Novel Approach to Automated Flow Titration for the Determination of Fe(III)


Abstract
1. Introduction
2. Results and Discussion
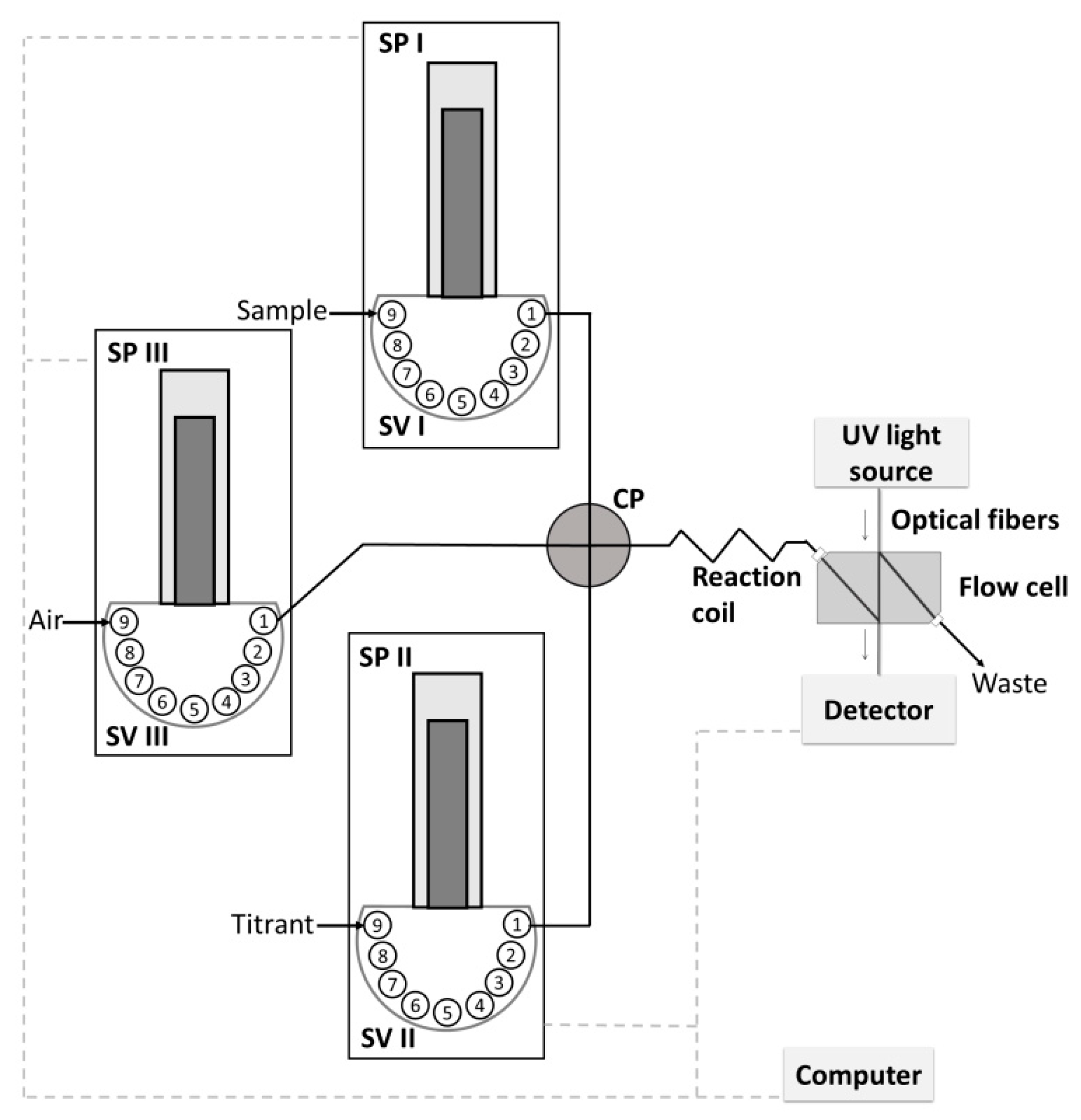
2.1. Flow System Developed for Titration
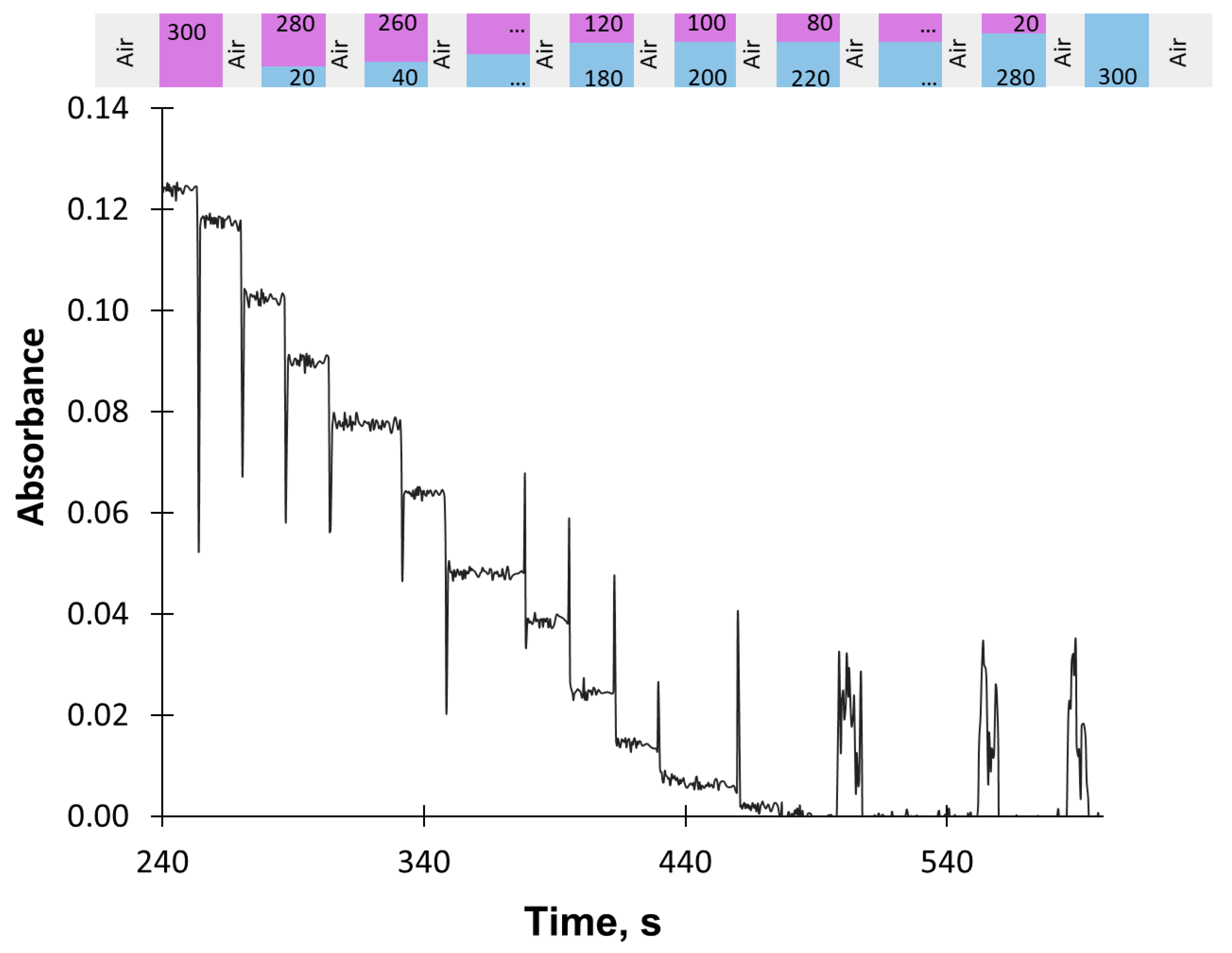
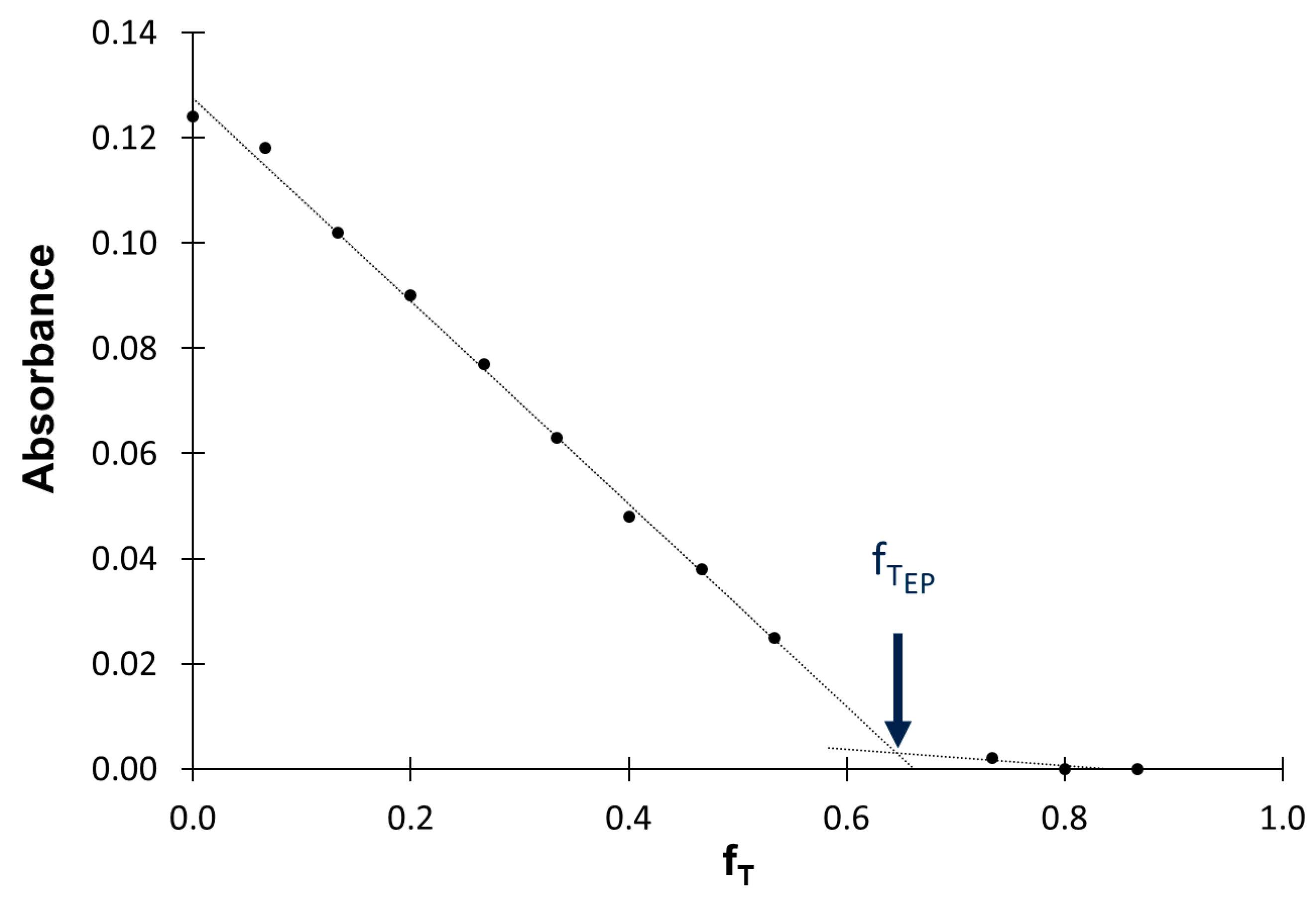
2.2. Procedure of Titration
Procedure Verification
2.3. Analysis of Real Samples
3. Methodology
3.1. Reagents and Solutions
3.2. Instrumentation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pasquini, C.; de Aquino, E.V.; das Virgens Reboucas, M.; Gonzaga, F.B. Robust flow-batch coulometric/biamperometric titration system: Determination of bromine index and bromine number of petrochemicals. Anal. Chim. Acta 2007, 600, 84–89. [Google Scholar] [CrossRef]
- Tan, A.; Zhang, L.; Xiao, C. Simultaneous and automatic determination of hydroxide and carbonate in aluminate solutions by a micro-titration method. Anal. Chim. Acta 1999, 388, 219–223. [Google Scholar] [CrossRef]
- Alerm, L.; Bartroli, J. Development of a sequential microtitration system. Anal. Chem. 1996, 68, 1394–1400. [Google Scholar] [CrossRef]
- Hoogendijk, R. A compact titration configuration for process analytical applications. Anal. Chim. Acta 1999, 378, 211–217. [Google Scholar] [CrossRef]
- Tan, A.; Xiao, C. An automatic back titration method for microchemical analysis. Talanta 1997, 44, 967–972. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, J.; Chen, Q.; Jiang, S.; Zhang, Y.; Fu, T. New intelligent photometric titration system and its method for constructing chemical oxygen demand based on micro-flow injection. Microchem. J. 2018, 143, 292–304. [Google Scholar] [CrossRef]
- Crispino, C.C.; Reis, B.F. Development of an automatic photometric titration procedure to determine olive oil acidity employing a miniaturized multicommuted flow-batch setup. Anal. Methods 2014, 6, 302–307. [Google Scholar] [CrossRef]
- Wójtowicz, M.; Kozak, J.; Kościelniak, P. Novel approaches to analysis by flow injection gradient titration. Anal. Chim. Acta 2007, 600, 78–83. [Google Scholar] [CrossRef]
- Marcos, J.; Ríos, A.; Valcárcel, M. Automatic titrations in unsegmented flow systems based on variable flow-rate patterns. Part 1. Principles and applications to acid-base titrations. Anal. Chim. Acta 1992, 261, 489–494. [Google Scholar] [CrossRef]
- Marcos, J.; Ríos, A.; Valcárcel, M. Automatic titrations in unsegmented flow systems based on variable flow-rate patterns. Part 2. Complexometric and redox titrations. Anal. Chim. Acta 1992, 261, 495–503. [Google Scholar] [CrossRef]
- Katsumata, H.; Teshima, N.; Kurihara, M.; Kawashima, T. Potentiometric flow titration of iron(II) and chromium(VI) based on flow rate ratio of a titrant to a sample. Talanta 1999, 48, 135–141. [Google Scholar] [CrossRef]
- Lopez García, I.; Viñas, P.; Campillo, N.; Hernandez Córdoba, M. Linear flow gradients for automatic titrations. Anal. Chim. Acta 1995, 308, 67–76. [Google Scholar] [CrossRef]
- Fuhrmann, B.; Spohn, U. Volumetric triangle-programmed flow titrations based on precisely generated concentration gradients. Anal. Chim. Acta 1993, 282, 397–406. [Google Scholar] [CrossRef]
- Dasgupta, P.K.; Tanaka, H.; Jo, K.D. Continuous on-line true titrations by feedback based flow ratiometry: Application to potentiometric acid-base titrations. Anal. Chim. Acta 2001, 435, 289–297. [Google Scholar] [CrossRef]
- Jo, K.D.; Dasgupta, P.K. Continuous on-line feedback based flow titrations. Complexometric titrations of calcium and magnesium. Talanta 2003, 60, 131–137. [Google Scholar] [CrossRef]
- Tanaka, H.; Baba, T. High throughput continuous titration based on a flow ratiometry controlled with feedback-based variable triangular waves and subsequent fixed triangular waves. Talanta 2005, 67, 848–853. [Google Scholar] [CrossRef]
- Dakashev, A.D.; Dimitrova, V.T. Pulse coulometric titration in continuous flow. Analyst 1994, 119, 1835–1838. [Google Scholar] [CrossRef]
- He, Z.K.; Fuhrmann, B.; Spohn, U. Coulometric microflow titrations with chemiluminescent and amperometric equivalence point detection. Bromimetric titration of low concentrations of hydrazine and ammonium. Anal. Chim. Acta 2000, 409, 83–91. [Google Scholar] [CrossRef]
- Guenat, O.T.; van der Schoolt, B.H.; Morf, W.E.; de Rooij, N.F. Triangle-programmed coulometric nanotitrations completed by continuous flow with potentiometric detection. Anal. Chem. 2000, 72, 1585–1590. [Google Scholar] [CrossRef]
- Fuhrmann, B.; Spohn, U. A PC-based titrator for flow gradient titrations. J. Autom. Chem. 1993, 15, 209–216. [Google Scholar] [CrossRef]
- Martelli, P.B.; Reis, B.F.; Korn, M.; Lima, J.L.F.C. Automatic potentiometric titration in monosegmented flow system exploiting binary search. Anal. Chim. Acta 1999, 387, 165–173. [Google Scholar] [CrossRef]
- Aquino, E.V.; Rohwedder, J.J.R.; Pasquini, C. Monosegmented flow titrator. Anal. Chim. Acta 2001, 438, 67–74. [Google Scholar] [CrossRef]
- Borges, E.P.; Martelli, P.B.; Reis, B.F. Automatic stepwise potentiometric titration in a monosegmented flow system. Mikrochim. Acta 2000, 135, 179–184. [Google Scholar] [CrossRef]
- Assali, M.; Raimundo, I.M., Jr.; Facchin, I. Simultaneous multiple injection to perform titration and standard addition in monosegmented flow analysis. J. Autom. Methods Manag. Chem. 2001, 23, 83–89. [Google Scholar] [CrossRef]
- Honorato, R.S.; Araújo, M.C.U.; Veras, G.; Zagatto, E.A.G.; Lapa, R.A.S.; Lima, J.L.F.C. A monosegmented flow titration for the spectrophotometric determination of total acidity in vinegars. Anal. Sci. 1999, 15, 665–668. [Google Scholar] [CrossRef][Green Version]
- Vidal de Aquino, E.; Rohwedder, J.J.R.; Pasquini, C. A new approach to flow-batch titration. A monosegmented flow titrator with coulometric reagent generation and potentiometric or biamperometric detection. Anal. Bioanal. Chem. 2006, 386, 1921–1930. [Google Scholar] [CrossRef]
- Jakmunee, J.; Pathimapornlert, L.; Hartwell, S.K.; Grudpan, K. Novel approach for mono-segmented flow micro-titration with sequential injection using a lab-on-valve system: A model study for the assay of acidity in fruit juices. Analyst 2005, 130, 299–303. [Google Scholar] [CrossRef]
- Kozak, J.; Wójtowicz, M.; Gawenda, N.; Kościelniak, P. An automatic system for acidity determination based on sequential injection titration and the monosegmented flow approach. Talanta 2011, 84, 1379–1383. [Google Scholar] [CrossRef]
- Almeida, C.M.N.V.; Lapa, R.A.S.; Lima, J.L.F.C.; Zagatto, E.A.G.; Araújo, M.C.U. An automatic titrator based on a multicommutated unsegmented flow system. Its application to acid-base titrations. Anal. Chim. Acta 2000, 407, 213–223. [Google Scholar] [CrossRef]
- Almeida, C.M.N.V.; Araújo, M.C.U.; Lapa, R.A.S.; Lima, J.L.F.C.; Reis, B.F.; Zagatto, E.A.G. Precipitation titrations using an automatic titrator based on a multicommutated unsegmented flow system. Analyst 2000, 125, 333–340. [Google Scholar] [CrossRef]
- Almeida, C.M.N.V.; Lapa, R.A.S.; Lima, J.L.F.C. Automatic flow titrator based on a multicommutated unsegmented flow system for alkalinity monitoring in wastewater. Anal. Chim. Acta 2001, 438, 291–298. [Google Scholar] [CrossRef]
- Paim, A.P.S.; Almeida, C.M.N.V.; Reis, B.F.; Lapa, R.A.S.; Zagatto, E.A.G.; Lima, J.L.F.C. Automatic potentiometric flow titration procedure for ascorbic acid determination in pharmaceutical formulations. J. Pharm. Biomed. Anal. 2002, 28, 1221–1225. [Google Scholar] [CrossRef]
- Wang, X.D.; Cardwell, T.J.; Cattrall, R.W.; Dyson, R.P.; Jenkins, G.E. Time-division multiplex technique for producing concentration profiles in flow analysis. Anal. Chim. Acta 1998, 368, 105–110. [Google Scholar] [CrossRef]
- Lima, M.J.A.; Reis, B.F. Fully automated photometric titration procedure employing a multicommuted flow analysis setup for acidity determination in fruit juice, vinegar, and wine. Microchem. J. 2017, 135, 207–212. [Google Scholar] [CrossRef]
- Sasaki, M.K.; Rocha, D.L.; Rocha, F.R.P.; Zagatto, E.A.G. Tracer-monitored flow titrations. Anal. Chim. Acta 2016, 902, 123–128. [Google Scholar] [CrossRef]
- Martz, T.R.; Dickson, A.G.; DeGrandpre, M.D. Tracer monitored titrations: Measurement of total alkalinity. Anal. Chem. 2006, 78, 1817–1826. [Google Scholar] [CrossRef]
- Kochana, J.; Parczewski, A. New method of simultaneous determination of Fe (II) and Fe (III) by two-component photometric titration. Chem. Anal. 1997, 42, 411–416. [Google Scholar]
- Kozak, J.; Gutowski, J.; Kozak, M.; Wieczorek, M.; Kościelniak, P. New method for simultaneous determination of Fe(II) and Fe(III) in water using flow injection technique. Anal. Chim. Acta 2010, 668, 8–12. [Google Scholar] [CrossRef]
- Paluch, J.; Kozak, J.; Wieczorek, M.; Kozak, M.; Kochana, J.; Widurek, K.; Konieczna, M.; Kościelniak, P. Novel approach to two-component speciation analysis. Spectrophotometric flow-based determinations of Fe(II)/Fe(III) and Cr(III)/Cr(VI). Talanta 2017, 171, 275–282. [Google Scholar] [CrossRef]
- Water Quality—Sampling—Part 3: Guidance on the Preservation and Handling of Water Samples; PN-EN ISO 5667-3:2005; Polish Committee for Standardization: Warsaw, Poland, 2005.
- Water Quality—Determination of Selected Elements by Inductively Coupled Optical Emission Spectrometry (ICP-OES); ISO 11885:2007(E); International Organization for Standardization: Geneva, Switzerland, 2007.
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
| Step | SV Position | SP Flow Rate, µL s−1 | Volume, µL | Action | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| I | II | III | I | II | III | I | II | III | ||
| 1 | 9 | 9 | 9 | 100 | 100 | 100 | 1000 | 1000 | 1000 | Aspiration of sample, titrant, and air into syringes |
| 2 | 1 | 1 | 1 | 0 | 0 | 100 | 0 | 0 | 100 | Introduction of air into reaction coil |
| 3 | 1 | 1 | 1 | 100 | 0 | 0 | 300 | 0 | 0 | Formation of zone I in reaction coil |
| 4 | 1 | 1 | 1 | 0 | 0 | 100 | 0 | 0 | 100 | Introduction of air into reaction coil |
| 5 | 1 | 1 | 1 | 94 | 10 | 0 | 280 | 20 | 0 | Formation of zone II in reaction coil |
| 6 | 1 | 1 | 1 | 0 | 0 | 100 | 0 | 0 | 100 | Introduction of air into reaction coil |
| 7 | 1 | 1 | 1 | 86 | 20 | 0 | 260 | 40 | 0 | Formation of zone III in reaction coil |
| 8 | 1 | 1 | 1 | 0 | 0 | 100 | 0 | 0 | 100 | Introduction of air into reaction coil |
| 9 | 9 | 9 | 9 | 100 | 0 | 100 | 840 | 0 | 400 | Aspiration of sample and air into syringes |
| 10 | 1 | 1 | 1 | 80 | 30 | 0 | 240 | 60 | 0 | Formation of zone IV in reaction coil |
| 11 | 1 | 1 | 1 | 0 | 0 | 100 | 0 | 0 | 100 | Introduction of air into reaction coil |
| 12 | 1 | 1 | 1 | 74 | 40 | 0 | 220 | 80 | 0 | Formation of zone V in reaction coil |
| 13 | 1 | 1 | 1 | 0 | 0 | 100 | 0 | 0 | 100 | Introduction of air into reaction coil |
| 14 | 1 | 1 | 1 | 67 | 50 | 0 | 200 | 100 | 0 | Formation of zone VI in reaction coil |
| 15 | 1 | 1 | 1 | 0 | 0 | 100 | 0 | 0 | 100 | Introduction of air into reaction coil |
| 16 | 1 | 1 | 1 | 60 | 60 | 0 | 180 | 120 | 0 | Formation of zone VII in reaction coil |
| 17 | 1 | 1 | 1 | 0 | 0 | 100 | 0 | 0 | 100 | Introduction of air into reaction coil |
| 18 | 1 | 1 | 1 | 54 | 70 | 0 | 160 | 140 | 0 | Formation of zone VIII in reaction coil |
| 19 | 1 | 1 | 1 | 0 | 0 | 100 | 0 | 0 | 100 | Introduction of air into reaction coil |
| 20 | 9 | 9 | 9 | 100 | 0 | 100 | 560 | 0 | 500 | Aspiration of sample and air into syringes |
| 21 | 1 | 1 | 1 | 47 | 80 | 0 | 140 | 160 | 0 | Formation of zone IX in reaction coil |
| 22 | 1 | 1 | 1 | 0 | 0 | 100 | 0 | 0 | 100 | Introduction of air into reaction coil |
| 23 | 1 | 1 | 1 | 40 | 90 | 0 | 120 | 180 | 0 | Formation of zone X in reaction coil |
| 24 | 1 | 1 | 1 | 0 | 0 | 100 | 0 | 0 | 100 | Introduction of air into reaction coil |
| 25 | 9 | 9 | 9 | 0 | 100 | 100 | 0 | 900 | 200 | Aspiration of titrant and air into syringes |
| 26 | 1 | 1 | 1 | 34 | 100 | 0 | 100 | 200 | 0 | Formation of zone XI in reaction coil |
| 27 | 1 | 1 | 1 | 0 | 0 | 100 | 0 | 0 | 100 | Introduction of air into reaction coil |
| 28 | 1 | 1 | 1 | 27 | 100 | 0 | 80 | 220 | 0 | Formation of zone XII in reaction coil |
| 29 | 1 | 1 | 1 | 0 | 0 | 100 | 0 | 0 | 100 | Introduction of air into reaction coil |
| 30 | 1 | 1 | 1 | 20 | 100 | 0 | 60 | 240 | 0 | Formation of zone XIII in reaction coil |
| 31 | 1 | 1 | 1 | 0 | 0 | 100 | 0 | 0 | 100 | Introduction of air into reaction coil |
| 32 | 1 | 1 | 1 | 14 | 100 | 0 | 40 | 260 | 0 | Formation of zone XIV in reaction coil |
| 33 | 1 | 1 | 1 | 0 | 0 | 100 | 0 | 0 | 100 | Introduction of air into reaction coil |
| 34 | 9 | 9 | 9 | 0 | 100 | 100 | 0 | 900 | 400 | Aspiration of titrant and air into syringes |
| 35 | 1 | 1 | 1 | 7 | 100 | 0 | 20 | 280 | 0 | Formation of zone XV in reaction coil |
| 36 | 1 | 1 | 1 | 0 | 0 | 100 | 0 | 0 | 100 | Introduction of air into reaction coil |
| 37 | 1 | 1 | 1 | 0 | 100 | 0 | 0 | 300 | 0 | Formation of zone XVI in reaction coil |
| 38 | 1 | 1 | 1 | 0 | 0 | 100 | 0 | 0 | 300 | Introduction of air into mixing coil and transport of zones to detector |
| No. | Fe(III), mg L−1 | CV, % | |RE|, % | Fe(III) + Fe(II), mg L−1 | Fe(II), mg L−1 | CV, % | |RE|, % | ||
|---|---|---|---|---|---|---|---|---|---|
| Expected | Determined | Determined | Expected | Determined | |||||
| 1 | 0.50 | 0.50 | 1.8 | 0.9 | 1.01 | 0.50 | 0.51 | 1.8 | 2.3 |
| 2 | 0.50 | 0.51 | 2.4 | 1.4 | 2.53 | 2.00 | 2.02 | 2.3 | 1.0 |
| 3 | 1.00 | 0.99 | 2.9 | 1.4 | 1.98 | 1.00 | 0.99 | 3.4 | 0.5 |
| 4 | 1.00 | 1.02 | 0.1 | 2.3 | 3.54 | 2.50 | 2.51 | 3.7 | 0.6 |
| 5 | 1.00 | 1.03 | 1.5 | 3.3 | 4.10 | 3.00 | 3.07 | 0.7 | 2.3 |
| 6 | 2.50 | 2.49 | 1.1 | 0.3 | 3.49 | 1.00 | 1.00 | 2.8 | 0.2 |
| Parameter | Value |
|---|---|
| Accuracy, , % | 3.3 |
| Precision, CV, % (n = 6) | 1.7 |
| Sample consumption, mL | 2.4 |
| Titrant consumption, mL | 2.4 |
| Time of single titration, min | 6 |
| Sample | Fe(III), mg L−1 | Fe(II), mg L−1 | Fe (Total), mg L−1 | |
|---|---|---|---|---|
| Developed Procedure | 1 ICP–OES/ 2 Certified Value | |||
| Water 1 | 0.63 ± 0.01 | 0.94 ± 0.04 | 1.56 ± 0.04 | 1 1.56 ± 0.01 |
| Water 2 | 0.35 ± 0.01 | 2.40 ± 0.04 | 2.75 ± 0.04 | 1 2.73 ± 0.04 |
| Water 3 | 0.57 ± 0.03 | 0.70 ± 0.03 | 1.27 ± 0.01 | 1 1.26 ± 0.02 |
| WWater | 0.36 ± 0.04 | 0.13 ± 0.05 | 0.49 ± 0.03 | 2 0.49 ± 0.02 |
| *WWater | 0.85 ± 0.04 | 0.13 ± 0.05 | 0.98 ± 0.04 | 2 0.99 ± 0.02 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozak, J.; Paluch, J.; Kozak, M.; Duracz, M.; Wieczorek, M.; Kościelniak, P. Novel Approach to Automated Flow Titration for the Determination of Fe(III). Molecules 2020, 25, 1533. https://doi.org/10.3390/molecules25071533
Kozak J, Paluch J, Kozak M, Duracz M, Wieczorek M, Kościelniak P. Novel Approach to Automated Flow Titration for the Determination of Fe(III). Molecules. 2020; 25(7):1533. https://doi.org/10.3390/molecules25071533
Chicago/Turabian StyleKozak, Joanna, Justyna Paluch, Marek Kozak, Marta Duracz, Marcin Wieczorek, and Paweł Kościelniak. 2020. "Novel Approach to Automated Flow Titration for the Determination of Fe(III)" Molecules 25, no. 7: 1533. https://doi.org/10.3390/molecules25071533
APA StyleKozak, J., Paluch, J., Kozak, M., Duracz, M., Wieczorek, M., & Kościelniak, P. (2020). Novel Approach to Automated Flow Titration for the Determination of Fe(III). Molecules, 25(7), 1533. https://doi.org/10.3390/molecules25071533