Synthesis, Biological Evaluation and Stability Studies of Some Novel Aza-Acridine Aminoderivatives

 ,
,  and
and 
Abstract
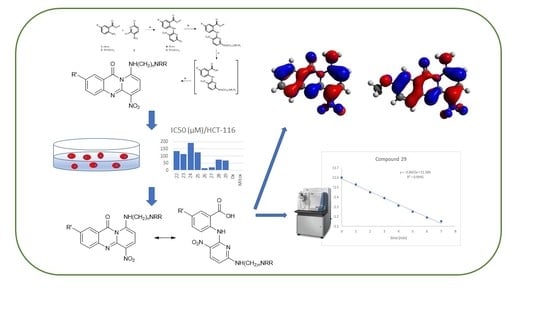
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Evaluation
2.3. Mass Spectrometry
2.4. Computational Chemistry
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.3. Biological Assays and Experiments
3.4. Computational Chemistry
3.5. Chemical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Belmont, P.; Bosson, J.; Godet, T.; Tiano, M. Acridine and Acridone Derivatives, Anticancer Properties and Synthetic Methods: Where Are We Now? Anticancer Agents Med. Chem. 2007, 7, 139–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, X.; Li, B.; Gao, C.; Jiang, Y. Acridine and its derivatives: A patent review (2009–2013). Expert Opin. Ther. Pat. 2014, 24, 647–664. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Singh, P. Acridine derivatives: A patent review (2009–2010). Expert Opin. Ther. Pat. 2011, 21, 437–454. [Google Scholar] [CrossRef] [PubMed]
- Demeunynck, M. Antitumour acridines. Expert Opin. Ther. Pat. 2004, 14, 55–70. [Google Scholar] [CrossRef]
- Pommier, Y. DNA Topoisomerases and Cancer; Springer Science & Business Media: Berlin, Germany, 2011. [Google Scholar]
- Gunaratnam, M.; Greciano, O.; Martins, C.; Reszka, A.P.; Schultes, C.M.; Morjani, H.; Riou, J.-F.; Neidle, S. Mechanism of acridine-based telomerase inhibition and telomere shortening. Biochem. Pharmacol. 2007, 74, 679–689. [Google Scholar] [CrossRef]
- Yuan, Z.; Chen, S.; Chen, C.; Chen, J.; Chen, C.; Dai, Q.; Gao, C.; Jiang, Y. Design, synthesis and biological evaluation of 4-amidobenzimidazole acridine derivatives as dual PARP and Topo inhibitors for cancer therapy. Eur. J. Med. Chem. 2017, 138, 1135–1146. [Google Scholar] [CrossRef]
- Ebead, Y.; Roshal, A.D.; Wroblewska, A.; Doroshenko, A.O.; Błazejowski, J. Tautomerism of acridin-9-amines substituted at the exocyclic nitrogen atom: Spectroscopic investigations and theoretical studies. Spectrochim. Acta A 2007, 66, 1016–1023. [Google Scholar] [CrossRef]
- Capomacchia, C.; Casper, J.; Schulman, S.G. Valence Tautomerism of Singly Protonated 9-Aminoacridine and Its Implications for Intercalative Interactions with Nucleic Acids. J. Pharm. Sci. 1974, 63, 1272–1276. [Google Scholar] [CrossRef]
- Bu, X.; Chen, J.; Deady, L.W.; Denny, W.A. Synthesis and cytotoxicity of potential anticancer derivatives of pyrazolo[3,4,5-kl]acridine and indolo[2,3-a]acridine. Tetrahedron 2002, 58, 175–181. [Google Scholar] [CrossRef]
- Cholody, W.M.; Martelli, S.; Paradziej-Lukowicz, J.; Konopa, J. 5-[(Aminoalkyl)amino]imidazo[4,5,1-de]acridin-6-ones as a novel class of antineoplastic agents. Synthesis and biological activity. J. Med. Chem. 1990, 33, 49–52. [Google Scholar] [CrossRef]
- Antonini, I.; Polucci, P.; Magnano, A.; Cacciamani, D.; Konieczny, M.T.; Paradziej-Łukowicz, J.; Martelli, S. Rational design, synthesis and biological evaluation of thiadiazinoacridines: A new class of antitumor agents. Bioorg. Med. Chem. 2003, 11, 399–405. [Google Scholar] [CrossRef]
- Zalupski, M.M.; Shields, A.F.; Philip, P.A.; Kraut, M.; LoRusso, P.; Heilbrun, L.K.; Vaitkevicius, V. Evaluation of pyrazoloacridine in patients with advanced pancreatic carcinoma. Invest. New Drugs 1998, 16, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, B.; Mrozek, E.; Kuebler, J.P.; Bekaii-Saab, T.; Kraut, E.H. Phase II trial of pyrazoloacridine (NSC#366140) in patients with metastatic breast cancer. Invest. New Drugs 2011, 29, 347–351. [Google Scholar] [PubMed]
- Kostakis, I.K.; Magiatis, P.; Pouli, N.; Marakos, P.; Skaltsounis, A.-L.; Pratsinis, H.; Léonce, S.; Pierré, A. Design, Synthesis, and Anti-proliferative Activity of Some New Pyrazole-Fused Amino Derivatives of the Pyranoxanthenone, Pyranothioxanthenone, and Pyranoacridone Ring Systems: A New Class of Cytotoxic Agents. J. Med. Chem. 2002, 45, 2599–2609. [Google Scholar] [CrossRef] [PubMed]
- Lemke, K.; Poindessous, V.; Skladanowski, A.; Larsen, A.K. The Antitumor Triazoloacridone C-1305 Is a Topoisomerase II Poison with Unusual Properties. Mol. Pharmacol. 2004, 66, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Mazerska, Z.; Sowiński, P.; Konopa, J. Molecular mechanism of the enzymatic oxidation investigated for imidazoacridinone antitumor drug, C-1311. Biochem. Pharmacol. 2003, 66, 1727–1736. [Google Scholar] [CrossRef]
- Wiśniewska, A.; Chrapkowska, A.; Kot-Wasik, A.; Konopa, J.; Mazerska, Z. Metabolic transformations of antitumor imidazoacridinone, C-1311, with microsomal fractions of rat and human liver. Acta Biochim. Pol. 2007, 54, 831–838. [Google Scholar] [CrossRef]
- Guilbaud, N.; Léonce, S.; Tillequin, F.; Koch, M.; Hickman, J.A.; Pierré, A. Acronycine derivatives as promising antitumor agents. Anticancer Drugs 2002, 13, 445–449. [Google Scholar] [CrossRef]
- Elomri, A.; Skaltsounis, A.L.; Michel, S.; Tillequin, F.; Koch, M.; Rolland, Y.; Pierré, A.; Atassi, G. Synthesis and cytotoxic activity of acronycine derivatives modified at the pyran ring. Chem. Pharm. Bull. 1996, 44, 2165–2168. [Google Scholar] [CrossRef]
- Kostakis, I.K.; Pouli, N.; Marakos, P.; Skaltsounis, A.L.; Pratsinis, H.; Kletsas, D. Design and synthesis of novel amino-substituted xanthenones and benzo[b]xanthenones: Evaluation of their antiproliferative activity and their ability to overcome multidrug resistance toward MES-SA/Dx5 cells. Bioorg. Med. Chem. 2006, 14, 2910–2934. [Google Scholar] [CrossRef]
- Kostakis, I.K.; Tenta, R.; Pouli, N.; Marakos, P.; Skaltsounis, A.L.; Pratsinis, H.; Kletsas, D. Design, synthesis, and antiproliferative activity of some novel aminosubstituted xanthenones, able to overcome multidrug resistance toward MES-SA/Dx5 cells. Bioorg. Med. Chem. Lett. 2005, 15, 5057–5060. [Google Scholar] [CrossRef] [PubMed]
- Giannouli, V.; Kostakis, I.K.; Pouli, N.; Marakos, P.; Kousidou, O.C.; Tzanakakis, G.N.; Karamanos, N.K. Design, Synthesis, and Evaluation of the Antiproliferative Activity of a Series of Novel Fused Xanthenone Aminoderivatives in Human Breast Cancer Cells. J. Med. Chem. 2007, 50, 1716–1719. [Google Scholar] [CrossRef] [PubMed]
- Giannouli, V.; Kostakis, I.K.; Pouli, N.; Marakos, P.; Samara, P.; Tsitsilonis, O. Synthesis and anti-proliferative activity of some novel benzo-fused imidazo[1,8]naphthyridinones. Bioorg. Med. Chem. Lett. 2015, 25, 2621–2623. [Google Scholar] [CrossRef] [PubMed]
- Timári, G.; Hajós, G.; Messmer, A. Synthesis, alkylation and ring opening of two differently fused pyridoquinazolones. J. Heterocycl. Chem. 1990, 27, 2005–2009. [Google Scholar] [CrossRef]
- Samara, P.; Christoforidou, N.; Lemus, C.; Argyropoulou, A.; Ioannou, K.; Vougogiannopoulou, K.; Aligiannis, N.; Paronis, E.; Gaboriaud-Kolar, N.; Tsitsilonis, O.; et al. New semi-synthetic analogs of oleuropein show improved anticancer activity in vitro and in vivo. Eur. J. Med. Chem. 2017, 137, 11–29. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Citation | Gaussian.com; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Sánchez-Márquez, J.; Zorrilla, D.; Sánchez-Coronilla, A.; de los Santos, D.M.; Navas, J.; Fernández-Lorenzo, C.; Alcántara, R.; Martín-Calleja, J. Introducing “UCA-FUKUI” software: Reactivity-index calculations. J. Mol. Model. 2014, 20, 2492. [Google Scholar] [CrossRef]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 22–29 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (μM) a | |||
|---|---|---|---|---|
| HCT-116 | MES-SA | MES-SA/Dx5 | RF b | |
| 22 | 136.03 ± 4.80 | 72.10 ± 1.90 | 65.07 ± 3.93 | 0.90 |
| 23 | 114.93 ± 5.99 | 49.90 ± 3.03 | 50.30 ± 1.35 | 1.01 |
| 24 | 192.43 ± 10.47 | 188.10 ± 1.82 | 164.47 ± 4.20 | 0.87 |
| 25 | 126.30 ± 5.37 | 51.73 ± 3.47 | 47.93 ± 1.99 | 0.93 |
| 26 | 15.37 ± 2.34 | 37.03 ± 3.42 | 21.27 ± 0.76 | 0.57 |
| 27 | 20.47 ± 3.75 | 22.27 ± 1.60 | 10.33 ± 0.55 | 0.46 |
| 28 | 73.13 ± 3.18 | 47.43 ± 2.56 | 34.40 ± 1.41 | 0.73 |
| 29 | 67.67 ± 1.39 | 36.83 ± 1.37 | 19.47 ± 1.25 | 0.53 |
| Dx | 0.183 ± 0.016 | 0.037 + 0.011 | 2.680 ± 0.261 | 72.43 |
| Mitox | 0.020 ± 0.006 | 0.007 ± 0.003 | 0.081± 0.013 | 11.57 |
| Compound | Equation y = intercept (±Standard Error) + slope (±Standard Error) × t | Correlation Coefficient |
|---|---|---|
| 25 | y = 11.49 (±0.001) − 0.065 (±0.0001) × t | 0.992 |
| 29 | y = 11.59 (±0.008) − 0.0025 (±0.002) × t | 0.994 |
 |  | ||||||
| Compound 25 | Compound 29 | ||||||
| Bond Length (Å) | 9-10 | 4-10 | 10-16 * | 10-15 * | 8-9 | 4-5 | |
| 25 | 1.445 | 1.449 | 1.261 | 1.461 | 1.415 | ||
| 29 | 1.441 | 1.448 | 1.261 | 1.46493 | 1.416 | ||
| Angle (°) | 11-9-10 | 9-10-4 | 9-10-16 | 9-10-15 | 4-10-16 | 4-10-15 | 10-4-1 |
| 25 | 121.032 | 116.810 | 120.886 | 122.299 | 120.034 | ||
| 29 | 120.975 | 116.838 | 121.133 | 122.023 | 119.779 | ||
| Dihedral Angle (°) | 12-11-9-10 | 11-9-10-16 | 11-9-10-15 | 9-10-4-1 | 10-4-1-3 | ||
| 25 | 177.74735 | 2.686 | 179.446 | −178.913 | |||
| 29 | −177.38246 | −2.483 | −179.522 | 179.078 | |||
| Atomic Charges (−e) | N9 | C10 | C11 | C4 | O16 | O15 | C1 |
| 25 | −0.445 | 0.761 | 0.763 | −0.374 | −0.641 | 0.004 | |
| 29 | −0.522 | 0.828 | 0.797 | −0.319 | −0.635 | −0.283 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karelou, M.; Kourafalos, V.; Tragomalou, A.P.; Marakos, P.; Pouli, N.; Tsitsilonis, O.E.; Gikas, E.; Kostakis, I.K. Synthesis, Biological Evaluation and Stability Studies of Some Novel Aza-Acridine Aminoderivatives. Molecules 2020, 25, 4584. https://doi.org/10.3390/molecules25194584
Karelou M, Kourafalos V, Tragomalou AP, Marakos P, Pouli N, Tsitsilonis OE, Gikas E, Kostakis IK. Synthesis, Biological Evaluation and Stability Studies of Some Novel Aza-Acridine Aminoderivatives. Molecules. 2020; 25(19):4584. https://doi.org/10.3390/molecules25194584
Chicago/Turabian StyleKarelou, Maria, Vasileios Kourafalos, Athanasia P. Tragomalou, Panagiotis Marakos, Nicole Pouli, Ourania E. Tsitsilonis, Evangelos Gikas, and Ioannis K. Kostakis. 2020. "Synthesis, Biological Evaluation and Stability Studies of Some Novel Aza-Acridine Aminoderivatives" Molecules 25, no. 19: 4584. https://doi.org/10.3390/molecules25194584
APA StyleKarelou, M., Kourafalos, V., Tragomalou, A. P., Marakos, P., Pouli, N., Tsitsilonis, O. E., Gikas, E., & Kostakis, I. K. (2020). Synthesis, Biological Evaluation and Stability Studies of Some Novel Aza-Acridine Aminoderivatives. Molecules, 25(19), 4584. https://doi.org/10.3390/molecules25194584