The results of this study are presented below in four sections. The first section presents the targets chosen due to their association with DA and the interaction energy resulting from each target–DA complex. The second section presents the control variables of the design of the bioreceptors and its affinity with DA after each structural modification. The third section includes the description of the parameters that allowed three bioreceptor candidates to be selected for future synthesis as well as the molecular interaction distances between DA and each bioreceptor. Finally, the fourth section analyzes the molecular dynamics of each dopamine–bioreceptor complex
2.2. Design and Analysis of Bioreceptors that Interact with DA
The bioreceptors were designed to be miniature versions of DAT, the protein responsible for DA reuptake into the presynaptic neurons [
22], but with better affinity and molecular distances. Considering that this protein is activated or deactivated according to both the short- and long-term physiological demands of the neurons [
22], the bioreceptors must mimic its ability to recognize the DA present at the synaptic cleft when coupled to the biosensor system.
It is important to mention that miniaturization is a concept that has been defined as “the process of doing something on a very small scale using modern technology” [
23] or as “a version of something on a small scale or small size” [
24]. In addition, this miniaturization trend was first applied to electronic devices in the 1960s and later evolved to biological molecules [
25] and drug release mechanisms [
26].
It is important to keep in mind that human DAT (hDAT) has not been crystallized yet; therefore, there is no information about its crystal structure determined through experimental methods. However, the structure of the DATs of other species, such as
Drosophila melanogaster, has already been determined [
27] (the PDB code for this protein is 4XP1), and it has been described that the 4XP1 protein is homologous to hDAT. However, there are computational models that have simulated the structure and interactions of hDAT [
28], and this information will also be considered in the design of proposed bioreceptors.
Furthermore, the initial bioreceptors were designed based on the amino acids of the active site of the 5M8L, 4IIT, 2A3R, and 2E82 enzymes that exhibit a direct interaction with the ligand, also known as the active site’s hotspots. Subsequently, structural changes were sequentially made to these initial bioreceptors as per the provisions included in the methodology.
The first group of bioreceptors was made out of peptides built based on the amino acids from the hot spots of 5M8L(1.0), 4IIT (2.0), 2A3R (3.0), and 2E82(4.0).
Table 3 denotes their identification code, their amino acid sequence, their average interaction affinity after performing this calculation thrice, and the interaction affinity’s standard deviation.
As expected, the results indicated that the affinity of the three bioreceptors for DA was lower than the affinity of DAT for DA, which is −5.2 kcal/mol. As previously reported, these results were expected because the steric hindrance exhibited by a peptide within this design length range is not comparable with that of a target [
18].
Thus, two options were considered. The first was to consider the number of residues between the selected amino acids from each hot spot, and the second was to increase the steric volume of the bioreceptors in order to increase their affinity [
18].
Accordingly,
Table 4 shows a new group of bioreceptors where methylene bridges were added to the previous amino acid sequences from
Table 3 to preserve the distances presented within the target’s active site. This table also presents interaction affinity averages calculated as previously described with their respective standard deviation.
In this case, if the interaction energies of the bioreceptors composed solely of peptides are compared against the interaction energies from the bioreceptors to which the methylene bridges were added, no conclusive pattern may be observed. In some cases, the energy increased (bioreceptors 1.1 and 3.1,
Table 3, second and fourth row), whereas in others, it decreased (bioreceptor 2.1). However, it was considered that because DA is a small molecule and does not have many atoms that may allow it to interact with other molecules, the amino acids are distant. Therefore, for the bioreceptor 2.1, the possible interactions that existed before adding the methylene bridges were lost. This result contradicts that of a previous study [
18], but if it is considered that the substrate size in the study was much larger compared to DA, it could be deduced that DA can only generate a few interactions.
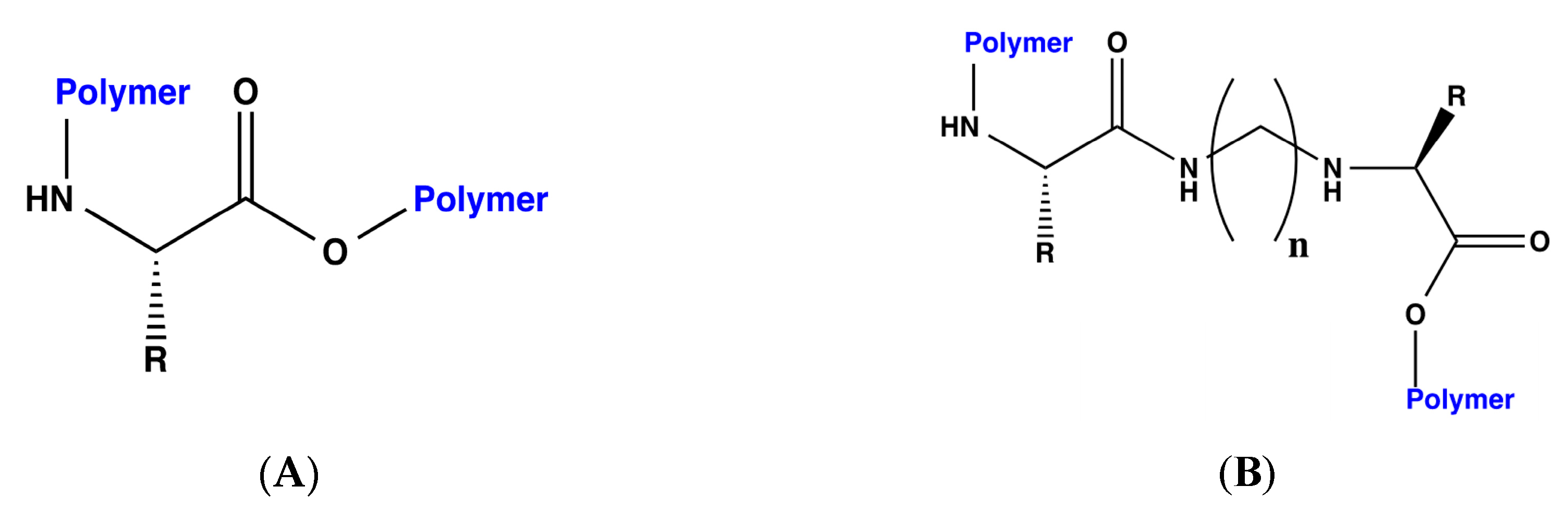
Considering that in previous studies, steric volume increased interaction energies [
18], each of the designed bioreceptors, those with and without the methylene bridges, were again modified by polymerizing the C and N terminals of each peptide. The general scheme for the polymerization is shown in
Figure 1.
The following two different polymers were used for this purpose: polyethylene and polystyrene. Once designed, the interaction energy for DA was calculated for each of the bioreceptors. The results of these dockings with polyethylene and polystyrene are presented in
Table 5, with their corresponding code and sequence.
According to the results presented in
Table 5 compared to those in
Table 3 and
Table 4, it is evident that affinity increases in all cases when the steric volume increases by means of polymerization. In this case particularly, there is a recognizable pattern. In addition, polymerization with polystyrene increases the affinity for DA in contrast to that of the bioreceptors polymerized with polyethylene. On the other hand, these results follow the pattern set out previously [
18], in which it has been stated that recognition interaction improves when polymerization is performed with polystyrene.
Continuing with the design of the bioreceptors and based on the above results, we decided to study which of the variables of the amino acids could influence affinity for DA. Thus, the first criterion was stereochemistry. Except for glycine, all other amino acids have a chiral carbon, which exhibits bonds with four different atoms or groups of atoms [
29], thereby generating an enantiomer pair of spatial isomers defined as non-superimposable mirror images of each other [
30]. It is important to keep in mind that the amino acids’ natural stereochemistry in the human body is the L configuration and not the D configuration [
29]. However, this section uses the
R and
S nomenclature, which applies to natural compounds and determines the stereochemistry based on the importance defined by the atomic number of the chiral carbon substituent. Using this information, amino acid stereochemistry was selected as the first variable to be analyzed to identify its influence on their interaction with DA.
To address the first variation, the NWR bioreceptor polymerized with polystyrene (
Table 5; code: 1.0.2) was selected because this was the polymerization that provided the best results and because its interaction with DA was restricted to only those three amino acids. In contrast, it cannot be accurately identified which of the amino acids from the larger peptides are responsible for their interaction with DA. Similarly, from the interactions between the computational model of hDAT and DA, it has been determined that such interaction occurs only due to two or four amino acids [
28].
Therefore, all possible combinations of the
R and
S amino acids of the NWR tripeptide polymerized with polystyrene were built.
Figure 2 presents the flat structure of this bioreceptor, where each amino acid whose stereochemistry will be modified is identified with a different color, and
Table 6 denotes the codes for the derived bioreceptors with the stereochemistry of each amino acid in its respective order and their average interaction energy in kcal/mol.
As seen in
Table 6, there are variations in the affinity results for each bioreceptor according to modifications in the stereochemistry of the amino acids that compose them. Hence, it was determined that the bioreceptor that best interacts with DA is the one with SSS stereochemistry (code 1.0.2), as shown in
Table 6. Although the other results were not considerably distant, this result was obtained because this is the natural stereochemistry of amino acids, and therefore, the other results exhibited decreased affinity. It is worth mentioning that the bioreceptor with SSS stereochemistry is the same one that was designed by polymerization with polystyrene, which is why the code did not change. The standard deviation of the data is generally reduced to one decimal or even becomes null in some cases, which means that the data dispersion is not very variable.
However, once we defined that we wanted to maintain the natural amino acid stereochemistry in the bioreceptor design, we proceeded to determine how many amino acids each bioreceptor should have.
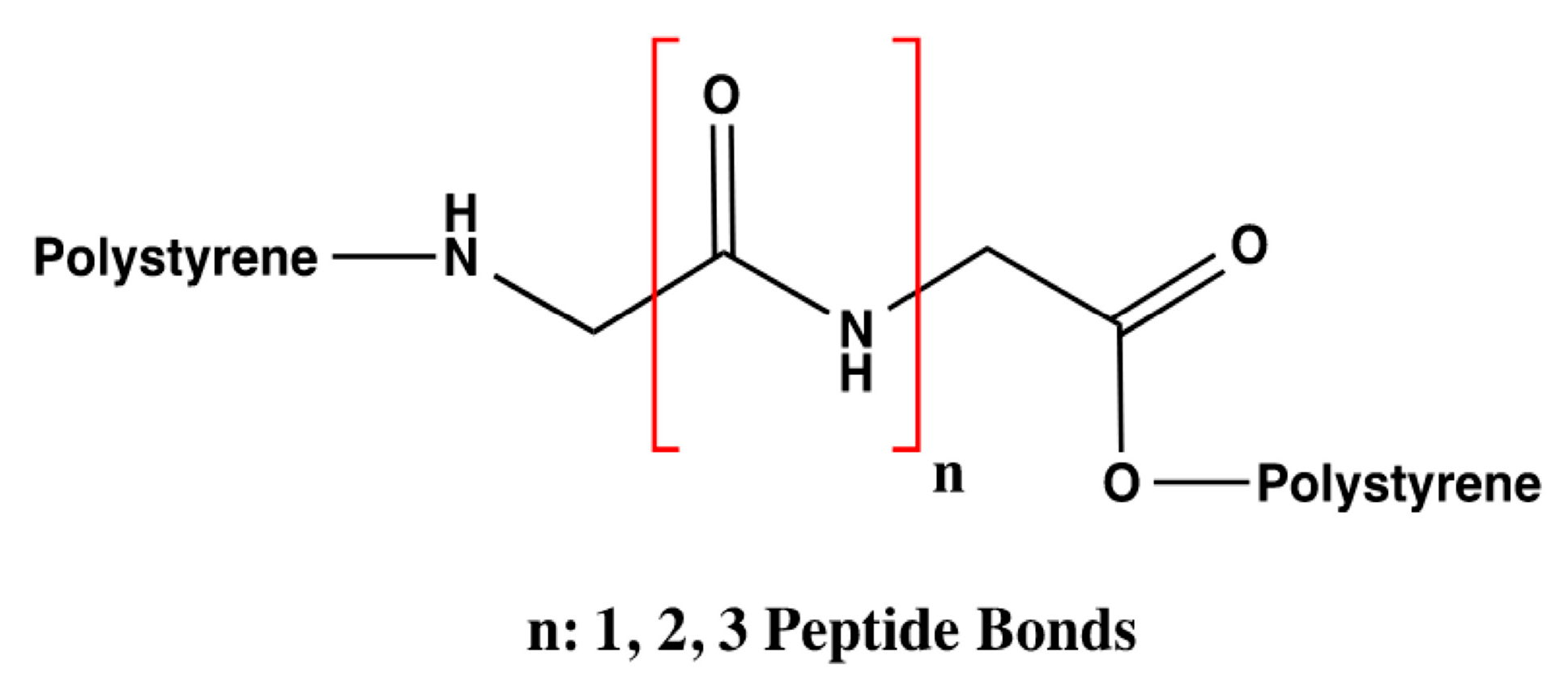
Based on the hDAT model described IN [
28] and the analysis of the number of interactions that DA can form, the influence of the number of amino acids was evaluated only with three styrene-polymerized bioreceptors according to the previous results. The design of these bioreceptors was carried out only with glycine so that only the interaction of DA with the number of peptide bonds could be assessed with no influence from the functional groups that compose the substituents of the other amino acids. The number of amino acids varied from two to four glycine molecules, as shown in
Figure 3.
Considering this,
Table 7 indicates the affinity results for the three glycine bioreceptors, specifying the amount of glycine they contain and their average interaction energy in kcal/mol.
As shown in
Table 7, the best results were obtained with the bioreceptor composed of three glycine molecules (4.1,
Table 7) with an affinity value of −5.1 kcal/mol. This value was taken as the parameter used to build the following bioreceptors in order to study the relationship of their affinity for DA according to the amino acids that compose them.
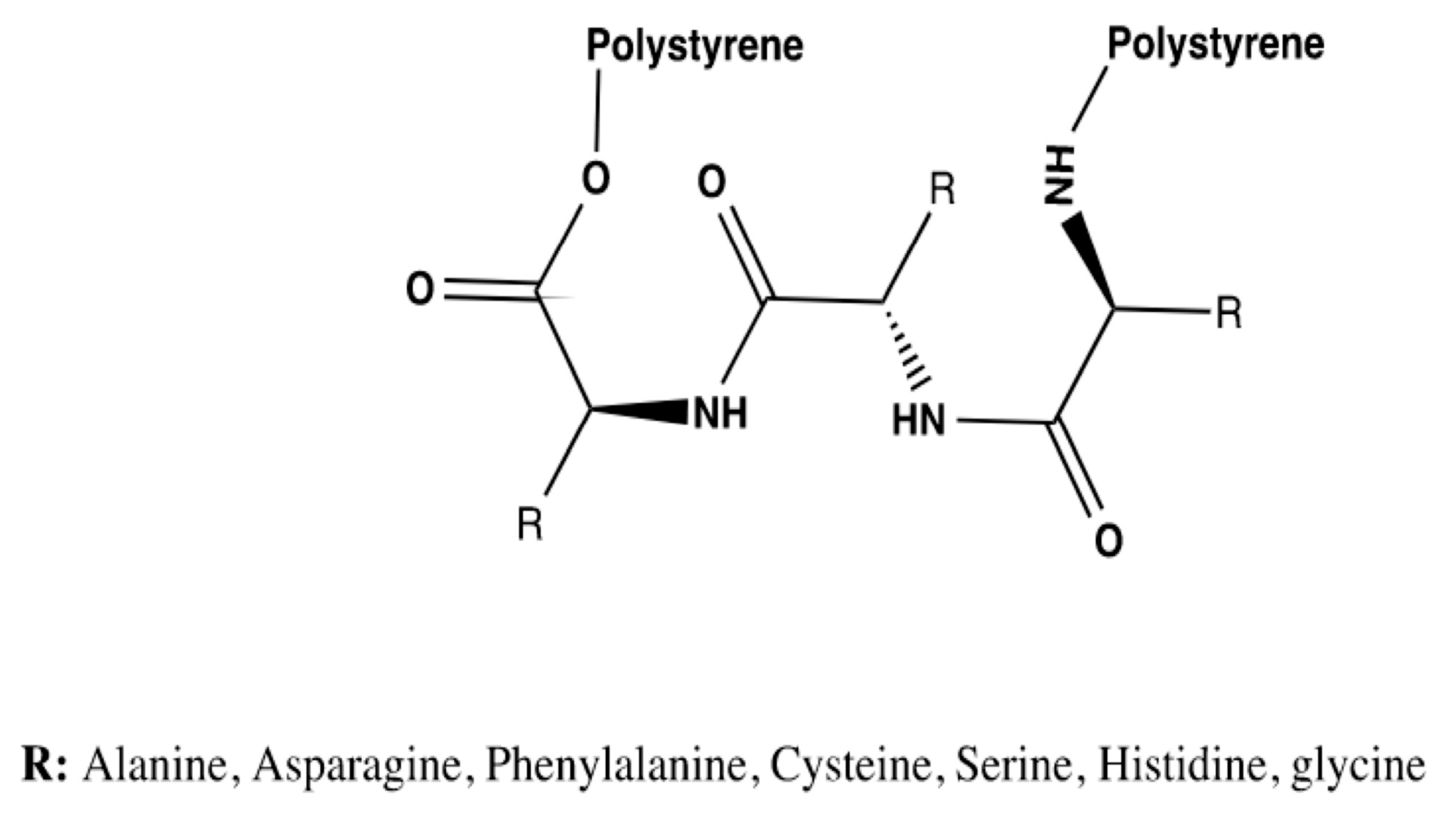
Thus, tripeptide bioreceptors were designed to study the influence of the amino acid’s nature on the interaction energies exhibited. A total of seven tripeptide bioreceptors made out of glycine, phenylalanine, alanine, asparagine, serine, cysteine, and histidine were modeled. The general structure for this group of bioreceptors is shown in
Figure 4.
Regarding the previous results, and as mentioned before, glycine was used to determine if the substituents of the amino acids that comprise the bioreceptors were essential or if the peptide bonds alone could generate enough affinity for DA. This glycine tripeptide’s affinity result is presented in the first row of
Table 8 and is the same one presented in
Table 7 under code 4.1.
Continuing with the variable evaluation and seeking to understand the influence of the amino acids’ nature on the non-covalent interactions of the bioreceptor, phenylalanine and alanine were initially selected as standards for the group of non-polar amino acids to simultaneously compare the influence of the amino acids with an aromatic substituent, which made it possible to analyze the π-π interaction that can occur between the amino acids themselves or with DA, as shown in
Table 8. We identified that this interaction can occur with phenylalanine [
31,
32]. Therefore, the bioreceptor results with phenylalanine can be compared with those that are formed only by alanine, which is also non-polar, but with an aliphatic and single-carbon substituent. This allows the result to be related to the type of interaction that can be formed and to the steric volume of the amino acid.
The result of the average affinity of the bioreceptor composed of a phenylalanine tripeptide is shown in
Table 8 line 2 under code 5. The alanine bioreceptor corresponds to code 6 and is presented in the third row of the same Table.
The group of polar amino acids was addressed by the bioreceptors designed with asparagine (code 7), serine (code 8), and cysteine (code 9). However, there are differences in the substituents of these three amino acids, which were considered during their selection.
Asparagine is an amino acid that, in addition to being polar, has the amide functional group (RCONH
2) in its substituent and has the capacity to accept three and donate two hydrogen bonds [
33]. On the other hand, serine has a hydroxyl group in its substituent and can donate three and accept four hydrogen bonds [
34]. Cysteine is a thiol [
35]. The results of these three bioreceptors are reported in rows four, five, and six, respectively, of
Table 8.
Histidine is a basic amino acid because of the chain in its substituent. It was selected because of its high reactivity and because it is an amino acid that plays an important role in the catalytic activity of proteins [
36]. The histidine bioreceptor was assigned code 10, and the result of its interaction energy with DA is reported in row seven of
Table 8.
As denoted in
Table 8, the results for this series of amino acids range from −4.3 to −5.1 kcal/mol, wherein only one of the bioreceptors has a standard deviation other than zero, which means that there was no variability between them and that in the case of bioreceptor 10, data dispersion decreased. Bioreceptor 6, composed of an alanine tripeptide, exhibited a more distant result than the others, as shown in row four of
Table 8. This may mean that the alanine substituent (CH
3) did not generate a significant affinity with DA, and this result is comparable with that of phenylalanine (row 3 of
Table 8), which is also non-polar and provides better results. Therefore, these π–π interactions are stronger than those formed by alanine, as mentioned in the amino acid selection criteria.
The next lowest result found was −4.5 kcal/mol, corresponding to bioreceptor 7, presented in row five of
Table 8. Here, the substituent was an amide which did not exhibit any worthwhile results despite being able to donate two and receive three hydrogen bonds. This may be because these protons possess a very weak acidic character; therefore, hydrogen bond interactions will probably be unlikely with, for example, the protons of aspartic acid [
37]. These results are comparable with those of the serine and cysteine bioreceptors which are also weak despite having acidic protons.
Based on the structural modifications made and that assessing the three variables mentioned above failed to achieve a bioreceptor with better affinity results, it was considered that mixtures between the different amino acids will potentiate the results, especially since it was observed that the expected results of the glycine bioreceptor were not attained by the other bioreceptors composed only of one amino acid. Then, to combine these amino acids, we decided to use the groups of amino acids that exhibited interaction in the models of hDAT and of DA receptors, which were taken from previous studies [
10,
28].
Before analyzing the results obtained for this group of bioreceptors, it was necessary to highlight that it had already been determined that the number of amino acids should not exceed four to ensure that interactions with DA were specifically occurring with the amino acids of interest. The existence of aromatic amino acids showed an increase in the interaction, and to add steric volume, the polymerization had to be performed with polystyrene.
Consequently, after testing the variables above, 13 additional bioreceptors were designed which correspond to the amino acid groups identified in the computational models both of the hDAT [
28] and the interaction mechanism of DA receptors [
10].
Overall, it has been reported that both intra and extracellular DA interactions with DATs and DA receptors involve groups of amino acids, with the number ranging from two to four. In fact, a study argues that aspartic acid is a very important amino acid and essential for DA reuptake. In addition, several aromatic interactions were also identified as playing a prominent role in the activity of DAT with DA [
28].
In total, 13 combinations of amino acids were designed. They were polymerized at the C and N terminals with polystyrene to give them steric volume, a characteristic that had already been proven to increase bioreceptor interaction.
Table 9 displays the code assigned to each bioreceptor, the peptide from which it was composed, and each of their average affinity results.
When observing the results obtained for the 13 bioreceptors presented in
Table 9, the affinity range obtained was determined to be ranging from −4.5 to −5.4 kcal/mol, with null or 0.1 standard deviations, indicating that data dispersion was insignificant. We were able to obtain bioreceptors with energies exceeded the interaction energy of the DAT previously stated as −5.2 kcal/mol. The two bioreceptors that showed an improved interaction with DA were 15 and 19, which are in rows five and ten in
Table 9.
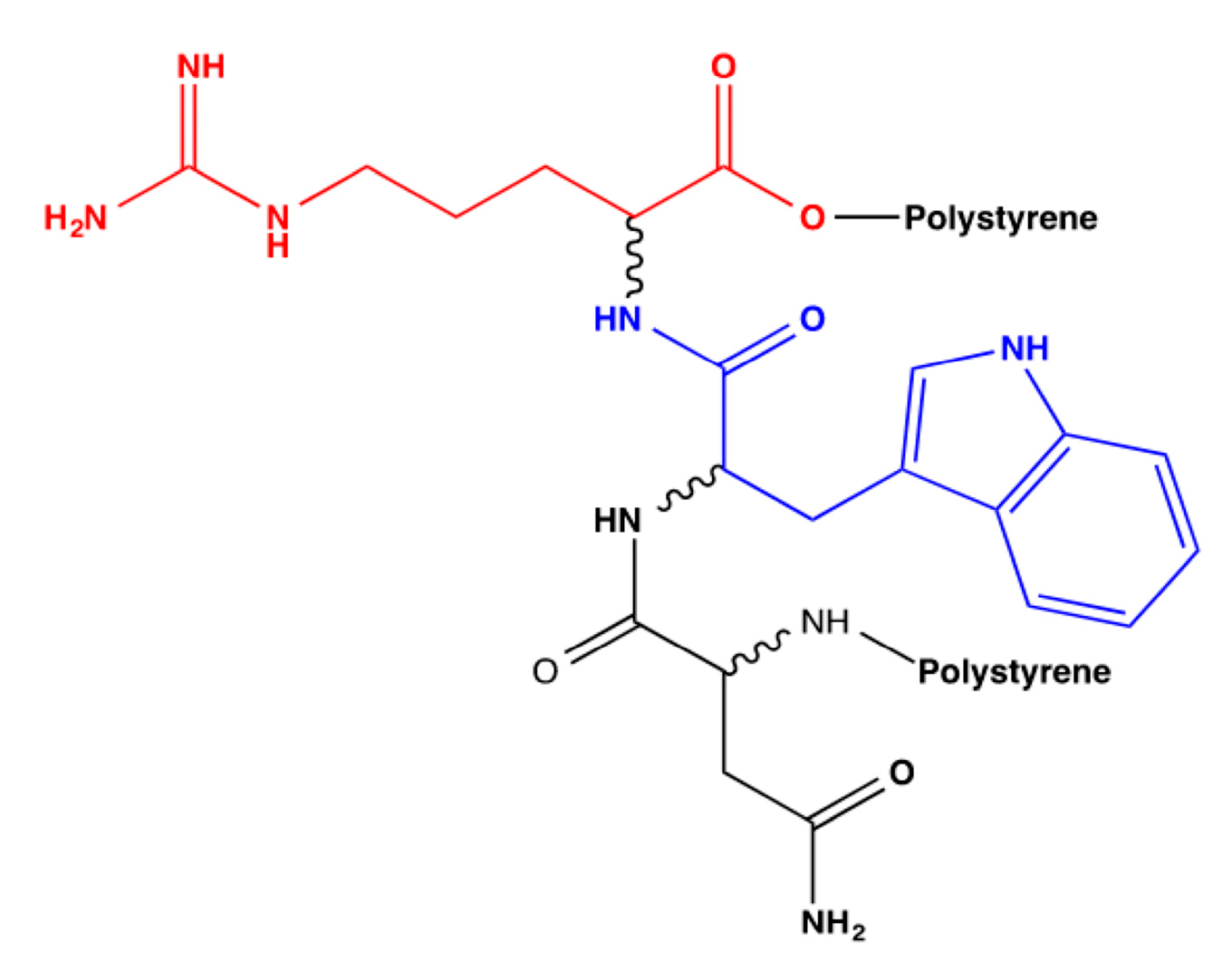
First of all, bioreceptor 15, denoted in row five of the previous table, will be discussed. This bioreceptor is composed of serine, aspartic acid, and tryptophan. As per the above-mentioned findings, serine is a polar amino acid because of its hydroxyl group, which has been described as playing a prominent role in the catalytic activity of enzymes [
34]. Conversely, aspartic acid was not addressed or considered in the tests; therefore, it is essential to emphasize its characteristics. This amino acid is acidic and can donate three and accept five hydrogen bonds; therefore, it could be said that it is the amino acid with the highest number of interactions to date [
31,
38]. The final one is tryptophan, which is part of the group of non-polar and aromatic amino acids. Its substituent has an indole functional group and can donate and accept three hydrogen bonds [
39].
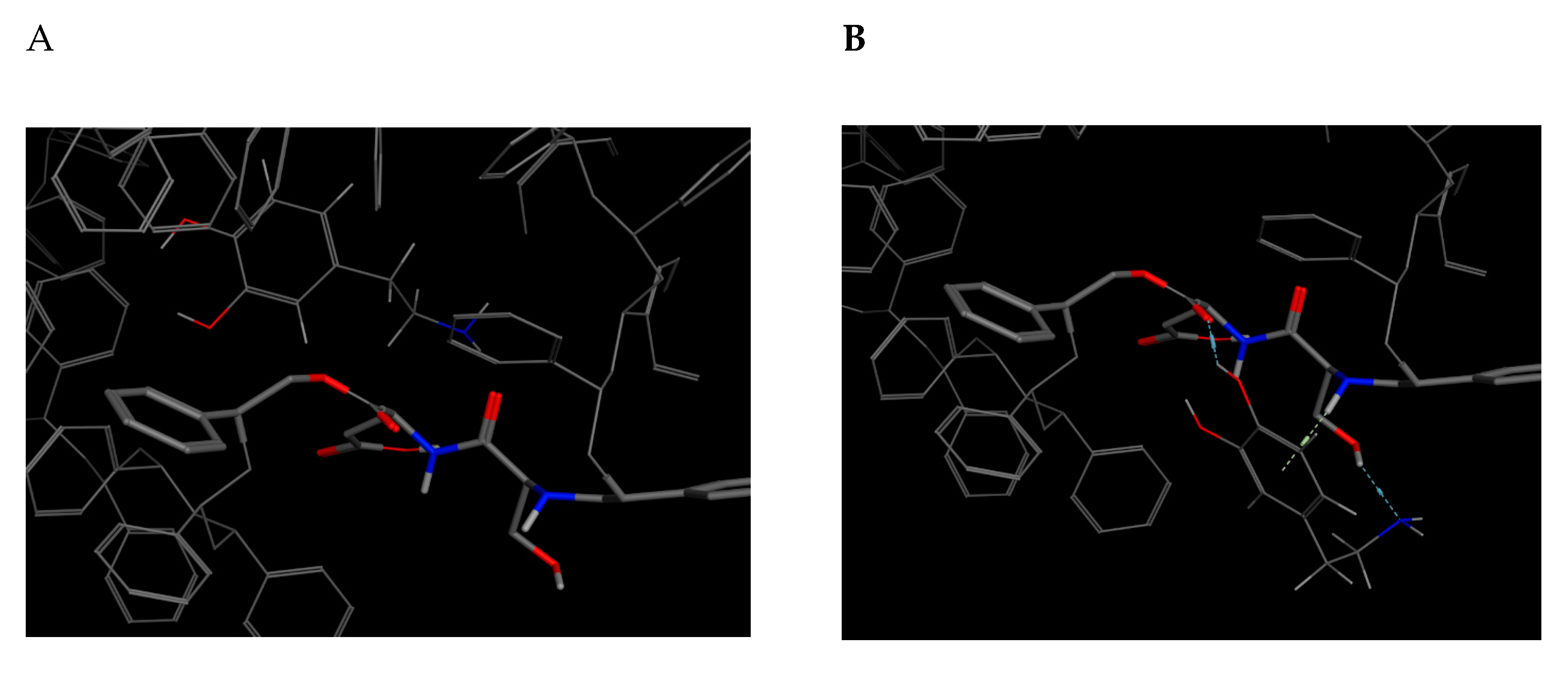
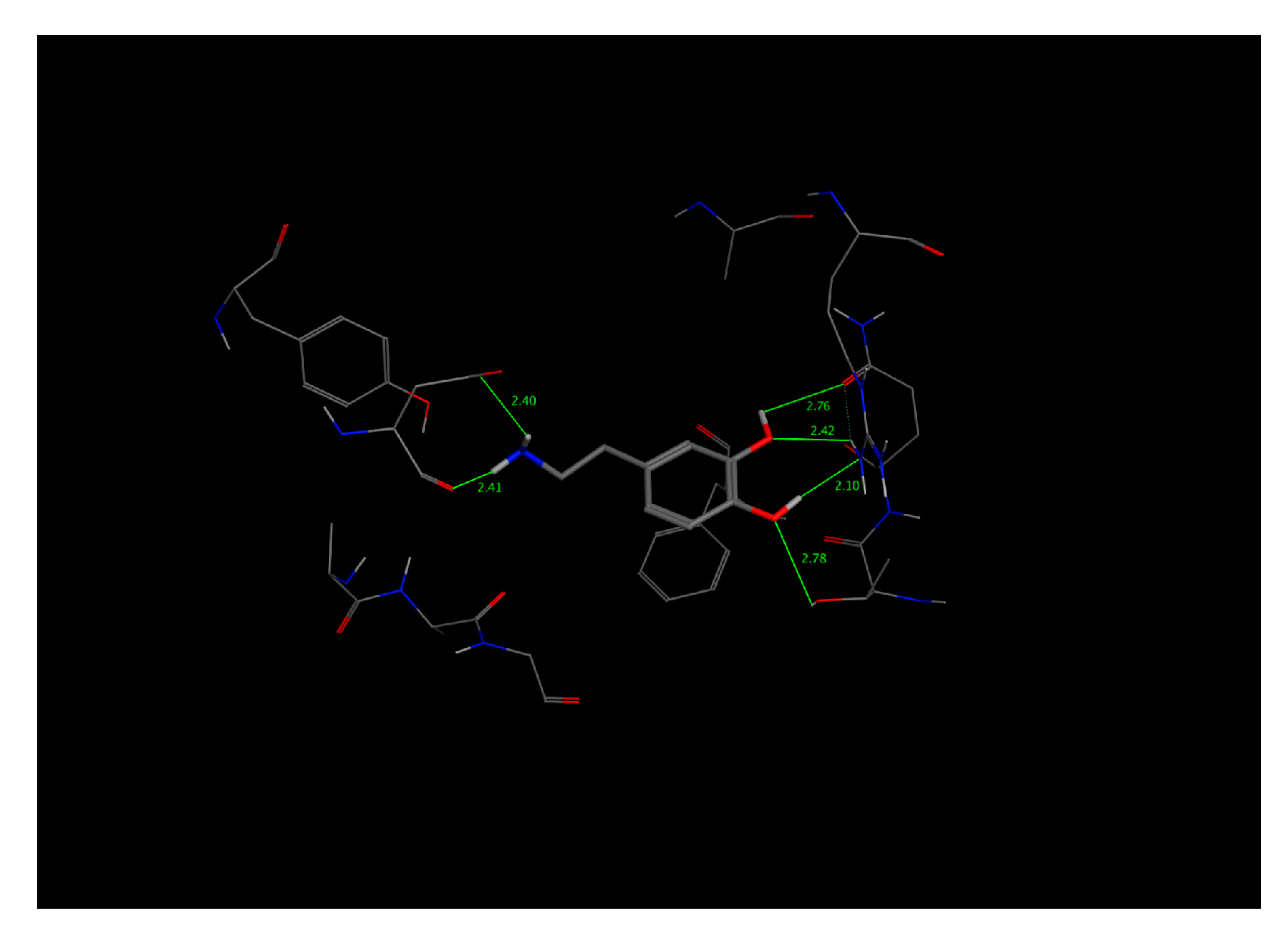
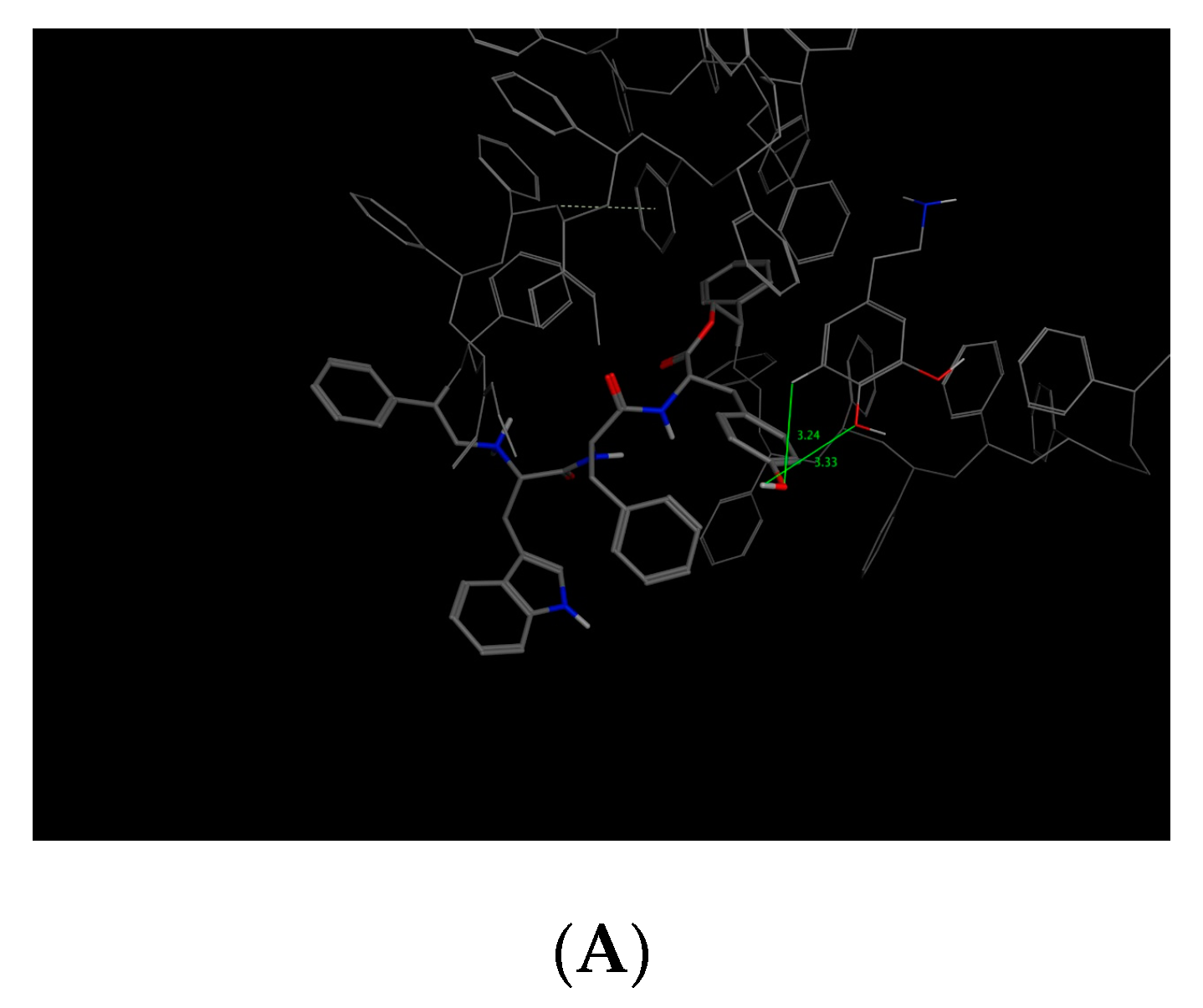
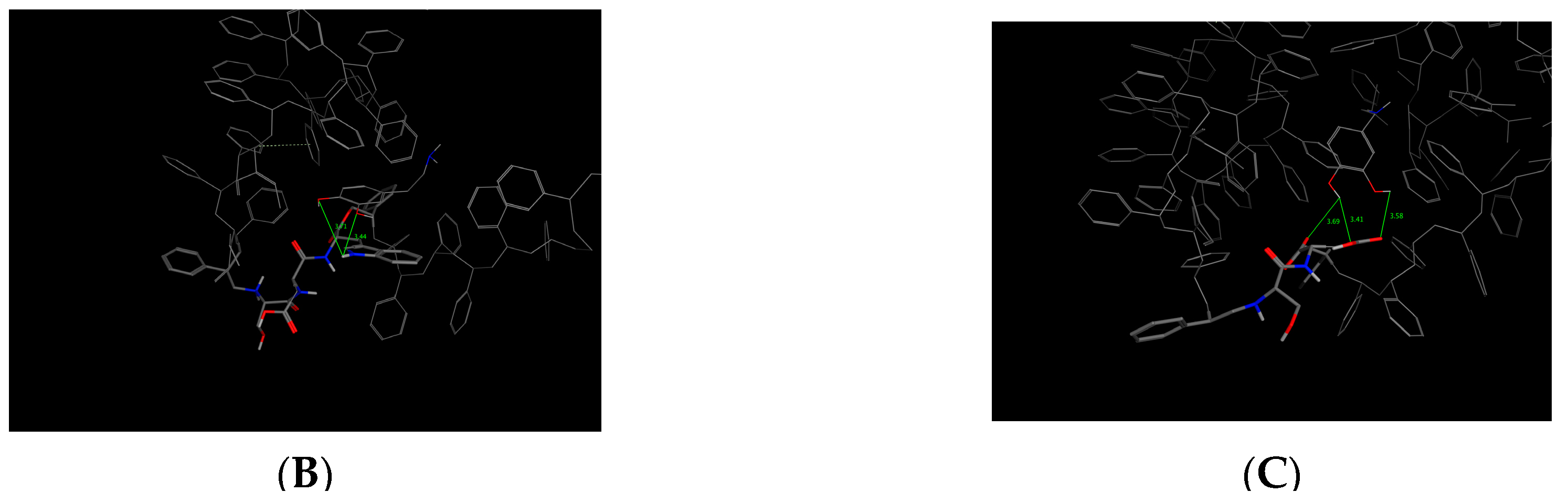
This bioreceptor exhibited an interaction energy of –5.3 kcal/mol. Its corresponding image shows how the amino acids were exposed, and the polymer provided steric volume creating a free pocket for its interaction with DA.
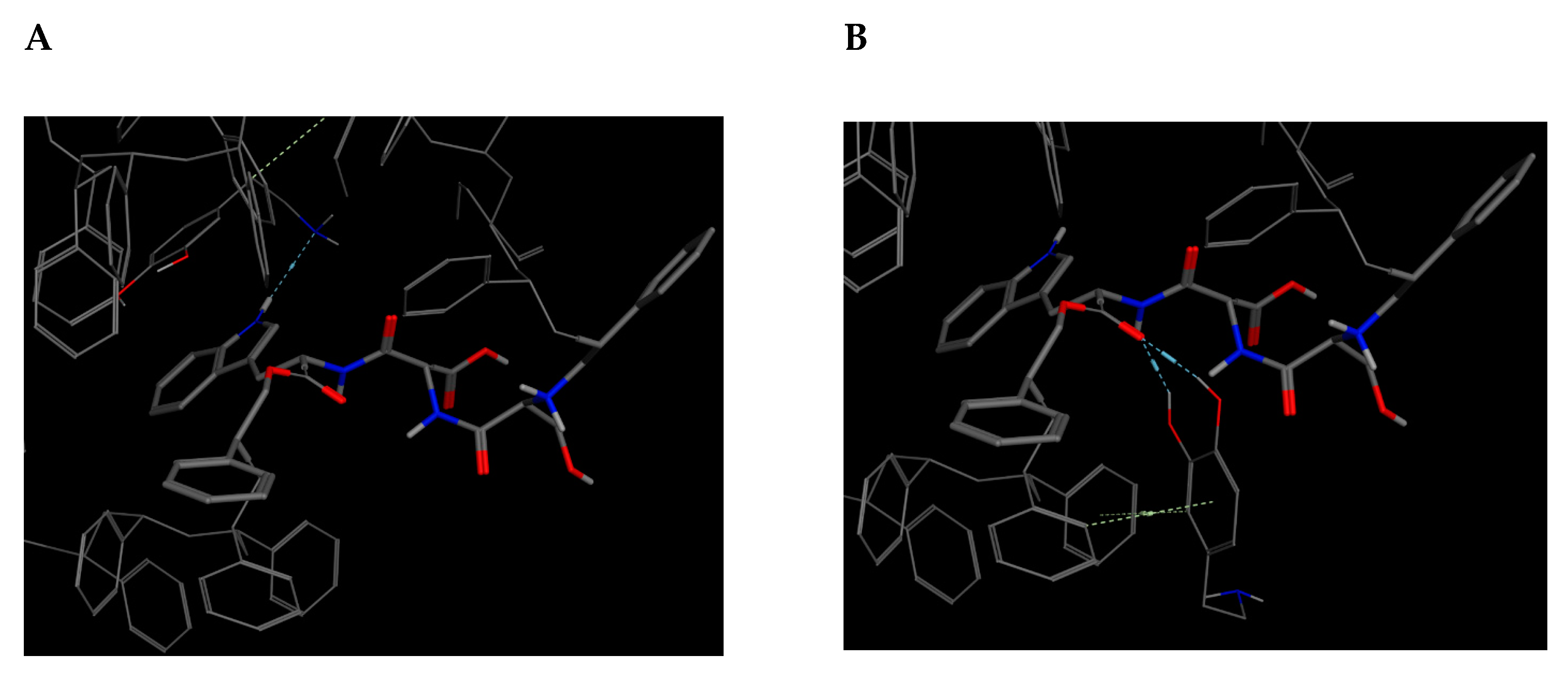
Bioreceptor 19 from
Table 9, composed of tryptophan, phenylalanine, and tyrosine obtained an affinity of –5.4 kcal/mol. Out of these three amino acids, tyrosine, an aromatic and polar amino acid capable of donating three and accepting four hydrogen bonds, was the only one not analyzed [
40]. It is among the amino acids found at the highest percentage of protein composition and has a phenol functional group in its substituent [
41].
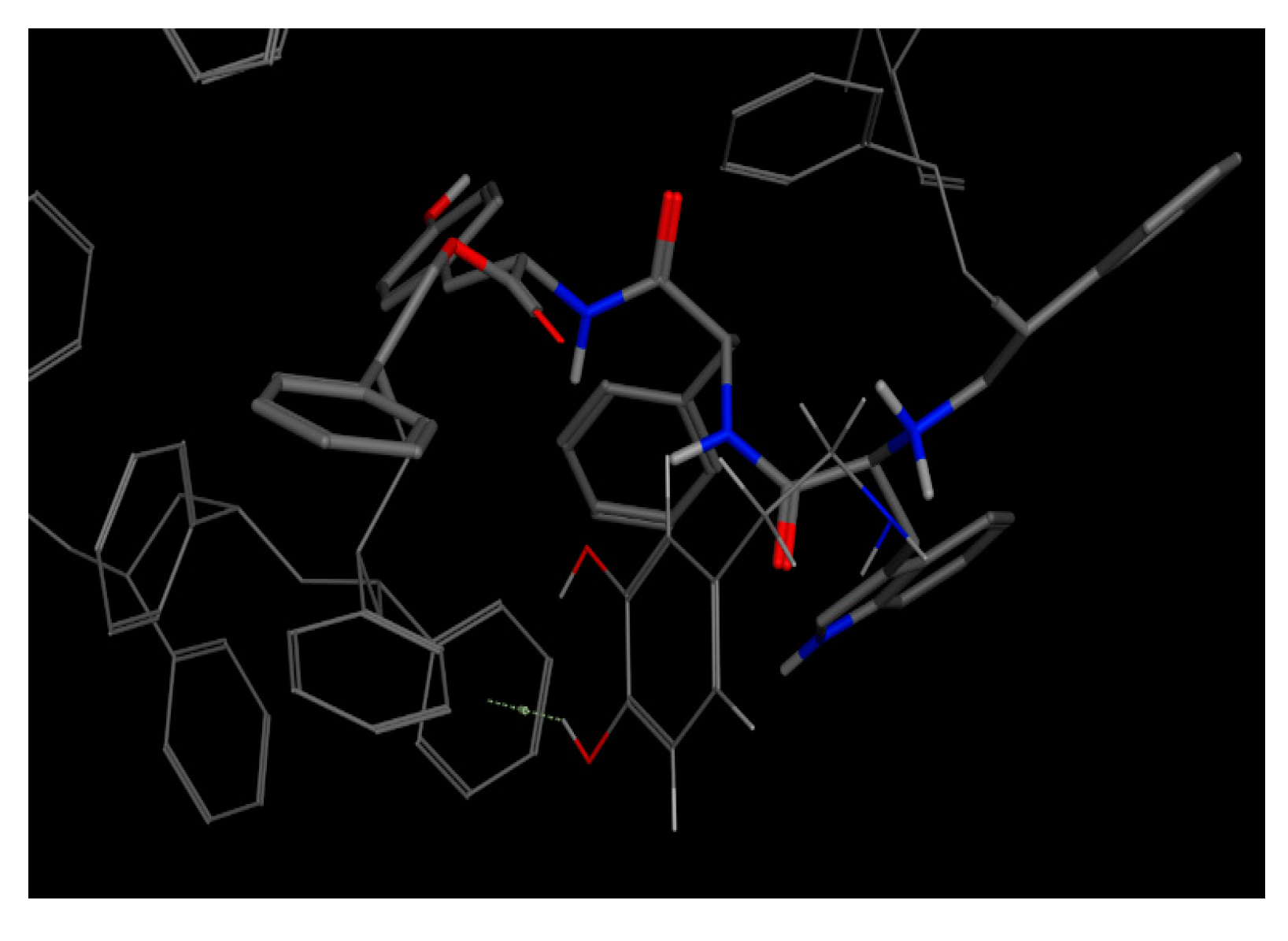
As previously described, this bioreceptor contains a tripeptide of aromatic amino acids, which reaffirms the idea that the π–π interaction is essential for DA recognition. However, it is evident that the aromatic group is not strong enough to interact with DA alone but, when supplemented by the hydroxyl group in the tyrosine ring and the benzo-fused substituent of tryptophan, they interact together to release more energy. The image depicting this bioreceptor also shows that the peptide bonds form a curve that exposes the amino acid substituents so that they can interact with DA.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


