
Friend or Foe: Lipid Droplets as Organelles for Protein and Lipid Storage in Cellular Stress Response, Aging and Disease

 , ,
, ,  and
and 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Special Features of LDs
2.1. LD Motility
2.2. LD-Organelle Interactions
3. The Role of LDs in Cellular Detoxification
3.1. Lipotoxicity
- (a)
- (b)
- DAGs and CEs are main molecular culprits involved in the development of insulin resistance [68]. DAGs activate protein kinase C (PKC) isoforms (among others PKCε), which phosphorylate the insulin receptor and inactivate its tyrosine kinase function.
- (c)
- CEs activate other PKC isoforms (PKCζ) which interfere with AKT translocation and signaling downstream of the insulin receptor. Furthermore, CEs activate protein phosphatase 2A, which dephosphorylates and inactivates AKT signaling [69]. CEs negatively affect the permeability of the OMM and form channels for the release of pro-apoptotic proteins in the mitochondrial intermembrane space. The pro-apoptotic protein Bax stabilizes the channel [70]. Different Cer species within a cell have specific and diverse effects on the translocation of Bax to the OMM. There is evidence that excess CEs need to be stored in LDs. The conversion of CEs to acylceramides is catalyzed by DGAT2 [71], and thus storage in LDs may have a protective function (see Figure 1). Failure in the production of omega-O-acylceramides occurs in neutral lipid storage diseases (NLSD).
3.2. Protein Homeostasis
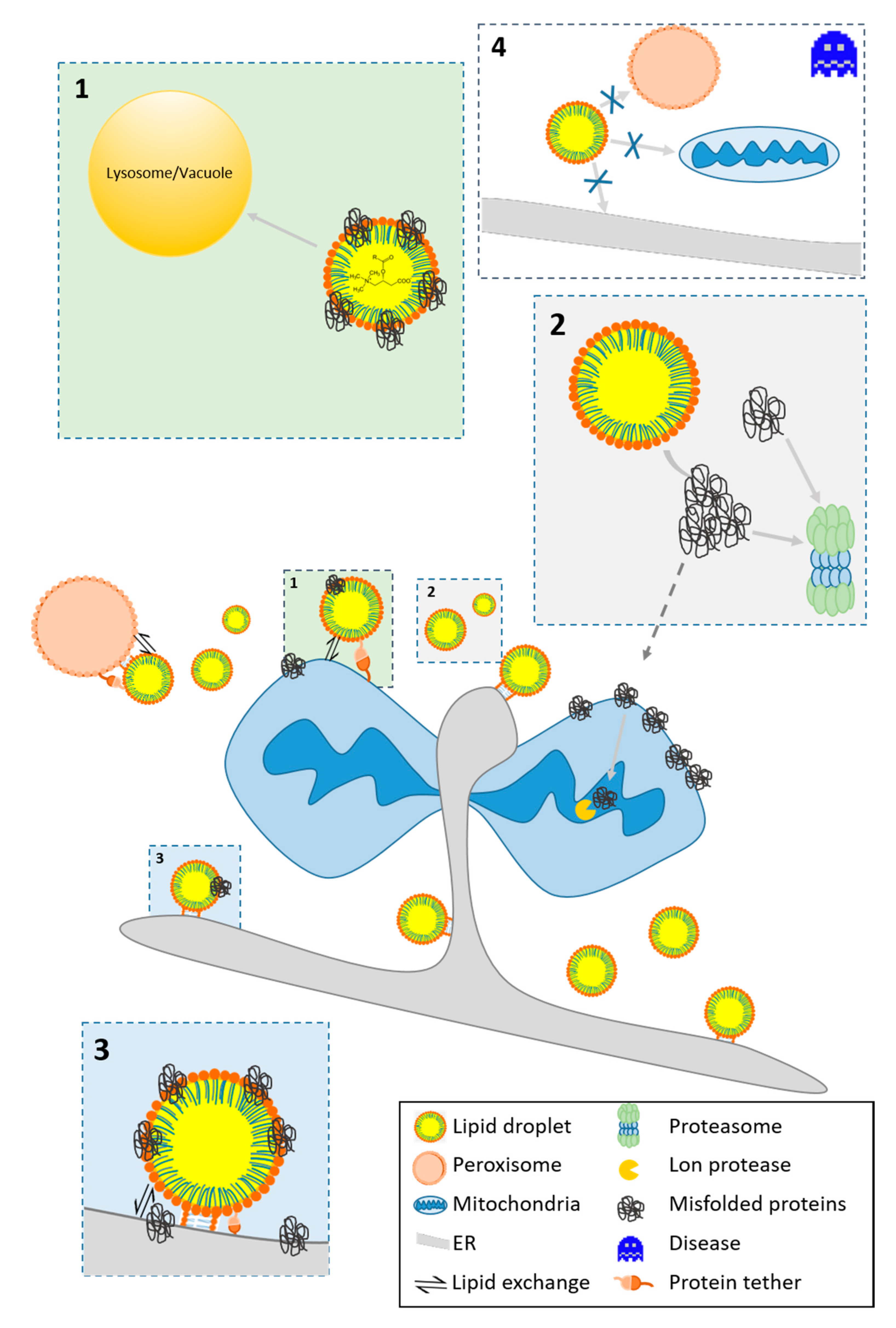
3.2.1. The Fate of Misfolded Proteins
3.2.2. LDs As A Protein Docking Site
3.2.3. Protein Motifs for LD Localization
4. The Role of LDs in Cellular Stress and Aging
4.1. The Function of LDs in Modulating Stress
- (a)
- The high energy demand during stress response causes fueling of FAs from LD to mitochondria for β-oxidation [130]. Reverse transfer of lipids occurs, and lipid relocation from mitochondria to LDs may protect from lipotoxic settings [131] and aberrant lipid signaling (see Section 3.1).
- (b)
- LDs are involved in protein homeostasis and assist in the removal of damaged and misfolded proteins from mitochondria (see Section 3.2)
- (c)
- LDs modulate the apoptotic program by redirecting acylcarnitines from mitochondria (see Section 3.1). Similarly, mitochondrial proteins involved in the apoptotic program translocate [9,81]. In several studies, the pro-apoptotic protein BAX was found as a bona fide member of the LD proteome [9,99,132].
- (d)
- LDs fulfill antioxidative functions, reduce ROS levels and prevent the peroxidation of PUFAs. The mechanism involves the removal of these lipids from membranes to the core LDs, where they are less accessible to ROS [129]. Consequently, a stimulated increase in LD number can augment cell survival upon the onset of stressors [9].
4.2. Emergence and Role of LDs During Aging
5. The Role of LDs in Disease
5.1. LDs and Neurodegenerative Diseases
5.2. Neutral Lipid Storage Disease (NLSD) and Rare Lipid Storage Diseases
5.3. Atherosclerosis
5.4. Obesity and Non-Alcoholic Fatty Liver Disease
5.5. LDs and Inflammation
5.6. The Role of LDs in Viral, Bacterial and Protozoan Infections
Author Contributions
Funding
Conflicts of Interest
References
- Gao, Q.; Goodman, J.M. The lipid droplet-a well-connected organelle. Front. Cell Dev. Biol. 2015, 3, 49. [Google Scholar] [CrossRef] [PubMed]
- Na, H.M.; Zhang, P.; Ding, Y.F.; Yang, L.; Wang, Y.; Zhang, H.N.; Xie, Z.S.; Yang, F.Q.; Cichello, S.; Liu, P.S. Proteomic Studies of Isolated Lipid Droplets from Bacteria, C-elegans, and Mammals. Method Cell Biol. 2013, 116, 1–14. [Google Scholar] [CrossRef]
- Penno, A.; Hackenbroich, G.; Thiele, C. Phospholipids and lipid droplets. BBA Mol. Cell Biol. Lipids 2013, 1831, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S. Lipid Droplets as Organelles. Int. Rev. Cell Mol. Bio. 2018, 337, 83–110. [Google Scholar] [CrossRef]
- Wolins, N.E.; Rubin, D.; Brasaemle, D.L. TIP47 associates with lipid droplets. J. Biol. Chem. 2001, 276, 5101–5108. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2018, 20, 137–155. [Google Scholar] [CrossRef]
- Reue, K. A thematic review series: Lipid droplet storage and metabolism: From yeast to man. J. Lipid Res. 2011, 52, 1865–1868. [Google Scholar] [CrossRef]
- Schuldiner, M.; Bohnert, M. A different kind of love - Lipid droplet contact sites. BBA Mol. Cell Biol. Lipids 2017, 1862, 1188–1196. [Google Scholar] [CrossRef]
- Bischof, J.; Salzmann, M.; Streubel, M.K.; Hasek, J.; Geltinger, F.; Duschl, J.; Bresgen, N.; Briza, P.; Haskova, D.; Lejskova, R.; et al. Clearing the outer mitochondrial membrane from harmful proteins via lipid droplets. Cell Death Discov. 2017, 3, 17016. [Google Scholar] [CrossRef] [PubMed]
- Nardi, F.; Fitchev, P.; Brooks, K.M.; Franco, O.E.; Cheng, K.; Hayward, S.W.; Welte, M.A.; Crawford, S.E. Lipid droplet velocity is a microenvironmental sensor of aggressive tumors regulated by V-ATPase and PEDF. Lab. Investig. 2019, 99, 1822–1834. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.L.; Fagarasanu, A.; Rachubinski, R.A. Peroxisomal peripheral membrane protein YlInp1p is required for peroxisome inheritance and influences the dimorphic transition in the yeast Yarrowia lipolytica. Eukaryot. Cell 2007, 6, 1528–1537. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Cyphersmith, A.; Zapata, J.A.; Kim, Y.J.; Payne, C.K. Lysosome Transport as a Function of Lysosome Diameter. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Welte, M.A. Fat on the move: Intracellular motion of lipid droplets. Biochem. Soc. Trans. 2009, 37, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Groebner, J.L.; Giron-Bravo, M.T.; Rothberg, M.L.; Adhikari, R.; Tuma, D.J.; Tuma, P.L. Alcohol-induced microtubule acetylation leads to the accumulation of large, immobile lipid droplets. Am. J. Physiol. Gastr. Liver 2019, 317, G373–G386. [Google Scholar] [CrossRef]
- Pfisterer, S.G.; Gateva, G.; Horvath, P.; Pirhonen, J.; Salo, V.T.; Karhinen, L.; Varjosalo, M.; Ryhanen, S.J.; Lappalainen, P.; Ikonen, E. Role for formin-like 1-dependent acto-myosin assembly in lipid droplet dynamics and lipid storage. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Veerabagu, M.; Paul, L.K.; Rinne, P.L.; van der Schoot, C. Plant Lipid Bodies Traffic on Actin to Plasmodesmata Motorized by Myosin XIs. Int. J. Mol. Sci. 2020, 21, 1422. [Google Scholar] [CrossRef]
- Knoblach, B.; Rachubinski, R.A. Transport and Retention Mechanisms Govern Lipid Droplet Inheritance in Saccharomyces cerevisiae. Traffic 2015, 16, 298–309. [Google Scholar] [CrossRef]
- Yang, H.J.; Osakada, H.; Kojidani, T.; Haraguchi, T.; Hiraoka, Y. Lipid droplet dynamics during Schizosaccharomyces pombe sporulation and their role in spore survival. Biol. Open 2017, 6, 217–222. [Google Scholar] [CrossRef]
- Yeshaw, W.M.; van der Zwaag, M.; Pinto, F.; Lahaye, L.L.; Faber, A.I.E.; Gomez-Sanchez, R.; Dolga, A.M.; Poland, C.; Monaco, A.P.; van IJzendoorn, S.C.D.; et al. Human VPS13A is associated with multiple organelles and influences mitochondrial morphology and lipid droplet motility. Elife 2019, 8. [Google Scholar] [CrossRef]
- Guimaraes, S.C.; Schuster, M.; Bielska, E.; Dagdas, G.; Kilaru, S.; Meadows, B.R.A.; Schrader, M.; Steinberg, G. Peroxisomes, lipid droplets, and endoplasmic reticulum "hitchhike" on motile early endosomes. J. Cell Biol. 2015, 211, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, M. Tethering Fat: Tethers in Lipid Droplet Contact Sites. Contact 2020, 3, 2515256420908142. [Google Scholar] [CrossRef]
- Lee, M.C.; Miller, E.A. Molecular mechanisms of COPII vesicle formation. Semin. Cell Dev. Biol. 2007, 18, 424–434. [Google Scholar] [CrossRef]
- Scorrano, L.; De Matteis, M.A.; Emr, S.; Giordano, F.; Hajnoczky, G.; Kornmann, B.; Lackner, L.L.; Levine, T.P.; Pellegrini, L.; Reinisch, K.; et al. Coming together to define membrane contact sites. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.G.; Weaver, D.; Shirihai, O.; Hajnoczky, G. Mitochondrial ‘kiss-and-run’: Interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. 2009, 28, 3074–3089. [Google Scholar] [CrossRef] [PubMed]
- Thiam, A.R.; Beller, M. The why, when and how of lipid droplet diversity. J. Cell Sci. 2017, 130, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Binns, D.; Januszewski, T.; Chen, Y.; Hill, J.; Markin, V.S.; Zhao, Y.M.; Gilpin, C.; Chapman, K.D.; Anderson, R.G.W.; Goodman, J.M. An intimate collaboration between peroxisomes and lipid bodies. J. Cell Biol. 2006, 173, 719–731. [Google Scholar] [CrossRef]
- Kohlwein, S.D.; Veenhuis, M.; van der Klei, I.J. Lipid Droplets and Peroxisomes: Key Players in Cellular Lipid Homeostasis or A Matter of Fat-Store’em up or Burn’em down. Genetics 2013, 193, 1–50. [Google Scholar] [CrossRef]
- Xu, N.Y.; Zhang, S.B.O.; Cole, R.A.; McKinney, S.A.; Guo, F.L.; Haas, J.T.; Bobba, S.; Farese, R.V.; Mak, H.Y. The FATP1-DGAT2 complex facilitates lipid droplet expansion at the ER-lipid droplet interface. J. Cell Biol. 2012, 198, 895–911. [Google Scholar] [CrossRef]
- Xu, D.J.; Li, Y.Q.; Wu, L.Z.; Li, Y.; Zhao, D.Y.; Yu, J.H.; Huang, T.Z.; Ferguson, C.; Parton, R.G.; Yang, H.Y.; et al. Rab18 promotes lipid droplet (LD) growth by tethering the ER to LDs through SNARE and NRZ interactions. J. Cell Biol. 2018, 217, 975–995. [Google Scholar] [CrossRef]
- Datta, S.; Liu, Y.; Hariri, H.; Bowerman, J.; Henne, W.M. Cerebellar ataxia disease-associated Snx14 promotes lipid droplet growth at ER-droplet contacts. J. Cell Biol. 2019, 218, 1335–1351. [Google Scholar] [CrossRef]
- Thomas, A.C.; Williams, H.; Seto-Salvia, N.; Bacchelli, C.; Jenkins, D.; O’Sullivan, M.; Mengrelis, K.; Ishida, M.; Ocaka, L.; Chanudet, E.; et al. Mutations in SNX14 cause a distinctive autosomal-recessive cerebellar ataxia and intellectual disability syndrome. Am. J. Hum. Genet. 2014, 95, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Liu, X.M.; Long, X.; Han, C.; Wan, F.; Fan, W.L.; Guo, X.F.; Ma, K.; Guo, S.Y.; Wang, L.X.; et al. Novel VPS13A Gene Mutations Identified in Patients Diagnosed with Chorea-acanthocytosis (ChAc): Case Presentation and Literature Review. Front. Aging Neuro Sci. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Boutant, M.; Kulkarni, S.S.; Joffraud, M.; Ratajczak, J.; Valera-Alberni, M.; Combe, R.; Zorzano, A.; Canto, C. Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 2017, 36, 1543–1558. [Google Scholar] [CrossRef]
- Escobar-Henriques, M.; Joaquim, M. Mitofusins: Disease Gatekeepers and Hubs in Mitochondrial Quality Control by E3 Ligases. Front. Physiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xue, L.J.; Xue, X.; Ou, Z.; Jiang, T.; Zhang, Y.D. MFN2 ameliorates cell apoptosis in a cellular model of Parkinson’s disease induced by rotenone. Exp. Ther. Med. 2018, 16, 3680–3685. [Google Scholar] [CrossRef]
- Freyre, C.A.C.; Rauher, P.C.; Ejsing, C.S.; Klemm, R.W. MIGA2 Links Mitochondria, the ER, and Lipid Droplets and Promotes De Novo Lipogenesis in Adipocytes. Mol. Cell 2019, 76, 811–825. [Google Scholar] [CrossRef]
- Pu, J.; Ha, C.W.; Zhang, S.Y.; Jung, J.P.; Huh, W.K.; Liu, P.S. Interactomic study on interaction between lipid droplets and mitochondria. Protein Cell 2011, 2, 487–496. [Google Scholar] [CrossRef]
- Chang, C.L.; Weigel, A.V.; Ioannou, M.S.; Pasolli, H.A.; Xu, C.S.; Peale, D.R.; Shtengel, G.; Freeman, M.; Hess, H.F.; Blackstone, C.; et al. Spastin tethers lipid droplets to peroxisomes and directs fatty acid trafficking through ESCRT-III. J. Cell Biol. 2019, 218, 2583–2599. [Google Scholar] [CrossRef]
- Van Zutphen, T.; Todde, V.; de Boer, R.; Kreim, M.; Hofbauer, H.F.; Wolinski, H.; Veenhuis, M.; van der Klei, I.J.; Kohlwein, S.D. Lipid droplet autophagy in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell 2014, 25, 290–301. [Google Scholar] [CrossRef]
- Wang, C.W.; Miao, Y.H.; Chang, Y.S. A sterol-enriched vacuolar microdomain mediates stationary phase lipophagy in budding yeast. J. Cell Biol. 2014, 206, 357–366. [Google Scholar] [CrossRef]
- Tsuji, T.; Fujimoto, M.; Tatematsu, T.; Cheng, J.; Orii, M.; Takatori, S.; Fujimoto, T. Niemann-Pick type C proteins promote microautophagy by expanding raft-like membrane domains in the yeast vacuole. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Garcia, E.J.; Vevea, J.D.; Pon, L.A. Lipid droplet autophagy during energy mobilization, lipid homeostasis and protein quality control. Front. Biosci. 2018, 23, 1552–1563. [Google Scholar] [CrossRef]
- Hariri, H.; Rogers, S.; Ugrankar, R.; Liu, Y.L.; Feathers, J.R.; Henne, W.M. Lipid droplet biogenesis is spatially coordinated at ER-vacuole contacts under nutritional stress. EMBO Rep. 2018, 19, 57–72. [Google Scholar] [CrossRef]
- Zhang, X.; Evans, T.D.; Jeong, S.J.; Razani, B. Classical and alternative roles for autophagy in lipid metabolism. Curr. Opin. Lipidol. 2018, 29, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Schulze, R.J.; Sathyanarayan, A.; Mashek, D.G. Breaking fat: The regulation and mechanisms of lipophagy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1178–1187. [Google Scholar] [CrossRef]
- Rui, Y.N.; Xu, Z.; Patel, B.; Chen, Z.H.; Chen, D.S.; Tito, A.; David, G.; Sun, Y.M.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 2015, 17, 759. [Google Scholar] [CrossRef]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V., Jr.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef]
- Arisawa, K.; Ichi, I.; Yasukawa, Y.; Sone, Y.; Fujiwara, Y. Changes in the phospholipid fatty acid composition of the lipid droplet during the differentiation of 3T3-L1 adipocytes. J. Biochem. 2013, 154, 281–289. [Google Scholar] [CrossRef]
- Bozza, P.T.; Bakker-Abreu, I.; Navarro-Xavier, R.A.; Bandeira-Melo, C. Lipid body function in eicosanoid synthesis: An update. Prostaglandins Leukot Essent Fatty Acids 2011, 85, 205–213. [Google Scholar] [CrossRef]
- Chen, Y.; Cruzat, V.F.; Newsholme, P. β-Cell Metabolism, Insulin Production and Secretion: Metabolic Failure Resulting in Diabetes. In Molecular Nutrition and Diabetes; Mauricio, D., Ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 29–40. [Google Scholar]
- Dyntar, D.; Eppenberger-Eberhardt, M.; Maedler, K.; Pruschy, M.; Eppenberger, H.M.; Spinas, G.A.; Donath, M.Y. Glucose and palmitic acid induce degeneration of myofibrils and modulate apoptosis in rat adult cardiomyocytes. Diabetes 2001, 50, 2105–2113. [Google Scholar] [CrossRef] [PubMed]
- Plotz, T.; Hartmann, M.; Lenzen, S.; Elsner, M. The role of lipid droplet formation in the protection of unsaturated fatty acids against palmitic acid induced lipotoxicity to rat insulin-producing cells. Nutr. Metab. 2016, 13, 16. [Google Scholar] [CrossRef] [PubMed]
- Plotz, T.; Krummel, B.; Laporte, A.; Pingitore, A.; Persaud, S.J.; Jorns, A.; Elsner, M.; Mehmeti, I.; Lenzen, S. The monounsaturated fatty acid oleate is the major physiological toxic free fatty acid for human beta cells. Nutr. Diabetes 2017, 7. [Google Scholar] [CrossRef]
- Jarc, E.; Petan, T. A twist of FATe: Lipid droplets and inflammatory lipid mediators. Biochimie 2020, 169, 69–87. [Google Scholar] [CrossRef]
- Najt, C.P.; Khan, S.A.; Heden, T.D.; Witthuhn, B.A.; Perez, M.; Heier, J.L.; Mead, L.E.; Franklin, M.P.; Karanja, K.K.; Graham, M.J.; et al. Lipid Droplet-Derived Monounsaturated Fatty Acids Traffic via PLIN5 to Allosterically Activate SIRT1. Mol. Cell 2020, 77, 810–824. [Google Scholar] [CrossRef] [PubMed]
- Nolan, C.J.; Larter, C.Z. Lipotoxicity: Why do saturated fatty acids cause and monounsaturates protect against it? J. Gastroen. Hepatol. 2009, 24, 703–706. [Google Scholar] [CrossRef]
- Hihi, A.K.; Michalik, L.; Wahli, W. PPARs: Transcriptional effectors of fatty acids and their derivatives. Cell Mol. Life Sci. 2002, 59, 790–798. [Google Scholar] [CrossRef]
- Mesilati-Stahy, R.; Argov-Argaman, N. Changes in lipid droplets morphometric features in mammary epithelial cells upon exposure to non-esterified free fatty acids compared with VLDL. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Mari, M.; Fernandez-Checa, J.C. Damage Mediated by Dysfunction of Organelles and Cellular Systems: Lysosomes. Pathobiol. Hum. Disease A Dyn. Encycl. Disease Mech. 2014, 97–107. [Google Scholar] [CrossRef]
- Makrecka-Kuka, M.; Sevostjanovs, E.; Vilks, K.; Volska, K.; Antone, U.; Kuka, J.; Makarova, E.; Pugovics, O.; Dambrova, M.; Liepinsh, E. Plasma acylcarnitine concentrations reflect the acylcarnitine profile in cardiac tissues. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Olzmann, J.A. Lipid droplets and lipotoxicity during autophagy. Autophagy 2017, 13, 2002–2003. [Google Scholar] [CrossRef]
- Petan, T.; Jarc, E.; Jusovic, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef]
- Wilfling, F.; Wang, H.J.; Haas, J.T.; Krahmer, N.; Gould, T.J.; Uchida, A.; Cheng, J.X.; Graham, M.; Christiano, R.; Frohlich, F.; et al. Triacylglycerol Synthesis Enzymes Mediate Lipid Droplet Growth by Relocalizing from the ER to Lipid Droplets. Dev. Cell 2013, 24, 384–399. [Google Scholar] [CrossRef]
- Thiam, A.R.; Antonny, B.; Wang, J.; Delacotte, J.; Wilfling, F.; Walther, T.C.; Beck, R.; Rothman, J.E.; Pincet, F. COPI buds 60-nm lipid droplets from reconstituted water-phospholipid-triacylglyceride interfaces, suggesting a tension clamp function. Proc. Natl. Acad. Sci. USA 2013, 110, 13244–13249. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Roh, Y.S.; Seki, E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J. Gastroen. Hepatol. 2013, 28, 38–42. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Mechanisms for Insulin Resistance: Common Threads and Missing Links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol. Sci. 2017, 38, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Siskind, L.J. Mitochondrial ceramide and the induction of apoptosis. J. Bioenerg. Biomembr. 2005, 37, 143–153. [Google Scholar] [CrossRef]
- Senkal, C.E.; Salama, M.F.; Snider, A.J.; Allopenna, J.J.; Rana, N.A.; Koller, A.; Hannun, Y.A.; Obeid, L.M. Ceramide Is Metabolized to Acylceramide and Stored in Lipid Droplets. Cell Metab. 2017, 25, 686–697. [Google Scholar] [CrossRef]
- Li, P.L.; Gulbins, E. Bioactive Lipids and Redox Signaling: Molecular Mechanism and Disease Pathogenesis. Antioxid. Redox Signal. 2018, 28, 911–915. [Google Scholar] [CrossRef]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef]
- Basu, S. F2-isoprostanes in human health and diseases: From molecular mechanisms to clinical implications. Antioxid. Redox Signal. 2008, 10, 1405–1434. [Google Scholar] [CrossRef] [PubMed]
- Gianazza, E.; Brioschi, M.; Fernandez, A.M.; Casalnuovo, F.; Altomare, A.; Aldini, G.; Banfi, C. Lipid Peroxidation in Atherosclerotic Cardiovascular Diseases. Antioxid. Redox Signal. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Li, L.; Shen, X.; Li, Q.; Xu, W.; Wang, X.; Tao, Y.; Yin, H. An update on lipid oxidation and inflammation in cardiovascular diseases. Free Radic. Biol. Med. 2019, 144, 266–278. [Google Scholar] [CrossRef]
- Farmer, B.C.; Walsh, A.E.; Kluemper, J.C.; Johnson, L.A. Lipid Droplets in Neurodegenerative Disorders. Front. Neurosci. Switz. 2020, 14. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.H.; Gao, P.P.; Shi, L.; Chen, L.; Liu, J.K.; Long, J.G. Central and Peripheral Metabolic Defects Contribute to the Pathogenesis of Alzheimer’s Disease: Targeting Mitochondria for Diagnosis and Prevention. Antioxid. Redox Signal. 2020, 32, 1188–1236. [Google Scholar] [CrossRef]
- Yang, J.; Fernandez-Galilea, M.; Martinez-Fernandez, L.; Gonzalez-Muniesa, P.; Perez-Chavez, A.; Martinez, J.A.; Moreno-Aliaga, M.J. Oxidative Stress and Non-Alcoholic Fatty Liver Disease: Effects of Omega-3 Fatty Acid Supplementation. Nutrients 2019, 11, 872. [Google Scholar] [CrossRef]
- Jarc, E.; Petan, T. Lipid Droplets and the Management of Cellular Stress. Yale J. Biol. Med. 2019, 92, 435–452. [Google Scholar]
- Geltinger, F.; Tevini, J.; Briza, P.; Geiser, A.; Bischof, J.; Richter, K.; Felder, T.; Rinnerthaler, M. The transfer of specific mitochondrial lipids and proteins to lipid droplets contributes to proteostasis upon stress and aging in the eukaryotic model system Saccharomyces cerevisiae. Geroscience 2020, 42, 19–38. [Google Scholar] [CrossRef]
- La Merrill, M.; Emond, C.; Kim, M.J.; Antignac, J.P.; Le Bizec, B.; Clement, K.; Birnbaum, L.S.; Barouki, R. Toxicological function of adipose tissue: Focus on persistent organic pollutants. Environ. Health Perspect. 2013, 121, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, S.L.; Kelly, J.W. Chemical and Biological Approaches for Adapting Proteostasis to Ameliorate Protein Misfolding and Aggregation Diseases-Progress and Prognosis. CSH Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, J.S.; Lindquist, S. Mechanisms of protein-folding diseases at a glance. Dis. Models Mech. 2014, 7, 9–14. [Google Scholar] [CrossRef]
- Rinnerthaler, M.; Bischof, J.; Streubel, M.K.; Trost, A.; Richter, K. Oxidative Stress in Aging Human Skin. Biomolecules 2015, 5, 545–589. [Google Scholar] [CrossRef]
- Kaganovich, D.; Kopito, R.; Frydman, J. Misfolded proteins partition between two distinct quality control compartments. Nature 2008, 454, 1088–1095. [Google Scholar] [CrossRef]
- Miller, S.B.M.; Ho, C.T.; Winkler, J.; Khokhrina, M.; Neuner, A.; Mohamed, M.Y.H.; Guilbride, D.L.; Richter, K.; Lisby, M.; Schiebel, E.; et al. Compartment-specific aggregases direct distinct nuclear and cytoplasmic aggregate deposition. EMBO J. 2015, 34, 778–797. [Google Scholar] [CrossRef] [PubMed]
- Gallina, I.; Colding, C.; Henriksen, P.; Beli, P.; Nakamura, K.; Offman, J.; Mathiasen, D.P.; Silva, S.; Hoffmann, E.; Groth, A.; et al. Cmr1/WDR76 defines a nuclear genotoxic stress body linking genome integrity and protein quality control. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Rothe, S.; Prakash, A.; Tyedmers, J. The Insoluble Protein Deposit (IPOD) in Yeast. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef]
- Parsell, D.A.; Kowal, A.S.; Singer, M.A.; Lindquist, S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature 1994, 372, 475–478. [Google Scholar] [CrossRef]
- Bagola, K.; Sommer, T. Protein quality control: On IPODs and other JUNQ. Curr. Biol. 2008, 18, R1019–R1021. [Google Scholar] [CrossRef]
- Kumar, R.; Nawroth, P.P.; Tyedmers, J. Prion Aggregates Are Recruited to the Insoluble Protein Deposit (IPOD) via Myosin 2-Based Vesicular Transport. PLoS Genet. 2016, 12, e1006324. [Google Scholar] [CrossRef]
- Moldavski, O.; Amen, T.; Levin-Zaidman, S.; Eisenstein, M.; Rogachev, I.; Brandis, A.; Kaganovich, D.; Schuldiner, M. Lipid Droplets Are Essential for Efficient Clearance of Cytosolic Inclusion Bodies. Dev. Cell 2015, 33, 603–610. [Google Scholar] [CrossRef]
- Lemus, L.; Goder, V. Regulation of Endoplasmic Reticulum-Associated Protein Degradation (ERAD) by Ubiquitin. Cells 2014, 3, 824–847. [Google Scholar] [CrossRef]
- Oslowski, C.M.; Urano, F. Measuring Er Stress and the Unfolded Protein Response Using Mammalian Tissue Culture System. Method EnzyMol. 2011, 490, 71–92. [Google Scholar] [CrossRef]
- Fei, W.; Wang, H.; Fu, X.; Bielby, C.; Yang, H. Conditions of endoplasmic reticulum stress stimulate lipid droplet formation in Saccharomyces cerevisiae. Biochem. J. 2009, 424, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Vevea, J.D.; Garcia, E.J.; Chan, R.B.; Zhou, B.W.; Schultz, M.; Di Paolo, G.; McCaffery, J.M.; Pon, L.A. Role for Lipid Droplet Biogenesis and Microlipophagy in Adaptation to Lipid Imbalance in Yeast. Dev. Cell 2015, 35, 584–599. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Sztalryd, C.; Lu, X.; Tansey, J.T.; Gan, J.; Dorward, H.; Kimmel, A.R.; Londos, C. Post-translational regulation of adipose differentiation-related protein by the ubiquitin/proteasome pathway. J. Biol. Chem. 2005, 280, 42841–42847. [Google Scholar] [CrossRef]
- Bersuker, K.; Peterson, C.W.H.; To, M.; Sahl, S.J.; Savikhin, V.; Grossman, E.A.; Nomura, D.K.; Olzmann, J.A. A Proximity Labeling Strategy Provides Insights into the Composition and Dynamics of Lipid Droplet Proteomes. Dev. Cell 2018, 44, 97–112. [Google Scholar] [CrossRef]
- Guerriero, C.J.; Brodsky, J.L. The Delicate Balance between Secreted Protein Folding and Endoplasmic Reticulum-Associated Degradation in Human Physiology. Physiol. Rev. 2012, 92, 537–576. [Google Scholar] [CrossRef]
- Callea, F.; Giovannoni, I.; Sari, S.; Guldal, E.; Dalgic, B.; Akyol, G.; Sogo, T.; Al-Hussaini, A.; Maggiore, G.; Bartuli, A.; et al. Fibrinogen Gamma Chain Mutations Provoke Fibrinogen and Apolipoprotein B Plasma Deficiency and Liver Storage. Int. J. Mol. Sci. 2017, 18, 2717. [Google Scholar] [CrossRef]
- Mazi, T.A.; Shibata, N.M.; Medici, V. Lipid and energy metabolism in Wilson disease. Liver Res. 2020, 4, 5–14. [Google Scholar] [CrossRef]
- Sobaniec-Lotowska, M.E.; Lebensztejn, D.M. Ultrastructure of Kupffer cells and hepatocytes in the Dubin-Johnson syndrome: A case report. World J. Gastroenterol. 2006, 12, 987–989. [Google Scholar] [CrossRef][Green Version]
- Ramakrishnan, S.; Narayanappa, G.; Christopher, R. Lipid storage myopathy with clinical markers of Marfan syndrome: A rare association. Ann. Indian Acad. Neurol. 2012, 15, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Thiel, K.; Thul, P.J.; Beller, M.; Kuhnlein, R.P.; Welte, M.A. Lipid droplets control the maternal histone supply of Drosophila embryos. Curr. Biol. 2012, 22, 2104–2113. [Google Scholar] [CrossRef]
- Tong, E.H.; Guo, J.J.; Huang, A.L.; Liu, H.; Hu, C.D.; Chung, S.S.; Ko, B.C. Regulation of nucleocytoplasmic trafficking of transcription factor OREBP/TonEBP/NFAT5. J. Biol. Chem. 2006, 281, 23870–23879. [Google Scholar] [CrossRef]
- Ueno, M.; Shen, W.J.; Patel, S.; Greenberg, A.S.; Azhar, S.; Kraemer, F.B. Fat-specific protein 27 modulates nuclear factor of activated T cells 5 and the cellular response to stress. J. Lipid Res. 2013, 54, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sreenivasan, U.; Hu, H.; Saladino, A.; Polster, B.M.; Lund, L.M.; Gong, D.W.; Stanley, W.C.; Sztalryd, C. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J. Lipid Res. 2011, 52, 2159–2168. [Google Scholar] [CrossRef]
- Gallardo-Montejano, V.I.; Saxena, G.; Kusminski, C.M.; Yang, C.; McAfee, J.L.; Hahner, L.; Hoch, K.; Dubinsky, W.; Narkar, V.A.; Bickel, P.E. Nuclear Perilipin 5 integrates lipid droplet lipolysis with PGC-1alpha/SIRT1-dependent transcriptional regulation of mitochondrial function. Nat. Commun. 2016, 7, 12723. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.D.; Puigserver, P.; Andersson, U.; Zhang, C.Y.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Soyal, S.M.; Zara, G.; Ferger, B.; Felder, T.K.; Kwik, M.; Nofziger, C.; Dossena, S.; Schwienbacher, C.; Hicks, A.A.; Pramstaller, P.P.; et al. The PPARGC1A locus and CNS-specific PGC-1alpha isoforms are associated with Parkinson’s Disease. Neurobiol. Dis. 2019, 121, 34–46. [Google Scholar] [CrossRef]
- Soyal, S.M.; Felder, T.K.; Auer, S.; Hahne, P.; Oberkofler, H.; Witting, A.; Paulmichl, M.; Landwehrmeyer, G.B.; Weydt, P.; Patsch, W.; et al. A greatly extended PPARGC1A genomic locus encodes several new brain-specific isoforms and influences Huntington disease age of onset. Hum. Mol. Genet. 2012, 21, 3461–3473. [Google Scholar] [CrossRef]
- Felder, T.K.; Soyal, S.M.; Oberkofler, H.; Hahne, P.; Auer, S.; Weiss, R.; Gadermaier, G.; Miller, K.; Krempler, F.; Esterbauer, H.; et al. Characterization of novel peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1alpha) isoform in human liver. J. Biol. Chem. 2011, 286, 42923–42936. [Google Scholar] [CrossRef] [PubMed]
- Vachharajani, V.T.; Liu, T.; Wang, X.; Hoth, J.J.; Yoza, B.K.; McCall, C.E. Sirtuins Link Inflammation and Metabolism. J. Immunol. Res. 2016, 2016, 8167273. [Google Scholar] [CrossRef] [PubMed]
- Kory, N.; Farese, R.V., Jr.; Walther, T.C. Targeting Fat: Mechanisms of Protein Localization to Lipid Droplets. Trends Cell Biol. 2016, 26, 535–546. [Google Scholar] [CrossRef]
- Olarte, M.J.; Kim, S.; Sharp, M.E.; Swanson, J.M.J.; Farese, R.V., Jr.; Walther, T.C. Determinants of Endoplasmic Reticulum-to-Lipid Droplet Protein Targeting. Dev. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Thiam, A.R.; Dugail, I. Lipid droplet-membrane contact sites - From protein binding to function. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [PubMed]
- Vieyres, G.; Pietschmann, T. HCV Pit Stop at the Lipid Droplet: Refuel Lipids and Put on a Lipoprotein Coat before Exit. Cells 2019, 8, 233. [Google Scholar] [CrossRef] [PubMed]
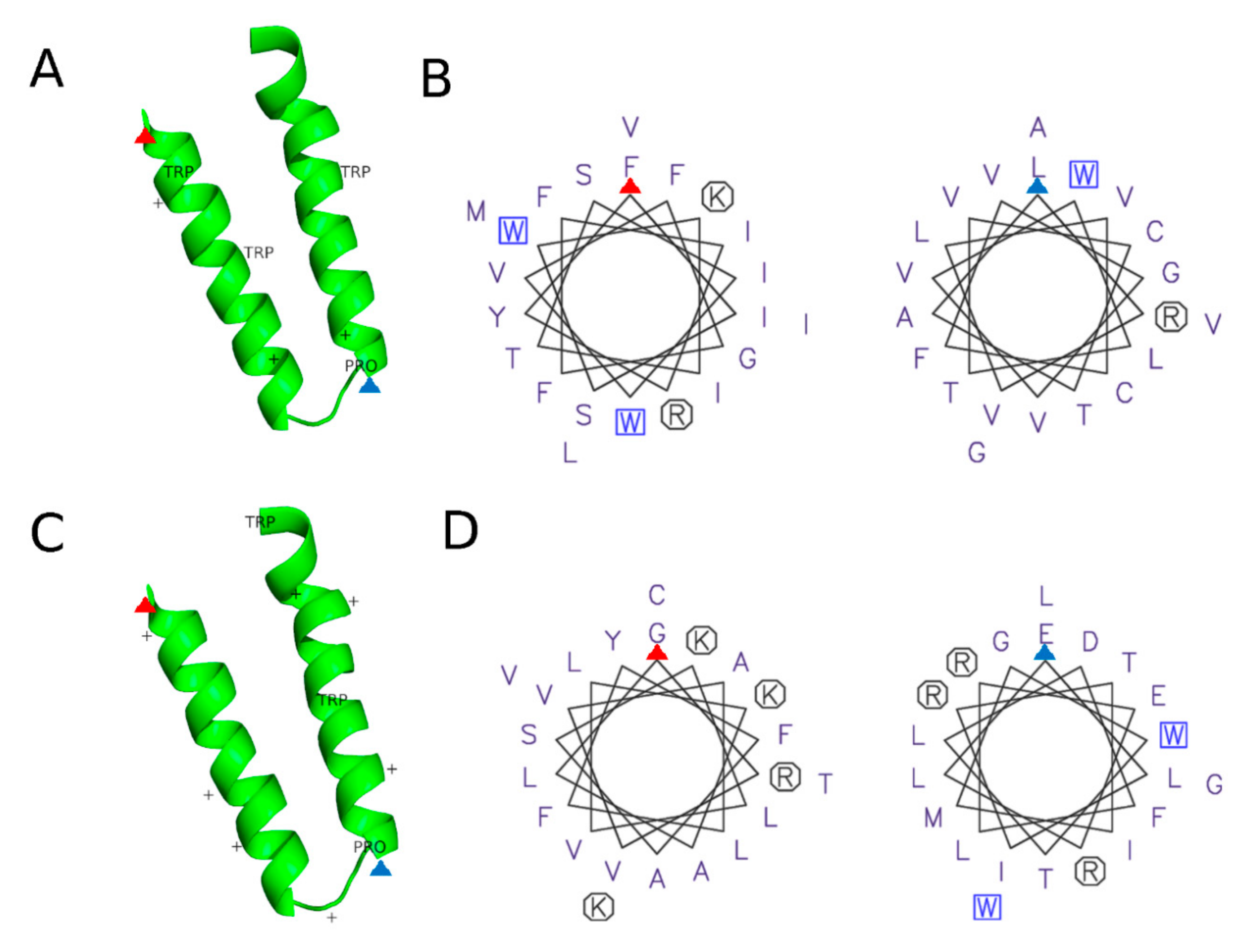
- Prevost, C.; Sharp, M.E.; Kory, N.; Lin, Q.Q.; Voth, G.A.; Farese, R.V.; Walther, T.C. Mechanism and Determinants of Amphipathic Helix-Containing Protein Targeting to Lipid Droplets. Dev. Cell 2018, 44, 73. [Google Scholar] [CrossRef]
- Itabe, H.; Yamaguchi, T.; Nimura, S.; Sasabe, N. Perilipins: A diversity of intracellular lipid droplet proteins. Lipids Health Dis. 2017, 16. [Google Scholar] [CrossRef]
- Rowe, E.R.; Mimmack, M.L.; Barbosa, A.D.; Haider, A.; Isaac, I.; Ouberai, M.M.; Thiam, A.R.; Patel, S.; Saudek, V.; Siniossoglou, S.; et al. Conserved Amphipathic Helices Mediate Lipid Droplet Targeting of Perilipins 1-3. J. Biol. Chem. 2016, 291, 6664–6678. [Google Scholar] [CrossRef]
- Favaloro, B.; Allocati, N.; Graziano, V.; Di Ilio, C.; De Laurenzi, V. Role of apoptosis in disease. Aging 2012, 4, 330–349. [Google Scholar] [CrossRef]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. BBA Bioenerg. 2012, 1817, 1833–1838. [Google Scholar] [CrossRef]
- Shutt, T.E.; McBride, H.M. Staying cool in difficult times: Mitochondrial dynamics, quality control and the stress response. BBA Mol. Cell Res. 2013, 1833, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Y.; Cui, L.; Deng, Y.; Xu, S.; Yu, J.; Cichello, S.; Serrero, G.; Ying, Y.; Liu, P. Morphologically and Functionally Distinct Lipid Droplet Subpopulations. Sci. Rep. 2016, 6, 29539. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, M. Wrapping up the fats-a structure of the lipid droplet biogenesis protein seipin. J. Cell Biol. 2018, 217, 4053–4054. [Google Scholar] [CrossRef] [PubMed]
- Herms, A.; Bosch, M.; Reddy, B.J.; Schieber, N.L.; Fajardo, A.; Ruperez, C.; Fernandez-Vidal, A.; Ferguson, C.; Rentero, C.; Tebar, F.; et al. AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat. Commun. 2015, 6, 7176. [Google Scholar] [CrossRef] [PubMed]
- Shaw, C.S.; Jones, D.A.; Wagenmakers, A.J.M. Network distribution of mitochondria and lipid droplets in human muscle fibres. Histochem. Cell Biol. 2008, 129, 65–72. [Google Scholar] [CrossRef]
- Bailey, A.P.; Koster, G.; Guillermier, C.; Hirst, E.M.; MacRae, J.I.; Lechene, C.P.; Postle, A.D.; Gould, A.P. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell 2015, 163, 340–353. [Google Scholar] [CrossRef]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty Acid Trafficking in Starved Cells: Regulation by Lipid Droplet Lipolysis, Autophagy, and Mitochondrial Fusion Dynamics. Dev. Cell 2015, 33, 489–490. [Google Scholar] [CrossRef]
- Wang, H.; Sztalryd, C. Oxidative tissue: Perilipin 5 links storage with the furnace. Trends Endocrinol. Metab. 2011, 22, 197–203. [Google Scholar] [CrossRef]
- Pataki, C.I.; Rodrigues, J.; Zhang, L.C.; Qian, J.Y.; Efron, B.; Hastie, T.; Elias, J.E.; Levitt, M.; Kopito, R.R. Proteomic analysis of monolayer-integrated proteins on lipid droplets identifies amphipathic interfacial alpha-helical membrane anchors. Proc. Natl. Acad. Sci. USA 2018, 115, Eb172–Eb180. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Beas, A.O.; Gordon, P.B.; Prentiss, C.L.; Olsen, C.P.; Kukurugya, M.A.; Bennett, B.D.; Parkhurst, S.M.; Gottschling, D.E. Independent regulation of age associated fat accumulation and longevity. Nat. Commun. 2020, 11, 2790. [Google Scholar] [CrossRef]
- Conte, M.; Vasuri, F.; Trisolino, G.; Bellavista, E.; Santoro, A.; Degiovanni, A.; Martucci, E.; D’Errico-Grigioni, A.; Caporossi, D.; Capri, M.; et al. Increased Plin2 Expression in Human Skeletal Muscle Is Associated with Sarcopenia and Muscle Weakness. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Flor, A.C.; Wolfgeher, D.; Wu, D.; Kron, S.J. A signature of enhanced lipid metabolism, lipid peroxidation and aldehyde stress in therapy-induced senescence. Cell Death Discov. 2017, 3, 17075. [Google Scholar] [CrossRef]
- Goldberg, A.A.; Bourque, S.D.; Kyryakov, P.; Boukh-Viner, T.; Gregg, C.; Beach, A.; Burstein, M.T.; Machkalyan, G.; Richard, V.; Rampersad, S.; et al. A novel function of lipid droplets in regulating longevity. Biochem. Soc. Trans. 2009, 37, 1050–1055. [Google Scholar] [CrossRef]
- Longo, V.D.; Shadel, G.S.; Kaeberlein, M.; Kennedy, B. Replicative and Chronological Aging in Saccharomyces cerevisiae. Cell Metab. 2012, 16, 18–31. [Google Scholar] [CrossRef]
- Laun, P.; Rinnerthaler, M.; Bogengruber, E.; Heeren, G.; Breitenbach, M. Yeast as a model for chronological and reproductive aging - A comparison. Exp. Gerontol. 2006, 41, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Arlia-Ciommo, A.; Leonov, A.; Beach, A.; Richard, V.R.; Bourque, S.D.; Burstein, M.T.; Kyryakov, P.; Gomez-Perez, A.; Koupaki, O.; Feldman, R.; et al. Caloric restriction delays yeast chronological aging by remodeling carbohydrate and lipid metabolism, altering peroxisomal and mitochondrial functionalities, and postponing the onsets of apoptotic and liponecrotic modes of regulated cell death. Oncotarget 2018, 9, 16163–16184. [Google Scholar] [CrossRef][Green Version]
- McCay, C.M.; Crowell, M.F.; Maynard, L.A. The effect of retarded growth upon the length of life span and upon the ultimate body size. J. Nutr. 1935, 10, 63–79. [Google Scholar] [CrossRef]
- Pifferi, F.; Terrien, J.; Marchal, J.; Dal-Pan, A.; Djelti, F.; Hardy, I.; Chahory, S.; Cordonnier, N.; Desquilbet, L.; Hurion, M.; et al. Caloric restriction increases lifespan but affects brain integrity in grey mouse lemur primates. Commun. Biol. 2018, 1. [Google Scholar] [CrossRef]
- Lin, S.J.; Kaeberlein, M.; Andalis, A.A.; Sturtz, L.A.; Defossez, P.A.; Culotta, V.C.; Fink, G.R.; Guarente, L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 2002, 418, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.D.; Wilson, M.A.; Zhu, M.; Wolkow, C.A.; de Cabo, R.; Ingram, D.K.; Zou, S.G. Dietary deprivation extends lifespan in Caenorhabditis elegans. Aging Cell 2006, 5, 515–524. [Google Scholar] [CrossRef]
- Katewa, S.D.; Demontis, F.; Kolipinski, M.; Hubbard, A.; Gill, M.S.; Perrimon, N.; Melov, S.; Kapahi, P. Intramyocellular Fatty-Acid Metabolism Plays a Critical Role in Mediating Responses to Dietary Restriction in Drosophila melanogaster. Cell Metab. 2012, 16, 97–103. [Google Scholar] [CrossRef]
- Palgunow, D.; Klapper, M.; Doring, F. Dietary Restriction during Development Enlarges Intestinal and Hypodermal Lipid Droplets in Caenorhabditis elegans. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Fernandez del Rio, L.; Gutierrez-Casado, E.; Varela-Lopez, A.; Villalba, J.M. Olive Oil and the Hallmarks of Aging. Molecules 2016, 21, 163. [Google Scholar] [CrossRef]
- Baati, T.; Bourasset, F.; Gharbi, N.; Njim, L.; Abderrabba, M.; Kerkeni, A.; Szwarc, H.; Moussa, F. The prolongation of the lifespan of rats by repeated oral administration of [60] fullerene. Biomaterials 2012, 33, 6292–6294. [Google Scholar] [CrossRef]
- Rohwedder, A.; Zhang, Q.F.; Rudge, S.A.; Wakelam, M.J.O. Lipid droplet formation in response to oleic acid in Huh-7 cells is mediated by the fatty acid receptor FFAR4. J. Cell Sci. 2014, 127, 3104–3115. [Google Scholar] [CrossRef]
- Bustos, V.; Partridge, L. Good Ol’ Fat: Links between Lipid Signaling and Longevity. Trends Biochem. Sci. 2017, 42, 812–823. [Google Scholar] [CrossRef]
- Lizardo, D.Y.; Lin, Y.L.; Gokcumen, O.; Atilla-Gokcumen, G.E. Regulation of lipids is central to replicative senescence. Mol. Biosyst. 2017, 13, 498–509. [Google Scholar] [CrossRef]
- Krahmer, N.; Farese, R.V.; Walther, T.C. Balancing the fat: Lipid droplets and human disease. EMBO Mol. Med. 2013, 5, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, K.; Sandoval, H.; Yamamoto, S.; Jaiswal, M.; Sanz, E.; Li, Z.H.; Hui, J.; Graham, B.H.; Quintana, A.; et al. Glial Lipid Droplets and ROS Induced by Mitochondrial Defects Promote Neurodegeneration. Cell 2015, 160, 177–190. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3. [Google Scholar] [CrossRef]
- Guerrero-Ferreira, R.; Taylor, N.M.I.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM structure of alpha-synuclein fibrils. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Sango, K.; Wada, K.; Nagai, Y. Pathological role of lipid interaction with alpha-synuclein in Parkinson’s disease. Neurochem. Int. 2018, 119, 97–106. [Google Scholar] [CrossRef]
- Halliday, G.M.; Ophof, A.; Broe, M.; Jensen, P.H.; Kettle, E.; Fedorow, H.; Cartwright, M.I.; Griffiths, F.M.; Shepherd, C.E.; Double, K.L. Alpha-synuclein redistributes to neuromelanin lipid in the substantia nigra early in Parkinson’s disease. Brain 2005, 128, 2654–2664. [Google Scholar] [CrossRef]
- Ross, B.M.; Mamalias, N.; Moszczynska, A.; Rajput, A.H.; Kish, S.J. Elevated activity of phospholipid biosynthetic enzymes in substantia nigra of patients with Parkinson’s disease. Neuroscience 2001, 102, 899–904. [Google Scholar] [CrossRef]
- Fanning, S.; Haque, A.; Imberdis, T.; Baru, V.; Barrasa, M.I.; Nuber, S.; Termine, D.; Ramalingam, N.; Ho, G.P.H.; Noble, T.; et al. Lipidomic Analysis of alpha-Synuclein Neurotoxicity Identifies Stearoyl CoA Desaturase as a Target for Parkinson Treatment. Mol. Cell 2018. [Google Scholar] [CrossRef]
- Han, X.; Zhu, J.; Zhang, X.; Song, Q.; Ding, J.; Lu, M.; Sun, S.; Hu, G. Plin4-Dependent Lipid Droplets Hamper Neuronal Mitophagy in the MPTP/p-Induced Mouse Model of Parkinson’s Disease. Front. Neurosci. 2018, 12, 397. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.M.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, U567–U574. [Google Scholar] [CrossRef]
- Karasinska, J.M.; Hayden, M.R. Cholesterol metabolism in Huntington disease. Nat. Rev. Neurol. 2011, 7, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Boussicault, L.; Alves, S.; Lamaziere, A.; Planques, A.; Heck, N.; Moumne, L.; Despres, G.; Bolte, S.; Hu, A.; Pages, C.; et al. CYP46A1, the rate-limiting enzyme for cholesterol degradation, is neuroprotective in Huntington’s disease. Brain 2016, 139, 953–970. [Google Scholar] [CrossRef] [PubMed]
- Aditi, K.; Shakarad, M.N.; Agrawal, N. Altered lipid metabolism in Drosophila model of Huntington’s disease. Sci. Rep. 2016, 6, 31411. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef]
- Hamilton, L.K.; Dufresne, M.; Joppe, S.E.; Petryszyn, S.; Aumont, A.; Calon, F.; Barnabe-Heider, F.; Furtos, A.; Parent, M.; Chaurand, P.; et al. Aberrant Lipid Metabolism in the Forebrain Niche Suppresses Adult Neural Stem Cell Proliferation in an Animal Model of Alzheime’s Disease. Cell Stem Cell 2015, 17, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Wani, W.Y.; Chatham, J.C.; Darley-Usmar, V.; McMahon, L.L.; Zhang, J. O-GlcNAcylation and neurodegeneration. Brain Res. Bull. 2017, 133, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Puglielli, L.; Tanzi, R.E.; Kovacs, D.M. Alzheimer’s disease: The cholesterol connection. Nat. Neurosci. 2003, 6, 345–351. [Google Scholar] [CrossRef]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef]
- Pralhada Rao, R.; Vaidyanathan, N.; Rengasamy, M.; Mammen Oommen, A.; Somaiya, N.; Jagannath, M.R. Sphingolipid metabolic pathway: An overview of major roles played in human diseases. J. Lipids 2013, 2013, 178910. [Google Scholar] [CrossRef] [PubMed]
- Siddique, M.M.; Li, Y.; Chaurasia, B.; Kaddai, V.A.; Summers, S.A. Dihydroceramides: From Bit Players to Lead Actors. J. Biol. Chem. 2015, 290, 15371–15379. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Risch, N.J.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C., Jr.; Rimmler, J.B.; Locke, P.A.; Conneally, P.M.; Schmader, K.E.; et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nature Genet. 1994, 7, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Rubio, J.P.; Danek, A.; Stone, C.; Chalmers, R.; Wood, N.; Verellen, C.; Ferrer, X.; Malandrini, A.; Fabrizi, G.M.; Manfredi, M.; et al. Chorea-acanthocytosis: Genetic linkage to chromosome 9q21. Am. J. Hum. Genet. 1997, 61, 899–908. [Google Scholar] [CrossRef]
- Blackstone, C. Converging cellular themes for the hereditary spastic paraplegias. Curr. Opin. Neurobiol. 2018, 51, 139–146. [Google Scholar] [CrossRef]
- Engelen, M.; Kemp, S.; de Visser, M.; van Geel, B.M.; Wanders, R.J.A.; Aubourg, P.; Poll-The, B.T. X-linked adrenoleukodystrophy (X-ALD): Clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J. Rare Dis. 2012, 7. [Google Scholar] [CrossRef]
- Fischer, J.; Lefevre, C.; Morava, E.; Mussini, J.M.; Laforet, P.; Negre-Salvayre, A.; Lathrop, M.; Salvayre, R. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat. Genet. 2007, 39, 28–30. [Google Scholar] [CrossRef]
- Lefevre, C.; Jobard, F.; Caux, F.; Bouadjar, B.; Karaduman, A.; Heilig, R.; Lakhdar, H.; Wollenberg, A.; Verret, J.L.; Weissenbach, J.; et al. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am. J. Hum. Genet. 2001, 69, 1002–1012. [Google Scholar] [CrossRef]
- Igal, R.A.; Rhoads, J.M.; Coleman, R.A. Neutral lipid storage disease with fatty liver and cholestasis. J. Pediatric Gastroenterol. Nutr. 1997, 25, 541–547. [Google Scholar] [CrossRef]
- Kaneko, K.; Kuroda, H.; Izumi, R.; Tateyama, M.; Kato, M.; Sugimura, K.; Sakata, Y.; Ikeda, Y.; Hirano, K.; Aoki, M. A novel mutation in PNPLA2 causes neutral lipid storage disease with myopathy and triglyceride deposit cardiomyovasculopathy: A case report and literature review. Neuromuscul. Disord. 2014, 24, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Laforet, P.; Stojkovic, T.; Bassez, G.; Carlier, P.G.; Clement, K.; Wahbi, K.; Petit, F.M.; Eymard, B.; Carlier, R.Y. Neutral lipid storage disease with myopathy: A whole-body nuclear MRI and metabolic study. Mol. Genet. Metab. 2013, 108, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Reilich, P.; Horvath, R.; Krause, S.; Schramm, N.; Turnbull, D.M.; Trenell, M.; Hollingsworth, K.G.; Gorman, G.S.; Hans, V.H.; Reimann, J.; et al. The phenotypic spectrum of neutral lipid storage myopathy due to mutations in the PNPLA2 gene. J. Neurol. 2011, 258, 1987–1997. [Google Scholar] [CrossRef]
- Pennisi, E.M.; Arca, M.; Bertini, E.; Bruno, C.; Cassandrini, D.; D’Amico, A.; Garibaldi, M.; Gragnani, F.; Maggi, L.; Massa, R.; et al. Neutral Lipid Storage Diseases: Clinical/genetic features and natural history in a large cohort of Italian patients. Orphanet J. Rare Dis. 2017, 12, 90. [Google Scholar] [CrossRef] [PubMed]
- Lass, A.; Zimmermann, R.; Haemmerle, G.; Riederer, M.; Schoiswohl, G.; Schweiger, M.; Kienesberger, P.; Strauss, J.G.; Gorkiewicz, G.; Zechner, R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006, 3, 309–319. [Google Scholar] [CrossRef]
- Durdu, M.; Missaglia, S.; Moro, L.; Tavian, D. Clinical and genetic characterization of a Chanarin Dorfman Syndrome patient born to diseased parents. BMC Med. Genet. 2018, 19, 88. [Google Scholar] [CrossRef]
- Nur, B.G.; Gencpinar, P.; Yuzbasioglu, A.; Emre, S.D.; Mihci, E. Chanarin-Dorfman syndrome: Genotype-Phenotype Correlation. Eur. J. Med. Genet. 2015, 58, 238–242. [Google Scholar] [CrossRef]
- Akman, H.O.; Davidzon, G.; Tanji, K.; Macdermott, E.J.; Larsen, L.; Davidson, M.M.; Haller, R.G.; Szczepaniak, L.S.; Lehman, T.J.; Hirano, M.; et al. Neutral lipid storage disease with subclinical myopathy due to a retrotransposal insertion in the PNPLA2 gene. Neuromuscul. Disord. 2010, 20, 397–402. [Google Scholar] [CrossRef]
- Fiorillo, C.; Brisca, G.; Cassandrini, D.; Scapolan, S.; Astrea, G.; Valle, M.; Scuderi, F.; Trucco, F.; Natali, A.; Magnano, G.; et al. Subclinical myopathy in a child with neutral lipid storage disease and mutations in the PNPLA2 gene. Biochem. Biophys. Res. Commun. 2013, 430, 241–244. [Google Scholar] [CrossRef]
- Hirano, K.; Ikeda, Y.; Zaima, N.; Sakata, Y.; Matsumiya, G. Triglyceride deposit cardiomyovasculopathy. N. Engl. J. Med. 2008, 359, 2396–2398. [Google Scholar] [CrossRef]
- Li, M.; Hirano, K.I.; Ikeda, Y.; Higashi, M.; Hashimoto, C.; Zhang, B.; Kozawa, J.; Sugimura, K.; Miyauchi, H.; Suzuki, A.; et al. Triglyceride deposit cardiomyovasculopathy: A rare cardiovascular disorder. Orphanet J. Rare Dis. 2019, 14, 134. [Google Scholar] [CrossRef] [PubMed]
- Missaglia, S.; Valadares, E.R.; Moro, L.; Faguntes, E.D.; Quintao Roque, R.; Giardina, B.; Tavian, D. Early onset of Chanarin-Dorfman syndrome with severe liver involvement in a patient with a complex rearrangement of ABHD5 promoter. BMC Med. Genet. 2014, 15, 32. [Google Scholar] [CrossRef]
- Ronchetti, A.; Prati, D.; Pezzotta, M.G.; Tavian, D.; Colombo, R.; Callea, F.; Colli, A. Severe steatohepatitis in a patient with a rare neutral lipid storage disorder due to ABHD5 mutation. J. Hepatol. 2008, 49, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, C.; Coleman, R.A.; Moro, L.; Dacou-Voutetakis, C.; Elsayed, S.M.; Prati, D.; Colli, A.; Mela, D.; Colombo, R.; Tavian, D. Clinical and genetic characterization of Chanarin-Dorfman syndrome patients: First report of large deletions in the ABHD5 gene. Orphanet J. Rare Dis. 2010, 5, 33. [Google Scholar] [CrossRef]
- Kien, B.; Grond, S.; Haemmerle, G.; Lass, A.; Eichmann, T.O.; Radner, F.P.W. ABHD5 stimulates PNPLA1-mediated omega-O-acylceramide biosynthesis essential for a functional skin permeability barrier. J. Lipid Res. 2018, 59, 2360–2367. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Holleran, W.M. Omega-O-acylceramide, a lipid essential for mammalian survival. J. Dermatol. Sci. 2008, 51, 77–87. [Google Scholar] [CrossRef]
- Uchida, Y.; Cho, Y.; Moradian, S.; Kim, J.; Nakajima, K.; Crumrine, D.; Park, K.; Ujihara, M.; Akiyama, M.; Shimizu, H.; et al. Neutral lipid storage leads to acylceramide deficiency, likely contributing to the pathogenesis of Dorfman-Chanarin syndrome. J. Investig. Dermatol. 2010, 130, 2497–2499. [Google Scholar] [CrossRef]
- Chung, J.; Wu, X.; Lambert, T.J.; Lai, Z.W.; Walther, T.C.; Farese, R.V., Jr. LDAF1 and Seipin Form a Lipid Droplet Assembly Complex. Dev. Cell 2019, 51, 551–563. [Google Scholar] [CrossRef]
- Bielecka-Dabrowa, A.; Ebner, N.; Dos Santos, M.R.; Ishida, J.; Hasenfuss, G.; von Haehling, S. Cachexia, muscle wasting, and frailty in cardiovascular disease. Eur. J. Heart Fail. 2020. [Google Scholar] [CrossRef]
- Das, S.K.; Eder, S.; Schauer, S.; Diwoky, C.; Temmel, H.; Guertl, B.; Gorkiewicz, G.; Tamilarasan, K.P.; Kumari, P.; Trauner, M.; et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 2011, 333, 233–238. [Google Scholar] [CrossRef]
- Gown, A.M.; Tsukada, T.; Ross, R. Human atherosclerosis. II. Immunocytochemical analysis of the cellular composition of human atherosclerotic lesions. Am. J. Pathol. 1986, 125, 191–207. [Google Scholar]
- Katsuda, S.; Boyd, H.C.; Fligner, C.; Ross, R.; Gown, A.M. Human atherosclerosis. III. Immunocytochemical analysis of the cell composition of lesions of young adults. Am. J. Pathol. 1992, 140, 907–914. [Google Scholar] [PubMed]
- Wang, Y.; Dubland, J.A.; Allahverdian, S.; Asonye, E.; Sahin, B.; Jaw, J.E.; Sin, D.D.; Seidman, M.A.; Leeper, N.J.; Francis, G.A. Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 876–887. [Google Scholar] [CrossRef]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef]
- Stegemann, C.; Drozdov, I.; Shalhoub, J.; Humphries, J.; Ladroue, C.; Didangelos, A.; Baumert, M.; Allen, M.; Davies, A.H.; Monaco, C.; et al. Comparative lipidomics profiling of human atherosclerotic plaques. Circ. Cardiovasc. Genet. 2011, 4, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Lara-Guzman, O.J.; Gil-Izquierdo, A.; Medina, S.; Osorio, E.; Alvarez-Quintero, R.; Zuluaga, N.; Oger, C.; Galano, J.M.; Durand, T.; Munoz-Durango, K. Oxidized LDL triggers changes in oxidative stress and inflammatory biomarkers in human macrophages. Redox Biol. 2018, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Goo, Y.H.; Son, S.H.; Kreienberg, P.B.; Paul, A. Novel lipid droplet-associated serine hydrolase regulates macrophage cholesterol mobilization. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 386–396. [Google Scholar] [CrossRef]
- Ouimet, M.; Franklin, V.; Mak, E.; Liao, X.; Tabas, I.; Marcel, Y.L. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011, 13, 655–667. [Google Scholar] [CrossRef]
- Wang, N.; Lan, D.; Chen, W.; Matsuura, F.; Tall, A.R. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 9774–9779. [Google Scholar] [CrossRef]
- Paul, A.; Lydic, T.A.; Hogan, R.; Goo, Y.H. Cholesterol Acceptors Regulate the Lipidome of Macrophage Foam Cells. Int. J. Mol. Sci. 2019, 20, 3784. [Google Scholar] [CrossRef]
- Zhao, X.; Gao, M.; He, J.; Zou, L.; Lyu, Y.; Zhang, L.; Geng, B.; Liu, G.; Xu, G. Perilipin1 deficiency in whole body or bone marrow-derived cells attenuates lesions in atherosclerosis-prone mice. PLoS ONE 2015, 10, e0123738. [Google Scholar] [CrossRef]
- Becker, L.; Gharib, S.A.; Irwin, A.D.; Wijsman, E.; Vaisar, T.; Oram, J.F.; Heinecke, J.W. A macrophage sterol-responsive network linked to atherogenesis. Cell Metab. 2010, 11, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Chang, B.H.; Li, L.; Yechoor, V.K.; Chan, L. Deficiency of adipose differentiation-related protein impairs foam cell formation and protects against atherosclerosis. Circ. Res. 2008, 102, 1492–1501. [Google Scholar] [CrossRef]
- Li, H.; Song, Y.; Li, F.; Zhang, L.; Gu, Y.; Zhang, L.; Jiang, L.; Dong, W.; Ye, J.; Li, Q. Identification of lipid droplet-associated proteins in the formation of macrophage-derived foam cells using microarrays. Int. J. Mol. Med. 2010, 26, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Son, S.H.; Goo, Y.H.; Chang, B.H.; Paul, A. Perilipin 2 (PLIN2)-deficiency does not increase cholesterol-induced toxicity in macrophages. PLoS ONE 2012, 7, e33063. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.L.; Yang, Z.H.; Wang, X.C.; Liu, Y.; Yang, Y.H.; Li, L.X.; Liang, W.C.; Zhou, W.B.; Hu, R.M. Adipophilin affects the expression of TNF-alpha, MCP-1, and IL-6 in THP-1 macrophages. Mol. Cell Biochem. 2010, 337, 193–199. [Google Scholar] [CrossRef]
- Langlois, D.; Forcheron, F.; Li, J.Y.; del Carmine, P.; Neggazi, S.; Beylot, M. Increased atherosclerosis in mice deficient in perilipin1. Lipids Health Dis. 2011, 10, 169. [Google Scholar] [CrossRef]
- Pereira, K.; Salsamendi, J.; Casillas, J. The Global Nonalcoholic Fatty Liver Disease Epidemic: What a Radiologist Needs to Know. J. Clin. Imaging Sci. 2015, 5, 32. [Google Scholar] [CrossRef]
- Walker, M.; El-Serag, H.B.; Sada, Y.; Mittal, S.; Ying, J.; Duan, Z.; Richardson, P.; Davila, J.A.; Kanwal, F. Cirrhosis is under-recognised in patients subsequently diagnosed with hepatocellular cancer. Aliment. Pharmacol. Ther. 2016, 43, 621–630. [Google Scholar] [CrossRef]
- Bosetti, C.; Turati, F.; La Vecchia, C. Hepatocellular carcinoma epidemiology. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 753–770. [Google Scholar] [CrossRef]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, T.X.; Huang, X.; Li, Y.; Sun, T.; Zang, S.; Guan, K.L.; Xiong, Y.; Liu, J.; Yuan, H.X. Targeting ferroptosis alleviates methionine-choline deficient (MCD)-diet induced NASH by suppressing liver lipotoxicity. Liver Int. 2020. [Google Scholar] [CrossRef] [PubMed]
- Senoo, H.; Kojima, N.; Sato, M. Vitamin A-storing cells (stellate cells). Vitam. Horm. 2007, 75, 131–159. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic fibrosis—Overview. Toxicology 2008, 254, 120–129. [Google Scholar] [CrossRef]
- O’Mahony, F.; Wroblewski, K.; O’Byrne, S.M.; Jiang, H.; Clerkin, K.; Benhammou, J.; Blaner, W.S.; Beaven, S.W. Liver X receptors balance lipid stores in hepatic stellate cells through Rab18, a retinoid responsive lipid droplet protein. Hepatology 2015, 62, 615–626. [Google Scholar] [CrossRef]
- Minehira, K.; Gual, P. Role of Lipid Droplet Proteins in the Development of NAFLD and Hepatic Insulin Resistance. In Non-Alcoholic Fatty Liver Disease—Molecular Bases, Prevention and Treatment; Baez, R.V., Ed.; Intech: Rijeka, Croatia, 2017. [Google Scholar]
- Gluchowski, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V., Jr. Lipid droplets and liver disease: From basic biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 343–355. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nature Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Su, W.; Wang, Y.; Jia, X.; Wu, W.; Li, L.; Tian, X.; Li, S.; Wang, C.; Xu, H.; Cao, J.; et al. Comparative proteomic study reveals 17beta-HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2014, 111, 11437–11442. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Y.; Liu, P. Omic studies reveal the pathogenic lipid droplet proteins in non-alcoholic fatty liver disease. Protein Cell 2017, 8, 4–13. [Google Scholar] [CrossRef]
- Wang, R.; Kong, X.; Cui, A.; Liu, X.; Xiang, R.; Yang, Y.; Guan, Y.; Fang, F.; Chang, Y. Sterol-regulatory-element-binding protein 1c mediates the effect of insulin on the expression of Cidea in mouse hepatocytes. Biochem. J. 2010, 430, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Xu, L.; Ye, J.; Li, D.; Wang, W.; Li, X.; Wu, L.; Wang, H.; Guan, F.; Li, P. Cidea promotes hepatic steatosis by sensing dietary fatty acids. Hepatology 2012, 56, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Pawella, L.M.; Hashani, M.; Eiteneuer, E.; Renner, M.; Bartenschlager, R.; Schirmacher, P.; Straub, B.K. Perilipin discerns chronic from acute hepatocellular steatosis. J. Hepatol. 2014, 60, 633–642. [Google Scholar] [CrossRef]
- Chen, F.J.; Yin, Y.; Chua, B.T.; Li, P. CIDE family proteins control lipid homeostasis and the development of metabolic diseases. Traffic 2020, 21, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Chen, F.J.; Zhou, L.; Su, L.; Xu, D.; Xu, L.; Li, P. Control of lipid droplet fusion and growth by CIDE family proteins. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1197–1204. [Google Scholar] [CrossRef]
- Greco, D.; Kotronen, A.; Westerbacka, J.; Puig, O.; Arkkila, P.; Kiviluoto, T.; Laitinen, S.; Kolak, M.; Fisher, R.M.; Hamsten, A.; et al. Gene expression in human NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1281–G1287. [Google Scholar] [CrossRef]
- Straub, B.K.; Stoeffel, P.; Heid, H.; Zimbelmann, R.; Schirmacher, P. Differential pattern of lipid droplet-associated proteins and de novo perilipin expression in hepatocyte steatogenesis. Hepatology 2008, 47, 1936–1946. [Google Scholar] [CrossRef]
- Magnusson, B.; Asp, L.; Bostrom, P.; Ruiz, M.; Stillemark-Billton, P.; Linden, D.; Boren, J.; Olofsson, S.O. Adipocyte differentiation-related protein promotes fatty acid storage in cytosolic triglycerides and inhibits secretion of very low-density lipoproteins. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1566–1571. [Google Scholar] [CrossRef]
- Carr, R.M.; Patel, R.T.; Rao, V.; Dhir, R.; Graham, M.J.; Crooke, R.M.; Ahima, R.S. Reduction of TIP47 improves hepatic steatosis and glucose homeostasis in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R996–R1003. [Google Scholar] [CrossRef]
- Hall, A.M.; Brunt, E.M.; Chen, Z.; Viswakarma, N.; Reddy, J.K.; Wolins, N.E.; Finck, B.N. Dynamic and differential regulation of proteins that coat lipid droplets in fatty liver dystrophic mice. J. Lipid Res. 2010, 51, 554–563. [Google Scholar] [CrossRef]
- Li, Z.; Li, Y.; Zhang, H.X.; Guo, J.R.; Lam, C.W.K.; Wang, C.Y.; Zhang, W. Mitochondria-Mediated Pathogenesis and Therapeutics for Non-Alcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2019, 63, e1900043. [Google Scholar] [CrossRef]
- Benador, I.Y.; Veliova, M.; Mahdaviani, K.; Petcherski, A.; Wikstrom, J.D.; Assali, E.A.; Acin-Perez, R.; Shum, M.; Oliveira, M.F.; Cinti, S.; et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics that Support Lipid Droplet Expansion. Cell Metab. 2018, 27, 869. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, A.R.; Sztalryd, C. Perilipin 5, a lipid droplet protein adapted to mitochondrial energy utilization. Curr. Opin. Lipidol. 2014, 25, 110–117. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef]
- Kanda, T.; Matsuoka, S.; Yamazaki, M.; Shibata, T.; Nirei, K.; Takahashi, H.; Kaneko, T.; Fujisawa, M.; Higuchi, T.; Nakamura, H.; et al. Apoptosis and non-alcoholic fatty liver diseases. World J. Gastroenterol. 2018, 24, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Beier, J.I.; Banales, J.M. Pyroptosis: An inflammatory link between NAFLD and NASH with potential therapeutic implications. J. Hepatol. 2018, 68, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, N.; Carter-Kent, C.; Feldstein, A.E. Apoptosis in nonalcoholic fatty liver disease: Diagnostic and therapeutic implications. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 201–212. [Google Scholar] [CrossRef]
- Fabregat, I. Dysregulation of apoptosis in hepatocellular carcinoma cells. World J. Gastroenterol. 2009, 15, 513–520. [Google Scholar] [CrossRef]
- Berndt, N.; Eckstein, J.; Heucke, N.; Gajowski, R.; Stockmann, M.; Meierhofer, D.; Holzhutter, H.G. Characterization of Lipid and Lipid Droplet Metabolism in Human HCC. Cells 2019, 8, 512. [Google Scholar] [CrossRef]
- Henne, W.M.; Reese, M.L.; Goodman, J.M. The assembly of lipid droplets and their roles in challenged cells (vol. 37, e98947, 2018). EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Bozza, P.T.; Magalhaes, K.G.; Weller, P.F. Leukocyte lipid bodies—Biogenesis and functions in inflammation. Biochim. Biophys. Acta 2009, 1791, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Libbing, C.L.; McDevitt, A.R.; Azcueta, R.P.; Ahila, A.; Mulye, M. Lipid Droplets: A Significant but Understudied Contributor of Host(-)Bacterial Interactions. Cells 2019, 8, 354. [Google Scholar] [CrossRef]
- Papackova, Z.; Cahova, M. Fatty acid signaling: The new function of intracellular lipases. Int. J. Mol. Sci. 2015, 16, 3831–3855. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Gartung, A.; Zhao, J.; Chen, S.; Mottillo, E.; VanHecke, G.C.; Ahn, Y.H.; Maddipati, K.R.; Sorokin, A.; Granneman, J.; Lee, M.J. Characterization of Eicosanoids Produced by Adipocyte Lipolysis: Implication of cyclooxygenase-2 in adipose inflammation. J. Biol. Chem. 2016, 291, 16001–16010. [Google Scholar] [CrossRef]
- Schlager, S.; Goeritzer, M.; Jandl, K.; Frei, R.; Vujic, N.; Kolb, D.; Strohmaier, H.; Dorow, J.; Eichmann, T.O.; Rosenberger, A.; et al. Adipose triglyceride lipase acts on neutrophil lipid droplets to regulate substrate availability for lipid mediator synthesis. J. Leukoc. Biol. 2015, 98, 837–850. [Google Scholar] [CrossRef]
- Sohn, J.H.; Lee, Y.K.; Han, J.S.; Jeon, Y.G.; Kim, J.I.; Choe, S.S.; Kim, S.J.; Yoo, H.J.; Kim, J.B. Perilipin 1 (Plin1) deficiency promotes inflammatory responses in lean adipose tissue through lipid dysregulation. J. Biol. Chem. 2018, 293, 13974–13988. [Google Scholar] [CrossRef]
- Schlager, S.; Vujic, N.; Korbelius, M.; Duta-Mare, M.; Dorow, J.; Leopold, C.; Rainer, S.; Wegscheider, M.; Reicher, H.; Ceglarek, U.; et al. Lysosomal lipid hydrolysis provides substrates for lipid mediator synthesis in murine macrophages. Oncotarget 2017, 8, 40037–40051. [Google Scholar] [CrossRef]
- Johnson, M.M.; Vaughn, B.; Triggiani, M.; Swan, D.D.; Fonteh, A.N.; Chilton, F.H. Role of arachidonyl triglycerides within lipid bodies in eicosanoid formation by human polymorphonuclear cells. Am. J. Respir. Cell Mol. Biol. 1999, 21, 253–258. [Google Scholar] [CrossRef]
- Triggiani, M.; Oriente, A.; Marone, G. Differential roles for triglyceride and phospholipid pools of arachidonic acid in human lung macrophages. J. Immunol. 1994, 152, 1394–1403. [Google Scholar]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Maxfield, F.R.; Tabas, I. Role of cholesterol and lipid organization in disease. Nature 2005, 438, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Bozza, P.T.; Melo, R.C.; Bandeira-Melo, C. Leukocyte lipid bodies regulation and function: Contribution to allergy and host defense. Pharmacol. Ther. 2007, 113, 30–49. [Google Scholar] [CrossRef]
- Accioly, M.T.; Pacheco, P.; Maya-Monteiro, C.M.; Carrossini, N.; Robbs, B.K.; Oliveira, S.S.; Kaufmann, C.; Morgado-Diaz, J.A.; Bozza, P.T.; Viola, J.P. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res. 2008, 68, 1732–1740. [Google Scholar] [CrossRef]
- Chen, X.; Xu, S.; Wei, S.; Deng, Y.; Li, Y.; Yang, F.; Liu, P. Comparative Proteomic Study of Fatty Acid-treated Myoblasts Reveals Role of Cox-2 in Palmitate-induced Insulin Resistance. Sci. Rep. 2016, 6, 21454. [Google Scholar] [CrossRef]
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef]
- Chitraju, C.; Mejhert, N.; Haas, J.T.; Diaz-Ramirez, L.G.; Grueter, C.A.; Imbriglio, J.E.; Pinto, S.; Koliwad, S.K.; Walther, T.C.; Farese, R.V., Jr. Triglyceride Synthesis by DGAT1 Protects Adipocytes from Lipid-Induced ER Stress during Lipolysis. Cell Metab. 2017, 26, 407–418. [Google Scholar] [CrossRef]
- Schreiber, R.; Xie, H.; Schweiger, M. Of mice and men: The physiological role of adipose triglyceride lipase (ATGL). Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 880–899. [Google Scholar] [CrossRef]
- Khan, S.A.; Sathyanarayan, A.; Mashek, M.T.; Ong, K.T.; Wollaston-Hayden, E.E.; Mashek, D.G. ATGL-catalyzed lipolysis regulates SIRT1 to control PGC-1alpha/PPAR-alpha signaling. Diabetes 2015, 64, 418–426. [Google Scholar] [CrossRef]
- Chen, X.; Lu, Y.; Zhang, Z.; Wang, J.; Yang, H.; Liu, G. Intercellular interplay between Sirt1 signalling and cell metabolism in immune cell biology. Immunology 2015, 145, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Zechner, R.; Madeo, F.; Kratky, D. Cytosolic lipolysis and lipophagy: Two sides of the same coin. Nat. Rev. Mol. Cell Biol. 2017, 18, 671–684. [Google Scholar] [CrossRef]
- Granneman, J.G.; Moore, H.P.; Krishnamoorthy, R.; Rathod, M. Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl). J. Biol. Chem. 2009, 284, 34538–34544. [Google Scholar] [CrossRef] [PubMed]
- Chiurchiu, V.; Leuti, A.; Maccarrone, M. Bioactive Lipids and Chronic Inflammation: Managing the Fire Within. Front. Immunol. 2018, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Guijas, C.; Rodriguez, J.P.; Rubio, J.M.; Balboa, M.A.; Balsinde, J. Phospholipase A2 regulation of lipid droplet formation. Biochim. Biophys. Acta 2014, 1841, 1661–1671. [Google Scholar] [CrossRef]
- Ogawa, K.; Hishiki, T.; Shimizu, Y.; Funami, K.; Sugiyama, K.; Miyanari, Y.; Shimotohno, K. Hepatitis C virus utilizes lipid droplet for production of infectious virus. Proc. Jpn. Acad. B Phys. 2009, 85, 217–228. [Google Scholar] [CrossRef]
- McLauchlan, J.; Lemberg, M.K.; Hope, G.; Martoglio, B. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 2002, 21, 3980–3988. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.; Herker, E.; Farese, R.V.; Ott, M. Hepatitis C Virus Core protein decreases lipid droplet turnover a mechanism for core-induced steatosis. J. Biol. Chem. 2011, 286, 42615–42625. [Google Scholar] [CrossRef]
- Senecal, M.; Brisson, M.; Lebel, M.H.; Yaremko, J.; Wong, R.; Gallant, L.A.; Garfield, H.A.; Ableman, D.J.; Ward, R.L.; Samjalis, J.S.; et al. Measuring the Impact of Rotavirus Acute Gastroenteritis Episodes (MIRAGE): A prospective community-based study. Can. J. Infect. Dis. Med. 2008, 19, 397–404. [Google Scholar] [CrossRef][Green Version]
- Contin, R.; Arnoldi, F.; Mano, M.; Burrone, O.R. Rotavirus replication requires a functional proteasome for effective assembly of viroplasms. J. Virol. 2011, 85, 2781–2792. [Google Scholar] [CrossRef]
- Cheung, W.; Gill, M.; Esposito, A.; Kaminski, C.F.; Courousse, N.; Chwetzoff, S.; Trugnan, G.; Keshavan, N.; Lever, A.; Desselberger, U. Rotaviruses associate with cellular lipid droplet components to replicate in viroplasms, and compounds disrupting or blocking lipid droplets inhibit viroplasm formation and viral replication. J. Virol. 2010, 84, 6782–6798. [Google Scholar] [CrossRef] [PubMed]
- Gaunt, E.R.; Zhang, Q.F.; Cheung, W.; Wakelam, M.J.O.; Lever, A.M.L.; Desselberger, U. Lipidome analysis of rotavirus-infected cells confirms the close interaction of lipid droplets with viroplasms. J. Gen. Virol. 2013, 94, 1576–1586. [Google Scholar] [CrossRef]
- York, A. Oiling the Flavivirus replication machinery. Nat. Rev. Microbiol. 2018, 16, 455. [Google Scholar] [CrossRef]
- Leier, H.C.; Weinstein, J.B.; Kyle, J.E.; Lee, J.Y.; Bramer, L.M.; Stratton, K.G.; Kempthorne, D.; Navratil, A.R.; Tafesse, E.G.; Hornemann, T.; et al. A global lipid map defines a network essential for Zika virus replication. Nat. Commun. 2020, 11, 3652. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geltinger, F.; Schartel, L.; Wiederstein, M.; Tevini, J.; Aigner, E.; Felder, T.K.; Rinnerthaler, M. Friend or Foe: Lipid Droplets as Organelles for Protein and Lipid Storage in Cellular Stress Response, Aging and Disease. Molecules 2020, 25, 5053. https://doi.org/10.3390/molecules25215053
Geltinger F, Schartel L, Wiederstein M, Tevini J, Aigner E, Felder TK, Rinnerthaler M. Friend or Foe: Lipid Droplets as Organelles for Protein and Lipid Storage in Cellular Stress Response, Aging and Disease. Molecules. 2020; 25(21):5053. https://doi.org/10.3390/molecules25215053
Chicago/Turabian StyleGeltinger, Florian, Lukas Schartel, Markus Wiederstein, Julia Tevini, Elmar Aigner, Thomas K. Felder, and Mark Rinnerthaler. 2020. "Friend or Foe: Lipid Droplets as Organelles for Protein and Lipid Storage in Cellular Stress Response, Aging and Disease" Molecules 25, no. 21: 5053. https://doi.org/10.3390/molecules25215053
APA StyleGeltinger, F., Schartel, L., Wiederstein, M., Tevini, J., Aigner, E., Felder, T. K., & Rinnerthaler, M. (2020). Friend or Foe: Lipid Droplets as Organelles for Protein and Lipid Storage in Cellular Stress Response, Aging and Disease. Molecules, 25(21), 5053. https://doi.org/10.3390/molecules25215053