partial molar volumes of electrolytes and electrostriction in water and non-aqueous solvents. Chem. Sci. 2017, 8, 7052–7065. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, V.; Liu, G.; Craig, V.S. Probing the Hofmeister series beyond water: Specific-ion effects in non-aqueous solvents. J. Chem. Phys. 2018, 148, 222805. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.D. Charge density-dependent strength of hydration and biological structure. Biophys. J. 1997, 72, 65. [Google Scholar] [CrossRef]
- Salis, A.; Ninham, B.W. Models and mechanisms of Hofmeister effects in electrolyte solutions, and colloid and protein systems revisited. Chem. Soc. Rev. 2014, 43, 7358–7377. [Google Scholar] [CrossRef]
- Mazzini, V.; Craig, V.S. Volcano Plots Emerge from a Sea of Nonaqueous Solvents: The Law of Matching Water Affinities Extends to All Solvents. ACS Cent. Sci. 2018, 4, 1056–1064. [Google Scholar] [CrossRef]
- Lytle, T.K.; Chang, L.W.; Markiewicz, N.; Perry, S.L.; Sing, C.E. Designing electrostatic interactions via polyelectrolyte monomer sequence. ACS Cent. Sci. 2019, 5, 709–718. [Google Scholar] [CrossRef]
- Sing, C.E. Development of the modern theory of polymeric complex coacervation. Adv. Colloid Interface Sci. 2017, 239, 2–16. [Google Scholar] [CrossRef]
- Yigit, C.; Heyda, J.; Ballauff, M.; Dzubiella, J. Like-charged protein-polyelectrolyte complexation driven by charge patches. J. Chem. Phys. 2015, 143, 064905. [Google Scholar] [CrossRef]
- Chudoba, R.; Heyda, J.; Dzubiella, J. Tuning the collapse transition of weakly charged polymers by ion-specific screening and adsorption. Soft Matter 2018, 14, 9631–9642. [Google Scholar] [CrossRef]
- Solis, F.J.; De La Cruz, M.O. Collapse of flexible polyelectrolytes in multivalent salt solutions. J. Chem. Phys. 2000, 112, 2030–2035. [Google Scholar] [CrossRef]
- Antila, H.S.; Van Tassel, P.R.; Sammalkorpi, M. Repulsion between oppositely charged rod-shaped macromolecules: Role of overcharging and ionic confinement. J. Chem. Phys. 2017, 147, 124901. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.D.; Olvera de la Cruz, M. Manipulation of confined polyelectrolyte conformations through dielectric mismatch. ACS Nano 2019, 13, 9298–9305. [Google Scholar] [CrossRef] [PubMed]
- Smiatek, J. Aqueous ionic liquids and their influence on protein conformations: An overview on recent theoretical and experimental insights. J. Phys. Condens. Matter 2017, 29, 233001. [Google Scholar] [CrossRef] [PubMed]
- Ben-Naim, A.Y. Statistical Thermodynamics for Chemists and Biochemists; Springer: Berlin, Germany, 1992. [Google Scholar]
- Pierce, V.; Kang, M.; Aburi, M.; Weerasinghe, S.; Smith, P.E. Recent Applications of Kirkwood-Buff Theory to Biological Systems. Cell. Biochem. Biophys. 2008, 50, 1–22. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smiatek, J. Theoretical and Computational Insight into Solvent and Specific Ion Effects for Polyelectrolytes: The Importance of Local Molecular Interactions. Molecules 2020, 25, 1661. https://doi.org/10.3390/molecules25071661
Smiatek J. Theoretical and Computational Insight into Solvent and Specific Ion Effects for Polyelectrolytes: The Importance of Local Molecular Interactions. Molecules. 2020; 25(7):1661. https://doi.org/10.3390/molecules25071661
Chicago/Turabian StyleSmiatek, Jens. 2020. "Theoretical and Computational Insight into Solvent and Specific Ion Effects for Polyelectrolytes: The Importance of Local Molecular Interactions" Molecules 25, no. 7: 1661. https://doi.org/10.3390/molecules25071661
APA StyleSmiatek, J. (2020). Theoretical and Computational Insight into Solvent and Specific Ion Effects for Polyelectrolytes: The Importance of Local Molecular Interactions. Molecules, 25(7), 1661. https://doi.org/10.3390/molecules25071661