
Visible Light-Induced Homolytic Cleavage of Perfluoroalkyl Iodides Mediated by Phosphines

 , and
, and
Abstract
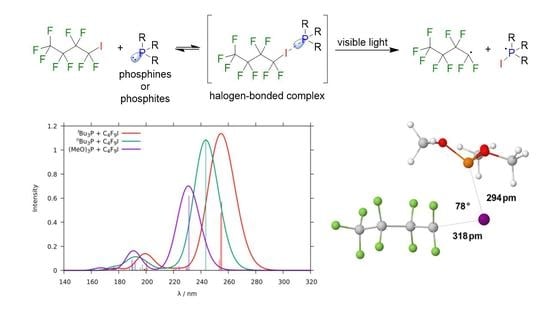
1. Introduction
2. Results
2.1. Quantum Chemical Characterization of the Compounds in the Franck–Condon Region
2.1.1. Perfluorobutyl Iodide
2.1.2. Phosphines
2.1.3. Phosphine–Perfluorobutyl Iodide Adducts
2.2. Relaxed Excited-State Geometries of the Phosphine–Perfluorobutyl Iodide Adducts
2.2.1. The First Excited Triplet State
2.2.2. The First Excited Singlet State
3. Discussion
4. Materials and Methods
4.1. Computational Methods
4.2. Experimental Procedures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATRA | atom transfer radical addition |
| CT | charge transfer |
| DCM | dichloromethane |
| DFT | density functional theory |
| EDA | electron donor–acceptor |
| HOMO | highest occupied molecular orbital |
| IC | internal conversion |
| ISC | intersystem crossing |
| KS | Kohn–Sham |
| LED | light-emitting diode |
| LUMO | lowest unoccupied molecular orbital |
| MRCI | multi-reference configuration interaction |
| MRSOCI | multi-reference spin–orbit configuration interaction |
| PES | potential energy surface |
| QDPT | quasi-degenerate perturbation theory |
| SOC | spin–orbit coupling |
| SOCME | spin–orbit coupling matrix element |
| SOMO | singly occupied molecular orbital |
| TDDFT | time-dependent density functional theory |
| TDA | Tamm–Dancoff approximation |
References
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef]
- Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, UK, 2009. [Google Scholar] [CrossRef]
- Ma, J.A.; Cahard, D. Strategies for nucleophilic, electrophilic, and radical trifluoromethylations. J. Fluor. Chem. 2007, 128, 975–996. [Google Scholar] [CrossRef]
- Ma, J.A.; Cahard, D. Update 1 of: Asymmetric Fluorination, Trifluoromethylation, and Perfluoroalkylation Reactions. Chem. Rev. 2008, 108, PR1–PR43. [Google Scholar] [CrossRef] [PubMed]
- Tomashenko, O.A.; Grushin, V.V. Aromatic Trifluoromethylation with Metal Complexes. Chem. Rev. 2011, 111, 4475–4521. [Google Scholar] [CrossRef] [PubMed]
- Barata-Vallejo, S.; Postigo, A. Metal-mediated radical perfluoroalkylation of organic compounds. Coord. Chem. Rev. 2013, 257, 3051–3069. [Google Scholar] [CrossRef]
- Studer, A. A “Renaissance” in Radical Trifluoromethylation. Angew. Chem. Int. Ed. 2012, 51, 8950–8958. [Google Scholar] [CrossRef]
- Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar] [CrossRef]
- Liu, S.Q.; Wang, S.W.; Qing, F.L. The perfluoroallylation of alkynes and transformation of the products. J. Fluor. Chem. 2005, 126, 771–778. [Google Scholar] [CrossRef]
- Erdbrink, H.; Peuser, I.; Gerling, U.I.M.; Lentz, D.; Koksch, B.; Czekelius, C. Conjugate hydrotrifluoromethylation of α,β-unsaturated acyl-oxazolidinones: Synthesis of chiral fluorinated amino acids. Org. Biomol. Chem. 2012, 10, 8583–8586. [Google Scholar] [CrossRef]
- Brace, N.O. l-Iodo-2-(perfluoroalkyl)cycloalkanes by the Free Radical Addition of Iodoperfluoroalkanes to Cyclohexane and Cyclopentene. J. Org. Chem. 1963, 28, 3093–3102. [Google Scholar] [CrossRef]
- Dolbier, W.R. Structure, Reactivity, and Chemistry of Fluoroalkyl Radicals. Chem. Rev. 1996, 96, 1557–1584. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.S.; Dixneuf, P.H.; Soulé, J.F. Photoredox Catalysis for Building C–C Bonds from C(sp2)–H Bonds. Chem. Rev. 2018, 118, 7532–7585. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, C.; Yoon, T.; MacMillan, D.W.C. Visible Light Photocatalysis in Organic Chemistry; Wiley-VCH: Weinheim, Germany, 2018. [Google Scholar] [CrossRef]
- Nagib, D.A.; Scott, M.E.; MacMillan, D.W.C. Enantioselective α-Trifluoromethylation of Aldehydes via Photoredox Organocatalysis. J. Am. Chem. Soc. 2009, 131, 10875–10877. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef]
- Rawner, T.; Lutsker, E.; Kaiser, C.A.; Reiser, O. The Different Faces of Photoredox Catalysts: Visible-Light-Mediated Atom Transfer Radical Addition (ATRA) Reactions of Perfluoroalkyl Iodides with Styrenes and Phenylacetylenes. ACS Catal. 2018, 8, 3950–3956. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, M.; Peng, Q.; Huo, Y.; Liu, Q.; Wang, R.; Xu, Z. Dual-Functional Chiral Cu-Catalyst- Induced Photoredox Asymmetric Cyanofluoroalkylation of Alkenes. ACS Catal. 2019, 9, 4470–4476. [Google Scholar] [CrossRef]
- Li, Z.L.; Fang, G.C.; Gu, Q.S.; Liu, X.Y. Recent advances in copper-catalysed radical-involved asymmetric 1,2-difunctionalization of alkenes. Chem. Soc. Rev. 2020, 49, 32–48. [Google Scholar] [CrossRef]
- Reckenthäler, M.; Griesbeck, A.G. Photoredox Catalysis for Organic Syntheses. Adv. Synth. Catal. 2013, 355, 2727–2744. [Google Scholar] [CrossRef]
- Hari, D.P.; König, B. Synthetic applications of eosin Y in photoredox catalysis. Chem. Commun. 2014, 50, 6688–6699. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A. Electron Donor-Acceptor Complexes in Perfluoroalkylation Reactions. Eur. J. Org. Chem. 2018, 2018, 6391–6404. [Google Scholar] [CrossRef]
- Lima, C.G.S.; de M. Lima, T.; Duarte, M.; jurberg, I.D.; Paixão, M.W. Organic Synthesis Enabled by Light-Irradiation of EDA Complexes: Theoretical Background and Synthetic Applications. ACS Catal. 2016, 6, 1389–1407. [Google Scholar] [CrossRef]
- Marzo, L.; Pagire, S.K.; Reiser, O.; König, O. Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis? Angew. Chem. Int. Ed. 2018, 57, 10034–10072. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, W.; Li, Y.; Ma, J.; Yu, S. Halogen-Bond-Promoted Double Radical Isocyanide Insertion under Visible-Light Irradiation: Synthesis of 2-Fluoroalkylated Quinoxalines. Org. Lett. 2016, 18, 4638–4641. [Google Scholar] [CrossRef]
- Sun, X.; He, Y.; Yu, S. Halogen-bond-mediated atom transfer radical addition of perfluoroalkyl iodides to alkynes under visible light irradiation. J. Photochem. Photobiol. A 2018, 355, 326–331. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Li, G.X.; He, G.; Chen, G. Halogen-Bond-Promoted Photoactivation of Perfluoroalkyl Iodides: A Photochemical Protocol for Perfluoroalkylation Reactions. Org. Lett. 2017, 19, 1442–1445. [Google Scholar] [CrossRef]
- Tang, X.; Studer, A. Alkene 1,2-Difunctionalization by Radical Alkenyl Migration. Angew. Chem. Int. Ed. 2018, 57, 814–817. [Google Scholar] [CrossRef]
- Yajima, T.; Ikegami, M. Metal-Free Visible-Light Radical Iodoperfluoroalkylation of Terminal Alkenes and Alkynes. Eur. J. Org. Chem. 2017, 2126–2129. [Google Scholar] [CrossRef]
- Rosso, C.; Williams, J.D.; Filippini, G.; Prato, M.; Kappe, C.O. Visible-Light-Mediated Iodoperfluoroalkylation of Alkenes in Flow and Its Application to the Synthesis of a Key Fulvestrant Intermediate. Org. Lett. 2019, 21, 5341–5345. [Google Scholar] [CrossRef]
- Zhu, E.; Liu, X.X.; Wang, A.J.; Mao, T.; Zhao, L.; Zhang, X.; He, C.Y. Visible light promoted fluoroalkylation of alkenes and alkynes using 2-bromophenol as a catalyst. Chem. Commun. 2019, 55, 12259–12262. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, Ł.; Murphy, J.J.; Melchiorre, P. Photo-organocatalytic Enantioselective Perfluoroalkylation of β-Ketoesters. J. Am. Chem. Soc. 2015, 137, 5678–5681. [Google Scholar] [CrossRef] [PubMed]
- Mao, T.; Ma, M.J.; Zhao, L.; Xue, D.P.; Yu, Y.; Gu, J.; He, C.Y. A general and green fluoroalkylation reaction promoted via noncovalent interactions between acetone and fluoroalkyl iodides. Chem. Commun. 2020. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.Y.; Zhang, H.Z. Reaction of perfluoroalkyl iodides with alkenes initiated by organophosphine and related compounds. J. Fluor. Chem. 1990, 50, 133–140. [Google Scholar] [CrossRef]
- Lumbierres, M.; Moreno-Mañas, M.; Vallribera, A. Addition of perfluorooctyl iodide to alkenes. Catalysis by triphenylphosphane. Tetrahedron 2002, 58, 4061–4065. [Google Scholar] [CrossRef]
- Zhao, L.; Huang, Y.; Wang, Z.; Zhu, E.; Mao, T.; Jia, J.; Gu, J.; Li, X.F.; He, C.Y. Organophosphine-Catalyzed Difluoroalkylation of Alkenes. Org. Lett. 2019, 21, 6705–6709. [Google Scholar] [CrossRef]
- Helmecke, L.; Spittler, M.; Baumgarten, K.; Czekelius, C. Metal-Free Activation of C–I Bonds and Perfluoroalkylation of Alkenes with Visible Light Using Phosphine Catalysts. Org. Lett. 2019, 21, 7823–7827. [Google Scholar] [CrossRef]
- Lu, H.; Wang, D.; Zhang, A. Visible Light-Promoted Phosphine-Catalyzed Difluoroalkylation of Arenes and Heterocycles. J. Org. Chem. 2020, 85, 942–951. [Google Scholar] [CrossRef]
- Behrends, I.; Bähr, S.; Czekelius, C. Perfluoroalkylation of Alkenes by Frustrated Lewis Pairs. Chem. Eur. J. 2016, 22, 17177–17181. [Google Scholar] [CrossRef]
- Spittler, M.; Helmecke, L.; Czekelius, C. Mechanistic Insights into FLP-Catalyzed Iodoperfluoroalkylations. Eur. J. Org. Chem. 2019, 458–468. [Google Scholar] [CrossRef]
- Bulfield, D.; Huber, S.M. Halogen Bonding in Organic Synthesis and Organocatalysis. Chem. Eur. J. 2016, 22, 14434–14450. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Rabie, U.M.A. A review on electronic spectral studies of charge transfer complexes. J. Mol. Struct. 2013, 1034, 393–403. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Chudzinski, M.G.; Taylor, M.S. Correlations between Computation and Experimental Thermodynamics of Halogen Bonding. J. Org. Chem. 2012, 77, 3483–3491. [Google Scholar] [CrossRef] [PubMed]
- Angarov, V.; Kozuch, S. On the σ, π and δ hole interactions: A molecular orbital overview. New J. Chem. 2018, 42, 1413–1422. [Google Scholar] [CrossRef]
- Manning, H.C.; Bai, M.; Anderson, B.M.; Lisiak, R.; Samuelson, L.E.; Bornhop, D.J. Expeditious synthesis of ‘P’-protected macrocycles en route to lanthanide chelate metal complexes. Tetrahedron Lett. 2005, 46, 4707–4710. [Google Scholar] [CrossRef]
- Bietti, M.; Calcagni, A.; Salamone, M. The Role of Structural Effects on the Reactions of Alkoxyl Radicals with Trialkyl and Triaryl Phosphites. A Time-Resolved Kinetic Study. J. Org. Chem. 2010, 75, 4514–4520. [Google Scholar] [CrossRef]
- Mark, V.; Wazer, J.R.V. Tri-t-butyl Phosphite and Some of Its Reactions. J. Org. Chem. 1964, 29, 1006–1008. [Google Scholar] [CrossRef]
- Treutler, O.; Ahlrichs, R. Efficient molecular numerical integration schemes. J. Chem. Phys. 1995, 102, 346–354. [Google Scholar] [CrossRef]
- von Arnim, M.; Ahlrichs, R. Performance of Parallel TURBOMOLE for Density Functional Calculations. J. Comput. Chem. 1998, 19, 1746–1757. [Google Scholar] [CrossRef]
- TURBOMOLE, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, Since 2007. Available online: http://www.turbomole.com (accessed on 30 March 2020).
- Becke, A.D. Density-functional thermochemistry. 3. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S. Density functional theory with London dispersion corrections. WIREs Comput. Mol. Sci. 2011, 1, 211–228. [Google Scholar] [CrossRef]
- Klamt, A.; Schüürmann, G. COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and its Gradient. J. Chem. Soc. Perkin Trans. 2 1993, 5, 799–805. [Google Scholar] [CrossRef]
- Schäfer, A.; Klamt, A.; Sattel, D.; Lohrenz, J.C.W.; Eckert, F. COSMO Implementation in TURBOMOLE: Extension of an Efficient Quantum Chemical Code Towards Liquid Systems. Phys. Chem. Chem. Phys. 2000, 2, 2187–2193. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis sets for the post-d group 16–18 elements. J. Chem. Phys. 2003, 119, 11113. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M.; Patzelt, H.; Ahlrichs, R. RI-MP2: Optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143–152. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Furche, F.; Ahlrichs, R. Adiabatic Time-Dependent Density Functional Methods for Excited State Properties. J. Chem. Phys. 2002, 117, 7433–7447. [Google Scholar] [CrossRef]
- Hirata, S.; Head-Gordon, M. Time-dependent density functional theory within the Tamm–Dancoff approximation. Chem. Phys. Lett. 1999, 314, 291–299. [Google Scholar] [CrossRef]
- Weymuth, T.; Haag, M.P.; Kiewisch, K.; Luber, S.; Schenk, S.; Jacob, C.R.; Herrmann, C.; Neugebauer, J.; Reiher, M. MOVIPAC: Vibrational Spectroscopy with a Robust Meta-Program for Massively Parallel Standard and Inverse Calculations. J. Comput. Chem. 2012, 33, 2186–2198. [Google Scholar] [CrossRef]
- Pohler, L.; Kleinschmidt, M.; Etinski, M.; Marian, C.M. In search of the dark state of 5-methyl-2- hydroxypyrimidine using a numerical DFT/MRCI gradient. Mol. Phys. 2012, 110, 2429–2438. [Google Scholar] [CrossRef]
- Grimme, S.; Waletzke, M. A combination of Kohn–Sham density functional theory and multi-reference configuration interaction methods. J. Chem. Phys. 1999, 111, 5645–5655. [Google Scholar] [CrossRef]
- Marian, C.M.; Heil, A.; Kleinschmidt, M. The DFT/MRCI Method. WIREs Comp. Mol. Sci. 2019, 9, e1394. [Google Scholar] [CrossRef]
- Heil, A.; Kleinschmidt, M.; Marian, C.M. On the performance of DFT/MRCI Hamiltonians for electronic excitations in transition metal complexes: The role of the damping function. J. Chem. Phys. 2018, 149, 164106. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Vahtras, O.; Almlöf, J.; Feyereisen, M. Integral approximations for LCAO-SCF calculations. Chem. Phys. Lett. 1993, 213, 514–518. [Google Scholar] [CrossRef]
- Hellweg, A.; Hättig, C.; Höfener, S.; Klopper, W. Optimized accurate auxiliary basis sets for RI-MP2 and RI-CC2 calculations for the atoms Rb to Rn. Theoret. Chem. Acc. 2007, 117, 587–597. [Google Scholar] [CrossRef]
- Kleinschmidt, M.; Tatchen, J.; Marian, C.M. Spin-Orbit Coupling of DFT/MRCI Wavefunctions: Method, Test Calculations, and Application to Thiophene. J. Comput. Chem. 2002, 23, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmidt, M.; Marian, C.M. Efficient Generation of Matrix Elements for One-Electron Spin–Orbit Operators. Chem. Phys. 2005, 311, 71–79. [Google Scholar] [CrossRef]
- Kleinschmidt, M.; van Wüllen, C.; Marian, C.M. Intersystem-crossing and phosphorescence rates in Fac-IrIII(ppy)3: A Theor. Study Involv. Multi-Ref. Interact. Wavefunct. J. Chem. Phys. 2015, 142, 094301. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.A.; Marian, C.M.; Wahlgren, U.; Gropen, O. A Mean-Field Spin-orbit Method Applicable to Correlated Wavefunctions. Chem. Phys. Lett. 1996, 251, 365–371. [Google Scholar] [CrossRef]
- Schimmelpfennig, B. AMFI Is An Atomic Mean-Field Spin-Orbit Integral Program; University of Stockholm: Stockholm, Sweden, 1996. [Google Scholar]
- Kleinschmidt, M.; Tatchen, J.; Marian, C.M. SPOCK.CI: A Multireference Spin–Orbit Configuration Interaction Method for Large Molecules. J. Chem. Phys. 2006, 124, 124101. [Google Scholar] [CrossRef]
- Cole, J.R.; Dellinger, M.E.; Johnson, T.J.; Reinecke, B.A.; Pike, R.D.; Pennington, W.T.; Krawiec, M.; Rheingold, A.L. Caged phosphite complexes of copper(I) halides. J. Chem. Crystallogr. 2003, 33, 341–347. [Google Scholar] [CrossRef]
- Taira, K.; Mock, W.L.; Gorenstein, D.G. Experimental Tests of the Stereoelectronic Effect at Phosphorus: Nucleophilic Reactivity of Phosphite Esters. J. Am. Chem. Soc. 1984, 106, 7831–7835. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | P–I Bond Length/pm | I–C Bond Length/pm | P–I–C Bond Angle/ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| S | T | S | S | T | S | S | T | S | |||
| CFI | 216 | ||||||||||
| BuP-ICF | 297 | 303 | 343 | 225 | 309 | 270 | 179 | 110 | 104 | ||
| BuP-ICF | 297 | 308 | 331 | 224 | 292 | 281 | 177 | 76 | 96 | ||
| (MeO)P-ICF | 316 | 294 | 320 | 220 | 318 | 294 | 179 | 78 | 96 | ||
| Compound | T | S | |||||||
|---|---|---|---|---|---|---|---|---|---|
| BuP-ICF | 3.70 | 335 | 1.42 | 870 | 4.84 | 256 | 1.70 | 729 | |
| BuP-ICF | 3.91 | 317 | 1.46 | 850 | 5.07 | 245 | 1.82 | 681 | |
| (MeO)P-ICF | 4.48 | 276 | 1.62 | 765 | 5.34 | 232 | 1.87 | 663 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bracker, M.; Helmecke, L.; Kleinschmidt, M.; Czekelius, C.; Marian, C.M. Visible Light-Induced Homolytic Cleavage of Perfluoroalkyl Iodides Mediated by Phosphines. Molecules 2020, 25, 1606. https://doi.org/10.3390/molecules25071606
Bracker M, Helmecke L, Kleinschmidt M, Czekelius C, Marian CM. Visible Light-Induced Homolytic Cleavage of Perfluoroalkyl Iodides Mediated by Phosphines. Molecules. 2020; 25(7):1606. https://doi.org/10.3390/molecules25071606
Chicago/Turabian StyleBracker, Mario, Lucas Helmecke, Martin Kleinschmidt, Constantin Czekelius, and Christel M. Marian. 2020. "Visible Light-Induced Homolytic Cleavage of Perfluoroalkyl Iodides Mediated by Phosphines" Molecules 25, no. 7: 1606. https://doi.org/10.3390/molecules25071606
APA StyleBracker, M., Helmecke, L., Kleinschmidt, M., Czekelius, C., & Marian, C. M. (2020). Visible Light-Induced Homolytic Cleavage of Perfluoroalkyl Iodides Mediated by Phosphines. Molecules, 25(7), 1606. https://doi.org/10.3390/molecules25071606