
Carbon Anode in Carbon History


Abstract
1. Introduction
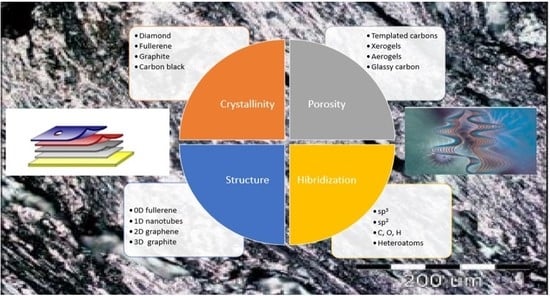
2. Structure in Carbons and Carbon Forms
3. Catalysis of Carbon Oxidation Reactions
- The metal concerned,
- The gasification reaction being studied, and thermal conditions employed,
- The size of the catalyst particles and their state of dispersion throughout the carbon,
- The chemical state of the catalyst,
- The relative amounts of catalyst.
4. Nanotechnology
5. Electrocatalysis
5.1. Particle Size Effect on Phase Transformations
5.2. Particle Size Effect on Catalytic Activity
6. Aluminium Smelter Technology
7. Electrochemical Kinetics
7.1. Hydrogen Reactions
7.2. Oxygen Reactions
7.3. Hydrogen Peroxide Production
7.4. Carbon Dioxide Reduction Reaction
7.5. Nitrogen Reduction Reaction
7.6. Borohydride Oxidation Reaction
8. Direct Carbon Fuel Cell
- -
- Temperature of operation is currently 800–1000 °C;
- -
- OCVs obtained are in the range 0.95–1.10 V;
- -
- Power densities achieved of up to 150 mW/cm2 over a short testing period;
- -
- Electrical efficiency >80%;
- -
- CHP efficiency >90%;
- -
- Heat output 500–800 J;
- -
- High thermal insulation.
9. Neurochemical Monitoring
10. Lithium Ion Batteries
11. Electrochemical Capacitors
12. Conclusions
Funding
Conflicts of Interest
References
- Brodd, R.J. Electrochemistry in Industry: New Directions; Plenum Press: New York, NY, USA, 1980. [Google Scholar]
- Thonstad, J.; Fellner, P.; Haarberg, G.M.; Hives, J.; Kvande, H.; Sterten, A. Aluminium Electrolysis; Aluminium-Verlag: Dusseldorf, Germany, 2001. [Google Scholar]
- Grjotheim, K.; Welch, B.J. Aluminium Smelter Technology–A Pure and Applied Approach, 2nd ed.; Aluminium Verlag: Dusseldorf, Germany, 1988. [Google Scholar]
- Grjotheim, K.; Kvande, H. (Eds.) Introduction to Aluminium Electrolysis; Aluminium-Verlag: Dusseldorf, Germany, 1993. [Google Scholar]
- Radenović, A. Svojstva komponenti ugljične anode za proizvodnju aluminija. Nafta 2012, 63, 111–114. [Google Scholar]
- Wei, M.; Zhang, F.; Wang, W.; Alexandridis, P.; Zhou, C.; Wu, G. 3D direct writing fabrication of electrodes for electrochemical storage devices. J. Power Sources 2017, 354, 134–147. [Google Scholar] [CrossRef]
- Alkire, R.C.; Bartlett, P.N.; Lijkowski, J. (Eds.) Electrochemistry of Carbon Electrodes; Wiley-VCH Verlag: Weinheim, Germany, 2016. [Google Scholar]
- McDermott, M.T.; Bélanger, D.; Zaghib, K. (Eds.) Electrochemistry of Carbon Materials; The Electrochemical Society, Inc.: Pennington, NJ, USA, 2004. [Google Scholar]
- Khaji, K.; Al Qassemi, M. The role of anode manufacturing processes in net carbon consumption. Metals 2016, 6, 128. [Google Scholar] [CrossRef]
- Hazen, R.M.; Bundy, F.P. The Diamond Makers. Phys. Today 2000, 53, 58–59. [Google Scholar] [CrossRef][Green Version]
- Bundy, F.P.; Hall, H.T.; Strong, H.M.; Wentorf, R.H. Man-made diamonds. Nature 1955, 176, 51–55. [Google Scholar] [CrossRef]
- Sirk, A.H.C.; Sadoway, D.R. Electrochemical synthesis of diamondlike carbon films. J. Electrochem. Soc. 2008, 155, E49–E55. [Google Scholar] [CrossRef]
- Zeng, A.; Neto, V.F.; Gracio, J.J.; Fan, Q.H. Diamond-like carbon (DLC) films as electrochemical electrodes. Diam. Relat. Mater. 2014, 43, 12–22. [Google Scholar] [CrossRef]
- Shindo, A. Tanso sen’i no kenkyu—Netsu shori ni tomonau kesshoshi no seicho (Study of carbon fiber—Growth of crytallite in heat treatment). Osaka Kogyo Gijitsuo Shikenjo Koho 1961, 12, 110–119. [Google Scholar]
- Otani, S. On the carbon fiber from the molten pyrolysis products. Carbon 1965, 3, 31–38. [Google Scholar] [CrossRef]
- Bacon, R. Carbon Fibres from Rayon Percursors. In Chemistry and Physics of Carbon; Walker, P.L., Thrower, P.A., Eds.; Marcel Dekker: New York, NY, USA, 1975; Volume 5, pp. 1–101. [Google Scholar]
- Roberts, T. The Carbon Fibre Industry Worldwide 2011–2020; Materials Technology Publications: London, UK, 2012. [Google Scholar]
- Marsh, H.; Diez, M.A. Mesophase of Gnaphitizable Carbons. In Liquid Crystalline and Mesomorphic Polymers; Shibaev, V.P., Lam, L., Eds.; Springer: New York, NY, USA, 1994; pp. 231–257. [Google Scholar]
- Kroto, H.W. Symmetry, space, stars and C60. Rev. Mod. Phys. 1997, 69, 703–722. [Google Scholar] [CrossRef]
- Smalley, R.E. Discovering the fullerenes. Rev. Mod. Phys. 1997, 69, 723–730. [Google Scholar] [CrossRef]
- Ball, P. The perfect nanotube. Nature 1996, 382, 207–208. [Google Scholar] [CrossRef]
- Fukunaga, A.; Chu, S.Y.; McHenry, M.E. Synthesis, structure, and superconducting properties of tantalum carbide nanorods and nanoparticles. J. Mater. Res. 1998, 13, 2465–2471. [Google Scholar] [CrossRef]
- Serp, P.; Figueiredo, J.L. (Eds.) Carbon Materials for Catalysis; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Gullapalli, S.; Wong, M.S. Nanotechnology: A guide to nano-objects. Chem. Eng. Prog. 2011, 107, 28–32. [Google Scholar]
- Srivastava, D.; Menon, M.; Cho, K. Computational nanotechnology with carbon nanotubes and fullerenes. Comput. Sci. Eng. 2001, 3, 42–55. [Google Scholar] [CrossRef]
- Harris, P.J.F. Carbon Nanotubes and Related Structures; Cambridge University Press: Cambridge, UK, 1999. [Google Scholar]
- Trogadas, P.; Fuller, T.F.; Strasser, P. Carbon as catalyst and support for electrochemical energy conversion. Carbon 2014, 75, 5–42. [Google Scholar] [CrossRef]
- Yang, N.J.; Swain, J.M.; Jiang, X. Nanocarbon electrochemistry and electroanalysis: Current status and future perspectives. Electroanal 2016, 28, 27–34. [Google Scholar] [CrossRef]
- Mao, X.W.; Rutledge, G.C.; Hatton, T.A. Nanocarbon-based electrochemical systems for sensing, electrocatalysis, and energy storage. Nano Today 2014, 9, 405–432. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. The role of nanostructure in improving the performance of electrodes for energy storage and conversion. Eur. J. Inorg. Chem. 2009, 26, 3851–3878. [Google Scholar] [CrossRef]
- Rios, G.; Centi, G.; Kanellopoulos, N. (Eds.) Nanoporous Materials for Energy and the Environment; Pan Stansford Pub: Singapure, 2012. [Google Scholar]
- Wang, Y.; Shao, Y.Y.; Matson, D.W.; Li, J.H.; Lin, Y.H. Nitrogen-doped graphene and its application in electrochemical biosensing. ACS Nano 2010, 4, 1790–1798. [Google Scholar] [CrossRef]
- Wilddgoose, G.G.; Banks, C.E.; Leventis, H.C.; Compton, R.G. Chemically modified carbon nanotubes for use in electroanalysis. Microchim. Acta 2006, 152, 187–214. [Google Scholar] [CrossRef]
- Yang, W.R.; Ratinac, K.R.; Ringer, S.P.; Thordarson, P.; Gooding, J.J.; Braet, E. Carbon nanomaterials in biosensors: Should you use nanotubes or graphene? Angew. Chem. Int. Edit. 2010, 49, 2114–2138. [Google Scholar] [CrossRef]
- Wu, S.X.; He, Q.Y.; Tan, C.L.; Wang, Y.D.; Zhang, H. Graphene-based electrochemical sensors. Small 2013, 9, 1160–1172. [Google Scholar] [CrossRef]
- Fernandes, D.M.; Freire, C. Carbon nanomaterial–phosphomolybdate composites for oxidative electrocatalysis. ChemElectroChem 2015, 2, 269–279. [Google Scholar] [CrossRef]
- Liang, Y.Y.; Li, Y.G.; Wang, H.L.; Dai, H.J. Strongly coupled inorganic/nanocarbon hybrid materials for advanced electrocatalysis. J. Am. Chem. Soc. 2013, 135, 2013–2036. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L. The Nature of the Chemical Bond, 3rd ed.; Cornell University Press: Ithaca, NY, USA, 1960. [Google Scholar]
- Honda, H. Carbonaceous mesophase: History and prospects. Carbon 1988, 26, 139–156. [Google Scholar] [CrossRef]
- Lewis, I.C.; Lewis, R.T. “Carbonaceous mesophase: History and prospects”—A reply. Carbon 1988, 26, 757–758. [Google Scholar] [CrossRef]
- Watt, W.; Perov, B.V. Strong Fibres; Elsevier Science Ltd.: Amsterdam, The Netherlands, 1985. [Google Scholar]
- Hirsch, P.B. X-ray scattering from coals. Proc. Roy. Soc. 1954, A226, 143–169. [Google Scholar]
- Bennett, S.C.; Johnson, D.J.; Johnson, W. Strength-structure relationships in PAN-based carbon fibres. J. Mater. Sci. 1983, 18, 3337–3347. [Google Scholar] [CrossRef]
- Feldman, D. Carbon fibers, by JP Donnet and RC Bansal, Marcel Dekker, New York, 1984, 291 pp. No price given. J. Polym. Sci. Polym. Lett. Ed. 1985. [Google Scholar] [CrossRef]
- Donnet, J.B.; Voet, A. Carbon Black; Marcel Dekker: New York, NY, USA, 1976. [Google Scholar]
- Honda, H.; Yamada, Y. Meso-carbon microbeads. J. Jpn. Petrol. Inst. 1973, 16, 392–397. [Google Scholar]
- Auguie, D.; Oberlin, M.; Oberlin, A.; Hyvermat, P. Microtexture of mesophase spheres as studied by high resolution conventional transmission electron microscopy (CTEM). Carbon 1980, 18, 337–346. [Google Scholar] [CrossRef]
- Kodama, M.; Esumi, K.; Maguro, K.; Honda, H. Adsorption of human serum globulin on mesocarbon microbeads. Carbon 1988, 26, 777–783. [Google Scholar] [CrossRef]
- Messier, R.; Spear, K.E.; Badzian, A.R.; Roy, R. The quest for diamond coatings. J. Mater. 1987, 39, 8–11. [Google Scholar] [CrossRef]
- Mantell, C.L. Carbon and Graphite Handbook; Interscience: New York, NY, USA, 1968. [Google Scholar]
- Mckee, D.W.; Spiro, C.L.; Lamby, E.J. The effects of boron additives on the oxidation behavior of carbons. Carbon 1984, 22, 507–511. [Google Scholar] [CrossRef]
- Long, F.J.; Sykes, K.W. The catalysis of the oxidation of carbon. J. Chim. Phys. 1950, 47, 361–378. [Google Scholar] [CrossRef]
- Amariglio, H.; Duval, X. Etude de la combustion catalytique du graphite. Carbon 1966, 4, 323–332. [Google Scholar] [CrossRef]
- Moulijn, J. Advanced study institute on carbon and coal gasification science and technology. Fuel 1985, 64, 1477–1478. [Google Scholar] [CrossRef]
- Cerfontain, M.B.; Agalianos, D.; Moulijn, J.A. CO2 step-response experiments during alkali catalyzed carbon gasification; evaluation of the so-called CO overshoot. Carbon 1987, 25, 351–359. [Google Scholar] [CrossRef]
- Cerfontain, M.B.; Kapteijn, P.; Moulijn, J.A. Characterization of alkali carbonate catalysts for carbon gasification with 18O labeled CO2. Carbon 1988, 26, 41–48. [Google Scholar] [CrossRef]
- Baker, R.T.K. Metal Catalysed Gasification of Graphites. In Carbon and Coal Gasification; Figueiredo, J., Moulijn, J.A., Eds.; Proc. NATO ASI: Alvor, Portugal, 1986; pp. 231–268. [Google Scholar]
- Jones, L.A. Catalysis of Carbon by Sodium. Ph.D. Thesis, University of Newcastle upon Tyne, Newcastle upon Tyne, UK, 1988. [Google Scholar]
- Ertl, G.; Knozinger, H.; Schuth, F.; Weitkamp, J. (Eds.) Infrared spectroscopy for the characterization of surface acidity and basicity. In Handbook of Heterogeneous Catalysis; Wiley-VCH Verlag: Weinheim, Germnay, 2008. [Google Scholar]
- Abu-Lebdeh, Y.; Davidson, I. (Eds.) Nanotechnology for Lithium-ion Batteries; Springer: New York, NY, USA, 2013. [Google Scholar]
- Ramsden, J. Applied Nanotechnology: The Conversion of Research Results to Products; Elsevier: London, UK, 2013. [Google Scholar]
- Luttge, R. Microfabrication for Industrial Applications; Elsevier: London, UK, 2011. [Google Scholar]
- Asmatulu, R.; Khan, W.S. (Eds.) Nanotechnology Safety; Elsevier: Oxford, UK, 2013. [Google Scholar]
- Alkhazov, T.G.; Lisovskii, A.E.; Gulakhmedova, T.K. Oxidative dehydrogenation of ethylbenzene over a charcoal catalyst. React. Kinet. Catal. Lett. 1979, 12, 189–193. [Google Scholar] [CrossRef]
- Byung, H.H.; Dae, H.S.; Sung, Y.C. Graphite catalyzed reduction of aromatic and aliphatic nitro compounds with hydrazine hydrate. Tetrahedron Lett. 1985, 26, 6233–6234. [Google Scholar] [CrossRef]
- Iijima, S. Helical microtubules of graphitic carbon. Nature 1991, 354, 56–58. [Google Scholar] [CrossRef]
- Kroto, H.W.; Heath, J.R.; O’Brien, S.C.; Curl, R.F.; Smalley, R.E. C60: Buckminsterfullerene. Nature 1985, 318, 162–163. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Blume, R.; Zhang, A.; Schlogl, R.; Su, D.S. Surface-modified carbon nanotubes catalyze oxidative dehydrogenation of n-butane. Science 2008, 322, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Frank, B.; Blume, R.; Rinaldi, A.; Trunschke, A.; Schlogl, R. Oxygen Insertion Catalysis by sp2 Carbon. Angew. Chem. Int. Ed. 2011, 50, 10226–10230. [Google Scholar] [CrossRef]
- Li, B.; Xu, Z. A nonmetal catalyst for molecular hydrogen activation with comparable catalytic hydrogenation capability to noble metal catalyst. J. Am. Chem. Soc. 2009, 131, 16380–16382. [Google Scholar] [CrossRef]
- Dreyer, D.R.; Jia, H.P.; Bielowski, C.W. Graphene oxide: A convenient carbocatalyst for facilitating oxidation and hydration reactions. Angew. Chem. Int. Ed. 2010, 49, 6813–6816. [Google Scholar] [CrossRef]
- Su, D.S.; Perathoner, S.; Centi, G. Nanocarbons for the development of advanced catalysts. Chem. Rev. 2013, 113, 5782–5816. [Google Scholar] [CrossRef] [PubMed]
- Sattler, K.D. (Ed.) Carbon Nanomaterials Sourcebook; CRC Press: Boca Raton, FL, USA, 2016; Volume II. [Google Scholar]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Norskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Santos, D.M.F.; Sequeira, C.A.C.; Figueiredo, J.L. Hydrogen production by alkaline water electrolysis. Química Nova 2013, 36, 1176–1193. [Google Scholar] [CrossRef]
- Sequeira, C.A.C.; Santos, D.M.F. Electrochemical routes for industrial synthesis. J. Braz. Chem. Soc. 2009, 20, 387–406. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X.; Chen, L.; Chen, C.; Wang, Q.; Sequeira, C.A.C. Electrochemical properties of rare-earth based hydrogen storage alloy for replacing Pt as the anode electrocatalyst in AFC. J. Alloys Compd. 2006, 421, 223–227. [Google Scholar] [CrossRef]
- Sequeira, C.A.C.; Santos, D.M.F. Electrochemical behaviour of oxygen reduction on polymer carbon electrodes in alkaline media. Russ. J. Electrochem. 2012, 48, 727–737. [Google Scholar] [CrossRef]
- Gonçalves, M.R.; Gomes, A.; Condeço, J.; Fernandes, T.R.C.; Pardal, T.; Sequeira, C.A.C.; Branco, J.B. Electrochemical conversion of CO2 to C2 hydrocarbons using different ex situ copper electrodeposits. Electrochim. Acta 2013, 102, 388–392. [Google Scholar] [CrossRef]
- Gonçalves, M.R.; Gomes, A.; Condeço, J.; Fernandes, T.R.C.; Pardal, T.; Sequeira, C.A.C.; Branco, J.B. Conversion of carbon dioxide into fuel by electrochemical reduction in aqueous solvents. Energy Convers. Mang. 2010, 51, 30–32. [Google Scholar] [CrossRef]
- van der Ham, C.J.M.; Koper, M.T.M.; Hetterschid, D.G.H. Challenges in reduction of dinitrogen by proton and electron transfer. Chem. Soc. Rev. 2014, 43, 5183–5191. [Google Scholar] [CrossRef]
- Bench, J.D.; Hellstern, T.R.; Kibsgaard, J.; Chakthranont, P.; Jaramillo, T.F. Catalyzing the hydrogen evolution reaction (HER) with molybdenum sulfide nanomaterials. ACS Catal. 2014, 4, 3957–3971. [Google Scholar] [CrossRef]
- Doyle, A.D.; Montoya, J.H.; Vojvodic, A. Improving oxygen electrochemistry through nanoscopic confinement. Chemcatchem 2015, 7, 738–742. [Google Scholar] [CrossRef]
- Peterson, A.A.; Norskov, J.K. Activity Descriptors for CO2 Electroreduction to Methane on Transition-Metal Catalysts. J. Phys. Chem. Lett. 2012, 3, 251–258. [Google Scholar] [CrossRef]
- Hong, X.; Chan, K.; Tsai, C.; Norskov, J.K. How Doped MoS2 Breaks Transition-Metal Scaling Relations for CO2 Electrochemical Reduction. ACS Catal. 2016, 6, 4428–4437. [Google Scholar] [CrossRef]
- Halck, N.B.; Petrykin, V.; Krtil, P.; Rossmeisl, J. Beyond the volcano limitations in electrocatalysis–oxygen evolution reaction. Phys. Chem. Chem. Phys. 2014, 16, 13682–13688. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Barisik, M. Size dependent surface charge properties of silica nano-channels: Double layer overlap and inlet/ outlet effects. Phys. Chem. Chem. Phys. 2018, 20, 16719–16728. [Google Scholar] [CrossRef]
- Spohre, E. Molecular simulation of the electrochemical double layer. Electrochim. Acta 1999, 44, 1697–1705. [Google Scholar] [CrossRef]
- Mitropoulos, A.C. The kelvin equation. J. Colloid. Interface Sci. 2008, 317, 643–648. [Google Scholar] [CrossRef]
- Sheng, W.H. Superheating and melting-point depression of Pb nanoparticles embedded in Al matrices. Phil. Mag. Lett. 1996, 73, 179–186. [Google Scholar] [CrossRef]
- Schafer, R. Melting of isolated tin nanoparticles. Phys. Rev. Lett. 2000, 85, 1250–1253. [Google Scholar]
- Barnard, A.S.; Zapol, P. A model for the phase stability of arbitrary nanoparticles as a function of size and shape. J. Chem. Phys. 2004, 121, 4276–4283. [Google Scholar] [CrossRef]
- Ram, S. Self-confined dimension of thermodynamic stability in Co-nanoparticles in fcc and bcc allotropes with a thin amorphous Al2O3 surface layer. Acta Mater. 2001, 49, 2297–2307. [Google Scholar] [CrossRef]
- Zhang, H.; Huang, F.; Gilbert, B.; Banfield, J.F. Molecular dynamics simulations, thermodynamic analysis, and experimental study of phase stability of zinc sulfide nanoparticles. J. Phys. Chem. B 2003, 107, 13051–13060. [Google Scholar] [CrossRef]
- Chon, C.H.; Kihm, K.D.; Lu, S.P.; Choi, S.U.S. Comment on “Electric-field effect on carbon nanotubes in a twisted nematic liquid crystal cell”. Appl. Phys. Lett. 2005, 87, 263110. [Google Scholar]
- Xie, H.; Wang, Y.; Xi, T.; Liu, Y.; Ai, F.; Wu, Q. Thermal conductivity enhancement of suspensions containing nanosized alumina particles. J. Appl. Phys. 2002, 91, 4568–4572. [Google Scholar] [CrossRef]
- Zhang, H.; Penn, R.L.; Harners, R.J.; Banfield, J.F. Enhanced adsorption of molecules on surfaces of nanocrystalline particles. J. Phys. Chem. B 1999, 103, 4656–4662. [Google Scholar] [CrossRef]
- Shao, M.-H.; Sasaki, K.; Adzic, R.R. Pd− Fe nanoparticles as electrocatalysts for oxygen reduction. J. Am. Chem. Soc. 2006, 128, 3526–3527. [Google Scholar] [CrossRef]
- Greely, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, J.; Norskov, J.K. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef]
- Conway, B.E.; Jerkiewicz, G. Relation of energies and coverages of underpotential and overpotential deposited H at Pt and other metals to the ’volcano curve’for cathodic H2 evolution kinetics. Electrochim. Acta 2000, 45, 4075–4083. [Google Scholar] [CrossRef]
- Hoolbaek, B.; Janssens, T.V.W.; Clausen, B.S.; Falsig, H.; Christensen, C.H.; Norskov, J.K. Catalytic activity of Au nanoparticles. Nanotoday 2007, 2, 14–18. [Google Scholar] [CrossRef]
- Corma, A.; Leyva-Perez, A.; Sabater, M.J. Gold-catalyzed carbon− heteroatom bond-forming reactions. Chem. Rev. 2011, 111, 1657–1712. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.P.; Seipenbusch, M.; Kasper, G. Size effects in the catalytic activity of unsupported metallic nanoparticles. J. Nanopart. Res. 2003, 5, 293–298. [Google Scholar] [CrossRef]
- Schultze, J.W.; Koppitz, F.D. Bond formation in electrosorbates—I correlation between the electrosorption valency and pauling’s electronegativity for aqueous solutions. Electrochim. Acta 1976, 21, 327–336. [Google Scholar] [CrossRef]
- Ying, T.-Y.; Yang, K.-L.; Yacoumi, S.; Tsouris, C. Electrosorption of ions from aqueous solutions by nanostructured carbon aerogel. J. Colloid Interface Sci. 2002, 250, 18–27. [Google Scholar] [CrossRef]
- Thiele, E.W. Relation between catalytic activity and size of particle. Ind. Eng. Chem. 1939, 31, 916–920. [Google Scholar] [CrossRef]
- Lopez, N.; Janssens, T.V.W.; Clausen, B.S.; Xu, Y.; Mavrikakis, M.; Blijaard, T.; Norskov, J.K. On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation. J. Catal. 2004, 223, 232–235. [Google Scholar] [CrossRef]
- Sequeira, C.A.C.; Santos, D.M.F. Mass transfer to microelectrodes and arrays. Z. Phys. Chem. 2010, 224, 1297–1336. [Google Scholar] [CrossRef]
- Stulfk, K.; Amatore, C.; Holub, K.; Marecek, V.; Kutner, W. Microelectrodes. Definitions, characterization, and applications. Pure Appl. Chem. 2000, 72, 1483–1492. [Google Scholar] [CrossRef]
- Madsen, D.N.; Molhave, K.; Mateia, R.; Rasmussen, A.M.; Brorson, M.; Jacobsen, C.Y.H.; Boggild, P. Soldering of nanotubes onto microelectrodes. Nano Lett. 2003, 3, 47–49. [Google Scholar] [CrossRef]
- Zoski, C.G. Ultramicroelectrodes: Design, fabrication, and characterization. Electroanalysis 2002, 14, 1041–1051. [Google Scholar] [CrossRef]
- Feenay, R.; Kounaves, S.P. Microfabricated ultramicroelectrode arrays: Developments, advances, and applications in environmental analysis. Electroanalysis 2000, 12, 677–684. [Google Scholar] [CrossRef]
- Wang, J. Study of electrode reactions and interfacial properties. In Analytical Electrochemistry; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- Chae, H.K.; Siberio-Pérez, D.Y.; Kim, J.; Go, Y.; Eddaoudi, M.; Matzger, A.J.; Keeffe, M.O.; Yaghi, O.M. A route to high surface area, porosity and inclusion of large molecules in crystals. Nature 2004, 427, 523–527. [Google Scholar] [CrossRef]
- Siegel, R.W.; Hu, E.; Cox, D.M.; Goronkin, H.; Jelinski, L.; Koch, C.C.; Mendel, J.; Roco, M.C.; Shaw, D.T. Nanostructure Science and Technology. A Worldwide Study, The Interagency Working Group on NanoScience, Engineering and Technology; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 1999; p. 116. [Google Scholar]
- Sinha, P.; Datar, A.; Jeong, C.; Deng, X.; Chung, Y.G.; Lin, L.-C. Surface area determination of porous materials using the BET method: Limitations and improvements. J. Phys. Chem. C 2019, 123, 20195–20209. [Google Scholar] [CrossRef]
- Schmidt, G. Clusters and Colloids: From Theory to Applications; Wiley-VCH: Weinheim, NY, USA, 1994. [Google Scholar]
- Kreibig, U.; Vollmer, M. Optical Properties of Metal Clusters. In Architectured Materials in Nature and Engineering; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 1995; p. 119. [Google Scholar]
- Feldheim, D.L.; Colby, A.F., Jr. (Eds.) Metal Nanoparticles-Synthesis, Characterization and Applications; Marcel Dekker: New York, NY, USA, 2002. [Google Scholar]
- Pellizzetti, G. (Ed.) Fine Particles Sciences and Technology–From Micro-to New Particles; Kluwer: Dordecht, The Netherlands, 1996. [Google Scholar]
- Fendler, J.H.; Tian, Y. Nanoparticles and Nanostructured Films: Current Accomplishments and Future Prospects. In Nanoparticles and Nanostructured Films; Wiley: Hoboken, NJ, USA, 2007; pp. 429–461. [Google Scholar]
- Thrower, P.A.; Radovic, L.R. (Eds.) Chemistry and Physics of Carbon; Moral Dekker, Inc.: New York, NY, USA, 1999. [Google Scholar]
- Paucirova, M.M. Malinovsky, and K. Matiasovsky. Rev. Roum. Chim. 1972, 17, 807–817. [Google Scholar]
- Sato, V.; Kojima, A.; Ejima, T. Density and electrical conductivity of naf-alf3 binary melts. J. Jpn. Inst. Met. 1977, 41, 1249–1256. [Google Scholar] [CrossRef][Green Version]
- Grjotheim, K.; Krohn, C.; Malinovsky, M.; Matiasovsky, K.; Thonstad, J. Aluminium Electrolysis–The Chemistry of the Hall-Heroult Process; Aluminium Verlag GmbH: Dusseldorf, Germany, 1977. [Google Scholar]
- Gilbert, B.; Mamantov, G.; Begun, G.M. Raman spectra of Al2O3 solutions in molten cryolite and other aluminum fluoride containing melts. Nucl. Chem. Lett. 1976, 12, 415–424. [Google Scholar] [CrossRef]
- Heintz, E. Influence of coke structure on the properties of the carbon-graphite artefact. Fuel 1985, 64, 1192–1196. [Google Scholar] [CrossRef]
- Rastovean, A.; Ugarkovic, D.; Legin-kolar, M. Investigation of the change in nickel content in ideal and real carbon systems at temperatures up to 2400 °C. Metalurgija 1992, 31, 27–30. [Google Scholar]
- Mochida, I.; Korai, Y.; Ku, C.H.; Watanabe, F.; Sakai, Y. Chemistry of synthesis, structure, preparation and application of aromatic-derived mesophase pitch. Carbon 2000, 38, 305–328. [Google Scholar] [CrossRef]
- Radenovic, A.; Legin-kolar, M. Influence of Composition and Structure on Coal-tar Pitch Quality. Kem. Insdustriji 2005, 54, 425–428. [Google Scholar]
- Thonstad, J.; Hove, E. On the anodic overvoltage in aluminum electrolysis. Can. J. Chem. 1964, 42, 1542–1550. [Google Scholar] [CrossRef]
- Blyholder, G.; Eyring, H. Kinetics of graphite oxidation. J. Phys. Chem. 1957, 61, 682–688. [Google Scholar] [CrossRef]
- Bourke, A.; Miller, M.A.; Lynch, R.P.; Gao, X.; Landon, J.; Wainright, J.S.; Savinell, R.F.; Buckley, D.N. Electrode kinetics of vanadium flow batteries: Contrasting responses of V(II)-V(III) and V(IV)-V(V) to electrochemical pretreatment of carbon. J Electrochem. Soc. 2016, 163, A5097–A5105. [Google Scholar] [CrossRef]
- Bourke, A.; Lynch, R.P.; Buckley, D.N. Effect of Pretreatment on the Rate of the VO2+/VO2+ and V2+/V3+ Reactions at a Carbon Electrode. ECS Trans. 2014, 61, 15–21. [Google Scholar] [CrossRef]
- Bourke, A.; Lynch, R.P.; Buckley, D.N. Effect of Electrode Pretreatment on the Cyclic Voltammetry of VO2+/VO2+ at a Glassy Carbon Electrode. ECS Trans. 2013, 53, 59–64. [Google Scholar] [CrossRef]
- di Blasi, A.; di Blasi, O.; Briguglio, N.; Arico, A.S.; Sebastián, D.; Lázaro, M.L.; Monforte, G.; Antonucci, V. Investigation of several graphite-based electrodes for vanadium redox flow cell. J. Power Sources 2013, 227, 15–23. [Google Scholar] [CrossRef]
- Li, W.; Liu, J.; Yan, C. Reduced graphene oxide with tunable C/O ratio and its activity towards vanadium redox pairs for an all vanadium redox flow battery. Carbon 2013, 55, 313–320. [Google Scholar] [CrossRef]
- Zhang, W.; Xi, J.; Li, Z.; Zhou, H.; Liu, L.; Wu, Z.; Qiu, X. Electrochemical activation of graphite felt electrode for VO2+/VO2+ redox couple application. Electrochim. Acta 2013, 89, 429–435. [Google Scholar] [CrossRef]
- Xi, J.; Zhang, W.; Li, Z.; Zhou, H.; Liu, L.; Wu, Z. Effect of electro-oxidation current density on performance of graphite felt electrode for vanadium redox flow battery. Int. J. Electrochim. Sci. 2013, 8, 4700–4711. [Google Scholar]
- Men, Y.; Sun, T. Carbon felts electrode treated in different weak acid solutions through electrochemical oxidation method for all vanadium redox flow battery. Int. J. Electrochem. Sci. 2012, 7, 3482–3488. [Google Scholar]
- Li, X.G.; Huang, K.L.; Liu, S.Q.; Tan, N.; Chen, L.Q. Characteristics of graphite felt electrode electrochemically oxidized for vanadium redox battery application. Trans. Nonferrous Met. Soc. China 2007, 17, 195–199. [Google Scholar] [CrossRef]
- Yue, L.; Li, W.; Sun, F.; Zhao, L.; Xing, L. Highly hydroxylated carbon fibres as electrode materials of all-vanadium redox flow battery. Carbon 2010, 48, 3079–3090. [Google Scholar] [CrossRef]
- Sun, B.; Skyllas-Kazakos, M. Modification of graphite electrode materials for vanadium redox flow battery application—I I.: Acid treatments. Electrochim. Acta 1991, 37, 2459–2465. [Google Scholar] [CrossRef]
- Friedl, J.; Bauer, C.; Rinaldi, A.; Stimming, U. Electron transfer kinetics of the VO2+/VO2+–Reaction on multi-walled carbon nanotubes. Carbon 2013, 63, 228–239. [Google Scholar] [CrossRef]
- Sun, B.; Skyllas-Kazakos, M. Modification of graphite electrode materials for vanadium redox flow battery application—I: Thermal treatment. Electrochim. Acta 1991, 37, 1253–1260. [Google Scholar] [CrossRef]
- Gattrel, M.; Qian, J.; Stewart, C.; Graham, P.; MacDougall, B. The electrochemical reduction of VO2+ in acidic solution at high overpotentials. Electrochim. Acta 2005, 51, 395–407. [Google Scholar] [CrossRef]
- Gattrell, M.; Park, J.; MacDougall, B.; Apte, J.; McCarthy, S.; Wu, C.W. Study of the mechanism of the vanadium 4+/5+ redox reaction in acidic solutions. J. Electrochem. Soc. 2004, 151, A123–A130. [Google Scholar] [CrossRef]
- Kaneko, M.; Nozaki, K.; Wada, Y.; Aoki, T.; Negishi, A.; Kamimoto, M. Vanadium redox reactions and carbon electrodes for vanadium redox flow battery. Electrochim. Acta 1991, 36, 1191–1196. [Google Scholar] [CrossRef]
- Parks, G.; Morina, M. Alma Mater. World Lit. Today 2001, 75, 163. [Google Scholar] [CrossRef]
- Li, W.; Liu, J.; Yan, C. The electrochemical catalytic activity of single-walled carbon nanotubes towards VO2+/VO2+ and V3+/V2+ redox pairs for an all vanadium redox flow battery. Electrochim. Acta 2012, 79, 102–108. [Google Scholar] [CrossRef]
- Zhong, S.; Skyllas-Mazacos, M. Electrochemical behaviour of vanadium (V)/vanadium (IV) redox couple at graphite electrodes. J. Power Sources 1992, 39, 1–9. [Google Scholar] [CrossRef]
- Thorogood, C.A.; Wildgoose, G.G.; Jones, J.H.; Compton, R.G. Identifying quinone-like species on the surface of graphitic carbon and multi-walled carbon nanotubes using reactions with 2,4-dinitrophenylhydrazine to provide a voltammetric fingerprint. New J. Chem. 2007, 31, 958–965. [Google Scholar] [CrossRef]
- Quill, N.; Lynch, R.P.; Gao, X.; Buckley, D.N. The Electrochemical Society Meeting Abstract. MA 2014-01 2014, 1, 389. [Google Scholar]
- Flox, C.; Rubio-Garcia, J.; Skoumal, M.; Andrew, T.; Morante, J.R. Thermo–chemical treatments based on NH3/O2 for improved graphite-based fiber electrodes in vanadium redox flow batteries. Carbon 2013, 60, 280–288. [Google Scholar] [CrossRef]
- Yamamura, X.W.W.T.; Ohta, S.; Zhang, Q.X.; Lu, F.C.; Liu, C.M.; Shirasaki, K.; Satoh, I.; Shikama, T.; Lu, D.; Liu, S.Q. Acceleration of the redox kinetics of VO2+/VO2+ and V3+/V2+ couples on carbon paper. J.Appl. Electrochem. 2011, 41, 1183–1190. [Google Scholar]
- Yamamura, T.; Watanabe, N.; Yano, T.; Shiokawa, Y. Electron-transfer kinetics of Np3+/Np4+, NpO2+/NpO2 2+, V2+/V3+, and VO2+/VO2+ at carbon electrodes. J. Electrochem. Soc. 2005, 152, A830–A836. [Google Scholar] [CrossRef]
- Sum, E.; Skyllas-Kazacos, M. A study of the V (II)/V (III) redox couple for redox flow cell applications. J. Power Sources 1985, 15, 179–190. [Google Scholar] [CrossRef]
- Lin, A.Y.; Gridley, G.; Tucker, M. Benign Anal Lesions and Anal Cancer. N. Engl. J. Med. 1995, 332, 190–191. [Google Scholar] [CrossRef]
- Origi, G.; Katayama, Y.; Miura, T. Investigations on V (IV)/V (V) and V (II)/V (III) redox reactions by various electrochemical methods. J. Power Sources 2005, 139, 321–324. [Google Scholar] [CrossRef]
- Sum, E.; Rychaik, M.; Skyllas-Kozacos, M. Investigation of the V (V)/V (IV) system for use in the positive half-cell of a redox battery. J. Power Sources 1985, 16, 85–95. [Google Scholar] [CrossRef]
- Aaron, D.; Sun, C.-N.; Bright, M.; Papandrew, A.B.; Mench, M.M.; Zawodzinski, T.A. In situ kinetics studies in all-vanadium redox flow batteries. ECS Electrochem. Lett. 2013, 2, A29–A31. [Google Scholar] [CrossRef]
- Sun, C.-N.; Delnick, F.M.; Aron, D.S.; Papandrew, A.B.; Mench, M.M.; Zawodzinski, T.A. Probing electrode losses in all-vanadium redox flow batteries with impedance spectroscopy. ECS Electrochem. Lett. 2013, 2, A43–A45. [Google Scholar] [CrossRef]
- Lee, J.W.; Hong, J.K.; Kjeang, E. Electrochemical characteristics of vanadium redox reactions on porous carbon electrodes for microfluidic fuel cell applications. Electrochim. Acta 2012, 83, 430–438. [Google Scholar] [CrossRef]
- Kinoshita, K. Carbon: Electrochemical and Physicochemical Properties; Wiley: New York, NY, USA, 1988. [Google Scholar]
- Engstrom, R.C. Electrochemical pretreatment of glassy carbon electrodes. Anal. Chem. 1982, 54, 2310–2314. [Google Scholar] [CrossRef]
- Kneten, K.R.; McCreery, R.L. Effects of redox system structure on electron-transfer kinetics at ordered graphite and glassy carbon electrodes. Anal. Chem. 1992, 64, 2518–2524. [Google Scholar] [CrossRef]
- Zoski, C.G. (Ed.) Handbook of Electrochemistry; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Cardoso, J.A.S.B.; Cardoso, D.S.P.; Amaral, L.; Metim, O.; Sevim, M.; Sener, T.; Sequeira, C.A.C.; Santos, D.M.F. Reduced graphene oxide assembled Pd-based nanoalloys for hydrogen evolution reaction. Int. J. Hydrogen Energy 2017, 42, 3916–3925. [Google Scholar] [CrossRef]
- Sliukié, B.; Santos, D.M.F.; Vujkovié, M.; Amaral, L.; Rocha, R.P.; Sequeira, C.A.C.; Figueiredo, J.L. Molybdenum Carbide Nanoparticles on Carbon Nanotubes and Carbon Xerogel: Low-Cost Cathodes for Hydrogen Production by Alkaline Water Electrolysis. ChemSusChem 2016, 9, 1200–1208. [Google Scholar]
- Sljukié, B.; Vujkovié, M.; Amaral, L.; Santos, D.M.F.; Rocha, R.P.; Sequeira, C.A.C.; Figueiredo, J.L. Carbon-supported Mo2C electrocatalysts for hydrogen evolution reaction. J. Mater. Chem. 2015, A3, 15505–15512. [Google Scholar] [CrossRef]
- Dresselhaus, M.S.; Thomas, I.L. Alternative energy technologies. Nature 2001, 414, 332–337. [Google Scholar] [CrossRef]
- Serramedan, D.; Marc, F.; Pereyre, M.; Filliatre, C.; Chabardes, P.; Delmond, B. Delta-pyronene: Epoxydation se’lective et acces aux cyclocitrals. Tetrahedron Lett. 1992, 33, 4457–4460. [Google Scholar] [CrossRef]
- Walter, M.G.; Warren, E.L.; McKone, J.R.; Boettcher, S.W.; Mi, Q.; Santori, E.A.; Lewis, N.S. Solar water splitting cells. Chem. Rev. 2010, 110, 6446–6473. [Google Scholar] [CrossRef] [PubMed]
- Conway, B.E.; Tilak, B.V. Interfacial processes involving electrocatalytic evolution and oxidation of H2, and the role of chemisorbed H. Electrochim. Acta 2002, 47, 3571–3594. [Google Scholar] [CrossRef]
- Jiao, Y.; Zheng, Y.; Jaronier, M.T.; Qiao, S.Z. Design of electrocatalysts for oxygen-and hydrogen-involving energy conversion reactions. Chem. Soc. Rev. 2015, 44, 2060–2086. [Google Scholar] [CrossRef]
- Parsons, R. The rate of electrolytic hydrogen evolution and the heat of adsorption of hydrogen. Trans. Faraday Soc. 1958, 54, 1053–1063. [Google Scholar] [CrossRef]
- Norskov, J.K.; Bligaard, T.; Logadóttir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U.; Kitchin, J.R. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 2005, 152, J23–J26. [Google Scholar] [CrossRef]
- Greely, J.; Mavrikakis, M. Alloy catalysts designed from first principles. Nat. Mater. 2004, 3, 810–815. [Google Scholar] [CrossRef]
- Quaino, P.; Juarez, F.; Santos, E.; Schmickler, W. Volcano plots in in hydrogen electrocatalysis—uses and abuses. Beilstein J. Nanotechnol. 2014, 5, 846–854. [Google Scholar] [CrossRef]
- Yang, T.-L.; Ni, S.-F.; Qin, P.; Dang, L. A mechanism study on the hydrogen evolution reaction catalyzed by molybdenum disulfide complexes. Chem. Commun. 2018, 54, 1113–1116. [Google Scholar] [CrossRef]
- Liu, G.; Wang, Z.; Zhang, L.Z.Y.; Feng, Y.; Yang, S.; Jia, Y.; Wang, S.; Zhang, C.; Yang, J. Hydrogen evolution reactions boosted by bridge bonds between electrocatalysts and electrodes. Nanoscale 2018, 10, 4068–4076. [Google Scholar] [CrossRef]
- Li, K.; Li, Y.; Wang, Y.; Ge, J.; Liu, C.; Xing, W. Enhanced electrocatalytic performance for the hydrogen evolution reaction through surface enrichment of platinum nanoclusters alloying with ruthenium in situ embedded in carbon. Energy Environ. Sci. 2018, 11, 1232–1239. [Google Scholar] [CrossRef]
- Ojha, K.; Saha, S.; Dagar, P.; Ganguli, A.K. Nanocatalysts for hydrogen evolution reactions. Phys. Chem. Chem. Phys. 2018, 20, 6777–6799. [Google Scholar] [CrossRef]
- Sarkar, S.; Peter, S.C. An overview on Pd-based electrocatalysts for the hydrogen evolution reaction. Inorg. Chem. Front. 2018, 5, 2060–2080. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Sequeira, C.A.C.; Macció, D.; Saccone, A.; Figueiredo, J.L. Platinum—rare earth electrodes for hydrogen evolution in alkaline water electrolysis. Int. J. Hydrogen Energy 2013, 38, 3137–3145. [Google Scholar] [CrossRef]
- Luo, W.; Gan, J.; Huang, Z.; Chen, W.; Qian, G.; Zhou, X.; Duan, X. Boosting HER performance of Pt-based catalysts immobilized on functionalized Vulcan carbon by atomic layer deposition. Front. Mater. 2019, 6, 251. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Amaral, L.; Sljukić, B.; Macció, D.; Saccone, A.; Sequeira, C.A.C. Electrocatalytic activity of nickel-cerium alloys for hydrogen evolution in alkaline water electrolysis. J. Electrochem. Soc. 2014, 161, F386–F390. [Google Scholar] [CrossRef]
- Oliveira, R.C.P.; Sevim, M.; Sljukic, B.; Sequeira, C.A.C.; Metin, O.; Santos, D.M.F. Mesoporous graphitic carbon nitride—supported binary MPt (M: Co, Ni, Cu) nanoalloys as electrocatalysts for borohydride oxidation and hydrogen evolution reaction. Catal. Today 2020, 357, 291–301. [Google Scholar] [CrossRef]
- Wang, J.; Kong, H.; Zhang, J.; Hao, Y.; Shao, Z.; Ciucci, F. Carbon based electrocatalysts for sustainable energy applications. Prog. Mater. Sci. 2020. [Google Scholar] [CrossRef]
- Cardoso, D.S.P.; Amaral, L.; Santos, D.M.F.; Sljukić, B.; Sequeira, C.A.C.; Macció, D.; Saccone, A. Enhancement of hydrogen evolution in alkaline water electrolysis by using nickel-rare earth alloys. Int. J. Hydrogen Energy 2015, 40, 4295–4302. [Google Scholar] [CrossRef]
- Cardoso, D.S.P.; Eugénio, S.; Silva, T.M.; Santos, D.M.F.; Sequeira, C.A.C.; Montemor, M.F. Hydrogen evolution on nanostructured Ni–Cu foams. RSC Adv. 2015, 5, 43456–43461. [Google Scholar] [CrossRef]
- Danilovic, N.; Subbaraman, R.; Strmcnik, D.; Kovic, V.R.S.; Markovic, N.M. Electrocatalysis of the HER in acid and alkaline media. J. Serb. Chem. Soc. 2013, 78, 2007–2015. [Google Scholar] [CrossRef]
- Huang, Y.; Nielsen, R.J.; Goddard, W.A.; Soriaga, M.P. The Reaction Mechanism with Free Energy Barriers for Electrochemical Dihydrogen Evolution on MoS2. J. Am. Chem. Soc. 2015, 137, 6692–6698. [Google Scholar] [CrossRef]
- Hinnemann, B.; Moses, P.G.; Bonde, J.; Jorgensen, K.P.; Nielsen, J.H.; Horch, S.; Chorkendorff, I.; Norskov, J.K. Biomimetic Hydrogen Evolution: MoS2 Nanoparticles as Catalyst for Hydrogen Evolution. J. Am. Chem. Soc. 2005, 127, 5308–5309. [Google Scholar] [CrossRef]
- Tributsch, H.; Bennett, J.C. Electrochemistry and photochemistry of MoS2 layer crystals. I. J. Electroanal. Chem. 1977, 81, 97–111. [Google Scholar] [CrossRef]
- Tsai, C.; Shan, K.R.; Norskov, J.K.; Abild-Pederson, F. Theoretical insights into the hydrogen evolution activity of layered transition metal dichalcogenides. Surf. Sci. 2015, 640, 133–140. [Google Scholar] [CrossRef]
- Jaramillo, T.F.; Jorgensen, K.P.; Bonde, J.; Nielsen, J.H.; Chorkendorff, I. Identification of active edge sites for electrochemical H2 evolution from MoS2 nanocatalysts. Science 2007, 317, 100–102. [Google Scholar] [CrossRef]
- Kibsgaard, J.; Chen, Z.; Reinecke, B.N.; Jaramillo, T.F. Engineering the surface structure of MoS2 to preferentially expose active edge sites for electrocatalysis. Nat. Mater. 2012, 11, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.B.; Cummins, D.; Reinecke, B.N.; Clark, E.; SunKara, M.K.; Jaramillo, T.F. Core–shell MoO3–MoS2 Nanowires for Hydrogen Evolution: A Functional Design for Electrocatalytic Materials. Nano Lett. 2011, 11, 4168–4175. [Google Scholar] [CrossRef]
- Kong, D.; Wang, H.; Cha, J.J.; Pasta, M.; Koski, K.J.; Yao, J.; Cui, Y. Synthesis of MoS2 and MoSe2 Films with Vertically Aligned Layers. Nano Lett. 2013, 13, 1341–1347. [Google Scholar] [CrossRef]
- Wang, H.; Lu, Z.; Xu, S.; Kong, D.; Cha, J.J.; Zheng, G.; Hsu, P.-C.; Yan, K.; Bradshaw, D.; Prinz, F.B.; et al. Electrochemical tuning of vertically aligned MoS2 nanofilms and its application in improving hydrogen evolution reaction. Proc. Natl. Acad. Sci. USA 2013, 110, 19701–19706. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Xie, L.; Liang, Y.; Hong, G.; Dai, H. MoS2 Nanoparticles Grown on Graphene: An Advanced Catalyst for the Hydrogen Evolution Reaction. J. Am. Chem. Soc. 2011, 133, 7296–7299. [Google Scholar] [CrossRef]
- Wang, H.; Cui, L.-F.; Yang, Y.; Casalonque, H.S.; Robinson, J.T.; Liang, Y.; Cui, Y.; Dai, H. Mn3O4−Graphene Hybrid as a High-Capacity Anode Material for Lithium Ion Batteries. J. Am. Chem. Soc. 2010, 132, 13978–13980. [Google Scholar] [CrossRef]
- Strmcnik, D.; Uchimura, M.; Wang, C.; Subbaraman, R.; Danilovic, N.; van der Vilet, D.; Paulikas, A.P.; Stamenkovic, V.R.; Markovic, N.M. Improving the hydrogen oxidation reaction rate by promotion of hydroxyl adsorption. Nat. Chem. 2013, 5, 300–306. [Google Scholar] [CrossRef]
- Sheng, C.; Gasteiger, H.A.; Shao-Horn, Y. Hydrogen oxidation and evolution reaction kinetics on platinum: Acid vs alkaline electrolytes. J. Electrochem. Soc. 2010, 157, B1529–B1536. [Google Scholar] [CrossRef]
- Skulason, E.; Tripkovic, V.; Bjorketun, M.E.; Gudmundsdóttir, S.; Karlberg, G.; Roesmeil, J.; Bligaard, T.; Jonsson, H.; Norskov, J.K. Modeling the electrochemical hydrogen oxidation and evolution reactions on the basis of density functional theory calculations. J. Phys. Chem. C 2010, 114, 18182–18197. [Google Scholar] [CrossRef]
- Zhang, L.; Xiao, J.; Wang, H.; Shao, M. Carbon-based electrocatalysts for hydrogen and oxygen evolution reactions. ACS Catal. 2017, 7, 7855–7865. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Choi, B.; Kim, Y.-B. Development of highly active bifunctional electrocatalyst using Co3O4. on carbon nanotubes for oxygen reduction and oxygen evolution. Sci. Rep. 2018, 8, 2543–2552. [Google Scholar] [CrossRef]
- Hu, C.; Dai, L. Oxygen evolution reaction at universal pHs. Angew Chem. Int. Ed. 2016, 55, 11736–11758. [Google Scholar] [CrossRef]
- Xuan, J.; Liu, Z. High-performance N-doped bifunctional carbon electrocatalysts derived from polymer waste for oxygen reduction and evolution reaction. Int. J. Electrochem. Sci. 2017, 12, 10471–10483. [Google Scholar] [CrossRef]
- Lei, Y.; Wei, L.; Zhai, S.; Wang, Y.; Karahan, H.E.; Chen, X.; Zhou, Z.; Wang, C.; Sui, X.; Chen, Y. Metal-free bifunctional carbon electrocatalysts derived from zeolitic imidazolate frameworks for efficient water splitting. Mater. Chem. Front. 2018, 2, 102–111. [Google Scholar] [CrossRef]
- Mantani, K.B.; Jain, D.; Co, A.C.; Ozkan, U.S. Investigation of coke quality variation between heat-recovery and byproduct Cokemaking technology. Energy Fuels 2017, 31, 2087–2094. [Google Scholar]
- Jia, N.; Weng, Q.; Shi, Y.; Shi, X.; Chen, X.; Chen, P.; An, Z.; Chen, Y. N-doped carbon nanocages: Bifunctional electrocatalysts for the oxygen reduction and evolution reactions. Nano Res. 2018, 11, 1905–1916. [Google Scholar] [CrossRef]
- Chi, J.; Yu, H.; Li, G.; Fu, L.; Jia, J.; Gao, X.; Yi, B.; Shao, Z. Nickel/cobalt oxide as a highly efficient OER electrocatalyst in an alkaline polymer electrolyte water electrolyzer. Rev. Adv. 2016, 6, 90397–90400. [Google Scholar] [CrossRef]
- Chen, S.; Duan, J.; Jaroniec, M.; Qiao, S.-Z. Nitrogen and oxygen dual-doped carbon hydrogel film as a substrate-free electrode for highly efficient oxygen evolution reaction. Adv. Mater. 2014, 26, 2925–2930. [Google Scholar] [CrossRef]
- Chen, S.; Duan, J.; Ran, J.; Qiao, S.-Z. Paper-Based N-Doped Carbon Films for Enhanced Oxygen Evolution Electrocatalysis. Adv. Sci. 2015, 2, 1400015. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B Environ. 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Markovic, N.M. Just a dream—Or future reality? Science 2009, 324, 48–49. [Google Scholar] [CrossRef] [PubMed]
- Siahrostami, S. Enabling direct H2O2 production through rational electrocatalyst design. Nat. Mater. 2013, 12, 1137–1143. [Google Scholar] [CrossRef]
- Norskov, J.K. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B. 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Sidik, R.A.; Anderson, A.B.; Subramanian, N.P.; Kumaraguru, S.P.; Popov, B.N. O2 Reduction on Graphite and Nitrogen-Doped Graphite: Experiment and Theory. J. Phys. Chem. B. 2006, 110, 1787–1793. [Google Scholar] [CrossRef]
- Wang, J.; Wu, Z.; Han, L.; Xuan, C.; Zhu, J.; Xiao, W.; Wu, J.; Xin, H.L.; Wang, D. A general approach for the direct fabrication of metal oxide-based electrocatalysts for efficient bifunctional oxygen electrodes. Sustain. Energy Fuels 2017, 1, 823–831. [Google Scholar] [CrossRef]
- Tao, G.; Zhang, L.; Chen, L.; Cui, X.; Hua, Z.; Wang, M.; Wang, J.; Chen, Y.; Shi, J. N-doped hierarchically macro/mesoporous carbon with excellent electrocatalytic activity and durability for oxygen reduction reaction. Carbon 2015, 86, 108–117. [Google Scholar] [CrossRef]
- Liu, Q.; Duan, Y.; Zhao, Q.; Pan, F.; Zhang, B.; Zhang, J. Direct synthesis of nitrogen-doped carbon nanosheets with high surface area and excellent oxygen reduction performance. Langmuir 2014, 30, 8238–8245. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H.; Liu, P.; Wang, Y.; Yang, H.; Li, Y.; Zhao, H. Self-supported bimodal-pore structured nitrogen-doped carbon fiber aerogel as electrocatalyst for oxygen reduction reaction. Electrochem. Commun. 2015, 51, 6–10. [Google Scholar] [CrossRef]
- Liu, F.; Peng, H.; You, C.; Fu, Z.; Huang, P.; Song, H.; Liao, S. High-performance doped carbon catalyst derived from nori biomass with melamine promoter. Electrochim. Acta 2014, 138, 353–359. [Google Scholar] [CrossRef]
- Pan, F.; Cao, Z.; Zhao, Q.; Liang, H.; Zhang, J. Nitrogen-doped porous carbon nanosheets made from biomass as highly active electrocatalyst for oxygen reduction reaction. J. Power Sources 2014, 272, 8–15. [Google Scholar] [CrossRef]
- Gao, S.; Chen, Y.; Fan, H.; Wei, X.; Hu, C.; Luo, H.; Qu, L. Large scale production of biomass-derived N-doped porous carbon spheres for oxygen reduction and supercapacitors. J. Mater. Chem. 2014, A2, 3317–3324. [Google Scholar] [CrossRef]
- Meng, Y.; Voiry, D.; Goswami, A.; Zou, X.; Huang, X.; Chhowalla, M.; Liu, Z.; Asefa, T. N-, O-, and S-tridoped nanoporous carbons as selective catalysts for oxygen reduction and alcohol oxidation reactions. J. Am. Chem. Soc. 2014, 136, 13554–13557. [Google Scholar] [CrossRef]
- Gavrilov, N.; Pasti, I.A.; Mitrić, M.; Travas-Sejdić, J.; Cirić-Marjanović, J.; Mentus, S.V. Electrocatalysis of oxygen reduction reaction on polyaniline-derived nitrogen-doped carbon nanoparticle surfaces in alkaline media. J. Power Sources 2012, 220, 306–316. [Google Scholar] [CrossRef]
- Nam, G.; Park, J.; Kim, S.T.; Shin, D.-B.; Park, N.; Lee, Y.K.J.-S. Metal-free Ketjenblack incorporated nitrogen-doped carbon sheets derived from gelatin as oxygen reduction catalysts. J. Cho Nano Lett. 2014, 14, 1870–1876. [Google Scholar] [CrossRef]
- Qu, K.; Zheng, Y.; Dai, S.; Qiao, S.Z. Graphene oxide-polydopamine derived N, S-codoped carbon nanosheets as superior bifunctional electrocatalysts for oxygen reduction and evolution. Nano Energy 2016, 19, 373–381. [Google Scholar] [CrossRef]
- Gao, S.; Liu, H.; Geng, K.; Wei, X. Honeysuckles-derived porous nitrogen, sulfur, dual-doped carbon as high-performance metal-free oxygen electroreduction catalyst. Nano Energy 2015, 12, 785–793. [Google Scholar] [CrossRef]
- Zhao, G.; Shi, L.; Xu, J.; Yan, X.; Zhao, T.S. Role of phosphorus in nitrogen, phosphorus dual-doped ordered mesoporous carbon electrocatalyst for oxygen reduction reaction in alkaline media. Int. J. Hydrogen Energy 2018, 43, 1470–1478. [Google Scholar] [CrossRef]
- Borghei, M.; Laocharoen, N.; Kibena-Poldseff, E.; Johansson, L.-S.; Campbell, J.; Kauppinen, E.; Tammeveski, K.; Rojas, O.J. Porous N, P-doped carbon from coconut shells with high electrocatalytic activity for oxygen reduction: Alternative to Pt-C for alkaline fuel cells. Appl. Catal. B Environ. 2017, 204, 394–402. [Google Scholar] [CrossRef]
- Jiang, H.; Wang, Y.; Hao, J.; Liu, Y.; Li, W.; Li, J. N and P co-functionalized three-dimensional porous carbon networks as efficient metal-free electrocatalysts for oxygen reduction reaction. Carbon 2017, 122, 64–73. [Google Scholar] [CrossRef]
- Hwang, S.-H.; Chun, S.-H. Five Artistic Values of the Apple Company: Focusing on Apple’s Logo. Asia-Pac. J. Multimed. Serv. Converg. Art Humanit. Sociol. 2017, 7, 177–186. [Google Scholar] [CrossRef][Green Version]
- Wang, J.; Wu, Z.-X.; Man, L.-L.; Liu, Y.-Y.; Guo, J.-P.; Xin, H.L.; Wang, D.-L. Rational design of three-dimensional nitrogen and phosphorus co-doped graphene nanoribbons/CNTs composite for the oxygen reduction. Chin. Chem. Lett. 2016, 27, 597–601. [Google Scholar] [CrossRef]
- Wu, M.; Wang, J.; Wu, Z.; Xin, H.L.; Wang, D. Synergistic enhancement of nitrogen and sulfur co-doped graphene with carbon nanosphere insertion for the electrocatalytic oxygen reduction reaction. J. Mater. Chem. A 2015, 3, 7727–7731. [Google Scholar] [CrossRef]
- Wang, J.; Wu, Z.; Han, L.; Lin, R.; Xiao, W.; Xuan, C.; Xin, H.L.; Wang, D. Nitrogen and sulfur co-doping of partially exfoliated MWCNTs as 3-D structured electrocatalysts for the oxygen reduction reaction. J. Mater. Chem. A 2016, 4, 5678–5684. [Google Scholar] [CrossRef]
- He, J.; He, Y.; Fan, Y.; Zhang, B.; Du, Y.; Wang, J.; Xu, P. Conjugated polymer-mediated synthesis of nitrogen-doped carbon nanoribbons for oxygen reduction reaction. Carbon 2017, 124, 630–636. [Google Scholar] [CrossRef]
- Oh, H.-S.; Oh, J.-G.; Lee, W.H.; Kim, H.-J. The influence of the structural properties of carbon on the oxygen reduction reaction of nitrogen modified carbon based catalysts. Int. J. Hydrogen Energy 2011, 36, 8181–8186. [Google Scholar] [CrossRef]
- Ferrero, G.A.; Preuss, K.; Fuertes, A.B.; Sevilla, M.; Titirici, M.M. The influence of pore size distribution on the oxygen reduction reaction performance in nitrogen doped carbon microspheres. J. Mater. Chem. 2016, A4, 2581–2589. [Google Scholar] [CrossRef]
- Zheng, X.; Cao, X.; Li, X.; Tian, J.; Jin, C.; Yang, R. Biomass lysine-derived nitrogen-doped carbon hollow cubes via a NaCl crystal template: An efficient bifunctional electrocatalyst for oxygen reduction and evolution reactions. Nanoscale 2017, 9, 1059–1067. [Google Scholar] [CrossRef]
- Sui, Z.-Y.; Li, X.; -Sun, Z.-Y.; Tao, H.-C.; Zhang, P.-Y.; Zhao, L.; Han, B.-H. Nitrogen-doped and nanostructured carbons with high surface area for enhanced oxygen reduction reaction. Carbon 2018, 126, 111–118. [Google Scholar] [CrossRef]
- Chatterjee, K.; Kumar, M.A.; Gullafalli, H.; Gong, Y.; Vajtai, R.; Thanikaivelan, P.; Ajayan, P.M. Nitrogen-rich carbon nano-onions for oxygen reduction reaction. Carbon 2018, 130, 645–651. [Google Scholar] [CrossRef]
- Compos-Martin, J.M.; Blanco-Brieva, G.; Fierro, J.L.G. Hydrogen peroxide synthesis: An outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 2006, 45, 6962–6984. [Google Scholar] [CrossRef]
- Vendaguer-Casadevall, A.; Hernandez-Fernandez, P.; Stephens, I.E.L.; Chorkendorff, I.; Dahl, S. The effect of ammonia upon the electrocatalysis of hydrogen oxidation and oxygen reduction on polycrystalline platinum. J. Power Sources 2012, 220, 205–210. [Google Scholar] [CrossRef]
- Blizanac, B.B.; Ross, P.N.; Markovic, N.M. Oxygen electroreduction on Ag (1 1 1): The pH effect. Electrochim. Acta 2007, 52, 2264–2271. [Google Scholar] [CrossRef]
- Jirkovsky, J.S.; Panas, I.; Ahlberg, E.; Halasa, M.; Romani, S.; Schiffrin, D.J. Single Atom Hot-Spots at Au–Pd Nanoalloys for Electrocatalytic H2O2 Production. J. Am. Chem. Soc. 2011, 133, 19432–19441. [Google Scholar] [CrossRef]
- Fellinger, T.P.; Hasche, F.; Strasser, P.; Antonietti, M. Mesoporous nitrogen-doped carbon for the electrocatalytic synthesis of hydrogen peroxide. J. Am. Chem. Soc. 2012, 134, 4072–4075. [Google Scholar] [CrossRef]
- Liu, Y.; Quan, X.; Fan, X.; Wang, H.; Chen, S. High-yield electrosynthesis of hydrogen peroxide from oxygen reduction by hierarchically porous carbon. Angew. Chem. Int. Ed. 2015, 54, 6837–6841. [Google Scholar] [CrossRef]
- Verdagreer-Casadevall, A. Trends in the Electrochemical Synthesis of H2O2: Enhancing Activity and Selectivity by Electrocatalytic Site Engineering. Nano Lett. 2014, 14, 1603–1608. [Google Scholar] [CrossRef]
- Rahman, M.A.; Won, M.S.; Shim, Y.B. Xanthine sensors based on anodic and cathodic detection of enzymatically generated hydrogen peroxide. Electroanalysis 2007, 19, 631–637. [Google Scholar] [CrossRef]
- Tan, X.C.; Tian, Y.X.; Cai, P.X.; Zou, X.Y. Glucose biosensor based on glucose oxidase immobilized in sol–gel chitosan/silica hybrid composite film on Prussian blue modified glass carbon electrode. Anal. Bioanal. Chem. 2005, 381, 500–507. [Google Scholar] [CrossRef]
- Nakatani, H.; Santos, L.; Pelegrine, C. Biosensor based on xanthine oxidase for monitoring hypoxanthine in fish meat. Amer. J. Biochem. Biotechnol. 2005, 1, 85–89. [Google Scholar] [CrossRef]
- Simeonov, L.; Chirila, E. (Eds.) Chemicals as Intentional and Accidental Global Environmental Threats; Springer: Dordrecht, The Netherlands, 2006. [Google Scholar]
- Dimcheva, N.; Horozova, E.; Jordanova, Z. A glucose oxidase immobilized electrode based on modified graphite. Z. Nat. 2002, 57, 705–711. [Google Scholar] [CrossRef]
- Kulesza, P.J.; Marassi, R.; Karnicka, K. Electrocatalysis and bioelectrocatalysis at nanostructured composite films. Rev. Adv. Mater. Sci. 2007, 15, 225–233. [Google Scholar]
- Sun, L.; Cao, D.; Wang, G. Pd–Ru/C as the electrocatalyst for hydrogen peroxide reduction. J. Appl. Electrochem. 2008, 38, 1415–1419. [Google Scholar] [CrossRef]
- Horozova, E.; Dodevyka, T.; Dimcheva, N. Modified graphites: Application to the development of enzyme-based amperometric biosensors. Bioelectrochemistry 2009, 74, 260–264. [Google Scholar] [CrossRef]
- Welch, C.M.; Banks, C.E.; Simm, A.O.; Compton, R.G. Silver nanoparticle assemblies supported on glassy-carbon electrodes for the electro-analytical detection of hydrogen peroxide. Anal. Bioanal. Chem. 2005, 382, 12–21. [Google Scholar] [CrossRef]
- Lu, K.C.; Herzig, D.O. Anal Fissure. In The ASCRS Textbook of Colon and Rectal Surgery; Springer: Cham, Switzerland, 2016; Volume 38, pp. 205–214. [Google Scholar] [CrossRef]
- Dodevska, T.; Horozova, E.; Dimcheva, N. Electrocatalytic reduction of hydrogen peroxide on modified graphite electrodes: Application to the development of glucose biosensors. Anal. Bioanal. Chem. 2006, 386, 1413–1418. [Google Scholar] [CrossRef]
- Duan, L.; Xu, Q.; Xie, F.; Wang, S. Hydrogen peroxide biosensor based on the bioelectrocatalysis of myoglobin incorporated in multi-walled carbon nanotubes/chitosan composite film. Int. J. Electrochem. Sci. 2008, 3, 118–124. [Google Scholar]
- Ermat, A.; Makowski, O.; Kowalewska, B.; Mieeznikowski, K.; Kuleza, P.J. Hybrid bioelectrocatalyst for hydrogen peroxide reduction: Immobilization of enzyme within organic–inorganic film of structured Prussian Blue and PEDOT. Bioelectrochem 2007, 71, 23–28. [Google Scholar]
- Yang, P.; Wei, W.; Tao, C.; Xie, B.; Chen, X. Nano-silver/multi-walled carbon nanotube composite films for hydrogen peroxide electroanalysis. Microchim. Acta 2008, 162, 51–56. [Google Scholar] [CrossRef]
- Niwa, O. Electroanalytical chemistry with carbon film electrodes and micro and nano-structured carbon film-based electrodes. Bull. Chem. Soc. Jpn. 2005, 78, 555–571. [Google Scholar] [CrossRef]
- World Metereological Organization. Available online: http://www.wmo.int/ (accessed on 1 May 2018).
- Centi, G.; Perathoner, S. Opportunities and prospects in the chemical recycling of carbon dioxide to fuels. Catal. Today 2009, 148, 191–205. [Google Scholar] [CrossRef]
- Lai, J.; Nsabimana, A.; Lugue, R.; Xu, G. 3D porous carbonaceous electrodes for electrocatalytic applications. Joule 2018, 2, 76–93. [Google Scholar] [CrossRef]
- Bevilacqua, M.; Filiffi, J.; Miller, H.A.; Vizza, F. Recent Technological Progress in CO2 Electroreduction to Fuels and Energy Carriers in Aqueous Environments. Energy Technol. 2015, 3, 197–210. [Google Scholar] [CrossRef]
- Hori, Y.; Murata, A.; Takahski, R. Formation of hydrocarbons in the electrochemical reduction of carbon dioxide at a copper electrode in aqueous solution. J. Chem. Soc. Faraday Trans. 1989, 85, 2309–2326. [Google Scholar] [CrossRef]
- Shi, C.; Hansen, M.A.; Lausche, A.C.; Norskov, J.K. Trends in electrochemical CO 2 reduction activity for open and close-packed metal surfaces. Phys. Chem. Chem. Phys. 2014, 16, 4720–4727. [Google Scholar] [CrossRef]
- Williams, S.D.; Ortuzar, N. 100 Issues of ChemMedChem. ChemMedChem 2014, 9, 675–676. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S.; Wine, G.; Gangeri, M. Electrocatalytic conversion of CO2 to long carbon-chain hydrocarbons. Green Chem. 2007, 9, 671–678. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Problems and perspectives in nanostructured carbon-based electrodes for clean and sustainable energy. Catal. Today 2010, 150, 151–162. [Google Scholar] [CrossRef]
- Li, W.; Seredych, M.; Rodriguez-Castellon, E.; Bandooz, T.J. Metal-free Nanoporous Carbon as a Catalyst for Electrochemical Reduction of CO2 to CO and CH4. ChemSusChem 2016, 9, 606–616. [Google Scholar] [CrossRef]
- Perez-Cadenas, A.F. Doped Carbon Material for the Electrocatalytic Conversion of CO2 into Hydrocarbons, Uses of the Material and Conversion Method Using Said Material. International Patent Application WO/2013/004882, 1 October 2013. [Google Scholar]
- Perez-Cadenas, A.F.; Ross, C.H.; Morales-Torres, S.; Perez-Cadenas, M.; Kooyman, P.J.; Moreno-Castilla, C.; Kajteijn, F. Metal-doped carbon xerogels for the electro-catalytic conversion of CO2 to hydrocarbons. Carbon 2013, 56, 324–331. [Google Scholar] [CrossRef]
- Schouten, K.J.P.; Kwon, Y.; van der Ham, C.J.M.; Qin, Z.; Koper, M.T.M. A new mechanism for the selectivity to C 1 and C 2 species in the electrochemical reduction of carbon dioxide on copper electrodes. Chem. Sci. 2011, 2, 1902–1909. [Google Scholar] [CrossRef]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef]
- Abdelwahab, A.; Castelo-Quibén, J.; Perez-Cadenas, M.; Morales-Torres, S.; Maldonado-Hódar, F.J.; Carrasco-Marin, F.; Perez-Cadenas, A.F. Cobalt-doped carbon gels as electro-catalysts for the reduction of CO2 to hydrocarbons. Catalysts 2017, 7, 25. [Google Scholar] [CrossRef]
- Castelo-Quiben, J.; Abdelwahab, A.; Perez-Cadenas, M.; Elmouwahidi, A.; Maldonado-Hodar, F.J.; Carrasco-Marim, F.; Perez-Cadenas, A.F. Carbon-iron electro-catalysts for CO2 reduction. The role of the iron particle size. J. CO2 Util. 2018, 24, 240–249. [Google Scholar] [CrossRef]
- Montoya, J.H.; Tsai, C.; Vajvovic, A.; Norskov, J.K. The challenge of electrochemical ammonia synthesis: A new perspective on the role of nitrogen scaling relations. ChemSusChem 2015, 8, 2180–2186. [Google Scholar] [CrossRef]
- Zheng, G.; Yan, J.-M.; Yu, G. Nitrogen reduction reaction. Small Methods 2019, 3, 1900070. [Google Scholar] [CrossRef]
- Skulason, E.; Bligaard, T.; Gudmundsdottir, S.; Studt, F.; Rossmeisl, J.; Abild-Pedersen, F.; Vegge, T.; Jonsson, H.; Norskov, J.K. A theoretical evaluation of possible transition metal electro-catalysts for N2 reduction. Phys. Chem. Chem. Phys. 2012, 14, 1235–1245. [Google Scholar] [CrossRef]
- Cukier, R.I.; Nocera, D.G. Proton-coupled electron transfer. Annu. Rev. Phys. Chem. 1998, 49, 337–369. [Google Scholar] [CrossRef]
- Weinberg, D.R.; Gagliardi, C.J.; Hull, J.F.; Murphy, C.F.; Kent, C.A.; Westlake, B.C.; Paul, A.; Ess, D.H.; McCafferty, D.G.; Meyer, T.J. Proton-coupled electron transfer. Chem. Rev. 2012, 112, 4016–4093. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Saveant, J.M. Concerted proton—electron transfers: Electrochemical and related approaches. Acc. Chem. Res. 2010, 43, 1019–1029. [Google Scholar] [CrossRef]
- MShipman, A.; Symes, M.D. Recent progress towards the electrosynthesis of ammonia from sustainable resources. Catal. Today 2017, 286, 57–68. [Google Scholar] [CrossRef]
- Yao, Y.; Zhu, S.Q.; Wang, H.J.; Li, H.; Shao, M.H. A spectroscopic study on the nitrogen electrochemical reduction reaction on gold and platinum surfaces. J. Am. Chem. Soc. 2018, 140, 1496–1501. [Google Scholar] [CrossRef]
- Seefeldt, L.C.; Hoffman, B.M.; Dean, D.R. Mechanism of Mo-dependent nitrogenase. Annu. Rev. Biochem. 2009, 78, 701–722. [Google Scholar] [CrossRef]
- Becker, J.Y.; Avraham, S.; Porin, B. Nitrogen fixation: Part I. Electrochemical reduction of titanium compounds in the presence of catechol and N2 in MeOH or THF. J. Electroanal. Chem. 1987, 230, 143–153. [Google Scholar] [CrossRef]
- Kim, K.; Lee, N.; Yoo, C.Y.; Kim, J.N.; Yoon, H.C.; Han, J.I. Communication—Electrochemical reduction of nitrogen to ammonia in 2-propanol under ambient temperature and pressure. J. Electrochem. Soc. 2016, 163, F610–F612. [Google Scholar] [CrossRef]
- Chen, S.M.; Perathoner, S.; Ampelli, C.; Mebrahtu, C.; Su, D.S.; Centi, G. Electrocatalytic synthesis of ammonia at room temperature and atmospheric pressure from water and nitrogen on a carbon-nanotube-based electrocatalyst. Angew. Chem. Int. Ed. 2017, 56, 2699–2703. [Google Scholar] [CrossRef] [PubMed]
- Nangle, S.N.; Sakimoto, K.K.; Silveri, P.A.; Nocera, D.G. Biological-inorganic hybrid systems as a generalized platform for chemical production. Curr. Opin. Chem. Biol. 2017, 41, 107–113. [Google Scholar] [CrossRef]
- Sakimoto, K.K.; Kornienko, N.; Cestellos-Blanco, S.; Lim, J.; Liu, C.; Yang, P.D. Physical biology of the materials–microorganism interface. J. Am. Chem. Soc. 2018, 140, 1978–1985. [Google Scholar] [CrossRef]
- Milton, R.D.; Cri, R.; Abdellaoni, S.; Leech, D.; de Lacey, A.L.; Pita, M.; Minteer, S.D. Bioelectrochemical Haber–Bosch Process: An Ammonia-Producing H2/N2 Fuel Cell. Angew. Chem. Int. Ed. 2017, 56, 2680–2683. [Google Scholar] [CrossRef]
- Schlesinger, I.; Brown, H.C.; Finholt, A.E. The Preparation of Sodium Borohydride by the High Temperature Reaction of Sodium Hydride with Borate Esters1. J. Am. Chem. Soc. 1953, 75, 205–209. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Sequeira, C.A.C. Sodium borohydride as a fuel for the future. Renew. Sust. Energ. Rev. 2011, 15, 3980–4001. [Google Scholar] [CrossRef]
- Buckner, W.; Niederprum, H. Sodium borohydride and amine-boranes, commercially important reducing agents. Pure Appl. Chem. 1977, 49, 733–743. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Sequeira, C.A.C. Zinc anode for direct borohydride fuel cells. J. Electrochem. Soc. 2010, 157, B13–B19. [Google Scholar] [CrossRef]
- de León, C.P.; Walsh, F.C.; Rose, A.; Lakeman, J.B.; Browning, D.J.; Reeve, R.W. A direct borohydride—Acid peroxide fuel cell. J. Power Sources 2007, 164, 441–448. [Google Scholar] [CrossRef]
- Cao, D.; Gao, Y.; Wang, G.; Miao, R.; Liu, Y. A direct NaBH4–H2O2 fuel cell using Ni foam supported Au nanoparticles as electrodes. Int. J. Hydrogen Energy 2010, 35, 2648–2651. [Google Scholar] [CrossRef]
- Concha, B.M.; Chatenet, M. Direct oxidation of sodium borohydride on Pt, Ag and alloyed Pt–Ag electrodes in basic media. Part I: Bulk Electrodes. Electrochim. Acta 2009, 54, 6119–6129. [Google Scholar]
- Yang, J.Q.; Liu, B.H.; Wu, S. Carbon-supported Pd catalysts: Influences of nanostructure on their catalytic performances for borohydride electrochemical oxidation. J. Power Sources 2009, 194, 824–829. [Google Scholar] [CrossRef]
- Duan, D.H.; Liu, S.B.; Sun, Y.P. Analysis of the kinetics of borohydride oxidation in Cu anode for direct borohydride fuel cell. J. Power Sources 2012, 210, 198–203. [Google Scholar] [CrossRef]
- Wei, J.; Wang, X.; Wang, Y.; Chen, Q.; Pei, F.; Wang, Y. Investigation of carbon-supported Au hollow nanospheres as electrocatalyst for electrooxidation of sodium borohydride. Int. J. Hydrogen Energy 2009, 34, 3360–3366. [Google Scholar] [CrossRef]
- Scott, R.W.J.; Wilson, O.M.; Oh, S.K.; Kenik, E.A.; Crooks, R.M. Bimetallic palladium− gold dendrimer-encapsulated catalysts. J. Am. Chem. Soc. 2004, 126, 15583–15591. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.B.; Duan, D.H.; Ma, Y.H.; Wei, G.Q.; Zhang, Z.L.; Hao, X.G.; Liu, S.B. Performance of nano-nickel core wrapped with Pt crystalline thin film for methanol electro-oxidation. J. Power Sources 2014, 245, 886–891. [Google Scholar] [CrossRef]
- Zhou, W.; Lee, J.Y. Highly active core–shell Au@ Pd catalyst for formic acid electrooxidation. Electrochem. Commun. 2007, 9, 1725–1729. [Google Scholar] [CrossRef]
- Grouchko, M.; Kamyshny, A.; Magdssi, S. Formation of air-stable copper–silver core–shell nanoparticles for inkjet printing. J. Mater. Chem. 2009, 19, 3057–3062. [Google Scholar] [CrossRef]
- Duan, D.H.; Liang, J.W.; Liu, H.H.; You, X.; Wei, H.K.; Wei, G.Q.; Liu, S.B. The effective carbon supported core–shell structure of Ni@ Au catalysts for electro-oxidation of borohydride. Int. J. Hydrogen Energy 2015, 40, 488–500. [Google Scholar] [CrossRef]
- He, P.Y.; Wang, Y.W.X.Y.; Pei, F.; Wang, H.; Liu, L.; Yi, L.H. Investigation of carbon supported Au–Ni bimetallic nanoparticles as electrocatalyst for direct borohydride fuel cell. J. Power Sources 2011, 196, 1042–1047. [Google Scholar] [CrossRef]
- Arevalo, R.L.; Escano, M.C.S.; Gyenge, E.; Kasai, H. A theoretical study of the structure and stability of borohydride on 3d transition metals. Surf. Sci. 2012, 606, 1954–1959. [Google Scholar] [CrossRef]
- Elumalai, M.; Rajasekaran, A.; Chinnaraja, B. Performance of Pt–Ru–Mo Ternary Catalysts for Borohydride Electro-Oxidation in Membraneless Fuel Cell. Int. J. Ind. Eng. 2018, 2, 108–118. [Google Scholar]
- Ferrari, A.C. Raman spectroscopy of graphene and graphite: Disorder, electron–phonon coupling, doping and nonadiabatic effects. Solid State Commun. 2007, 143, 47–57. [Google Scholar] [CrossRef]
- Fampiou, I.; Ramasubramaniam, A. Binding of Pt nanoclusters to point defects in graphene: Adsorption, morphology, and electronic structure. J. Phys. Chem. C. 2012, 116, 6543–6555. [Google Scholar] [CrossRef]
- Antolini, A. Carbon supports for low-temperature fuel cell catalysts. Appl. Catal. B 2009, 88, 1–24. [Google Scholar] [CrossRef]
- Rady, A.C.; Giddey, S.; Badwai, S.P.S.; Ladewig, B.P.; Bhattacharya, S. Review of fuels for direct carbon fuel cells. Energy Fuels 2012, 26, 1471–1488. [Google Scholar]
- Ettingshansen, F.; Klemann, Y.; Marcu, A.; Toth, G.; Fuess, H.; Roth, C. Dissolution and migration of platinum in PEMFCs investigated for start/stop cycling and high potential degradation. Fuel Cells 2011, 11, 238–245. [Google Scholar] [CrossRef]
- Gur, T.M.; Huggins, R.A. Direct electrochemical conversion of carbon to electrical energy in a high temperature fuel cell. J. Electrochem. Soc. 1992, 139, L95–L97. [Google Scholar] [CrossRef]
- Bai, Y.; Liu, Y.; Tang, Y.; Xie, Y.; Liu, J. Direct carbon solid oxide fuel cell—A potential high performance battery. Int. J. Hydrogen Energy 2011, 36, 9189–9194. [Google Scholar] [CrossRef]
- Dudek, M.; Tomczyk, P.; Socha, R.; Skrzypkiewicz, M.; Jewulski, J. Biomass fuels for direct carbon fuel cell with solid oxide electrolyte. Int. J. Electrochem. Sci. 2013, 8, 3229–3253. [Google Scholar]
- Hasegawa, S.; Ihara, M. Reaction mechanism of solid carbon fuel in rechargeable direct carbon SOFCs with methane for charging. J. Electrochem. Soc. 2008, 155, B58–B63. [Google Scholar] [CrossRef]
- Lee, S.; Li, A.C.; Mitchell, R.E.; Gür, T.M. Direct carbon conversion in a helium fluidized bed fuel cell. Electrochem. Sol. St. Lett. 2008, 11, B20–B23. [Google Scholar] [CrossRef]
- Ponchon, J.L.; Cespuglio, R.; Gonon, F.; Jouvet, M.; Pujol, J.F. Normal pulse polarography with carbon fiber electrodes for in vitro and in vivo determination of catecholamines. Anal. Chem. 1979, 51, 1483–1486. [Google Scholar] [CrossRef] [PubMed]
- Gonon, F.; Buda, M.; Cespuglio, R.; Jouvet, M.; Pujol, J.F. In vivo electrochemical detection of catechols in the neostriatum of anaesthetized rats: Dopamine or DOPAC? Nature 1980, 286, 902–904. [Google Scholar] [CrossRef] [PubMed]
- Gonon, F.; Fombarlet, C.H.; Buda, M.; Pujol, J.F. Electrochemical treatment of pyrolytic carbon fiber electrodes. Anal. Chem. 1981, 53, 1386–1389. [Google Scholar] [CrossRef]
- Wightman, R.M.; May, L.J.; Michael, A.C. Detection of dopamine dynamics in the brain. Anal. Chem. 1988, 60, 769A–779A. [Google Scholar] [CrossRef]
- Justice, J.B. Voltammetry in the Neurosciences; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 1987. [Google Scholar]
- Phillips, P.E.M.; Wightman, R.M. Critical guidelines for validation of the selectivity of in-vivo chemical microsensors. TrAC Trends Anal. Chem. 2003, 22, 509–514. [Google Scholar] [CrossRef]
- Jones, S.R.; Garris, P.A.; Wightman, R.M. Different effects of cocaine and nomifensine on dopamine uptake in the caudate-putamen and nucleus accumbens. J. Pharmacol. Exp. Ther. 1995, 274, 396–403. [Google Scholar] [PubMed]
- Greco, P.G.; Garris, P.A. In vivo interaction of cocaine with the dopamine transporter as measured by voltammetry. Eur. J. Pharmacol. 2003, 479, 117–125. [Google Scholar] [CrossRef]
- Gumar, S.M.; Porterfield, D.M.; Muller, K.J.; Smith, P.J.S.; Sahley, C.L. Nerve injury induces a rapid efflux of nitric oxide (NO) detected with a novel NO microsensor. J. Neurosci. 2001, 21, 215–220. [Google Scholar]
- Colliver, T.L.; Pyott, S.J.; Achalabun, M.; Ewing, A.G. VMAT-mediated changes in quantal size and vesicular volume. J. Neurosci. 2000, 20, 5276–5282. [Google Scholar] [CrossRef]
- Avshalumov, M.V.; Chen, B.T.; Marshall, S.P.; Pena, D.M.; Rice, M.E. Glutamate-dependent inhibition of dopamine release in striatum is mediated by a new diffusible messenger, H2O2. J. Neurosci. 2003, 23, 2744–2750. [Google Scholar] [CrossRef]
- Boudko, D.Y.; Moroz, L.L.; Linser, P.J.; Trimarchi, J.R.; Smith, P.J.S.; Itarvey, W.R. In situ analysis of pH gradients in mosquito larvae using non-invasive, self-referencing, pH-sensitive microelectrodes. J. Exp. Biol. 2001, 204, 691–699. [Google Scholar]
- Kennedy, R.T.; Jones, S.R.; Wightman, R.M. Simultaneous measurement of oxygen and dopamine: Coupling of oxygen consumption and neurotransmission. Neuroscience 1992, 47, 603–612. [Google Scholar] [CrossRef]
- Kulagina, N.V.; Michael, A.C. Monitoring hydrogen peroxide in the extracellular space of the brain with amperometric microsensors. Anal. Chem. 2003, 75, 4875–4881. [Google Scholar] [CrossRef]
- Rebee, G.V.; Wang, Z. Behavioral activation in rats requires endogenous ascorbate release in striatum. J. Neurosci. 2001, 21, 668–675. [Google Scholar]
- Clark, L.C.; Lyons, C. Electrode systems for continuous monitoring in cardiovascular surgery. J. Ann. N. Y. Acad. Sci. 1962, 102, 29–45. [Google Scholar] [CrossRef]
- Bartlett, P.N.; Cooper, J.M. A review of the immobilization of enzymes in electropolymerized films. J. Electroanal. Chem. 1993, 362, 1–12. [Google Scholar] [CrossRef]
- Campbell, J.K.; Sun, L.; Crooks, R.M. Electrochemistry using single carbon nanotubes. J. Am. Chem. Soc. 1999, 121, 3779–3780. [Google Scholar] [CrossRef]
- Chen, R.S.; Huang, W.H.; Tong, H.; Wang, Z.L.; Cheng, J.K. Carbon fiber nanoelectrodes modified by single-walled carbon nanotubes. Anal. Chem. 2003, 75, 6341–6345. [Google Scholar] [CrossRef]
- Valcarcel, M.; Cardenas, S.; Simonet, B.M. Role of carbon nanotubes in analytical science. Anal. Chem. 2007, 79, 4788–4797. [Google Scholar] [CrossRef]
- Huguruma, M.; Hitoshi, M.; Yasunori, S.; Shibayama, S. Carbon nanotube–plasma polymer-based amperometric biosensors: Enzyme-friendly platform for ultrasensitive glucose detection. Jpn. J. Appl. Phys. 2007, 46, 6078–6082. [Google Scholar] [CrossRef]
- Cheng, J.; Meziani, M.J.; Sun, Y.P.; Cheng, S.H. Poly (ethylene glycol)-conjugated multi-walled carbon nanotubes as an efficient drug carrier for overcoming multidrug resistance. Toxicol. Appl. Pharmacol. 2011, 250, 184–193. [Google Scholar] [CrossRef]
- Bianco, A.; Kostarelos, K.; Prato, M. Applications of carbon nanotubes in drug delivery. Curr. Opin. Chem. Biol. 2005, 9, 674–679. [Google Scholar] [CrossRef]
- Berber, S.; Kwon, Y.K.; Tomanek, D. Unusually high thermal conductivity of carbon nanotubes. Phys. Rev. 2000, 84, 4613–4616. [Google Scholar] [CrossRef]
- Hone, J.; Llaguno, M.C.; Nemes, N.M.; Johnson, A.T.; Fisher, J.E.; Walters, D.A.; Casavant, M.J.; Schmidt, J.; Srualley, R.E. Electrical and thermal transport properties of magnetically aligned single wall carbon nanotube films. Appl. Phys. 2000, 77, 666–668. [Google Scholar] [CrossRef]
- Hone, J.; Whitney, M.; Piskoti, C.; Zettl, A. Thermal conductivity of single-walled carbon nanotubes. Phys. Rev. 1999, 59, 2514–2516. [Google Scholar] [CrossRef]
- Acquah, S.F.A.; Penkova, A.V.; Markelov, D.A.; Semisapova, A.S.; Leonhardt, B.E.; Magi, J.M. The beautiful molecule: 30 years of C60 and its derivatives. ECS J. Sol. St. Sci. Tech. 2017, 6, M3155–M3162. [Google Scholar]
- Azam, M.A.; Zulkapli, N.N.; Dorah, N.; Seman, R.N.A.R.; Ani, M.H.; Sirat, M.S.; Ismail, E.; Fauzi, F.B.; Mohamed, M.A.; Majlis, B.Y. Critical considerations of high quality graphene synthesized by plasma-enhanced chemical vapor deposition for electronic and energy storage devices. ECS J. Sol. St. Sci. Tech. 2017, 6, M3035–M3048. [Google Scholar] [CrossRef]
- Krueger, A. Carbon Materials and Nanotechnology; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Heister, F.; Neves, V. Drug loading, dispersion stability, and therapeutic efficacy in targeted drug delivery with carbon nanotubes. Carbon 2006, 128, 10568–10571. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, K.; Davis, C. Drug delivery with carbon nanotubes for in vivo cancer treatment. Cancer Res. 2008, 68, 6652–6660. [Google Scholar] [CrossRef]
- Lay, C.L.; Liu, H.Q.; Tan, H.R.; Liu, Y. Carbon nanotubes-the holy grail in anticancer therapy. Nanotechnology 2010, 21, 214–256. [Google Scholar]
- Liu, X.; Tao, H.; Yang, K.; Zhang, S.; Lee, S.T.; Liu, A. Optimization of surface chemistry on single-walled carbon nanotubes for in vivo photothermal ablation of tumors. Biomaterials 2011, 32, 144–151. [Google Scholar] [CrossRef]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Lai, C.H.; Kuo, K.H. The critical component to establish in vitro BBB model: Pericyte. Brain Res. Rev. 2005, 50, 258–265. [Google Scholar] [CrossRef]
- Hampel, S.; Kumze, D.; Haase, D. Carbon nanotubes filled with a chemotherapeutic agent: A nanocarrier mediates inhibition of tumor cell growth. Nanomedicine 2008, 3, 175–182. [Google Scholar] [CrossRef]
- Hossen, S.; Hossain, M.K.; Basher, M.K.; Mia, M.N.H.; Rahman, M.T.; Uddin, M.J. Smart nanocarrier-based drug delivery systems for cancer therapy and toxicity studies: A review. J. Adv. Res. 2019, 15, 1–18. [Google Scholar] [CrossRef]
- Firouzi, B.B.; Abarghooee, R.A. Battery energy storage system for frequency support in microgrids and with enhanced control features for uninterruptible supply of local loads. Int. J. Electr. Power Energy Syst. 2014, 54, 432–441. [Google Scholar]
- Bates, A.; Mukerjee, S.; Lee, S.E.; Lee, D.H.; Park, S. An analytical study of a lead-acid flow battery as an energy storage system. J. Power Sources 2014, 249, 207–218. [Google Scholar] [CrossRef]
- Babakhani, A.; Rashchi, F.; Zakeri, A.; Vahieli, E. Selective separation of nickel and cadmium from sulfate solutions of spent nickel–cadmium batteries using mixtures of D2EHPA and Cyanex 302. J. Power Sources 2014, 247, 127–133. [Google Scholar] [CrossRef]
- Huang, J.; Cao, D.; Lei, T.; Yang, S.; Zhou, X.; Xu, P.; Wang, G. Structural and electrochemical performance of Al-substituted β-Ni (OH) 2 nanosheets electrodes for nickel metal hydride battery. Electrochim. Acta 2013, 111, 713–719. [Google Scholar] [CrossRef]
- Separi, S.; Ghorbani, R.; Liaw, B. A novel on-board state-of-charge estimation method for aged Li-ion batteries based on model adaptive extended Kalman filter. J. Power Sources 2014, 245, 337–344. [Google Scholar]
- Mónconduit, L.; Croguennec, L. (Eds.) Prospects for Lithium Batteries and Emerging Energy Electrochemical Systems; World Scientific Publ. Co. Inc.: Hackensack, NJ, USA, 2018. [Google Scholar]
- Birke, K.P. Modern Battery Engineering; World Scientific Pub Co Pte Ltd.: Hackensack, NJ, USA, 2019; p. 372. [Google Scholar]
- Crawley, G.M. (Ed.) Energy Storage; World Scientific Publ. Co. Inc.: Hackensack, NJ, USA, 2008. [Google Scholar]
- Crawley, G.M. The World Scientific Handbook of Energy; World Scientific Pub Co Pte Ltd.: Hackensack, NJ, USA, 2013; Volume 3, p. 374. [Google Scholar]
- Dudney, N.J.; West, W.C.; Nanda, J. Handbook of Solid State Batteries; World Scientific Pub Co Pte Ltd.: Hackensack, NJ, USA, 2015. [Google Scholar]
- Qu, K.; Wang, Y.; Vasileff, A.; Jiao, Y.; Chen, H.; Zheng, Y. Polydopamine-inspired nanomaterials for energy conversion and storage. J. Mater. Chem. A 2018, 6, 21827–21846. [Google Scholar] [CrossRef]
- Demir-Cakan, R. (Ed.) Li-S Batteries. The Challenges, Chemistry, Materials and Future Perspectives; World Scientific Publ. Co. Inc.: Hackensack, NJ, USA, 2017. [Google Scholar]
- Borchardt, L.; Althues, L.B.H.; Kaskel, S. Carbon nanocomposites for lithium—sulphur batteries. Curr. Opin. Green Sustain. Chem. 2017, 4, 64–71. [Google Scholar] [CrossRef]
- Zang, J.; Xia, Z.; Dai, L. Carbon-based electrocatalysts for advanced energy conversion and storage. Sci. Adv. 2015, 1, e1500564. [Google Scholar] [CrossRef]
- Balbuena, P.B.; Wang, Y. (Eds.) Lithium-Ion Batteries, Solid Electrolyte Interphase; World Scientific Publ. Co. Inc.: Hackensack, NJ, USA, 2004. [Google Scholar]
- Kotobuki, M.; Song, S.; Chen, C.; Lu, L. Ceramic Electrolytes for All-Solid-State Li Batteries; World Scientific Pub Co Pte Ltd.: Hackensack, NJ, USA, 2018. [Google Scholar]
- Dusastre, V. (Ed.) Materials for Sustainable Energy; World Scientific Publ. Co. Inc.: Hackensack, NJ, USA, 2011. [Google Scholar]
- Liu, S.; Yang, J.; Yin, L.; Li, Z.; Wang, J.; Nuli, Y. Lithium-rich Li2.6BMg0.05 alloy as an alternative anode to metallic lithium for rechargeable lithium batteries. Electrochim. Acta 2011, 56, 8900–8905. [Google Scholar] [CrossRef]
- Whittingham, M.S. Electrical energy storage and intercalation chemistry. Science 1976, 192, 1126–1127. [Google Scholar] [CrossRef]
- Ivanovskaya, V.V.; Seifert, G. Tubular structures of titanium disulfide TiS2. Solid State Commun. 2004, 130, 175–180. [Google Scholar] [CrossRef]
- Tatsumi, K.; Kawamura, T.; Higuchi, S.; Hosotubo, T.; Nakajima, H.; Sawada, A. Anode characteristics of non-graphitizable carbon fibers for rechargeable lithium-ion batteries. J. Power Sources 1997, 68, 263–266. [Google Scholar] [CrossRef]
- Yazami, R.; Touzain, P. A reversible graphite-lithium negative electrode for electrochemical generators. J. Power Sources 1983, 9, 365–371. [Google Scholar] [CrossRef]
- Kumar, R.V.; Iwahara, H. Solid electrolytes. In Handbook on the Physics and Chemistry of Rare Earths; Elsevier: Amsterdam, The Netherlands, 2000; Volume 28, pp. 131–185. [Google Scholar]
- Casas, C.; Li, W. A review of application of carbon nanotubes for lithium ion battery anode material. J. Power Sources 2012, 208, 74–85. [Google Scholar] [CrossRef]
- Jacques, E.; Kjell, M.H.; Zenkert, D.; Lindbergh, G.; Behn, M.; Willgert, M. Impact of electrochemical cycling on the tensile properties of carbon fibres for structural lithium-ion composite batteries. Compos. Sci. Technol. 2012, 72, 792–798. [Google Scholar] [CrossRef]
- Kang, K.S.; Choi, S.; Song, J.; Woo, S.; Jo, Y.N.; Choi, J.; Yim, T.; Yu, J.; Kim, Y. Effect of additives on electrochemical performance of lithium nickel cobalt manganese oxide at high temperature. J. Power Sources 2014, 253, 48–54. [Google Scholar] [CrossRef]
- Vu, A.; Stein, A. Lithium iron phosphate spheres as cathode materials for high power lithium ion batteries. J. Power Sources 2014, 245, 48–58. [Google Scholar] [CrossRef]
- Xu, J.; Dou, S.; Liu, H.; Dai, L. Cathode materials for next generation lithium ion batteries. Nano Energy 2013, 2, 439–442. [Google Scholar] [CrossRef]
- Joshi, P.; Vedarajan, R.; Sheelam, A.; Ramanujam, K.; Malaman, B.; Matsumi, N. An all solid-state Li ion battery composed of low molecular weight crystalline electrolyte. RSC Adv. 2020, 10, 8780–8789. [Google Scholar] [CrossRef]
- Yoshio, M.; Brodd, R.J.; Kozawa, A. Lithium Ion Batteries: Science and Technologies; Springer: New York, NY, USA, 2009. [Google Scholar]
- Yoshino, A. The birth of the lithium-ion battery. Anqewandte Chem. 2012, 51, 5798–5800. [Google Scholar] [CrossRef]
- Goodnough, J.B.; Park, K.S. The Li-ion rechargeable battery: A perspective. J. Am. Chem. Soc. 2013, 135, 1167–1176. [Google Scholar] [CrossRef]
- Pham, V.H.; Kim, K.H.; Jung, D.W.; Singh, K.; Oh, C.S.; Chung, J.S. Liquid phase co-exfoliated MoS2–graphene composites as anode materials for lithium ion batteries. J. Power Sources 2013, 244, 280–286. [Google Scholar] [CrossRef]
- Bahceci, S.; Esat, B. A polyacetylene derivative with pendant TEMPO group as cathode material for rechargeable batteries. J. Power Sources 2013, 242, 33–40. [Google Scholar] [CrossRef]
- Ui, K.; Fujii, D.; Niwata, Y.; Karougi, T.; Shibata, Y.; Kadoma, Y.; Shimada, K.; Kumagai, N. Analysis of solid electrolyte interface formation reaction and surface deposit of natural graphite negative electrode employing polyacrylic acid as a binder. J. Power Sources 2014, 247, 981–990. [Google Scholar] [CrossRef]
- Teragawa, S.; Aso, K.; Tadanaga, K.; Hayashi, A.; Tatsumisago, M. Preparation of Li2S–P2S5 solid electrolyte from N-methylformamide solution and application for all-solid-state lithium battery. J. Power Sources 2014, 248, 939–942. [Google Scholar] [CrossRef]
- Xiong, M.; Tang, H.; Wang, Y.; Pan, M. Ethylcellulose-coated polyolefin separators for lithium-ion batteries with improved safety performance. Carbohydr. Polym. 2014, 101, 1140–1146. [Google Scholar] [CrossRef]
- Cho, E.; Mum, J.; Chae, O.B.; Kwon, O.M.; Kim, H.T.; Ryu, J.; Kim, Y.G.; Seung, M. Corrosion/passivation of aluminum current collector in bis (fluorosulfonyl) imide-based ionic liquid for lithium-ion batteries. Electrochem. Commun. 2012, 22, 1–3. [Google Scholar] [CrossRef]
- Hao, J.; Lei, G.; Li, Z.; Wu, L.; Xiao, Q.; Wang, L. A novel polyethylene terephthalate nonwoven separator based on electrospinning technique for lithium ion battery. J. Memb. Sci. 2013, 428, 11–16. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L.C.; Cheng, J.; Ding, C.; Chen, C. Watermelon used as a novel carbon source to improve the rate performance of iron oxide electrodes for lithium ion batteries. Electrochim. Acta 2013, 102, 306–311. [Google Scholar] [CrossRef]
- Rojas, D.M.; Cabanas, M.C.; Baudrin, E. Effect of particle size and cell parameter mismatch on the lithium insertion/deinsertion processes into RuO2. Solid State Ion. 2010, 181, 536–544. [Google Scholar] [CrossRef]
- Zang, J.; Zhao, Y. Silicon nanowire reinforced by single-walled carbon nanotube and its applications to anti-pulverization electrode in lithium ion battery. Compos. Part B Eng. 2012, 43, 76–82. [Google Scholar] [CrossRef]
- Sangare, M.; Fodjouong, G.J.; Huang, X. Graphene-encapsulated silicon nanoparticles as an anode material for lithium-ion batteries. Mendeleev Commun. 2013, 23, 284–285. [Google Scholar] [CrossRef]
- Schalkwijk, W.; Scrosati, B. Advances in lithium ion batteries introduction. In Advances in Lithium-Ion Batteries; Academic Plenum: New York, NY, USA, 2002. [Google Scholar]
- Kucinski, G.; Bajars, G.; Kleperis, J. Graphene in lithium ion battery cathode materials: A review. J. Power Sources 2013, 240, 66–79. [Google Scholar] [CrossRef]
- Chiao, T.; Lien, P.; Morgan, R.; Reifsnider, K.; Crossman, F. ADV. J. Compos. Technol. Res. 1979, 1, 8. [Google Scholar] [CrossRef]
- Wu, Y.P.; Rahm, E.; Holze, R. Carbon anode materials for lithium ion batteries. J. Power Sources 2003, 114, 228–236. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, S.; Sun, Y.; Parvez, K.; Feng, X.; Mullen, K. 3D Nitrogen-Doped Graphene Aerogel-Supported Fe3O4 Nanoparticles as Efficient Electrocatalysts for the Oxygen Reduction Reaction. J. Am. Chem. Soc. 2012, 134, 9082–9085. [Google Scholar] [CrossRef]
- Wu, Z.S.; Ren, W.; Xu, L.; Li, F.; Cheng, H.M. Doped graphene sheets as anode materials with superhigh rate and large capacity for lithium ion batteries. ACS Nano 2011, 5, 5463–5471. [Google Scholar] [CrossRef]
- Cui, S.; Mao, S.; Lu, G.; Chen, J. Graphene coupled with nanocrystals: Opportunities and challenges for energy and sensing applications. J. Phys. Chem. Lett 2013, 4, 2441–2454. [Google Scholar] [CrossRef]
- Liu, C.-Z.; Luo, Y.; Limbu, S.M.; Chen, L.; Du, Z.-Y. IGF-1 induces SOCS-2 but not SOCS-1 and SOCS-3 transcription in juvenile Nile tilapia (Oreochromis niloticus). J. Exp. Biol. 2018, 221, jeb179291. [Google Scholar] [CrossRef]
- Chen, H.; Xiao, Y.; Wang, L.; Yang, Y. Silicon nanowires coated with copper layer as anode materials for lithium-ion batteries. J. Power Sources 2011, 196, 6657–6662. [Google Scholar] [CrossRef]
- Há, J.; Paik, U. Hydrogen treated, cap-opened Si nanotubes array anode for high power lithium ion battery. J. Power Sources 2013, 244, 463–468. [Google Scholar] [CrossRef]
- Wu, P.; Du, N.; Zhang, H.; Yu, J.; Yang, D. CNTs@SnO2@C Coaxial Nanocables with Highly Reversible Lithium Storage. J. Phys. Chem. C 2010, 114, 22535–22538. [Google Scholar] [CrossRef]
- Yao, Y.; Matthew, T.; Wu, M.D.I.R.H.; Liu, N.; Hu, L.; Nix, W.D.; Cui, Y. Interconnected silicon hollow nanospheres for lithium-ion battery anodes with long cycle life. Nano Lett. 2011, 11, 2949–2954. [Google Scholar] [CrossRef]
- Zhou, X.; Yin, Y.; Cao, A.; Wan, L.; Guo, Y. Efficient 3D conducting networks built by graphene sheets and carbon nanoparticles for high-performance silicon anode. ACS Appl. Matter. Interfaces 2012, 4, 2824–2828. [Google Scholar] [CrossRef]
- Chou, S.; Wang, J.; Choucair, M.; Liu, H.; Stride, J.A.; Dou, S. Enhanced reversible lithium storage in a nanosize silicon/graphene composite. Electrochem. Commun. 2010, 12, 303–306. [Google Scholar] [CrossRef]
- Wang, J.; Zhong, C.; Chou, S.; Liu, H. Flexible free-standing graphene-silicon composite film for lithium-ion batteries. Electrochem. Commun. 2010, 12, 1467–1470. [Google Scholar] [CrossRef]
- Lee, J.K.; Smith, K.B.; Hayner, C.M.; Kung, H.H. Silicon nanoparticles–graphene paper composites for Li ion battery anodes. Chem. Commun. 2010, 12, 2025–2027. [Google Scholar] [CrossRef]
- Lai, S.Y.; Maehlen, J.P.; Preston, T.J.; Skare, M.O.; Nagell, M.U.; Ulvestad, A.; Lemordant, D.; Koposov, A.Y. Morphology Engineering of silicon nanoparticles for better performance in Li-ion battery anodes. Nanoscale Adv. 2020. [Google Scholar] [CrossRef]
- Xiang, H.; Zhang, K.; Ji, G.; Lee, J.Y.; Zou, C.; Chen, X.; Wu, J. Graphene/nanosized silicon composites for lithium battery anodes with improved cycling stability. Carbon 2011, 49, 1787–1796. [Google Scholar] [CrossRef]
- Yang, S.; Feng, X.; Ivanovici, S.; Mullen, K. Fabrication of graphene-encapsulated oxide nanoparticles: Towards high-performance anode materials for lithium storage. Chem. Int. Ed. 2010, 49, 8408–8411. [Google Scholar] [CrossRef]
- Sheng, Y.Y.; Xie, X.; Rick, J.; Chang, F.; Hwang, B. Improved anode materials for lithium-ion batteries comprise non-covalently bonded graphene and silicon nanoparticles. J. Power Sources 2014, 247, 991–998. [Google Scholar]
- Zhu, X.; Ye, J.; Lu, Y.; Jia, X. 3D graphene nanostructure composed of porous carbon sheets and interconnected nanocages for high-performance lithium-ion battery anodes and lithium sulphur batteries. ACS Sustain. Chem. Eng. 2019, 7, 11241–11249. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kumar, K.; Fisher, F.T.; Yang, E.H. Out-of-plane growth of CNTs on graphene for supercapacitor applications. Nanotechnology 2012, 23, 015301. [Google Scholar] [CrossRef]
- Bre, S.; Karthikeyan, K.; Lee, Y.; I-Kwon, O.I. Microwave self-assembly of 3D graphene-carbon nanotube-nickel nanostructure for high capacity anode material in lithium ion battery. Carbon 2013, 64, 527–536. [Google Scholar]
- Smith, A.J.; Burns, J.C.; Trassler, S.; Dahn, J.R. Precision measurements of the coulombic efficiency of lithium-ion batteries and of electrode materials for lithium-ion batteries. J. Electrochem Soc. 2010, 157, 196–202. [Google Scholar] [CrossRef]
- Russo, P.; Hul, A.; Compagnini, G. Synthesis, properties and potential applications of porous graphene: A review. Nano-Micro Lett. 2013, 5, 260–273. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, D.; Wang, H.G.; Wu, Z.; Zhang, X. In Situ Fabrication of Porous Graphene Electrodes for High-Performance Energy Storage. Nano 2013, 7, 2422–2430. [Google Scholar] [CrossRef]
- Blomgren, G.E. The development and future of lithium ion batteries. J. Electrochem. Soc. 2017, 164, A5019–A5025. [Google Scholar] [CrossRef]
- Conway, B.E. Electrochemical Supercapacitons, Scientific Fundamentals and Technological Applications; Kluwer Academic/Plennum Publishers: New York, NY, USA, 1999. [Google Scholar]
- Simon, P.; Burke, A. Nanostructured carbons: Double-layer capacitance and more. Interface 2008, 17, 38–43. [Google Scholar]
- Béguin, F.; Frackowiak, E. (Eds.) Carbon Materials for Electrochemical Energy Storage Systems; CRC Press: Baton Rouge, LA, USA, 2009. [Google Scholar]
- Zhou, M.; Gallegoa, A.; Liu, K.; Dai, S.; Wu, J. Insights from machine learning of carbon electrodes for electric double layer capacitors. Carbon 2019. [Google Scholar] [CrossRef]
- Ratajazak, P.; Suss, M.E.; Kaasic, F.; Béguin, F. Carbon electrodes for capacitive technologies. Energy Storage Mater. 2019, 16, 126–145. [Google Scholar] [CrossRef]
- Janes, A.; Kurig, H.; Lust, E. Characterisation of activated nanoporous carbon for supercapacitor electrode materials. Carbon 2007, 45, 1226–1233. [Google Scholar] [CrossRef]
- Alonso, A.; Ruiz, V.; Blanco, C.; Santamaria, R.; Granda, M.; Menendez, R.; Jager, S.G.E. Activated carbon produced from Sasol-Lurgi gasifier pitch and its application as electrodes in supercapacitors. Carbon 2006, 44, 441–446. [Google Scholar] [CrossRef]
- Rufford, T.E.; Hulicova-Jurcakova, D.; Zhu, Z.; Lu, G.Q. Nanoporous carbon electrode from waste coffee beans for high performance supercapacitors. Electrochem. Commun. 2008, 10, 1594–1597. [Google Scholar] [CrossRef]
- Ito, E.; Mozia, S.; Okuda, M.; Nakano, T.; Toyoda, M.; Inagaki, M. Nanoporous carbons from cypress II. Application to electric double layer capacitors. New Carbon Mater. 2007, 22, 321–326. [Google Scholar] [CrossRef]
- Kim, Y.; Abe, Y.; Yanagiura, T.; Park, K.; Shimizu, M.; Iwazaki, T.; Nakagawa, S.; Endo, M.; Dresselhaus, M.S. Easy preparation of nitrogen-enriched carbon materials from peptides of silk fibroins and their use to produce a high volumetric energy density in supercapacitors. Carbon 2007, 45, 2116–2125. [Google Scholar] [CrossRef]
- Xu, B.; Wu, F.; Chen, R.; Cao, G.; Chen, S.; Wang, G.; Yang, Y. Room temperature molten salt as electrolyte for carbon nanotube-based electric double layer capacitors. J. Power Sources 2006, 158, 773–778. [Google Scholar] [CrossRef]
- Shiraishi, S.; Miyauchi, T.; Sasaki, R.; Nishina, N.; Oya, A.; Hagiwara, R. Electric double layer capacitance of activated carbon nanofibers in ionic liquid: EMImBF4. Electrochemistry 2007, 75, 619–621. [Google Scholar] [CrossRef]
- Snyder, J.F.; Wong, E.L.; Hubbard, C.W. Evaluation of commercially available carbon fibers, fabrics, and papers for potential use in multifunctional energy storage applications. J. Electrochem. Soc. 2009, 156, A215–A224. [Google Scholar] [CrossRef]
- Toyoda, M.; Inagaki, M. Exfoliation of carbon fibers☆☆ Keynote Lecture. J. Phys. Chem. Solids 2004, 65, 109–117. [Google Scholar] [CrossRef]
- Toyoda, M.; Tani, Y.; Soneda, Y. Exfoliated carbon fibers as an electrode for electric double layer capacitors in a 1 mol/dm3 H2SO4 electrolyte. Carbon 2004, 42, 2833–2837. [Google Scholar] [CrossRef]
- Soneda, Y.; Yamashita, J.; Kodama, M.; Hatori, H.; Inagaki, M.T.M. Pseudo-capacitance on exfoliated carbon fiber in sulfuric acid electrolyte. Appl. Phys. A 2006, 82, 575–578. [Google Scholar] [CrossRef]
- Nishihara, H.; Itoi, H.; Kogure, T.; Hore, P.; Touhara, H.; Okino, F.; Kyotani, T. Investigation of the ion storage/transfer behavior in an electrical double-layer capacitor by using ordered microporous carbons as model materials. Chem. Eur. J. 2009, 15, 5355–5363. [Google Scholar] [CrossRef]
- Portet, C.; Korenblit, Z.; Gogotsi, Y.; Mokaya, R.; Yushin, G. Electrical double-layer capacitance of zeolite-templated carbon in organic electrolyte. J. Electrochem. Soc. 2009, 156, A1–A6. [Google Scholar] [CrossRef]
- Wang, H.; Gao, Q.; Hu, J.; Chen, Z. High performance of nanoporous carbon in cryogenic hydrogen storage and electrochemical capacitance. Carbon 2009, 47, 2259–2268. [Google Scholar] [CrossRef]
- Sevilla, M.; Alvarez, S.; Centeno, T.A.; Fuertes, A.B.; Stoeckli, F. Performance of templated mesoporous carbons in supercapacitors. Electrochim. Acta 2007, 52, 3207–3215. [Google Scholar] [CrossRef]
- Shiraishi, S.; Kurihara, H.; Tsubota, H.; Soneda, A.; Yamada, Y. Electric double layer capacitance of highly porous carbon derived from lithium metal and polytetrafluoroethylene. Electrochem. Solid-State Lett. 2001, 4, AA8. [Google Scholar] [CrossRef]
- Yamada, Y.; Tanaike, O.; Liang, T.-T.; Hatori, H.; Shiraishi, S.; Oya, A. Electric double layer capacitance performance of porous carbons prepared by defluorination of polytetrafluoroethylene with potassium. Electrochem. Solid-State Lett. 2002, 5, A283–A285. [Google Scholar] [CrossRef]
- Chmiola, J.; Yushim, G.; Dash, R.; Gogotsi, Y. Effect of pore size and surface area of carbide derived carbons on specific capacitance. J. Power Sources 2006, 158, 765–772. [Google Scholar] [CrossRef]
- Chmiola, J.; Yushim, G.; Gogotsi, Y.; Porter, C.; Simon, P.; Taberna, P.L. Anomalous increase in carbon capacitance at pore sizes less than 1 nanometer. Science 2006, 313, 1760–1763. [Google Scholar] [CrossRef]
- Lin, R.; Taberna, P.L.; Chmiola, J.; Guay, D.; Gogotsi, Y.; Simon, P. Microelectrode study of pore size, ion size, and solvent effects on the charge/discharge behavior of microporous carbons for electrical double-layer capacitors. J. Electrochem. Soc. 2009, 156, A7–A12. [Google Scholar] [CrossRef]
- Wang, H.; Gao, Q. Synthesis characterization and energy-related applications of carbide-derived carbons obtained by the chlorination of boron carbide. Carbon 2009, 47, 820–828. [Google Scholar] [CrossRef]
- Tao, Y.; Endo, H.; Kaneko, K. A review of synthesis and nanopore stuctures of organic polymer aerogels and carbon aerogels. Recent Pat. Chem. Eng. 2008, 1, 192–200. [Google Scholar] [CrossRef]
- Fang, B.; Wei, Y.-Z.; Maruyama, K.; Kumagai, M. High capacity supercapacitors based on modified activated carbon aerogel. J. Appl. Electrochem. 2005, 35, 229–233. [Google Scholar] [CrossRef]
- Frackowiak, E.; Jurewicz, K.; Deljeu, S.; Béguim, F. Nanotubular materials for supercapacitors. J. Power Sources 2001, 97–98, 822–825. [Google Scholar] [CrossRef]
- Liu, G.C.; Fang, H.T.; Li, F.; Liu, M.; Cheng, H.M. Single-walled carbon nanotubes modified by electrochemical treatment for application in electrochemical capacitors. J. Power Sources 2006, 160, 758–761. [Google Scholar] [CrossRef]
- Tanaike, O.; Futaba, D.N.; Hata, K.; Hatori, H. Supercapacitors using pure single-walled carbon nanotubes. Carbon Lett. 2009, 10, 90–93. [Google Scholar] [CrossRef]
- Miraoka, T.; Najafabadi, A.I.; Yamada, T.; Futaba, D.N.; Yasuda, S.; Tanaike, O.; Matori, H.; Yumura, M.; Iijama, S.; Hata, K. Compact and Light Supercapacitor Electrodes from a Surface-Only Solid by Opened Carbon Nanotubes with 2 200 m2 g−1 Surface Area. Adv. Funct. Mater. 2009, 20, 422–428. [Google Scholar]
- Yamada, Y.; Kimizuka, O.; Tanaika, O.; Mochida, K.; Suematsu, S.; Tamamitsu, K.; Saeki, S.; Hatori, H. Capacitor properties and pore structure of single-and double-walled carbon nanotubes. Electrochem. Solid-State Lett. 2009, 12, K14–K16. [Google Scholar] [CrossRef]
- Emmenegger, C.; Mauron, P.; Sudan, P.; Wenger, P.; Hermann, V.; Gallay, R.; Zuttel, A. Investigation of electrochemical double-layer (ECDL) capacitors electrodes based on carbon nanotubes and activated carbon materials. J. Power Sources 2003, 124, 321–329. [Google Scholar] [CrossRef]
- Honda, Y.; Haramoto, T.; Takeshige, M.; Shiazoki, H.; Kitamura, T.; Ishikawa, M. Aligned MWCNT sheet electrodes prepared by transfer methodology providing high-power capacitor performance. Electrochem. Solid-State Lett. 2007, 10, A106–A110. [Google Scholar] [CrossRef]
- Lu, W.; Qu, L.; Henry, K.; Dai, L. High performance electrochemical capacitors from aligned carbon nanotube electrodes and ionic liquid electrolytes. J. Power Sources 2009, 189, 1270–1277. [Google Scholar] [CrossRef]
- Conway, B.E.; Birss, V.; Wojtowicz, J. The role and utilization of pseudocapacitance for energy storage by supercapacitors. J. Power Sources 1997, 66, 1–14. [Google Scholar] [CrossRef]
- Ruiz, V.; Blanco, C.; Pinero, E.R.; Kohmenko, V.; Béguin, F.; Santamaria, R. Effects of thermal treatment of activated carbon on the electrochemical behaviour in supercapacitors. Electrochim. Acta 2007, 52, 4969–4973. [Google Scholar] [CrossRef]
- Okajima, K.; Ohta, K.; Sudoh, M. Capacitance behavior of activated carbon fibers with oxygen-plasma treatment. Electrochim Acta 2005, 50, 2227–2231. [Google Scholar] [CrossRef]
- Oda, H.; Yamashita, A.; Minoura, S.; Okamoto, M.; Morimoto, T. Modification of the oxygen-containing functional group on activated carbon fiber in electrodes of an electric double-layer capacitor. J. Power Sources 2006, 158, 1510–1516. [Google Scholar] [CrossRef]
- Kodame, M.; Yamashita, J.; Soneda, Y.; Hatori, H.; Kamegawa, K. Preparation and electrochemical characteristics of N-enriched carbon foam. Carbon 2007, 45, 1105–1107. [Google Scholar] [CrossRef]
- Jurewicz, K.; Pietrzak, R.; Nowicki, P.; Wachowska, H. Capacitance behaviour of brown coal based active carbon modified through chemical reaction with urea. Electrochim. Acta 2008, 53, 5469–5475. [Google Scholar] [CrossRef]
- Hulicova, D.; Kodama, M.; Hatori, H. Electrochemical performance of nitrogen-enriched carbons in aqueous and non-aqueous supercapacitors. Chem. Mater. 2006, 18, 2318–2326. [Google Scholar] [CrossRef]
- Hulicova, D.; Yamashita, J.; Soneda, Y.; Hatori, H.; Kodama, M. Supercapacitors prepared from melamine-based carbon. Chem. Mater. 2005, 17, 1241–1247. [Google Scholar] [CrossRef]
- Yamada, Y.; Tanaike, O.; Shiraishi, S. Preparation of porous carbon by defluorination of PTFE and its application to electric double layer capacitor. TANSO 2004, 215, 285–294. [Google Scholar] [CrossRef]
- Kodama, M.; Yamashita, J.; Soneda, Y.; Hatori, H.; Kamegawa, K.; Moriguchi, I. Structure and electrochemical capacitance of nitrogen-enriched mesoporous carbon. Chem. Lett. 2006, 35, 680–681. [Google Scholar] [CrossRef]
- Li, W.; Chen, D.; Li, Z.; Shi, Y.; Wang, Y.; Huang, J.; Zhao, D.; Jiang, Z. Nitrogen enriched mesoporous carbon spheres obtained by a facile method and its application for electrochemical capacitor. Electrochim. Commun. 2007, 9, 569–573. [Google Scholar] [CrossRef]
- Konno, H.; Onishi, H.; Yoshizawa, N.; Azumi, K. MgO-templated nitrogen-containing carbons derived from different organic compounds for capacitor electrodes. J. Power Sources 2010, 195, 667–673. [Google Scholar] [CrossRef]
- Seredych, M.; Hulicova-Jurcakova, D.; Lu, G.Q.; Bandosz, T.J. Surface functional groups of carbons and the effects of their chemical character, density and accessibility to ions on electrochemical performance. Carbon 2008, 46, 1475–1488. [Google Scholar] [CrossRef]
- Du, M.P.G.F.; Xia, Z.H.; Durstock, M.; Dai, L.M. Nitrogen-doped carbon nanotube arrays with high electrocatalytic activity for oxygen reduction. Science 2009, 323, 760–764. [Google Scholar]
- Tang, C.; Wang, H.F.; Chen, X.; Li, B.Q.; Hou, T.Z.; Zhang, B.; Zhang, Q.; Titirici, M.M.; Wei, F. Topological defects in metal-free nanocarbon for oxygen electrocatalysis. Adv. Mater. 2016, 28, 6845–6851. [Google Scholar] [CrossRef]
- Li, H.B.; Kang, W.J.; Wang, L.; Yue, Q.L.; Xu, S.L.; Wang, H.S.; Liu, J.F. Synthesis of three-dimensional flowerlike nitrogen-doped carbons by a copyrolysis route and the effect of nitrogen species on the electrocatalytic activity in oxygen reduction reaction. Carbon 2013, 54, 249–257. [Google Scholar] [CrossRef]
- Liu, J.; Song, P.; Xu, W. Structure-activity relationship of doped-nitrogen (N)-based metal-free active sites on carbon for oxygen reduction reaction. Carbon 2017, 115, 763–772. [Google Scholar] [CrossRef]
- Guo, D.H.; Shibuya, R.; Akida, C.; Saji, S.; Kondo, T.; Nakamura, J. Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts. Science 2016, 351, 361–365. [Google Scholar] [CrossRef]
- Shiraishi, S.; Kibe, M.; Yokoyama, T.; Kurihara, H.; Patel, N.; Oya, A.; Kaburagi, Y.; Hishiyama, Y. Electric double layer capacitance of multi-walled carbon nanotubes and B-doping effect. Appl. Phys. A 2006, 82, 585–591. [Google Scholar] [CrossRef]
- Wang, D.-W.; Li, F.; Chen, Z.-G.; Lu, G.Q.; Cheng, H.-M. Synthesis and electrochemical property of boron-doped mesoporous carbon in supercapacitor. Chem. Mater. 2008, 20, 7195–7200. [Google Scholar] [CrossRef]
- Ito, T.; Ushiro, M.; Fushimi, K.; Azumi, K.; Konno, H. Preparation of boron-containing carbons from glucose–borate complexes and their capacitive performance. TANSO 2009, 239, 156–161. [Google Scholar] [CrossRef]
- Guo, H.; Gao, Q. Boron and nitrogen co-doped porous carbon and its enhanced properties as supercapacitor. J. Power Sources 2009, 186, 551–556. [Google Scholar] [CrossRef]
- Konno, H.; Ito, T.; Ushiro, M.; Fushimi, K.; Azumi, K. High capacitance B/C/N composites for capacitor electrodes synthesized by a simple method. J. Power Sources 2010, 195, 1739–1746. [Google Scholar] [CrossRef]
- Sefehri, S.; Garcia, B.B.; Zhang, Q.; Cao, G. Enhanced electrochemical and structural properties of carbon cryogels by surface chemistry alteration with boron and nitrogen. Carbon 2009, 47, 1436–1443. [Google Scholar]
- Chang, H.; Hu, C.C. Hydrothermal synthesis of hydrous crystalline RuO2 nanoparticles for supercapacitors. Electrochem. Solid-State Lett. 2004, 7, A466–A469. [Google Scholar] [CrossRef]
- Kim, H.; Popov, B.N. Characterization of hydrous ruthenium oxide/carbon nanocomposite supercapacitors prepared by a colloidal method. J. Power Sources 2002, 104, 52–61. [Google Scholar] [CrossRef]
- Wu, N.L.; Wang, S.Y.; Han, C.Y.; Wu, D.S.; Shiue, L.R. Electrochemical capacitor of magnetite in aqueous electrolytes. J. Power Sources 2003, 113, 173–178. [Google Scholar] [CrossRef]
- Ushiro, M.; Yoneda, A.; Iwasa, N.; Fushimi, K.; Konno, H. Capacitance behavior of carbonaceous materials derived from Kapton films containing small amounts of metallic compounds. TANSO 2009, 238, 121–125. [Google Scholar] [CrossRef]
- Wang, Z.; Sanchez, A.; Herkersdorf, A. SciSim: A software performance estimation framework using source code instrumentation. In Proceedings of the 7th International Workshop on Software and Performance—WOSP’08, Princeton, NJ, USA, 23–26 June 2008; Volume 46, p. 491. [Google Scholar] [CrossRef]
- Ruiz, V.; Blanco, C.; Granda, M.; Santamaria, R. Enhanced life-cycle supercapacitors by thermal treatment of mesophase-derived activated carbons. Electrochim. Acta 2008, 54, 305–310. [Google Scholar] [CrossRef]
- Aida, T.; Murayama, I.; Yamada, K.; Morita, M. High-energy-density hybrid electrochemical capacitor using graphitizable carbon activated with KOH for positive electrode. J. Power Sources 2007, 166, 462–470. [Google Scholar] [CrossRef]
- Khonmenko, V.; Raymundo-Pinero, E.; Bégiun, F. Optimisation of an asymmetric manganese oxide/activated carbon capacitor working at 2 V in aqueous medium. J. Power Sources 2006, 153, 183–190. [Google Scholar] [CrossRef]
- Wang, D.W.; Li, F.; Chang, H.M. Hierarchical porous nickel oxide and carbon as electrode materials for asymmetric supercapacitor. J. Power Sources 2008, 185, 1563–1568. [Google Scholar] [CrossRef]
- Machida, K.; Suematsu, S.; Ishimoto, S.; Tamamitsuo, K. High-voltage asymmetric electrochemical capacitor based on polyfluorene nanocomposite and activated carbon. J. Electrochem. Soc. 2008, 155, A970–A974. [Google Scholar] [CrossRef]
- Pasquier, A.D.; Plitz, I.; Menocal, S.; Amatucci, G.G. A comparative study of Li-ion battery, supercapacitor and nonaqueous asymmetric hybrid devices for automotive applications. J. Power Sources 2003, 115, 171–178. [Google Scholar] [CrossRef]
- Rolison, D.L.; Nazar, L.F. Electrochemical energy storage to power the 21st century. MRS Bull. 2011, 36, 486–493. [Google Scholar] [CrossRef]
- Numan, A.; Duraisamy, N.; Omar, F.S.; Goji, D.; Ramehi, K.; Ramesh, S. Sonochemical synthesis of nanostructured nickel hydroxide as an electrode material for improved electrochemical energy storage application. Prog. Nat. Sci. Mater. Int. 2017, 27, 416–423. [Google Scholar] [CrossRef]
- Conway, B.E. Electrochemical Supercapacitors: Scientific Fundamentals and Technological Applications; Springer: New York, NY, USA, 2013. [Google Scholar]
- Pina, A.C.; Amaya, A.; Marcuzzo, Y.S.; Rodrigues, A.C.; Baldan, M.R.; Tancredi, N.; Cuña, A. Supercapacitor electrode based on activated carbon wool felt. J. Carbon Res. C 2018, 4, 24. [Google Scholar] [CrossRef]
- Shen, P.K.; Wang, C.Y.; Jiang, S.P.; Sun, X.; Zhang, J. Electrochemical Energy: Advanced Materials and Technologies; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Shao, H.; Padmanathan, N.; McNulty, D.; Dwyer, C.O.; Razeeb, K.M. Supercapattery Based on Binder-Free Co3(PO4)2·8H2O Multilayer Nano/Microflakes on Nickel Foam. ACS Appl. Mater. Interfaces 2016, 8, 28592–28598. [Google Scholar] [CrossRef]
- Chen, G.Z. Supercapacitor and supercapattery as emerging electrochemical energy stores. Int. Mater. Rev. 2017, 62, 173–202. [Google Scholar] [CrossRef]
- Zhang, J.; Terrones, M.; Park, C.R.; Mukherjee, R.; Monthioux, M.; Koratkar, N.; Kim, Y.S.; Hurt, R.; Frackowiak, E.; Enoki, T. Carbon science in 2016: Status, challenges and perspectives. Carbon 2016, 98, 708–712. [Google Scholar] [CrossRef]
- Dubal, D.; Ayyad, O.; Ruiz, V.; Gomez-Romero, P. Hybrid energy storage: The merging of battery and supercapacitor chemistries. Chem. Soc. Rev. 2015, 44, 1777–1790. [Google Scholar] [CrossRef]
- Miller, E.E.; Hua, Y.; Tezel, F. Materials for energy storage: Review of electrode materials and methods of increasing capacitance for supercapacitors. J. Energy Storage 2018, 20, 30–40. [Google Scholar] [CrossRef]
- Wang, J.; Nie, P.; Ding, B.; Dong, S.; Hao, X.; Dou, H.; Zhang, X. Biomass derived carbon for energy storage devices. J Mater. Chem. 2017, 5, 2411–2428. [Google Scholar] [CrossRef]
- Liu, F.; Lee, C.W.; Im, J. Graphene-based carbon materials for electrochemical energy storage. J. Nanomater. 2013, 2013, 642915. [Google Scholar] [CrossRef]
- Siwal, S.S.; Zhang, Q.; Devi, N.; Thakur, V.K. Carbon-based polymer nanocomposite for high-performance energy storage applications. Polymers 2020, 12, 505. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lin, J.; Shen, Z.X. Polyaniline (PANi) based electrode materials for energy storage and conversion. J. Sci. Adv. Mater. Devices 2016, 1, 225–255. [Google Scholar] [CrossRef]
- Li, R.; Zhou, Y.; Li, W.; Zhu, J.; Huang, W. Structure enginnering in biomass-derived carbon materials for electrochemical energy storage. Research 2020, 2020, 8685436. [Google Scholar] [CrossRef]
- Castro-Gutiérrez, J.; Celzard, A.; Fierro, V. Energy storage in supercapacitors: Focus on tannin-derived carbon electrodes. Front. Mater. 2020, 7, 217. [Google Scholar] [CrossRef]
- Iqbal, S.; Khatoon, H.; Pandit, A.H.; Ahmad, S. REcent development of carbon based materials for energy storage devices. Mater. Sci. Energy Technol. 2019, 2, 417–428. [Google Scholar] [CrossRef]
- Dubey, R.; Guruviah, V. Review of carbon-based electrode materials for supercapacitor energy storage. Ionics 2019, 25, 1419–1445. [Google Scholar] [CrossRef]
- Liu, Z.; Fu, L.; Wang, F.; Chen, Y.; Wu, Y. Three-dimensional ordered porous electrode materials for electrochemical energy storage. NPG Asia Mater. 2019, 11, 12. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
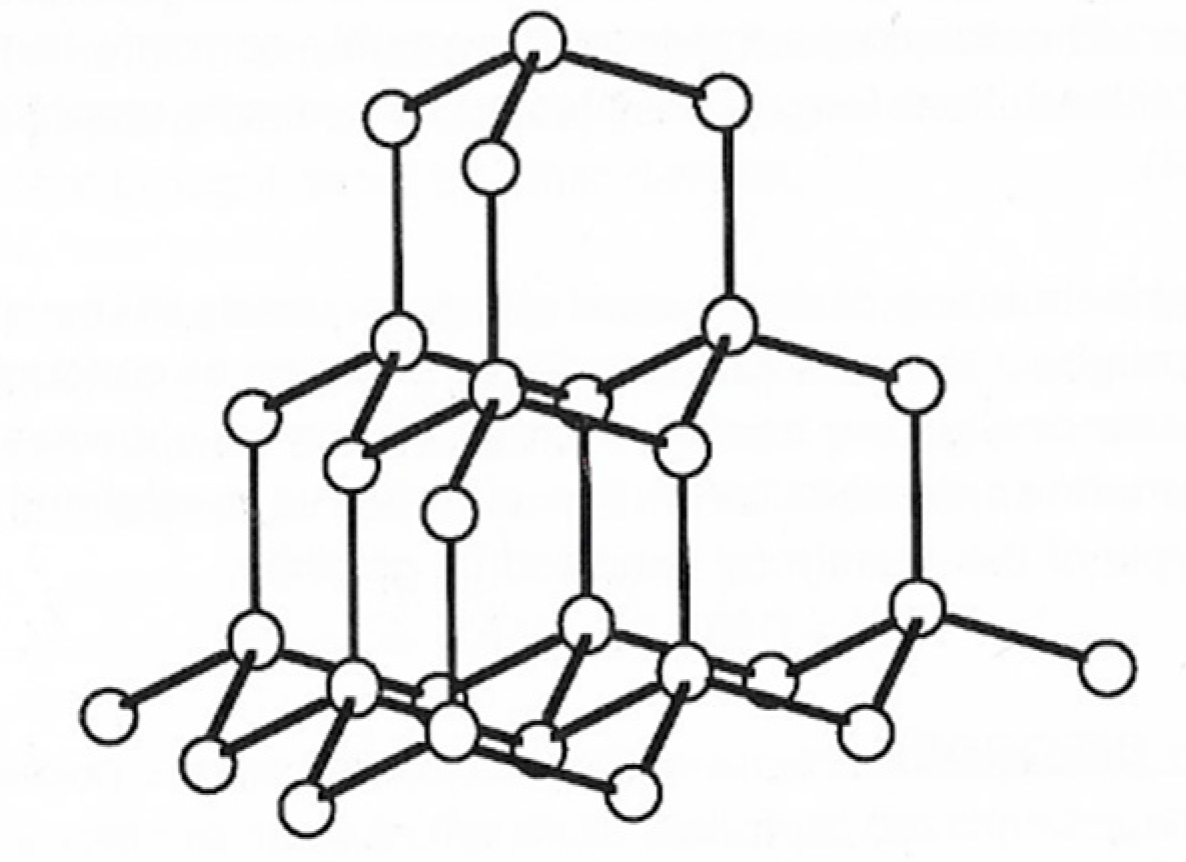{kind=link}
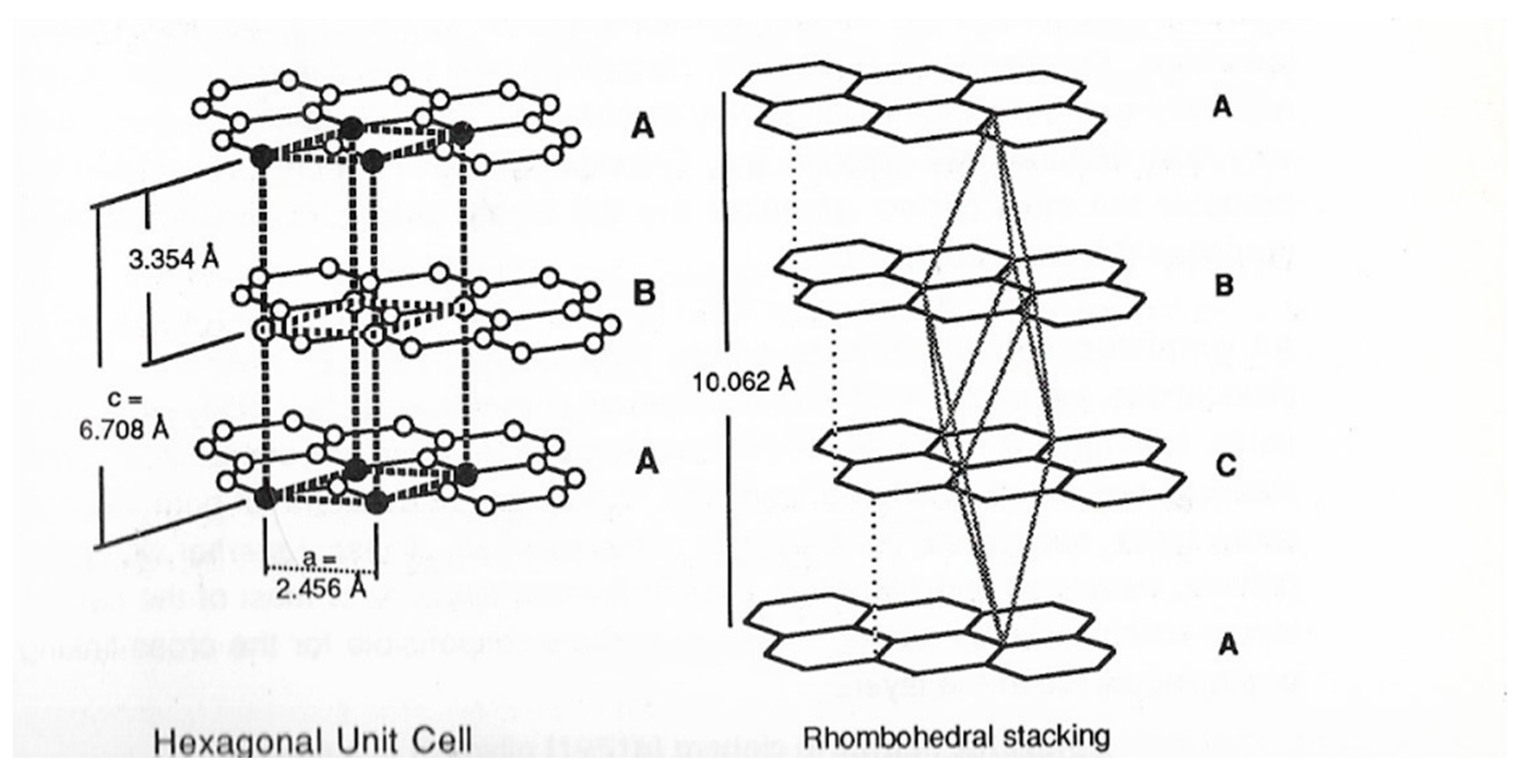{kind=link}
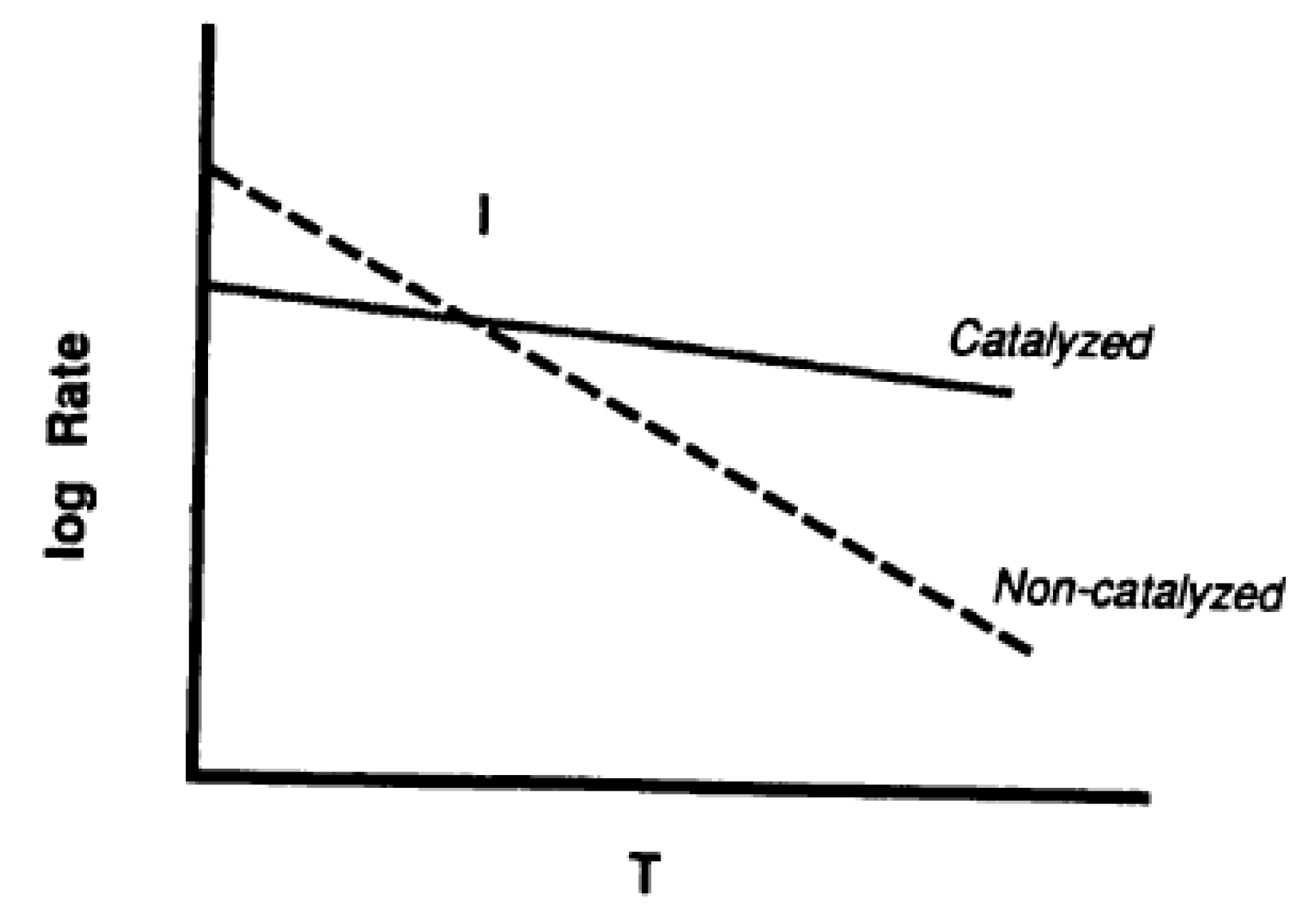{kind=link}
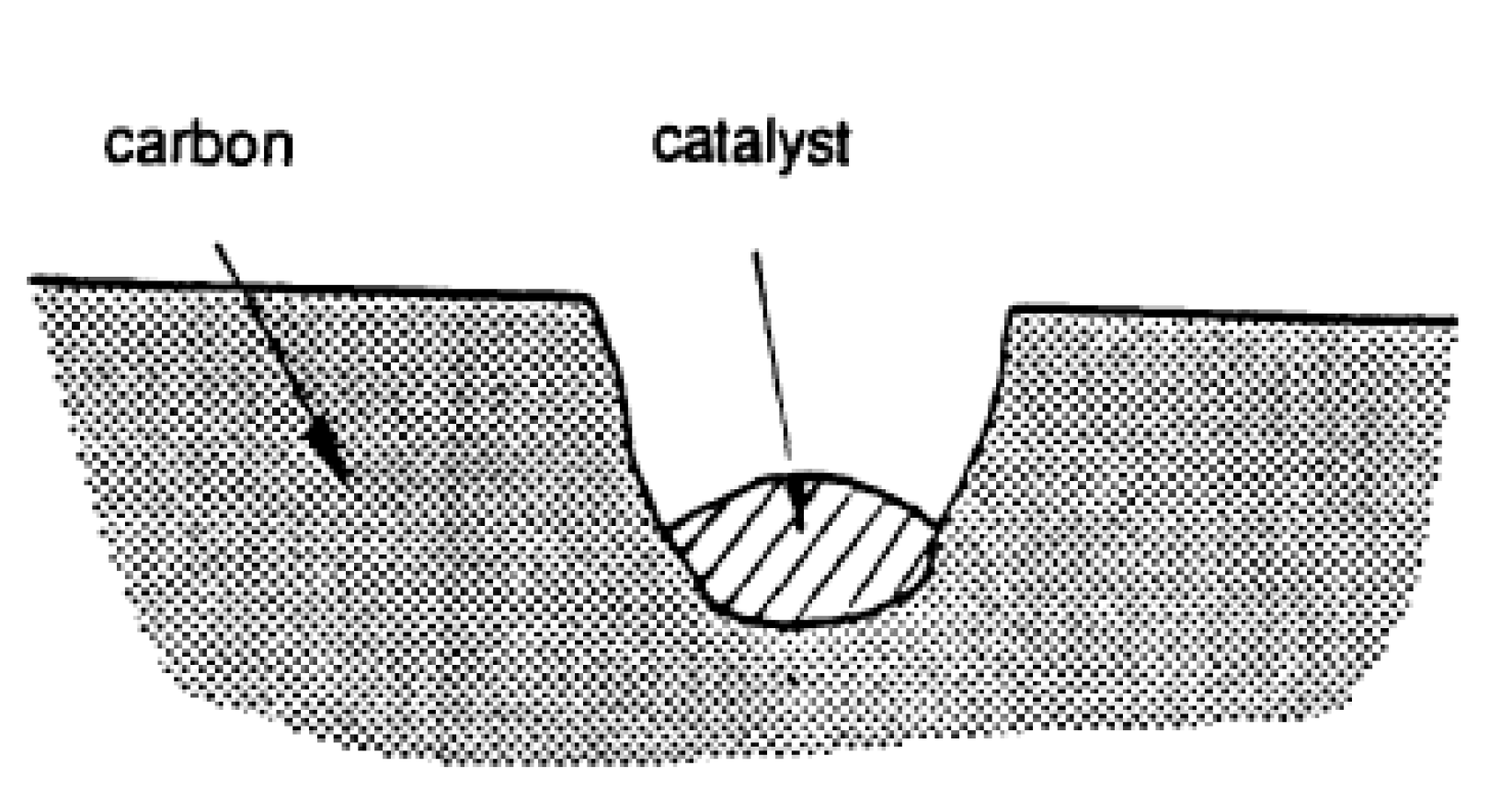{kind=link}
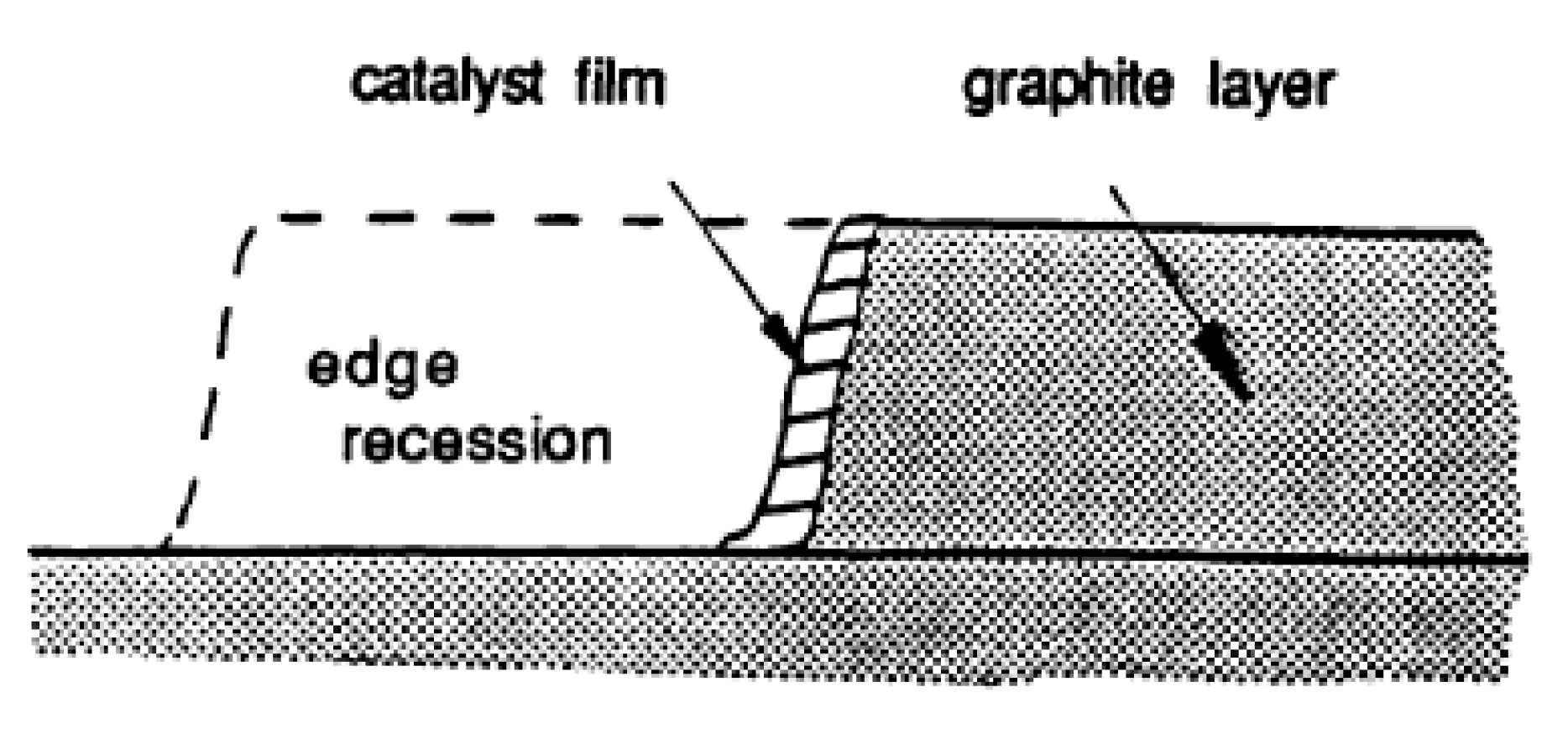{kind=link}
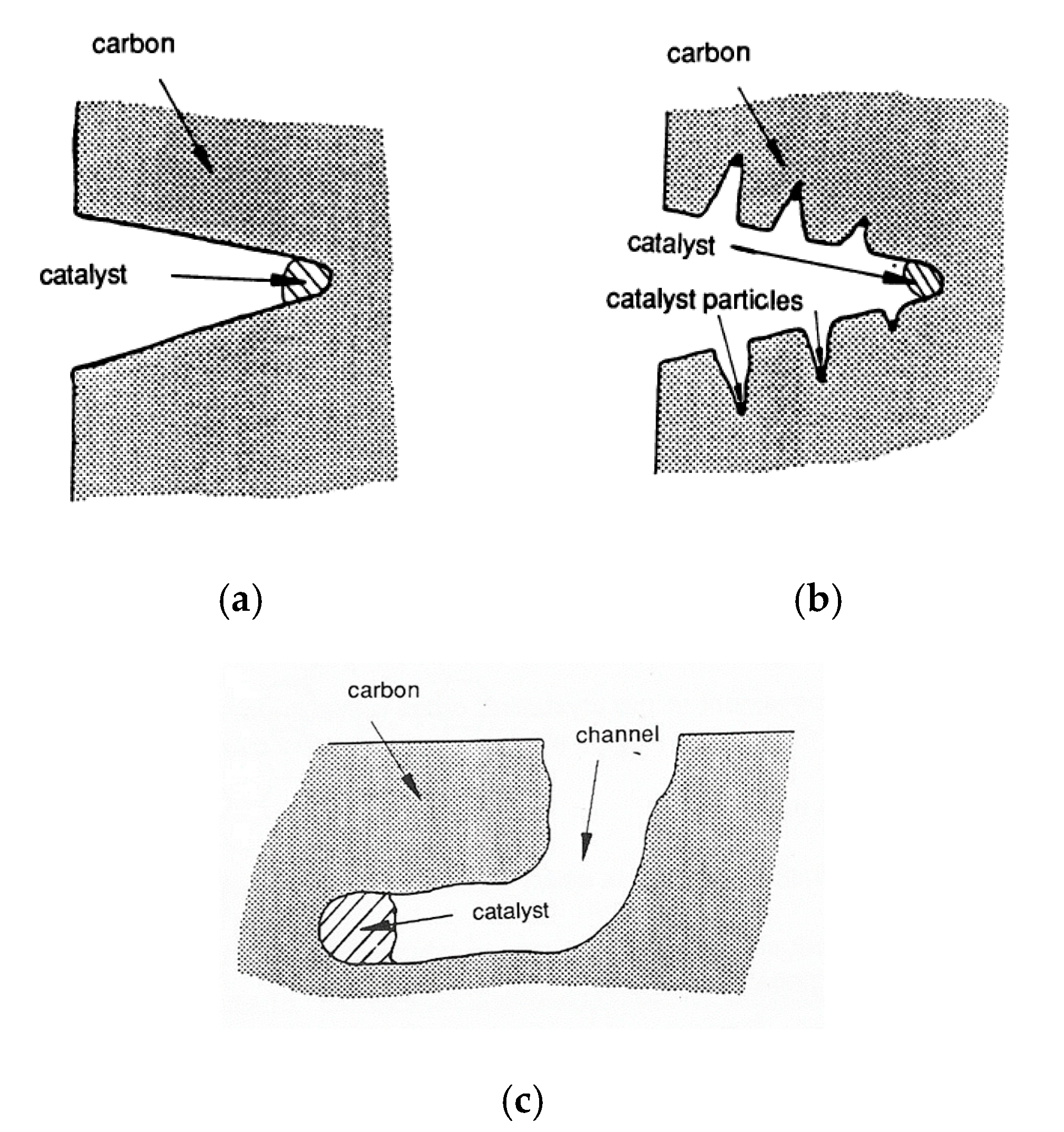{kind=link}
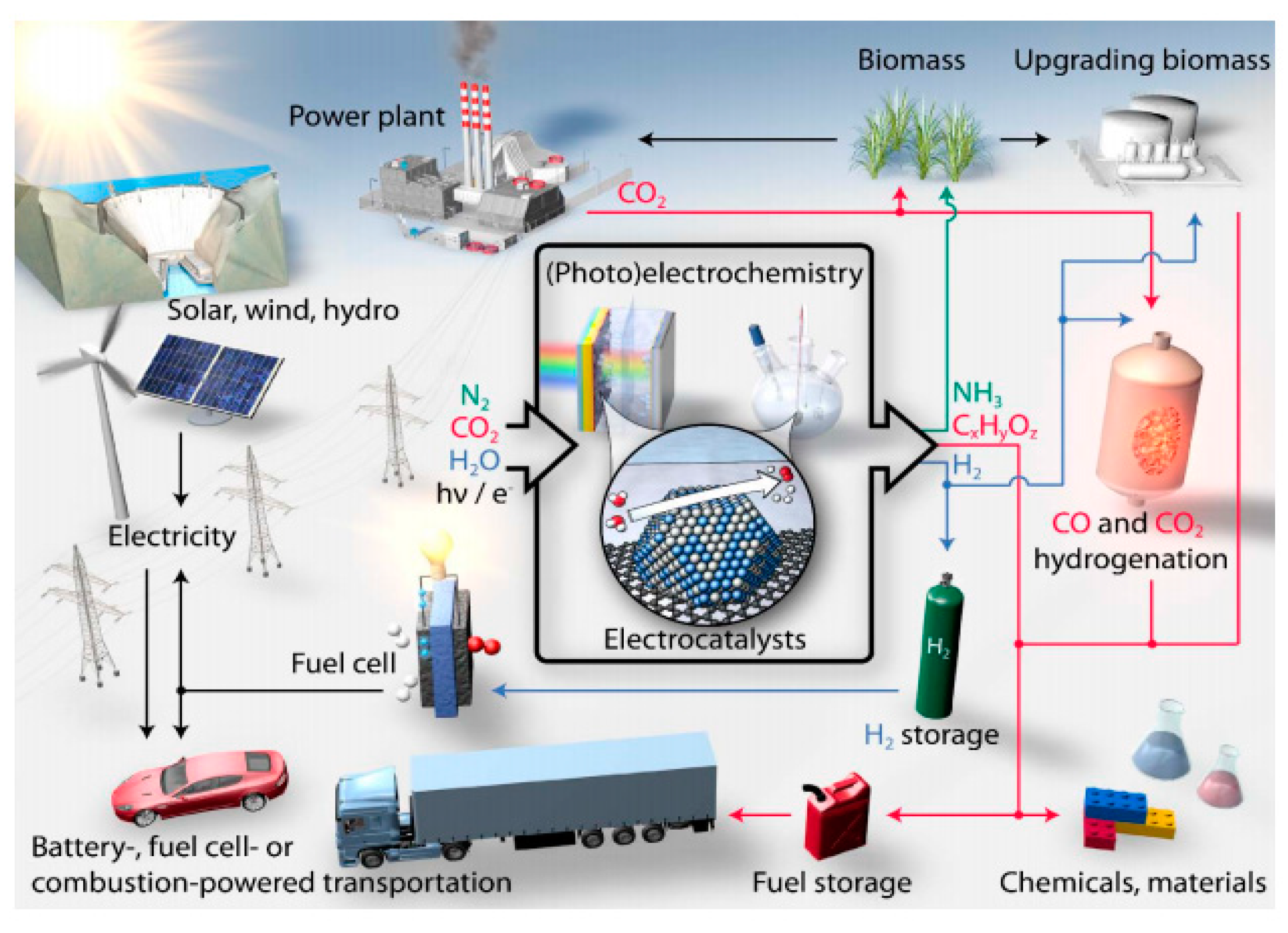{kind=link}
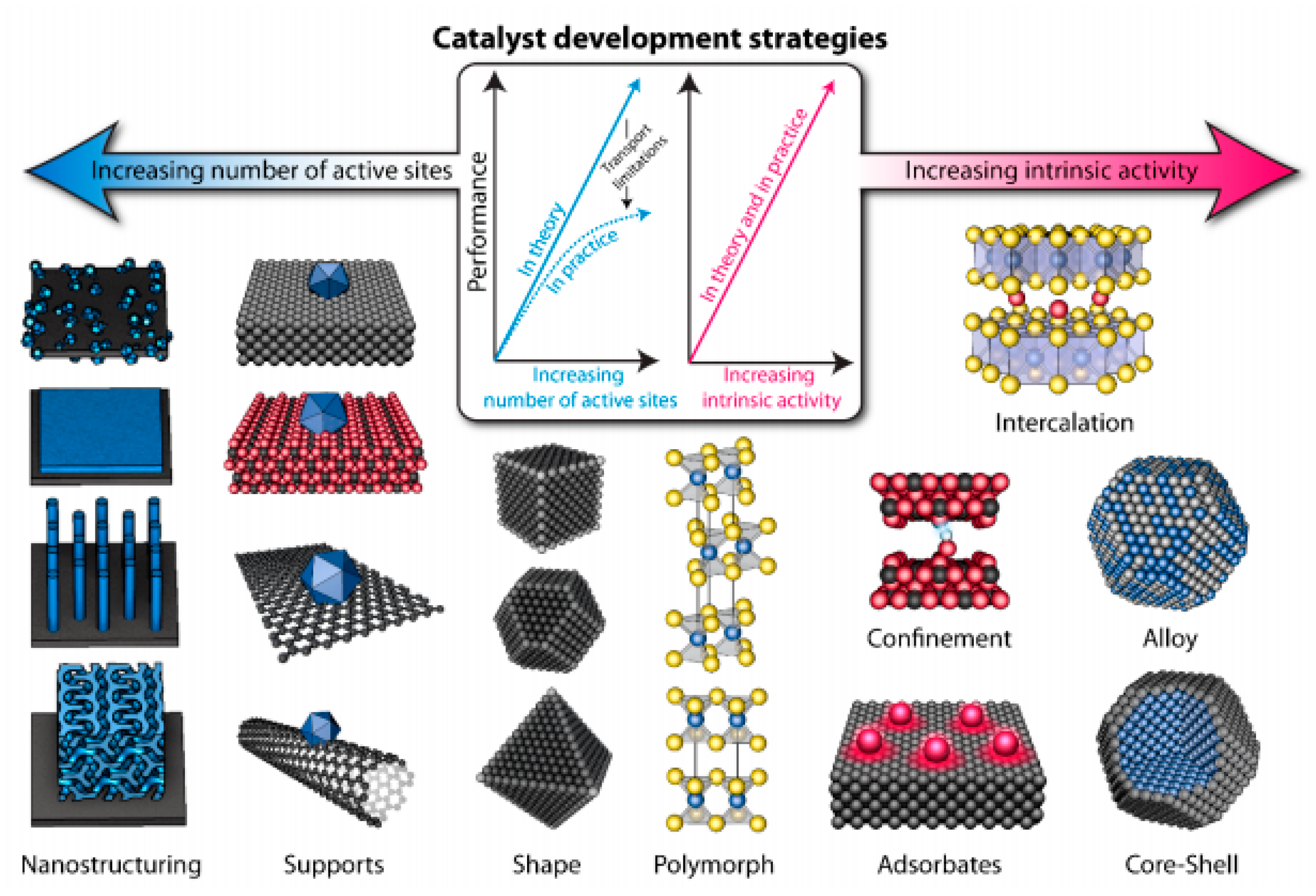{kind=link}
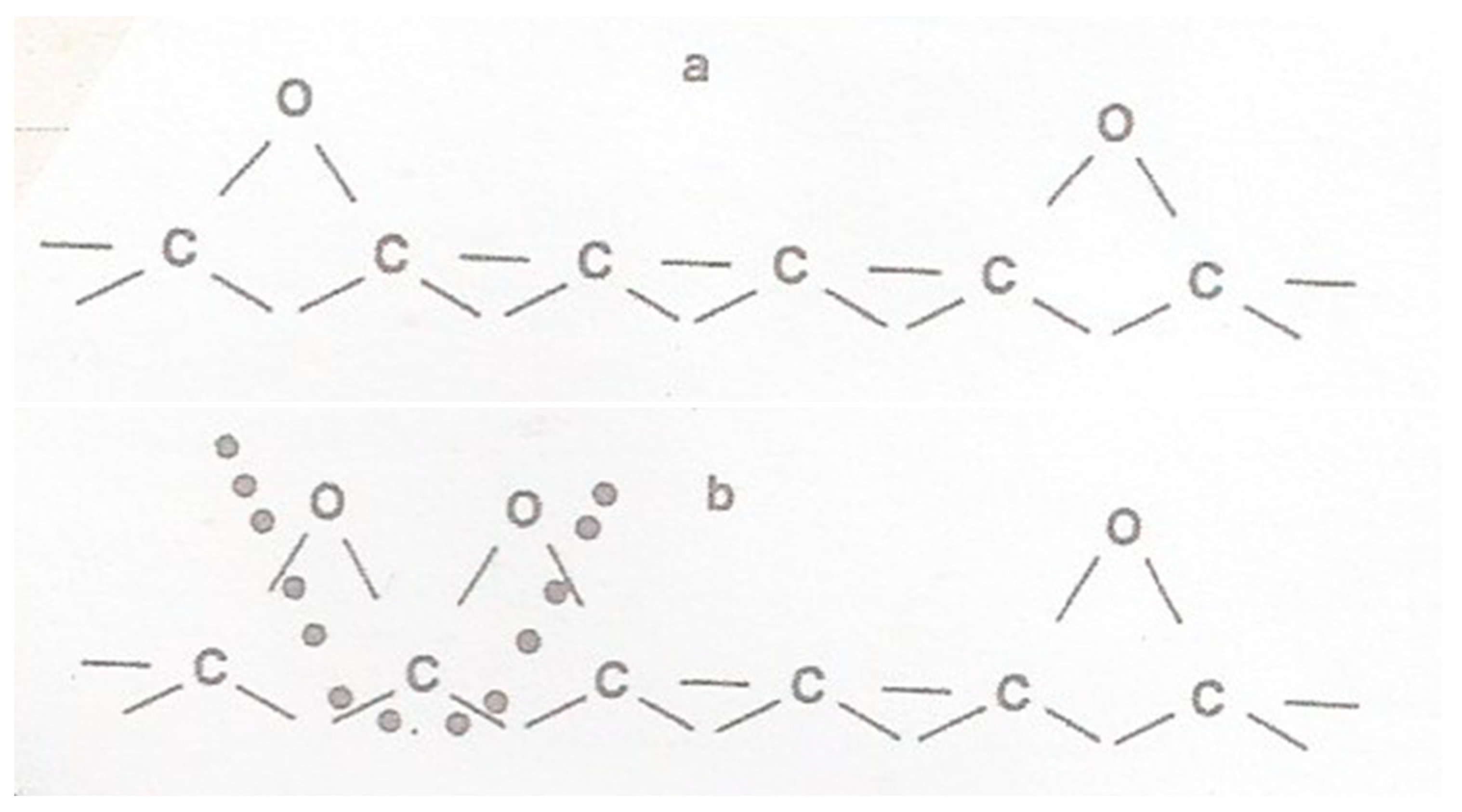{kind=link}
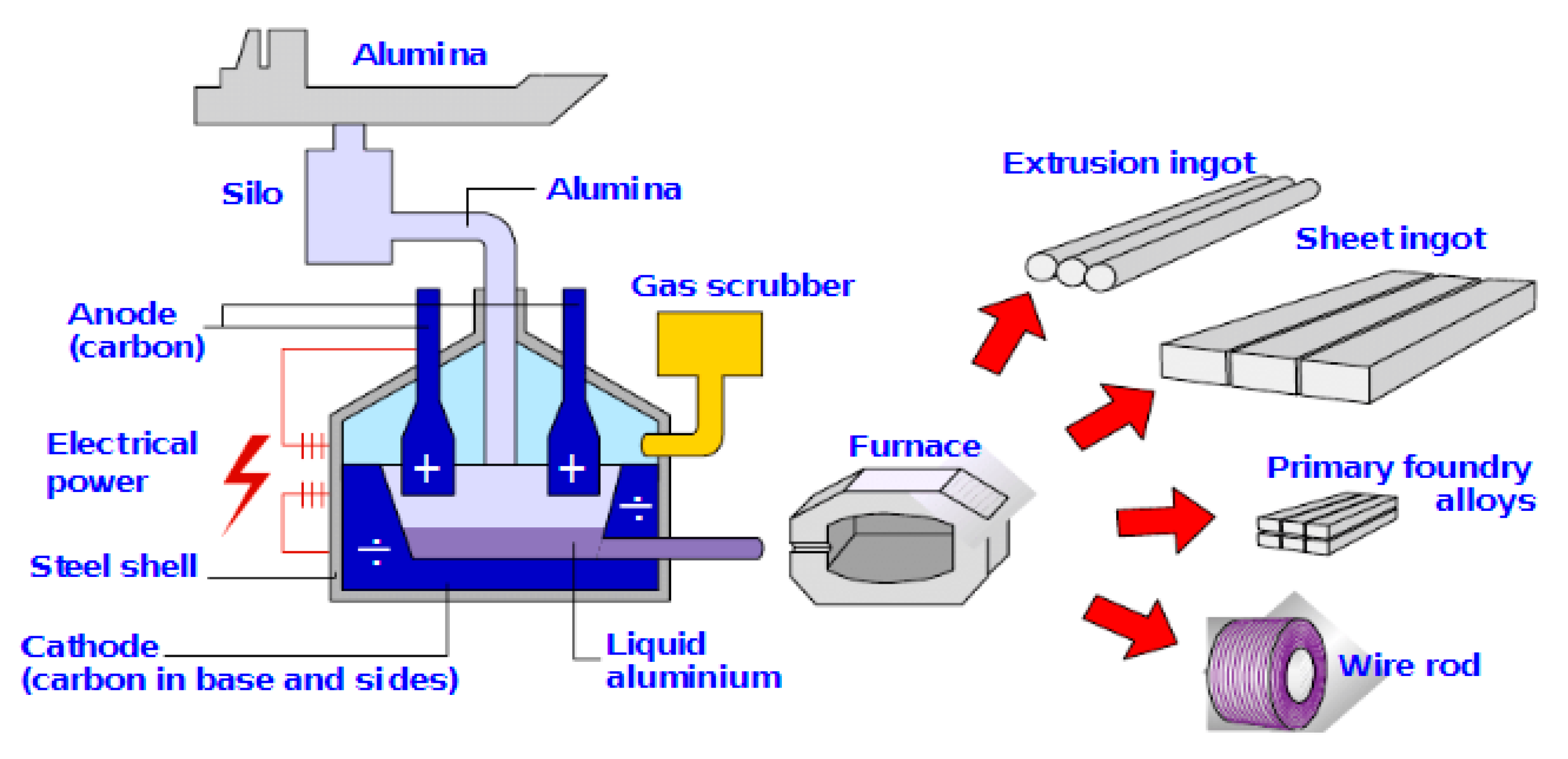{kind=link}
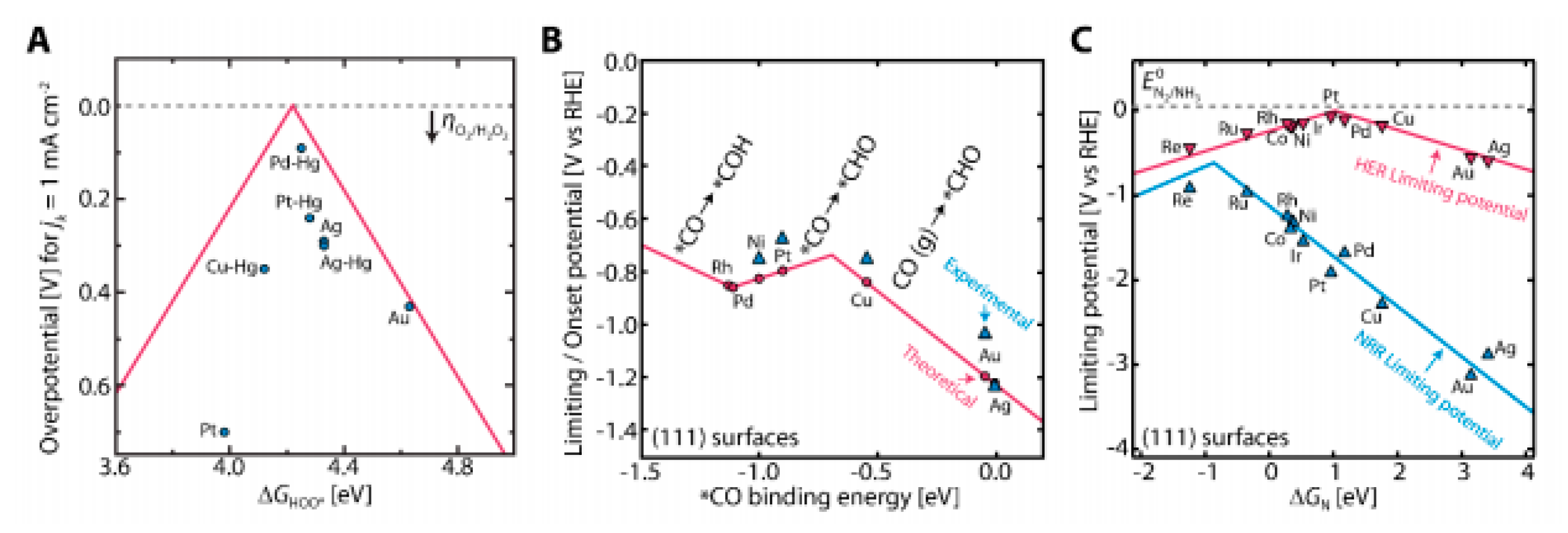{kind=link}
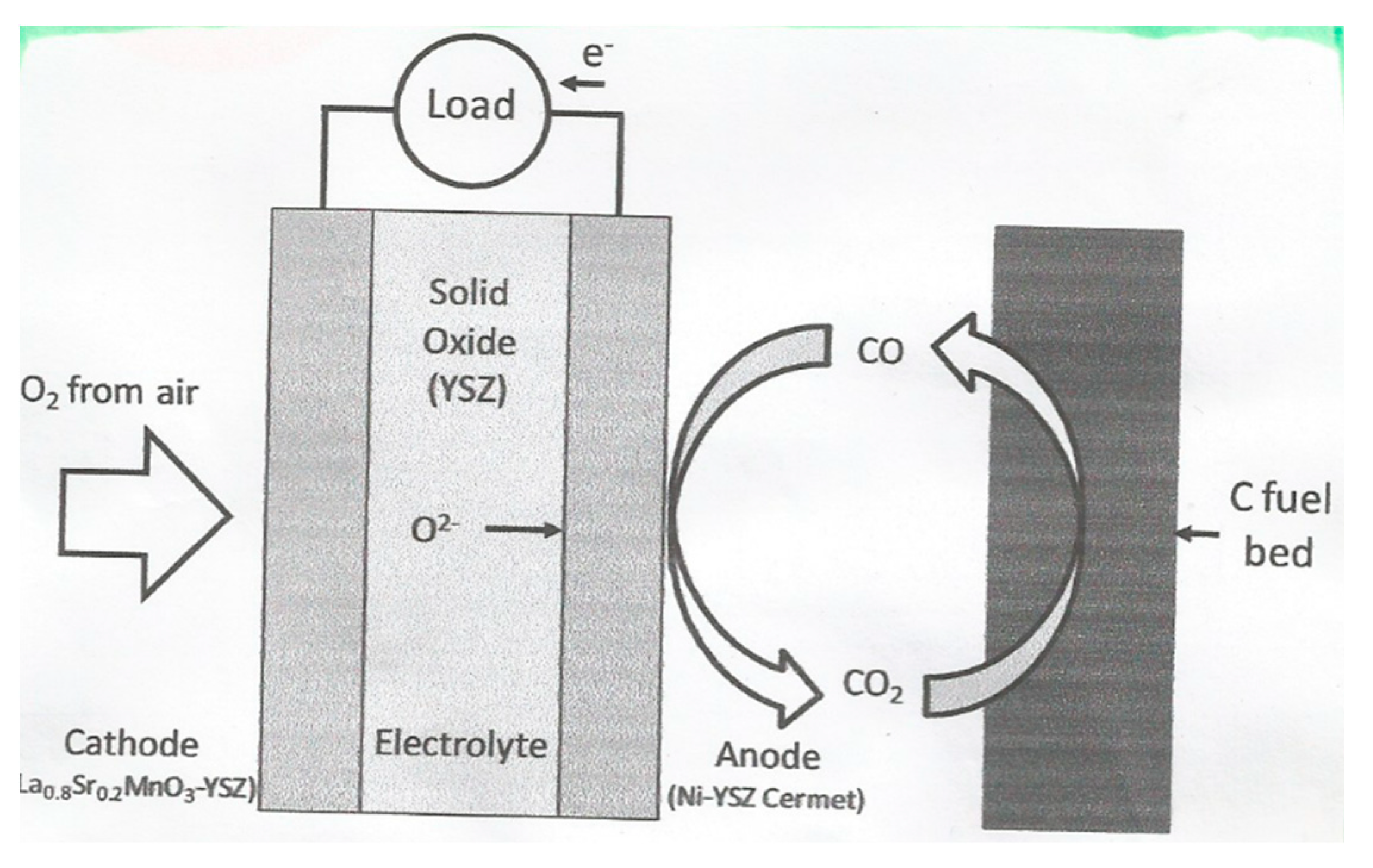{kind=link}
{kind=link}
| Fuel/Anode | Electrolyte | Cathode | T, °C |
|---|---|---|---|
| Solid graphite rod as fuel & anode C+4OH− = 2H2O + CO2 + 4e− | Molten Hydroxides OH− | Air as oxidant O2 + 2H2O + 4e− = 4OH− | 600 |
| Carbon particles as fuel in molten carbonate & anode C+2CO32− = 3CO2 + 4e− | Molten Carbonates CO32− | Air as oxidant O2 + 2CO2+4e− = 2CO32− | 800 |
| Carbon particles in a fluidized bed C + 2O2− = CO2 + 4e− | Oxygen ion conducting ceramic electrolyte O2− | Air as oxidant O2 + 4e− = 2O2− | 800 to 950 |
| Fuel in contact with molten tin Sn + 2O2− = SnO2 + 4e− | |||
| Carbon particles as fuel in molten carbonate & anode C + 2O2− = CO2 + 4e− |
| DCFC Technology | Status |
|---|---|
| Molten hydroxide | Average power densities of 40 mWcm−2 for over 540 h of operation with peak power density of 180 mWcm−2. The maximum efficiency achieved is 60%. |
| Molten carbonate | Power densities to 100–120 mWcm−2 and 80% efficiency. |
| -Solid Oxide -Solid carbon feed -Carbon mixed with molten metal -Carbon mixed with molten carbonate | The peak power density achieved is reported to be 140 mWcm−2 at 900 C. The peak power density achieved so far is about 160 mWcm−2 and 80 mWcm−2 respectively from hydrogen and liquid fuel JP-8. The peak power density achieved is 120 mWcm−2 using acetylene black as the fuel. SRI International has tested a 6 W 6-cell demonstration stack using different fuels. |
| Characteristics | Capacitor | Supercapacitor | Battery |
|---|---|---|---|
| Specific energy (W h kg−1) | <0.1 | 1–10 | 10–100 |
| Specific power (W kg−1) | >10.000 | 500–10,000 | <1000 |
| Discharge time | 10−6 to 10−3 | s to min | 0.3–3 h |
| Charge time | 10−6 to 10−3 | s to min | 1–5 h |
| Coulombic efficiency (%) | About 100 | 85–98 | 70–85 |
| Cycle-life | Almost infinite | >500,000 | About 1000 |
| Comparison Parameter | Battery | Supercapacitor |
|---|---|---|
| Storage mechanism | Chemical | Physical |
| Power limitation | Reaction kinetics, mass transport | Electrolyte conductivity |
| Energy storage | High (bulk) | Limited (surface area) |
| Charge rate | Kinetically limited | High, same as discharge |
| Cycle life limitations | Mechanical stability, chemical reversibility | Side reactions |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sequeira, C.A.C. Carbon Anode in Carbon History. Molecules 2020, 25, 4996. https://doi.org/10.3390/molecules25214996
Sequeira CAC. Carbon Anode in Carbon History. Molecules. 2020; 25(21):4996. https://doi.org/10.3390/molecules25214996
Chicago/Turabian StyleSequeira, César A. C. 2020. "Carbon Anode in Carbon History" Molecules 25, no. 21: 4996. https://doi.org/10.3390/molecules25214996
APA StyleSequeira, C. A. C. (2020). Carbon Anode in Carbon History. Molecules, 25(21), 4996. https://doi.org/10.3390/molecules25214996