
Reflection Absorption Infrared Spectroscopy Characterization of SAM Formation from 8-Mercapto-N-(phenethyl)octanamide Thiols with Phe Ring and Amide Groups


Abstract
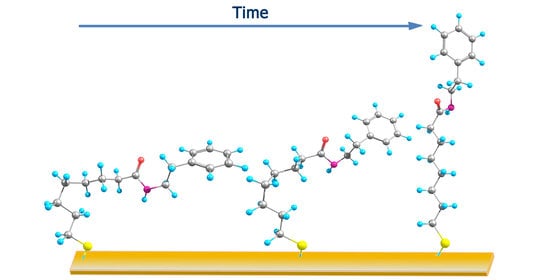
1. Introduction
2. Results and Discussion
2.1. Assignments of MOPHE Infrared Absorption bands
2.2. Reflection Absorption Infrared Spectroscopy (RAIRS) Analysis of Monolayer Formation
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Reflection-Absorption IR Spectroscopy (RAIRS)
4.3. Theoretical Modeling
4.4. Synthesis of MOPHE Compound
4.5. Synthesis of MOPHE-D5 Compound
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ulman, A. Formation and structure of self-assembled monolayers. Chem. Rev. 1996, 96, 1533–1554. [Google Scholar] [CrossRef]
- Vericat, C.; Vela, M.E.; Corthey, G.; Pensa, E.; Cortes, E.; Fonticelli, M.H.; Ibanez, F.; Benitez, G.E.; Carro, P.; Salvarezza, R.C. Self-assembled monolayers of thiolates on metals: A review article on sulfur-metal chemistry and surface structures. RSC Adv. 2014, 4, 27730–27754. [Google Scholar] [CrossRef]
- Valincius, G.; Niaura, G.; Kazakevičienė, B.; Talaikytė, Z.; Butkus, E.; Razumas, V. Anion effect of mediated electron transfer through ferrocene-terminated self-asembled monolayers. Langmuir 2004, 20, 6631–6638. [Google Scholar] [CrossRef]
- Matulaitienė, I.; Kuodis, Z.; Matijoška, A.; Eicher-Lorka, O.; Niaura, G. SERS of the positive charge bearing pyridinium ring terminated self-assembled monolayers: Structure and bonding spectral markers. J. Phys. Chem. C 2015, 119, 26481–26492. [Google Scholar] [CrossRef]
- Salonen, L.M.; Ellermann, M.; Diederich, F. Aromatic rings in chemical and biological recognition: Energetics and structures. Angew. Chem. Int. Ed. 2011, 50, 4808–4842. [Google Scholar] [CrossRef]
- Makwana, K.M.; Mahalakshmi, R. Implications of aromatic–aromatic interactions: From protein structures to peptide models. Protein Sci. 2015, 24, 1920–1933. [Google Scholar] [CrossRef]
- Dougherty, D.A. The cation-π interaction. Acc. Chem. Res. 2013, 46, 885–893. [Google Scholar] [CrossRef]
- Hunter, C.A.; Singh, J.; Thornton, J.M. π–π Interactions: The geometry and energetics of phenylalanine-phenylalanine interactions in proteins. J. Mol. Biol. 1991, 218, 837–846. [Google Scholar] [CrossRef]
- Santos, L.A.; Da Cunha, E.F.F.; Freitas, M.P.; Ramalho, T.C. Hydrophobic noncovalent interactions of inosine-phenylalanine: A theoretical model for investigating the molecular recognition of nucleobases. J. Phys. Chem. A 2014, 118, 5808–5817. [Google Scholar] [CrossRef]
- Kim, M.; Hohman, J.N.; Serino, A.C.; Weiss, P.S. Structural manipulation of hydrogen-bonding networks in amide-containing alkanethiolate monolayers via electrochemical procesing. J. Phys. Chem C 2010, 114, 19744–19751. [Google Scholar] [CrossRef]
- Lewis, P.A.; Smith, R.K.; Kelly, K.F.; Bumm, L.A.; Reed, S.M.; Clegg, R.S.; Gunderson, J.D.; Hutchison, J.E.; Weiss, P.S. The role of buried hydrogen bonds in self-assembled mixed composition thiols on Au{111}. J. Phys. Chem. B 2001, 105, 10630–10636. [Google Scholar] [CrossRef]
- Clegg, R.S.; Hutchison, J.E. Control of monolayer assembly structure by hydrogen bonding rather than adsorbate-substrate templating. J. Am. Chem. Soc. 1999, 121, 5319–5327. [Google Scholar] [CrossRef]
- Clegg, R.S.; Hutchison, J.E. Hydrogen-bonding, self-assembled monolayers: Ordered molecular films for study of through-peptide electron transfer. Langmuir 1996, 12, 5239–5243. [Google Scholar] [CrossRef]
- Malysheva, L.; Onipko, A.; Valiokas, R.; Liedberg, B. First-principles modeling of oligo(ethylene glycol)-terminated and amide group containing alkanethiolates. Appl. Surf. Sci. 2005, 246, 372–376. [Google Scholar] [CrossRef]
- Valiokas, R.; Ostblom, M.; Svedhem, S.; Svensson, S.C.T.; Liedberg, B. Thermal stability of self-assembled monolayers: Influence of lateral hydrogen bonding. J. Phys. Chem. B 2002, 106, 10401–10409. [Google Scholar] [CrossRef]
- Špandyreva, M.; Ignatjev, I.; Matulaitienė, I.; Kuodis, Z.; Niaura, G. Sum frequency generation spectroscopy probing of formation of self-assembled monolayers from thiols with terminal phenylalanine ring and intrachain amide. Chemija 2018, 29, 219–226. [Google Scholar] [CrossRef]
- Vericat, C.; Vela, M.E.; Benitez, G.; Carro, P.; Salvarezza, R.C. Self-assembled monolayers of thiols and dithiols on gold: New challengers for a well-known system. Chem. Soc. Rev. 2010, 39, 1805–1834. [Google Scholar] [CrossRef]
- Nuzzo, R.G.; Korenic, E.M.; Dubois, L.H. Studies on temperature-dependent phase behavior of long-chain normal-alkyl thiol monolayers on gold. J. Chem. Phys. 1990, 93, 767–773. [Google Scholar] [CrossRef]
- Shin, H.S.; Kim, J.H.; Kim, S.B.; Jung, Y.M. Thermal characterization of self-assembled monolayers of dialkyl disulfides containing the urea moiety. Langmuir 2007, 23, 10567–10572. [Google Scholar] [CrossRef]
- Stoycheva, S.; Himmelhaus, M.; Fick, J.; Korniakov, A.; Grunze, M.; Ulman, A. Spectroscopic characterization of ω-substituted biophenylthiolates on gold and their use as substrates for “on-top” siloxane SAM formation. Langmuir 2006, 22, 4170–4178. [Google Scholar] [CrossRef]
- Nuzzo, R.G.; Dubois, L.H.; Allara, D.L. Fundamental studies of microscopic wetting on organic surfaces. 1. Formation and structural characterization of a self-consistent series of polyfunctional organic monolayers. J. Am. Chem. Soc. 1990, 112, 558–569. [Google Scholar] [CrossRef]
- Porter, M.D.; Bright, T.B.; Allara, D.L.; Chidsey, C.E.D. Spontaneousy organized molecular assemblies. 4. Structural caracterization of n-alkyl thiol monolayers on gold by optical ellipsometry, infrared spectroscopy, and electrochemistry. J. Am. Chem. Soc. 1987, 109, 3559–3568. [Google Scholar] [CrossRef]
- Laibinis, P.E.; Whitesides, G.M.; Allara, D.L.; Tao, Y.-T.; Parikh, A.N.; Nuzzo, R.G. Comparison of the structures and wetting properties of self-assembled monolayersof n-alkanethiols on the coinage metal surfaces, copper, silver, and gold. J. Am. Chem. Soc. 1991, 113, 7152–7167. [Google Scholar] [CrossRef]
- Snyder, R.G.; Strauss, H.L.; Elliger, C.A. Carbon-hydrogen stretching modes and the structure of n-alkyl chains. 1. Long disordered chains. J. Phys. Chem. 1982, 86, 5145–5150. [Google Scholar] [CrossRef]
- Bensebaa, F.; Ellis, T.H.; Badia, A.; Lennox, R.B. Thermal treatment of n-alkanethiolate monolayers on gold, as observed by infrared spectroscopy. Langmuir 1998, 14, 2361–2367. [Google Scholar] [CrossRef]
- Greenler, R.G. Infrared study of adsorbed moleculeson metal surfaces by reflection techniques. J. Chem. Phys. 1966, 44, 310. [Google Scholar] [CrossRef]
- Ramin, M.A.; Le Bourdon, G.; Daugey, N.; Bennetau, B.; Vellutini, L.; Buffeteau, T. PM-IRRAS investigation of self-assembled monolayers grafted onto SiO2/Au substrates. Langmuir 2011, 27, 6076–6084. [Google Scholar] [CrossRef]
- Nogues, C.; Lang, P. Self-asembled monolayers on a Zn substrate: Structure and organization. Langmuir 2007, 23, 8385–8391. [Google Scholar] [CrossRef]
- Zaera, F. New advances in the use of infrared absorption spectroscopy for the characterization of heterogeneous catalytic reactions. Chem. Soc. Rev. 2014, 43, 7624–7663. [Google Scholar] [CrossRef]
- Silva, C.B.; da Silva Filho, J.G.; Pinheiro, G.S.; Teixeira, A.M.R.; Freire, P.T.C. Vibrational and structural properties of L-Alanyl-L-Phenylalanine dipeptide by Raman spectroscopy, infrared and DFT calculations. Vibr. Spectrosc. 2018, 98, 128–133. [Google Scholar] [CrossRef]
- Krimm, S.; Bandekar, J. Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. Adv. Protein Chem. 1986, 38, 181–364. [Google Scholar]
- Jackson, M.; Mantsch, H.H. The use and misuse of FTIR spectroscopy in the determination of protein structure. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 95–120. [Google Scholar] [CrossRef]
- Misiūnas, A.; Talaikytė, Z.; Niaura, G.; Razumas, V.; Nylander, T. Thermomyces lanuginosus lipase in the liquid-crystalline phases of aqueous phytantriol: X-ray diffraction and vibrational spectroscopy studies. Biophys. Chem. 2008, 134, 144–156. [Google Scholar] [CrossRef]
- Talaikis, M.; Strazdaitė, S.; Žiaunys, M.; Niaura, G. Far-of resonance: Multiwavelength Raman spectroscopy probing amide bands of amyloid-β-(37-42) peptide. Molecules 2020, 25, 3556. [Google Scholar] [CrossRef]
- Yatsyna, V.; Bakker, D.J.; Feifel, R.; Rijs, A.M.; Zhaunerchyk, V. Far-infrared amide IV-VI spectroscopy of isolated 2- and 4-methylacetanilide. J. Chem. Phys. 2016, 145, 104309. [Google Scholar] [CrossRef]
- Hernández, B.; Pflüger, F.; Adenier, A.; Kruglik, S.G.; Ghomi, M. Vibrational analysis of amino acids and short peptides in hydrated media. VIII. Amino acids with aromatic side chains: L-Phenylalanine, L-Tyrosine, and L-Tryptophan. J. Phys. Chem. B 2010, 114, 15319–15330. [Google Scholar] [CrossRef]
- Olsztynska, S.; Dupuy, N.; Vrielynck, L.; Komorowska, M. Water evaporation analysis of L-Phenylalanine from initial aqueous solutions to powder state by vibrational spectroscopy. Appl. Spectrosc. 2006, 60, 1040–1053. [Google Scholar] [CrossRef]
- Kocherbitov, V.; Latynis, J.; Misiūnas, A.; Barauskas, J.; Niaura, G. Hydration of lysozyme studied by Raman spectroscopy. J. Phys. Chem. B 2013, 117, 4981–4992. [Google Scholar] [CrossRef]
- Yokoyama, T.; Hirata, N.; Tsunoyama, H.; Eguchi, T.; Negishi, Y.; Nakajima, A. Vibrational spectra of thiolate-protected gold nanocluster with infrared reflection absorption spectroscopy: Size- and temperature-dependent ordering behavior of organic monolayer. J. Phys. Chem. C 2020, 124, 363–371. [Google Scholar] [CrossRef]
- Myshakina, N.S.; Ahmed, Z.; Asher, S.A. Dependence of amide vibrations on hydrogen bonding. J. Phys. Chem. B 2008, 112, 11873–11877. [Google Scholar] [CrossRef]
- Podstawka, E.; Niaura, G. Potential-dependent characterization of bombesin adsorbed states on roughened Ag, Au, and Cu electrode surfaces at physiological pH. J. Phys. Chem. B 2009, 113, 10974–10983. [Google Scholar] [CrossRef]
- Ignatjev, I.; Proniewicz, E.; Proniewicz, L.M.; Niaura, G. Effect of potential on temperature-dependent SERS spectra of neuromedin B on Cu electrode. Phys. Chem. Chem. Phys. 2013, 15, 807–815. [Google Scholar] [CrossRef]
- Clegg, R.S.; Reed, S.M.; Smith, R.K.; Barron, B.L.; Rear, J.A.; Hutchison, J.E. The interplay of lateral and tiered interactions in stratified self-assembled molecular assemblies. Langmuir 1999, 15, 8876–8883. [Google Scholar] [CrossRef]
- Sabapathy, R.C.; Bhattacharyya, S.; Leavy, M.C.; Cleland, W.E.; Hussey, C.L. Electrochemical and spectroscopic characterization of self-asembled monolayers of ferrocenylalkyl compounds with amide linkages. Langmuir 1998, 14, 124–136. [Google Scholar] [CrossRef]
- Cai, S.; Singh, B.R. Identification of b-turn and random coil amide III infrared bands for secondary structure estimation of proteins. Biophys. Chem. 1999, 80, 7–20. [Google Scholar] [CrossRef]
- Tam-Chang, S.-W.; Biebuyck, H.A.; Whitesides, G.M.; Jeon, N.; Nuzzo, R.A. Self-assembled monolayers on gold generated from alkanethiols with the structure RNHCOCH2SH. Langmuir 1995, 11, 4371–4382. [Google Scholar] [CrossRef]
- Blasi, D.; Sarcina, L.; Tricase, A.; Stefanachi, A.; Leonetti, F.; Alberga, D.; Mangiatordi, F.; Manoli, K.; Scamarcio, G.; Picca, R.A.; et al. Enhancing the sensitivity of biotynated surfaces by tailoring the design of the mixed self-assembled monolayer synthesis. ACS Omega 2020, 5, 16762–16771. [Google Scholar] [CrossRef]
- Magallanes, C.; Aguirre, B.M.; González, G.A.; Méndez De Leo, L.P. Interaction of aqueous Cu(II) with carboxylic acid and alcohol terminated self asembled monolayers: Surface and interfacial characterization. Surf. Sci. 2020, 692, 121529. [Google Scholar] [CrossRef]
- Snyder, R.G.; Maroncelli, M.; Strauss, H.L.; Hallmark, V.M. Temperature and phase behavior of infrared intensities: The poly(methylene) chain. J. Phys. Chem. 1986, 90, 5623–5630. [Google Scholar] [CrossRef]
- Troughton, E.B.; Bain, C.D.; Whitesides, G.M.; Nuzzo, R.G.; Allara, D.L.; Porter, M.D. Monolayer films prepared by the spontaneous self-assembly of symmetrical and unsymmetrical dialkyl sulfides from solution: Structure, properties, and reactivity of constituent functional groups. Langmuir 1988, 4, 365–385. [Google Scholar] [CrossRef]
- Kuttner, C. Plasmonic in sensing: From calorimetry to SERS analytics. In Plasmonics; Gric, T., Ed.; IntechOpen: London, UK, 2018; pp. 151–180. [Google Scholar] [CrossRef]
- Budvytytė, R.; Valincius, G.; Niaura, G.; Voiciuk, V.; Mickevičius, M.; Chapman, H.; Goh, H.-Z.; Shekhar, P.; Heinrich, F.; Shenoy, S.; et al. Structure and properties of tethered bilayer lipid membranes with unsaturated anchor molecules. Langmuir 2013, 29, 8645–8656. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Talaikis, M.; Eicher-Lorka, O.; Valincius, G.; Niaura, G. Water-induced structural changes in the membrane-anchoring monolayers revealed by isotope-edited SERS. J. Phys. Chem. C 2016, 120, 22489–22499. [Google Scholar] [CrossRef]
- Matulaitienė, I.; Pociutė, E.; Kuodis, Z.; Eicher-Lorka, O.; Niaura, G. Interaction of 4-imidazolemethanol with a copper electrode revealed by isotope-edited SERS and theoretical modeling. Phys. Chem. Chem. Phys. 2015, 17, 16483–16493. [Google Scholar] [CrossRef]
- Morrison, J.J.; Botting, N.P. The synthesis of [phenyl-2H5]glyconasturtiin and its metabolites for metabolic studies. J. Label. Compd. Radiopharm. 2005, 48, 897–907. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solid State | Calculated | Assignments | ||
|---|---|---|---|---|
| MOPHE | MOPHE-D5 | MOPHE | MOPHE-D5 | |
| 3309 vs | 3309 vs | 3568 | 3568 | ν(N−H) Amide-A |
| 3027 w | 2275 w | 3124 | 3124 | ν(=C–H) Phe |
| 2926 s | 2926 | 3004 | 3004 | νas(CH2) Chain |
| 2852 s | 2852 s | 2973 | 2973 | νs(CH2) Chain |
| 2558 w | 2258 v | 2635 | 2635 | ν(S−H) Thiol |
| 1640 vs | 1640 vs | 1713 | 1713 | ν(C=O) + δ(NH) Amide-I |
| 1546 vs | 1546 vs | 1525 | 1525 | ν(C−N) + δ(NH) Amide-II |
| 1496 m | 1383 m | 1481 | 1403 | ν(C=C) + β(CH) Phe (F19a) a |
| 1465 m | 1465 m | 1466 | 1466 | δ(CH2) Chain (scissoring) |
| 1454 w | 1383 w | − | − | ν(C=C) + β(CH) Phe (F19b) |
| 1418 w | 1418 w | 1392 | 1392 | δ(CH2) Chain (scissoring) |
| 1328 m | 1328 m | 1354 | 1354 | δ(CH2) Chain |
| 1257 w | 1257 w | 1262 | 1262 | δ(CH2) Chain |
| 1249 m | 1249 m | 1241 | 1241 | ν(C−N) + δ(CNH) Amide-III |
| 1195 m | 1195 m | 1206 | 1207 | δ(CH2) Chain |
| 1030 w | 821 w | 1048 | 837 | β(CH) Phe (F18a) |
| 749 s | 546 s | 763 | 554 | γ(CH) Phe (F11) |
| 707 br,m | 707 br, m | − | − | γ(NH) Amide V |
| 699 s | 447 s | 714 | 452 | γ(CH) Phe (F4) |
Sample Availability: Samples of the compounds are not available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuodis, Z.; Matulaitienė, I.; Špandyreva, M.; Labanauskas, L.; Stončius, S.; Eicher-Lorka, O.; Sadzevičienė, R.; Niaura, G. Reflection Absorption Infrared Spectroscopy Characterization of SAM Formation from 8-Mercapto-N-(phenethyl)octanamide Thiols with Phe Ring and Amide Groups. Molecules 2020, 25, 5633. https://doi.org/10.3390/molecules25235633
Kuodis Z, Matulaitienė I, Špandyreva M, Labanauskas L, Stončius S, Eicher-Lorka O, Sadzevičienė R, Niaura G. Reflection Absorption Infrared Spectroscopy Characterization of SAM Formation from 8-Mercapto-N-(phenethyl)octanamide Thiols with Phe Ring and Amide Groups. Molecules. 2020; 25(23):5633. https://doi.org/10.3390/molecules25235633
Chicago/Turabian StyleKuodis, Zenonas, Ieva Matulaitienė, Marija Špandyreva, Linas Labanauskas, Sigitas Stončius, Olegas Eicher-Lorka, Rita Sadzevičienė, and Gediminas Niaura. 2020. "Reflection Absorption Infrared Spectroscopy Characterization of SAM Formation from 8-Mercapto-N-(phenethyl)octanamide Thiols with Phe Ring and Amide Groups" Molecules 25, no. 23: 5633. https://doi.org/10.3390/molecules25235633
APA StyleKuodis, Z., Matulaitienė, I., Špandyreva, M., Labanauskas, L., Stončius, S., Eicher-Lorka, O., Sadzevičienė, R., & Niaura, G. (2020). Reflection Absorption Infrared Spectroscopy Characterization of SAM Formation from 8-Mercapto-N-(phenethyl)octanamide Thiols with Phe Ring and Amide Groups. Molecules, 25(23), 5633. https://doi.org/10.3390/molecules25235633