
Current Synthetic Routes to Peptidyl Mono-Fluoromethyl Ketones (FMKs) and Their Applications



Abstract
1. Introduction
2. Current Synthetic Routes to Peptidyl Mono-FMKs
2.1. Solution-Phase Routes
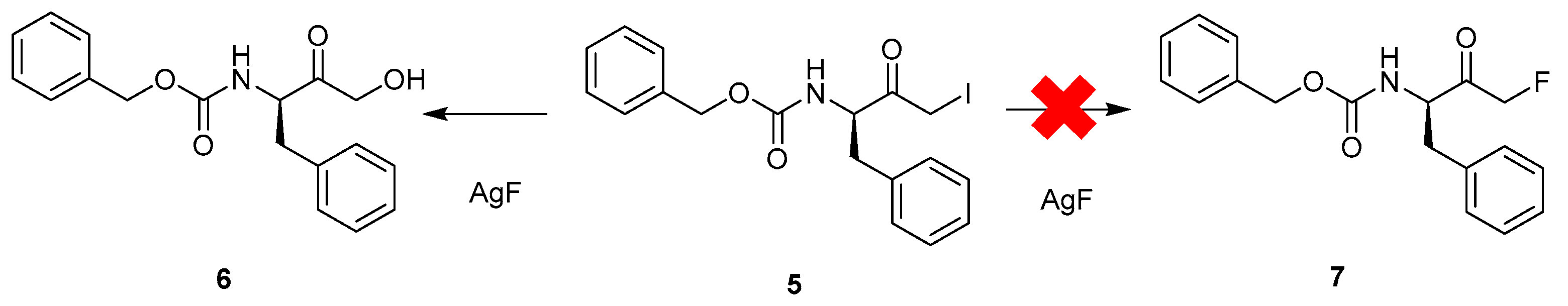
2.1.1. Halogen-Exchange
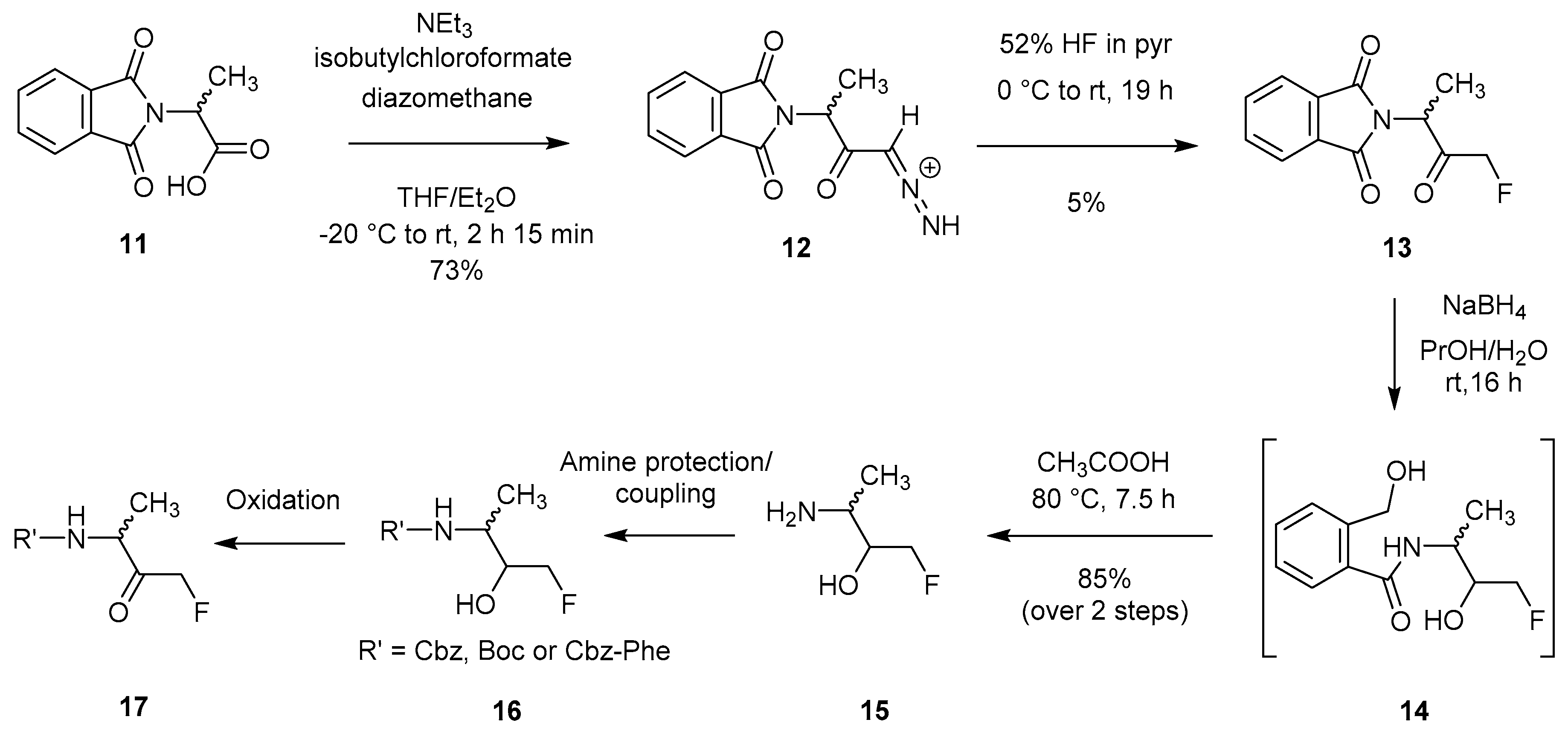
2.1.2. Synthesis via Direct Fluorination of a Diazo Intermediate
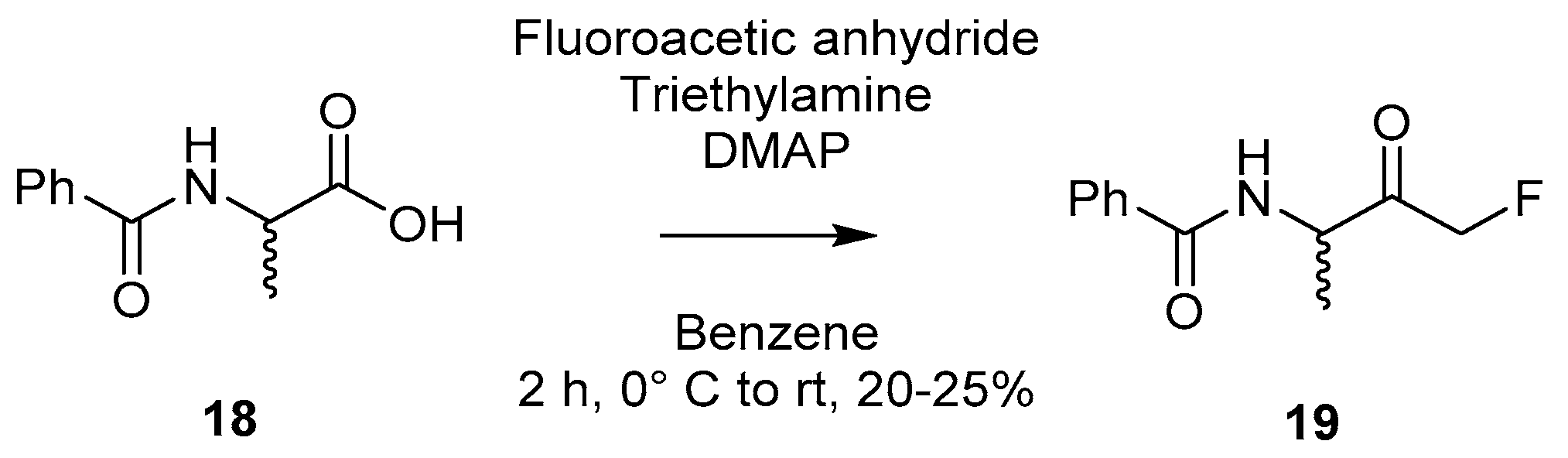
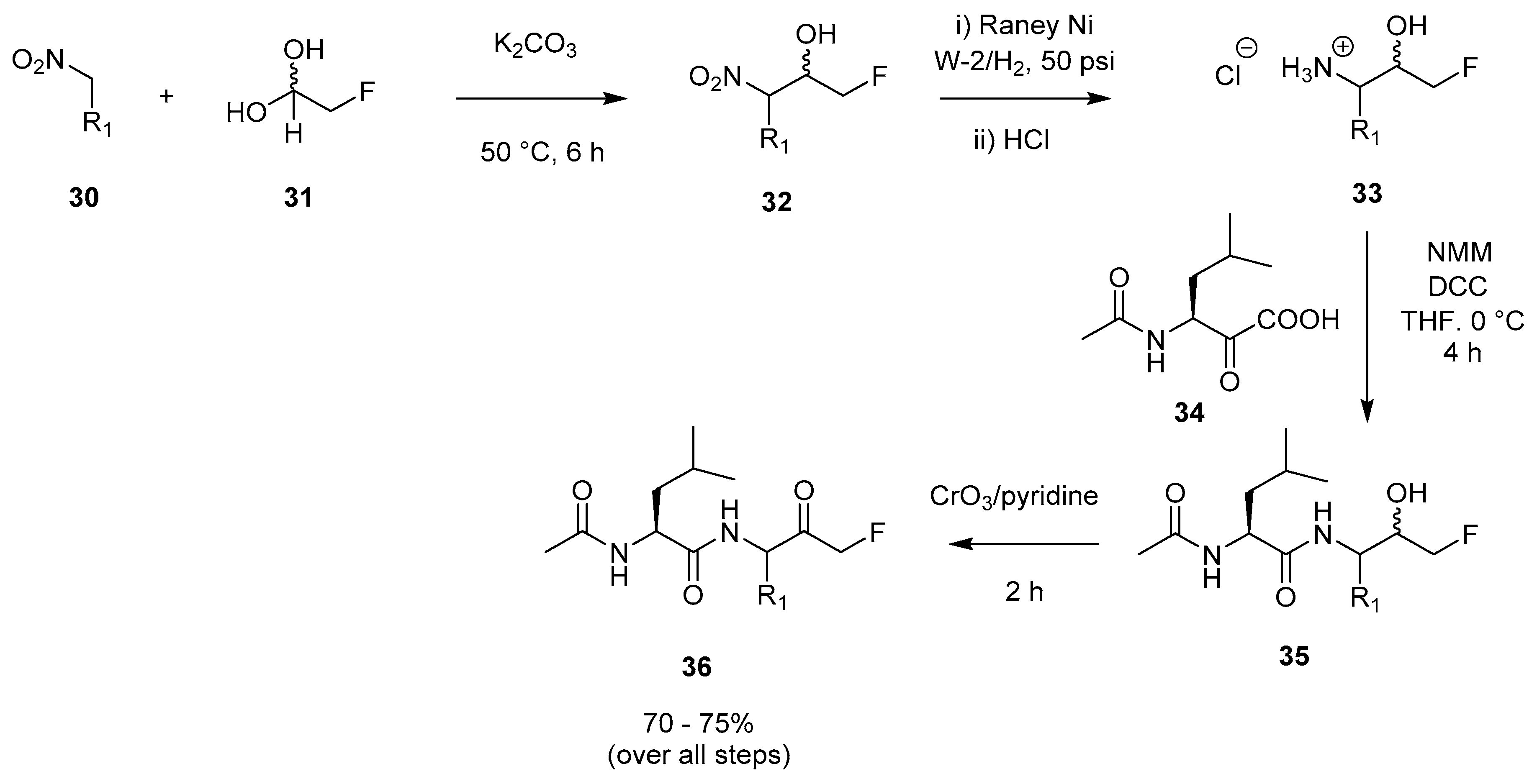
2.1.3. Dakin–West Modification
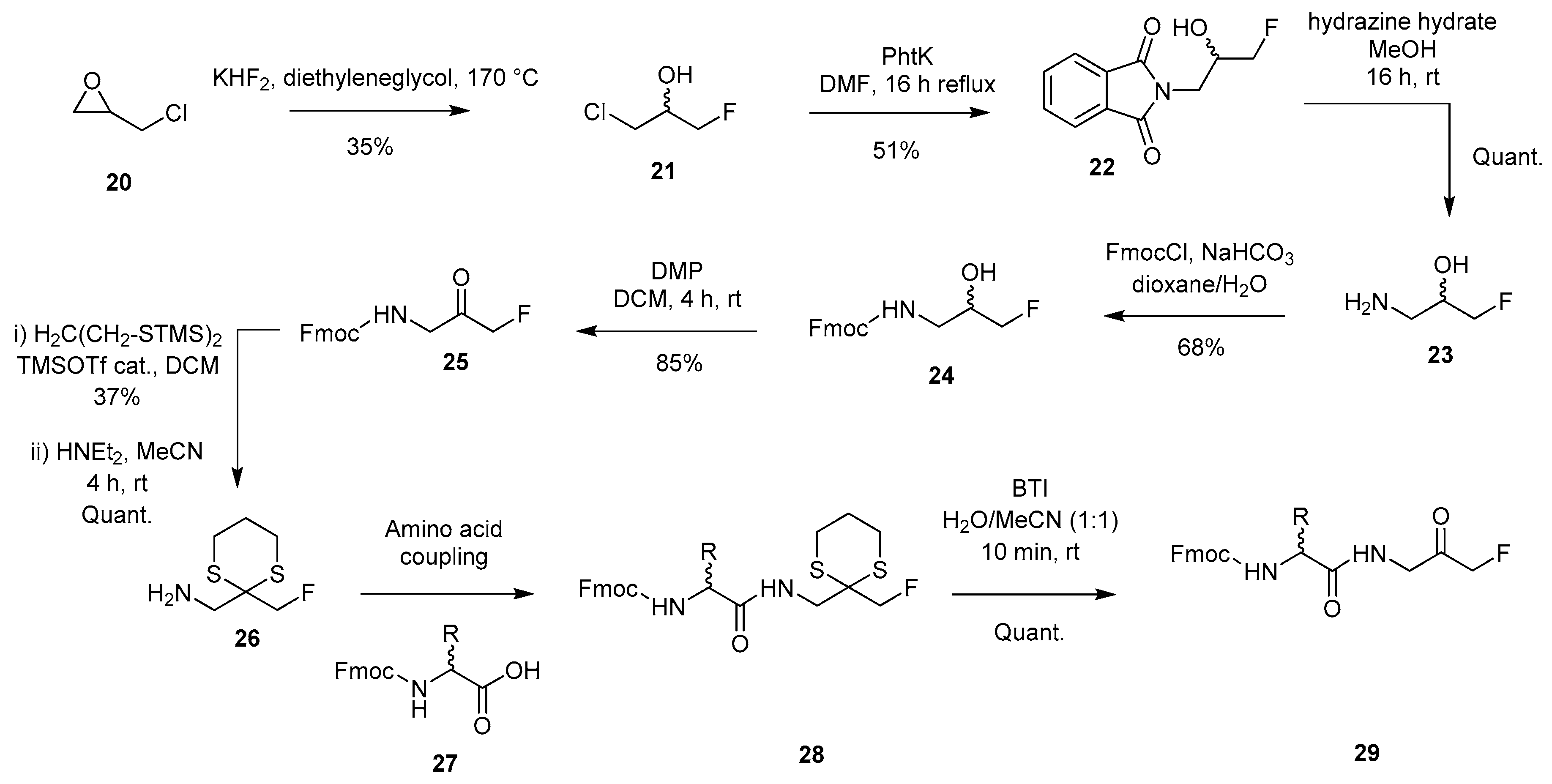
2.1.4. Epoxide Ring Opening Approach with Fluoride Source
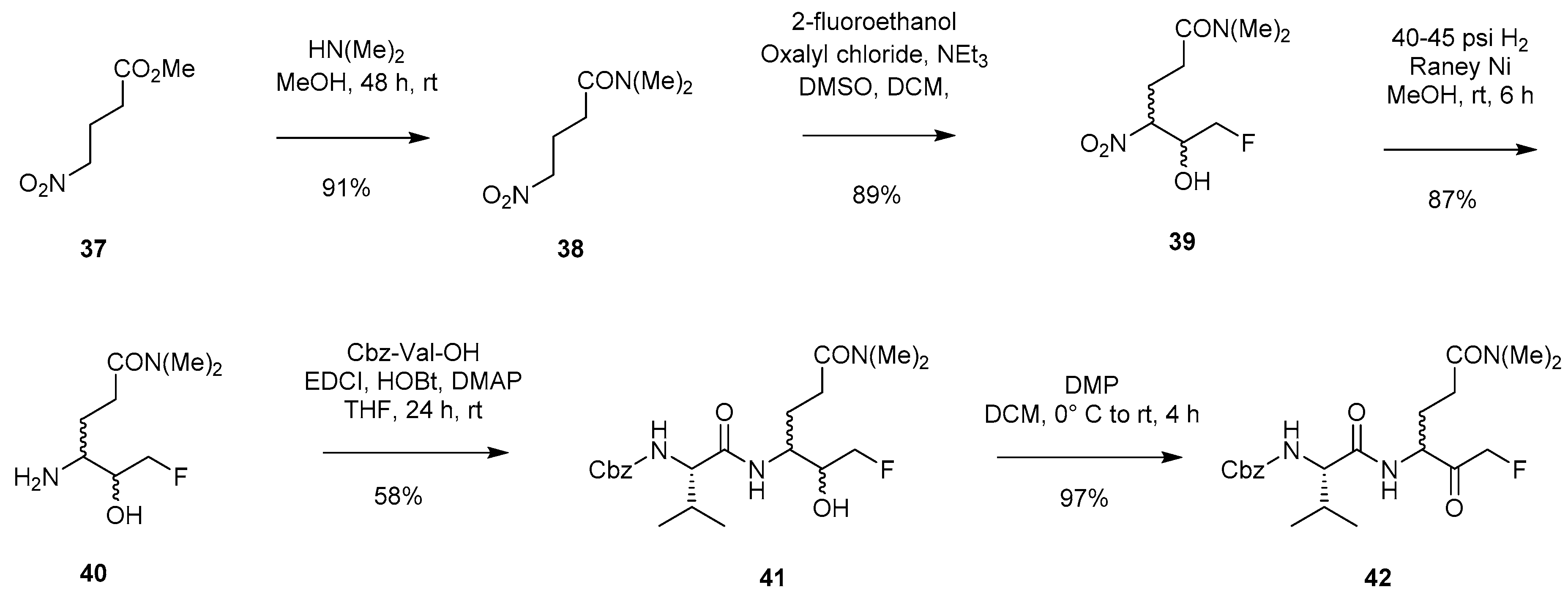
2.1.5. Synthetic Pathway Involving Utilisation of a Fluorinated Hemiacetal/Aldehyde
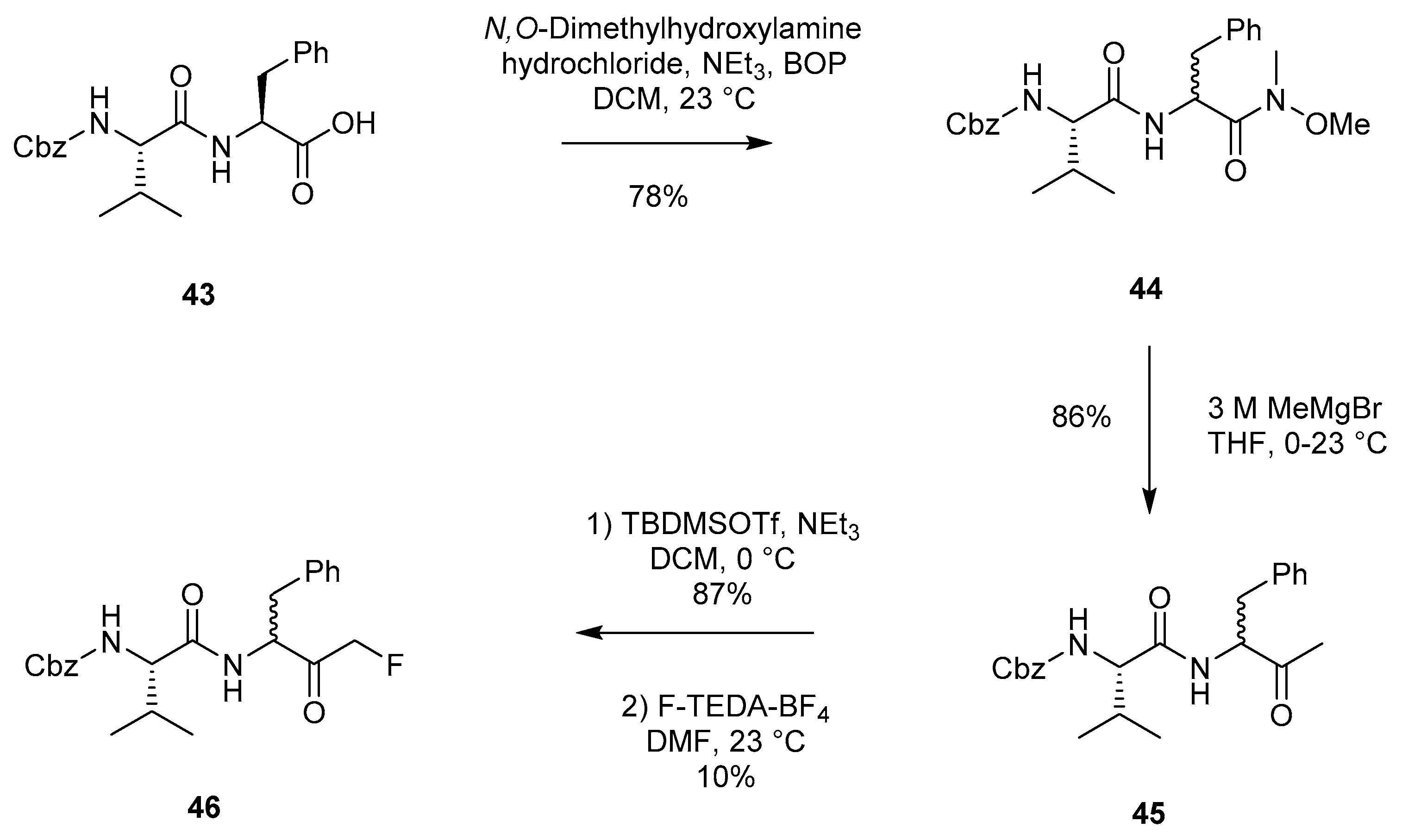
2.1.6. Synthetic Methodology Employing Silyl Enol Ether Fluorination
2.1.7. Synthetic Pathway Involving 1-amino-3-fluoro-propan-2-ol hydrochloride (48)
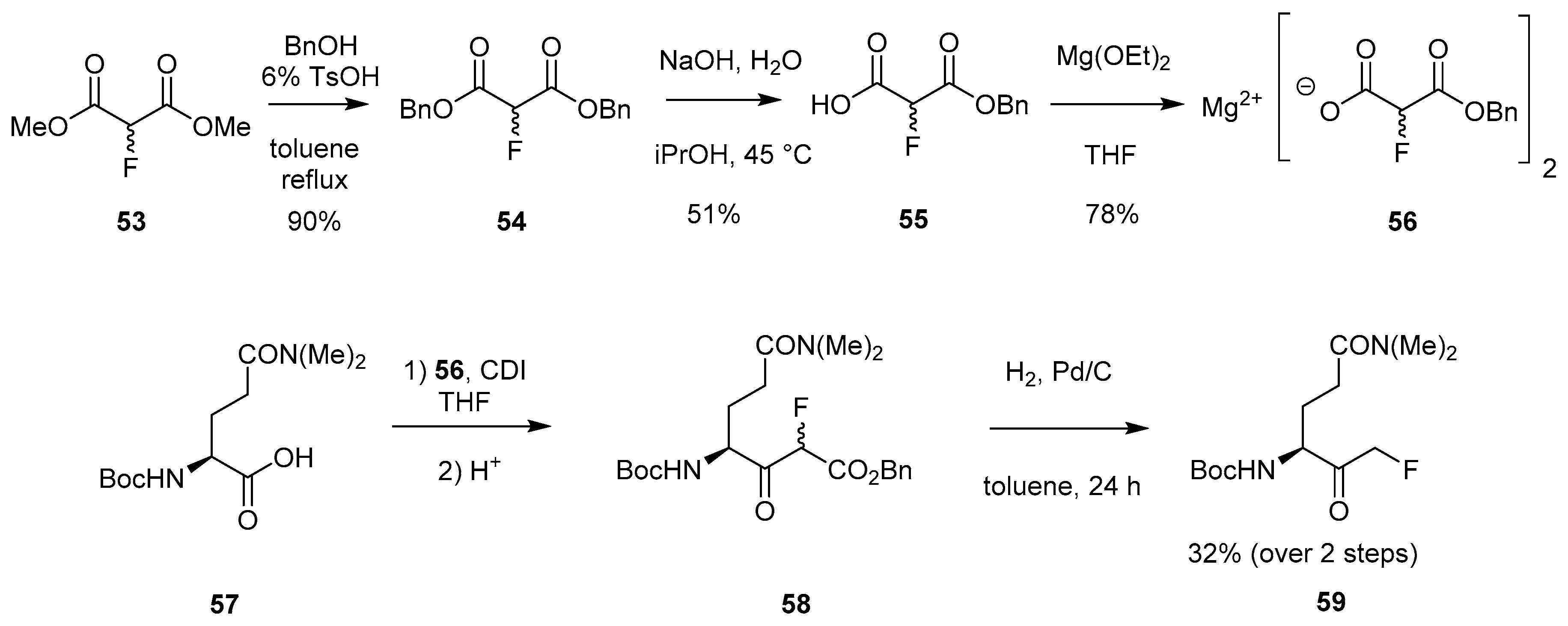
2.1.8. Synthetic Approach Involving Employment of a Magnesium Fluoromalonate
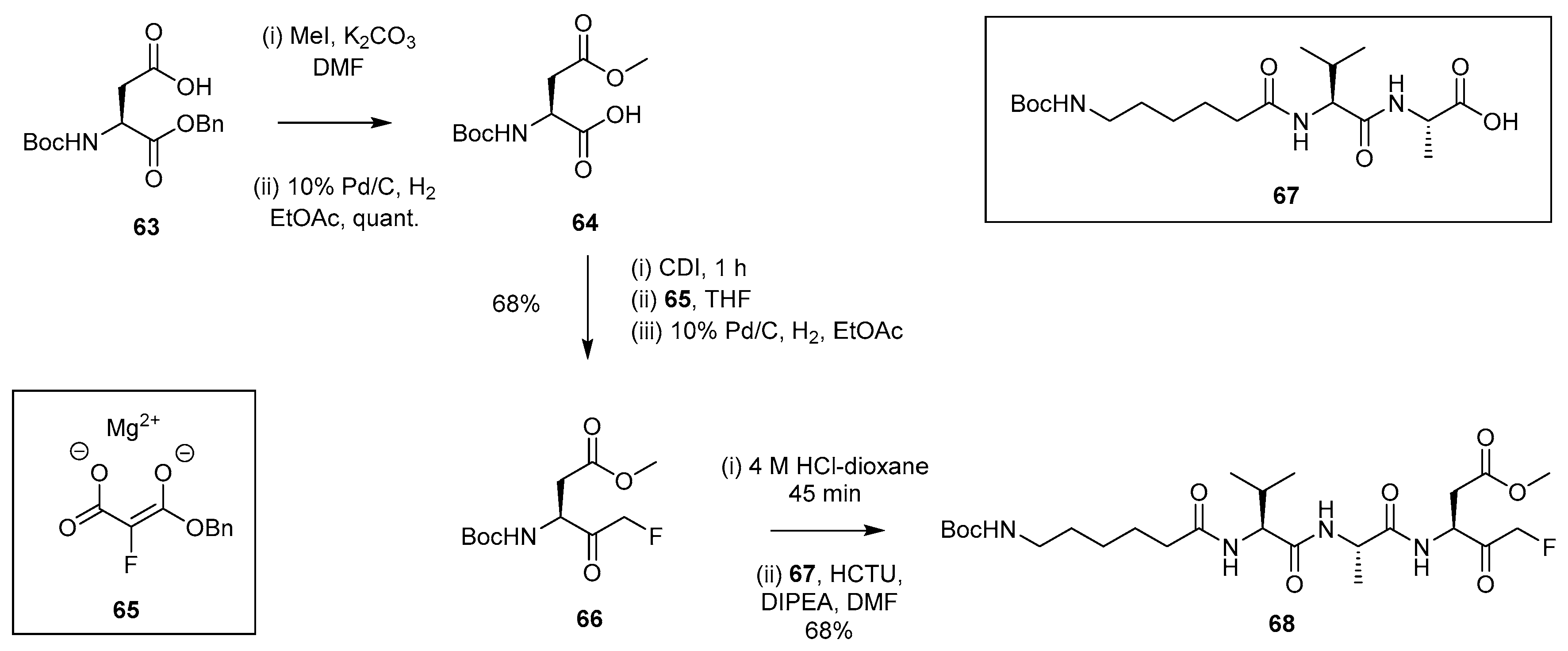
2.1.9. Synthetic Approach Involving Magnesium Monobenzyl Fluoromalonate Enolate 65
2.2. Solid-Phase Route
3. Applications and Uses of Peptidyl Mono-FMKs in Medicinal Chemistry
3.1. Drug Discovery
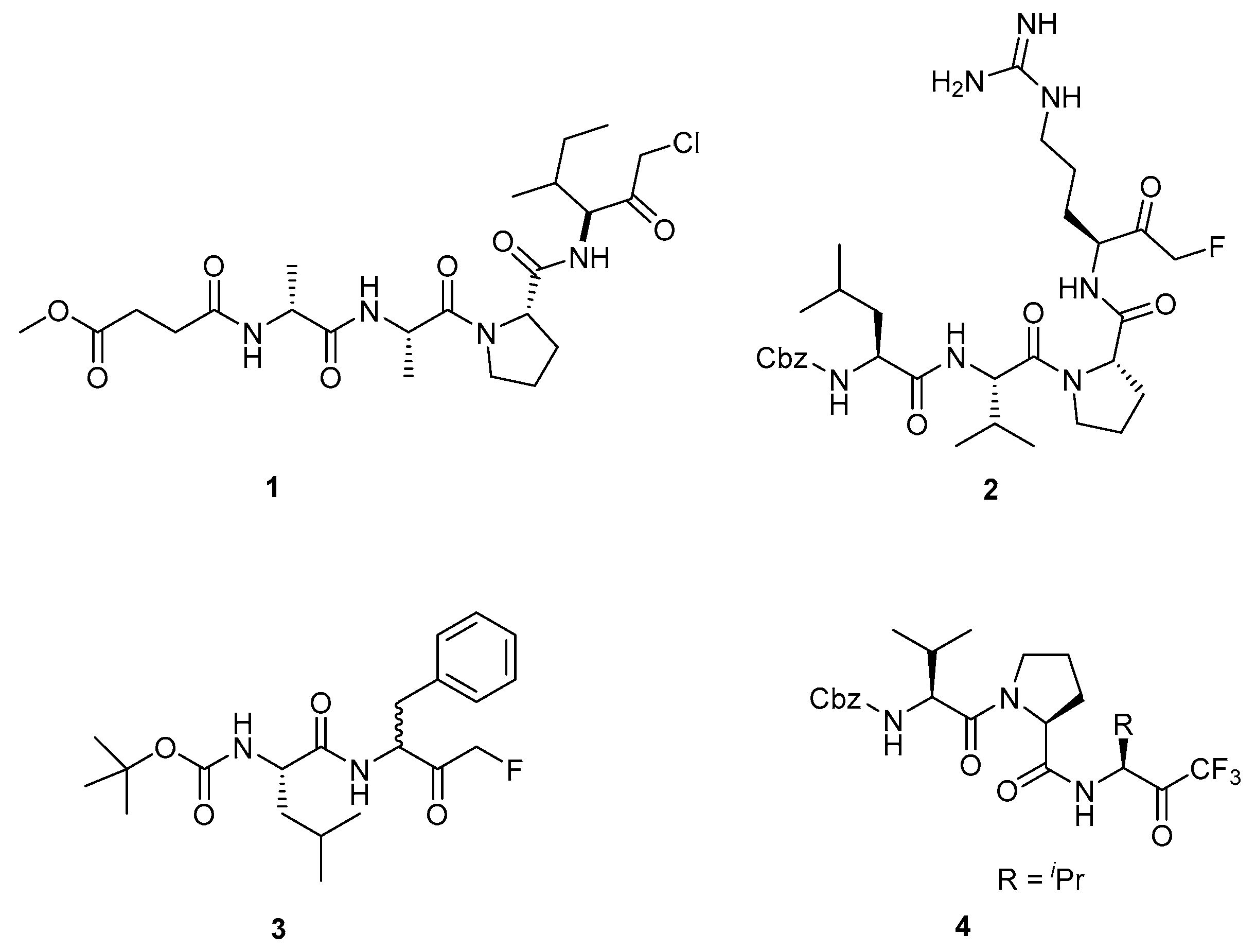
3.1.1. Peptidyl FMKs for the Treatment of Rheumatoid Arthritis
3.1.2. Peptidyl FMKs for the Treatment of Neurological Disorders
3.1.3. Peptidyl FMKs as Inhibitors of MALT1 Paracaspase
3.1.4. Dipeptidyl Glutaminyl FMKs as SARS-CoV Inhibitors
3.2. Probes for Cellular Interrogation
4. Conclusions
Funding
Conflicts of Interest
References
- Ahmed, N.K.; Martin, L.A.; Watts, L.M.; Palmer, J.; Thornburg, L.; Prior, J.; Esser, R.E. Peptidyl fluoromethyl ketones as inhibitors of cathepsin B. Biochem. Pharmacol. 1992, 44, 1201–1207. [Google Scholar] [CrossRef]
- Chatterjee, S.; Ator, M.A.; Bozyczko-coyne, D.; Josef, K.; Wells, G.; Tripathy, R.; Iqbal, M.; Bihovsky, R.; Senadhi, S.E.; Mallya, S.; et al. Synthesis and Biological Activity of a Series of Potent Fluoromethyl Ketone Inhibitors of Recombinant Human Calpain I. J. Med. Chem. 1997, 40, 3820–3828. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, J.M.; Du, G.; Fontán, L.; Us, I.; Qiao, Q.; Chennamadhavuni, S.; Shao, J.; Wu, H.; Melnick, A.; Gray, N.S.; et al. Peptide-based covalent inhibitors of MALT1 paracaspase. Bioorg. Med. Chem. Lett. 2019, 29, 1336–1339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-Z.; Zhang, H.; Kemnitzer, W.; Tseng, B.; Cinatl, J.; Michaelis, M.; Doerr, H.W.; Cai, S.X. Design and Synthesis of Dipeptidyl Glutaminyl Fluoromethyl Ketones as Potent Severe Acute Respiratory Syndrome Coronovirus (SARS-CoV) Inhibitors. J. Med. Chem. 2006, 49, 1198–1201. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Kuhn, B.; Aebi, J.; Lin, X.; Ding, H.; Zhou, Z.; Xu, Z.; Xu, D.; Han, L.; Liu, C.; et al. Discovery of Fluoromethylketone-Based Peptidomimetics as Covalent ATG4B (Autophagin-1) Inhibitors. ACS Med. Chem. Lett. 2016, 7, 802–806. [Google Scholar] [CrossRef]
- Dobrotǎ, C.; Fasci, D.; Hǎdade, N.D.; Roiban, G.D.; Pop, C.; Meier, V.M.; Dumitru, I.; Matache, M.; Salvesen, G.S.; Funeriu, D.P. Glycine Fluoromethylketones as SENP-Specific Activity Based Probes. ChemBioChem 2012, 13, 80–84. [Google Scholar] [CrossRef]
- Yepeng, L.; Yang, Q.; Xie, Y.; Duan, S.; Cai, S.; Forrest, M.L. A sensitive near-infrared fluorescent probe for caspase- mediated apoptosis: Synthesis and application in cell imaging. Drug Discov. Ther. 2012, 5, 220–226. [Google Scholar] [CrossRef]
- Coover, R.A.; Luzi, N.M.; Korwar, S.; Casile, M.E.; Lyons, C.E.; Peterson, D.L.; Ellis, K.C. Design, synthesis, and in vitro evaluation of a fluorescently labeled irreversible inhibitor of the catalytic subunit of cAMP-dependent protein kinase (PKACα). Org. Biomol. Chem. 2016, 14, 4576–4581. [Google Scholar] [CrossRef]
- Rasnick, D. Synthesis of Peptide Fluoromethyl Ketones and the inhibition of Human Cathepsin B. Anal. Biochem. 1985, 149, 461–465. [Google Scholar] [CrossRef]
- Hedstrom, L. Serine Protease Mechanism and Specificity. Chem. Rev. 2002, 102, 4501–4524. [Google Scholar] [CrossRef]
- Tsuda, Y.; Okada, Y.; Nagamatsu, Y.; Okamoto, U. Synthesis of peptide chloromethyl ketones and examination of their inhibitory effects on human spleen fibrinolytic proteinase(SFP) and human leukocyte elastase(LE). Chem. Pharm. Bull. 1987, 35, 3576–3584. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.; Kore, A.R.; Shanmugasundaram, M. Efficient Synthesis of New Peptidyl Chloromethyl Ketones for the Application of Proteinase K Inhibitors. Synth. Commun. 2012, 42, 40–45. [Google Scholar] [CrossRef]
- Sun, A.; Shoji, M.; Lu, Y.J.; Liotta, D.C.; Snyder, J.P. Synthesis of EF24—Tripeptide Chloromethyl Ketone: A Novel Curcumin-Related Anticancer Drug Delivery System. J. Med. Chem. 2006, 49, 3153–3158. [Google Scholar] [CrossRef] [PubMed]
- Imperiali, B.; Abeles, R.H. A versatile synthesis of peptidyl fluoromethyl ketones. Tetrahedron Lett. 1986, 27, 135–138. [Google Scholar] [CrossRef]
- Edwards, P.D. A Method For The Stereoselective Synthesis of Peptidyl Trifluoromethyl Ketones. Tetrahedron Lett. 1992, 33, 4279–4282. [Google Scholar] [CrossRef]
- Poupart, M.-A.; Fazal, G.; Goulet, S.; Mar, L.T. Solid-Phase Synthesis of Peptidyl Trifluoromethyl Ketones. J. Org. Chem. 1999, 64, 1356–1361. [Google Scholar] [CrossRef]
- Citarella, A.; Micale, N. Peptidyl Fluoromethyl Ketones and Their Applications in Medicinal Chemistry. Molecules 2020, 25, 4031. [Google Scholar] [CrossRef]
- Kolb, M.; Barth, J.; Neises, B. Synthesis of Fluorinated alpha-amino Ketones Part I: Alpha-Benzamidoalkyl mono- di- and Trifluoromethyl Ketones. Tetrahedron Lett. 1986, 27, 1579–1582. [Google Scholar] [CrossRef]
- Penke, B.; Czombos, J.; Balaspiri, L.; Petres, J.; Kovacs, K. Synthese von Diazoketonen aus Acylaminosauren unter Verwendung von gemischten Anhydriden bzw. N, N’ -Dicyclohexyl-carbodiimid. Helv. Chim. Acta 1970, 53, 1057–1061. [Google Scholar] [CrossRef]
- Becker, H. Organikum: Organisch-Chemisches Grundpraktikum, 8th ed.; Deutscher Verl. d. Wissenschaften: Berlin, Germany, 1968. [Google Scholar]
- Mancuso, A.J.; Huang, S.; Swern, D. Oxidation of Long-chain and Related Alcohols to Carbonyls by Dimethyl Sulfoxide “Activated” by Oxalyl Chloride. J. Org. Chem. 1978, 43, 2480–2482. [Google Scholar] [CrossRef]
- Kolb, M.; Neises, B.; Gerhart, F. Preparation of Fluorinated Ketone Analogues of Phenylalanine, Lysine, and p-(Guanidino)phenylalanine. Liebigs Ann. Chem. 1990, 1990, 1–6. [Google Scholar] [CrossRef]
- Morris, T.S.; Frormann, S.; Shechosky, S.; Lowe, C.; Lall, M.S.; Gauss-Muller, V.; Purcell, R.H.; Emerson, S.U.; Vederas, J.C.; Malcolm, B.A. In Vitro and Ex Vivo Inhibition of Hepatitis A Virus 3C Proteinase by a Peptidyl Monofluoromethyl Ketone. Bioorganic Med. Chem. 1997, 5, 797–807. [Google Scholar] [CrossRef]
- Rauber, P.; Angliker, H.; Walker, B.; Shaw, E. The synthesis of peptidylfluoromethanes and their properties inhibitors of serine proteinases and cysteine proteinases. Biochem. J. 1986, 239, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Olah, G.A.; Shih, G.J.; Prakash, G.K.S. Fluorine-containing reagents in organic synthesis. J. Fluor. Chem. 1986, 33, 377–396. [Google Scholar] [CrossRef]
- Vechia, L.D.; Octavio, R.; Alves, M.; Soter, L.; Mariz, D. The Dakin-West reaction: Past, present and future. Tetrahedron 2018, 74, 4359–4371. [Google Scholar] [CrossRef]
- Revesz, L.; Briswalter, C.; Heng, R.; Leutwiler, A.; Mueller, R.; Wuethrich, H.-J. Synthesis of Pl Aspartate-Based Peptide Acyloxymethyl and Fluoromethyl Ketones as Inhibitors of Interleukin-1 beta-Converting Enzyme. Tetrahedron Lett. 1994, 35, 9693–9696. [Google Scholar] [CrossRef]
- Palmer, J.T. Reagents and Methods for Stereospecific Fluoromethylation. EU Patent EP0442754A2, 1991. [Google Scholar]
- Witte, M.D.; Descals, C.V.; De Lavoir, S.V.P.; Florea, B.I.; Van der Marel, G.; Overkleeft, H.S. BODIPY-VAD-Fmk, a useful tool to study yeast peptide N-glycanase activity. Org. Biomol. Chem. 2007, 5, 3690–3697. [Google Scholar] [CrossRef]
- Palmer, J.T. Magnesium Fluoromalonates. US Patent US005101068A, 31 March 1992. [Google Scholar]
- Roiban, G.D.; Matache, M.; Hǎdade, N.D.; Funeriu, D.P. A general solid phase method for the synthesis of sequence independent peptidyl-fluoromethyl ketones. Org. Biomol. Chem. 2012, 10, 4516–4523. [Google Scholar] [CrossRef]
- La Manna, S.; Di Natale, C.; Florio, D.; Marasco, D. Peptides as Therapeutic Agents for Inflammatory-Related Diseases. Int. J. Mol. Sci. 2018, 19, 2714. [Google Scholar] [CrossRef]
- Eichhold, T.H.; Hookfin, E.B.; Taiwo, Y.O.; De, B.; Wehmeyer, K.R. Isolation and quantification of fluoroacetate in rat tissues, following dosing of Z-Phe-Ala-CH 2 -F, a peptidyl fluoromethyl ketone protease inhibitor. J. Pharm. Biomed. Anal. 1997, 16, 459–467. [Google Scholar] [CrossRef]
- Danckwardt, S.; Hentze, M.W.; Kulozik, A.E. Pathologies at the nexus of blood coagulation and inflammation: Thrombin in hemostasis, cancer, and beyond. J. Mol. Med. 2013, 91, 1257–1271. [Google Scholar] [CrossRef] [PubMed]
- Laurens, N.; Koolwijk, P.; De Maat, M.P. Fibrin structure and wound healing. J. Thromb. Haemost. 2006, 4, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Antalis, T.M.; Shea-Donohue, T.; Vogel, S.N.; Sears, C.; Fasano, A. Mechanisms of disease: Protease functions in intestinal mucosal pathobiology. Nat. Clin. Pract. Gastroenterol. Hepatol. 2007, 4, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Swanstrom, R.; Anderson, J.; Schiffer, C.; Lee, S.K. Viral protease inhibitors. Handb. Exp. Pharmacol. 2009, 189, 85–110. [Google Scholar] [CrossRef]
- Sani, M.; Sinisi, R.; Viani, F. Peptidyl Fluoro-Ketones as Proteolytic Enzyme Inhibitors. Curr. Top. Med. Chem. 2006, 6, 1545–1566. [Google Scholar] [CrossRef]
- Anand, K.; Anand, K.; Ziebuhr, J.; Wadhwani, P. Coronavirus Main Proteinase (3CL pro) Structure: Basis for Design of Anti-SARS Drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| R | % Inhibition in Liver Homogenates | % Inhibition in Kidney Homogenates | R | % Inhibition in Liver Homogenates | % Inhibition in Kidney Homogenates |
 | 70.5 | 39.0 |  | 81.8 | 75.4 |
 | 86.9 | 57.3 |  | 22.2 | 29.8 |
 | 28.3 | 24.2 |  | 91.2 | 78.8 |
 | |||||||
|---|---|---|---|---|---|---|---|
| EC50 (µM) | CC50 (µM) | SI | |||||
| Vero | CaCo2 | Vero | |||||
| Entry | Compound | R | FFM1 | 6109 | FFM1 | ||
| 1 | 80a |  | 3.6 ± 1.3 | 2.5 ± 0.4 | 2.4 ± 0.56 | >100 | >40 |
| 2 | 80b |  | 8.9 ± 2.9 | 5.3 ± 1.7 | 8.8 ± 2.5 | >100 | >18 |
| 3 | 80c |  | 6.2 ± 1.9 | 6.6 ± 3.0 | 12.6 ± 4.1 | >100 | >15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lloyd, C.M.; Colgin, N.; Cobb, S.L. Current Synthetic Routes to Peptidyl Mono-Fluoromethyl Ketones (FMKs) and Their Applications. Molecules 2020, 25, 5601. https://doi.org/10.3390/molecules25235601
Lloyd CM, Colgin N, Cobb SL. Current Synthetic Routes to Peptidyl Mono-Fluoromethyl Ketones (FMKs) and Their Applications. Molecules. 2020; 25(23):5601. https://doi.org/10.3390/molecules25235601
Chicago/Turabian StyleLloyd, Carissa M., Neil Colgin, and Steven L. Cobb. 2020. "Current Synthetic Routes to Peptidyl Mono-Fluoromethyl Ketones (FMKs) and Their Applications" Molecules 25, no. 23: 5601. https://doi.org/10.3390/molecules25235601
APA StyleLloyd, C. M., Colgin, N., & Cobb, S. L. (2020). Current Synthetic Routes to Peptidyl Mono-Fluoromethyl Ketones (FMKs) and Their Applications. Molecules, 25(23), 5601. https://doi.org/10.3390/molecules25235601