Ultrasound-Assisted Extraction Optimization of α-Glucosidase Inhibitors from Ceratophyllum demersum L. and Identification of Phytochemical Profiling by HPLC-QTOF-MS/MS

Abstract
1. Introduction
2. Results and Discussion
2.1. Effect of Solvent Polarity on the Recovery of AGIs
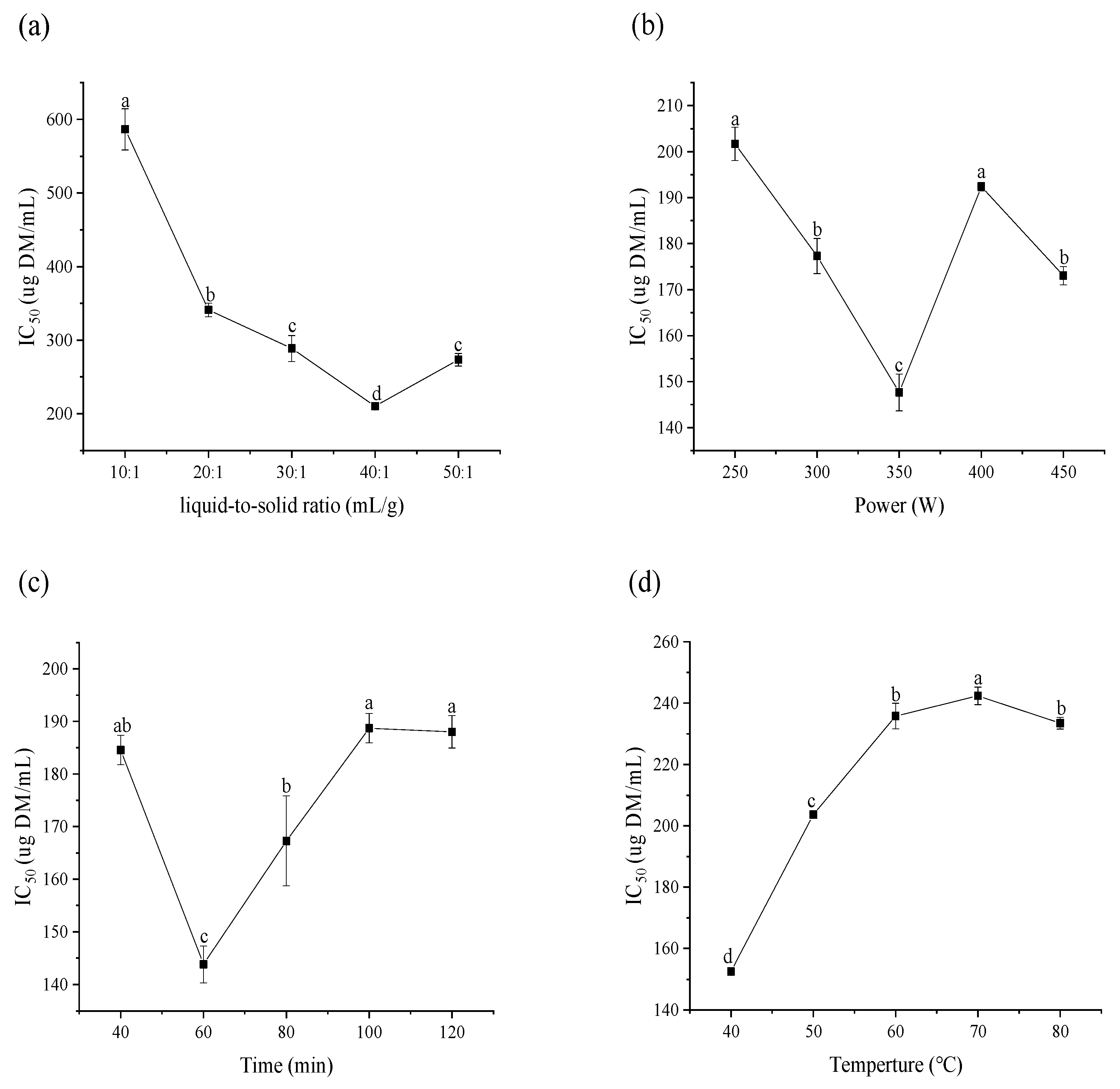
2.2. Preliminary Screening of Each Single Factor Analysis
2.3. Response Surface Analysis
2.4. Optimal Extraction Conditions Analysis
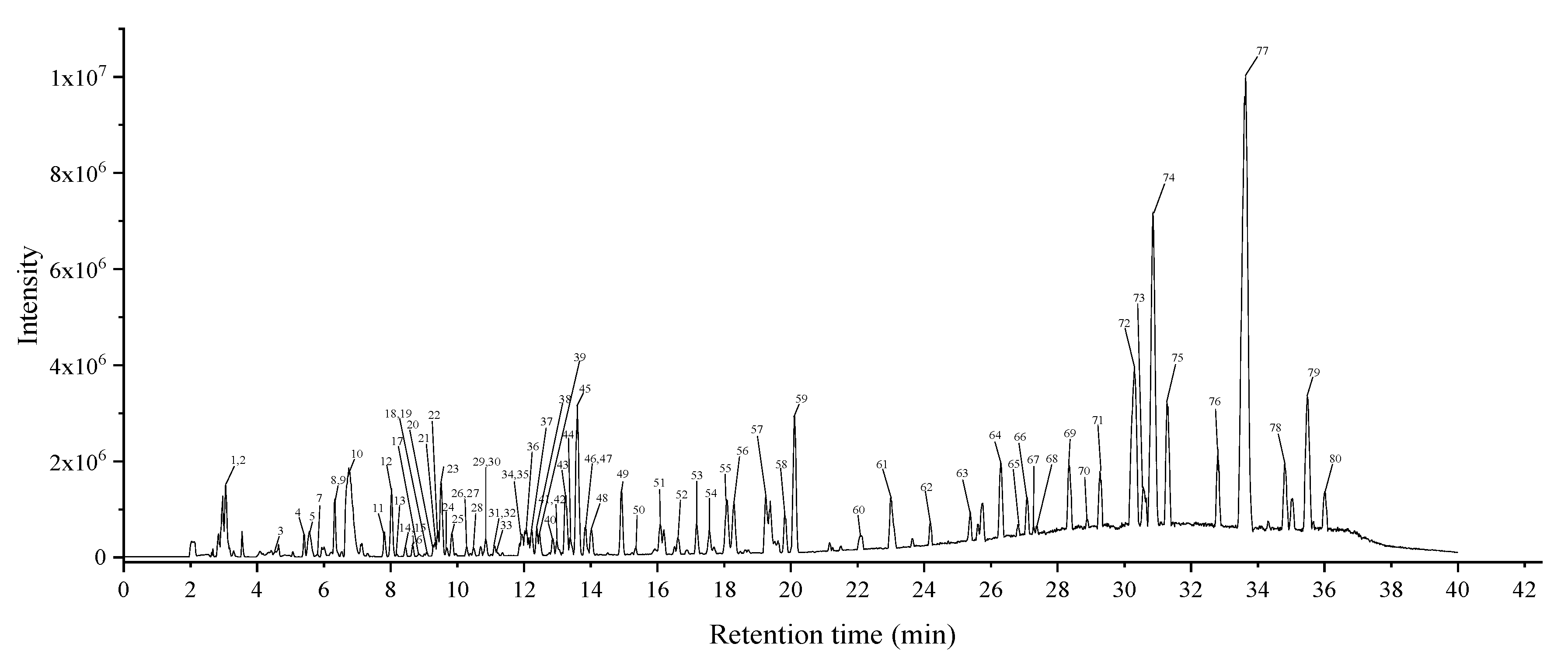
2.5. Analysis of Phytochemical Constituents
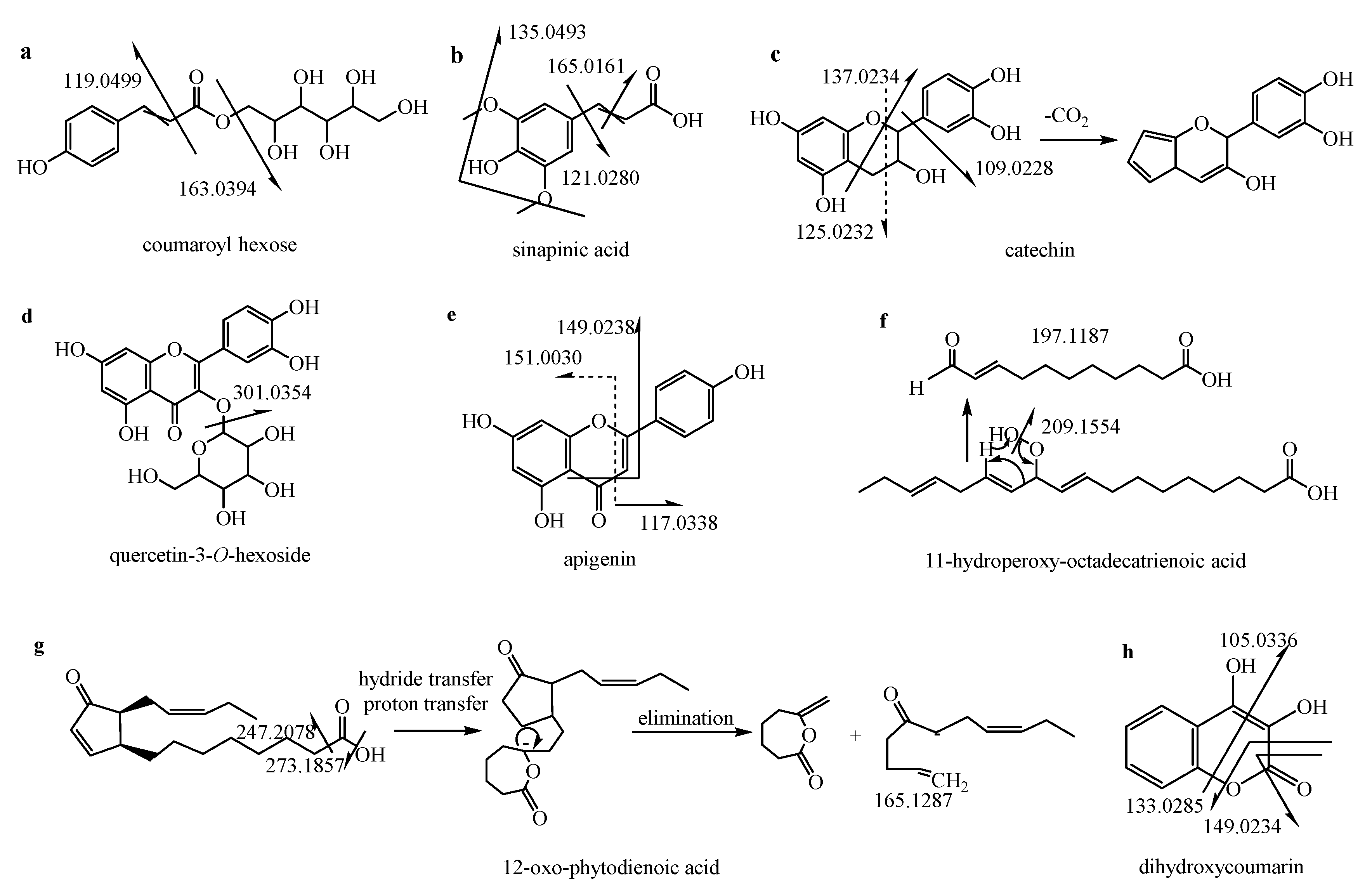
2.5.1. Organic Acids
2.5.2. Phenols and Derivatives
2.5.3. Flavonoids
2.5.4. Fatty Acids
2.5.5. Others
3. Material and Methods
3.1. Reagents
3.2. Preparation of Extracts
3.3. Determination of Total Phenolic and Flavonoid Content
3.4. Single Factor Experiments
3.5. α-Glucosidase Inhibition Assay
3.6. Statistical Optimization of UAE
3.7. HPLC-QTOF-MS/MS Analysis
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CDL | Ceratophyllum demersum L |
| AGIs | α-glucosidase inhibitors |
| RSM | response surface methodology |
| UAE | ultrasonic-assisted extraction |
| BBD | Box-Behnken design |
| TFC | total flavonoid content |
| TPC | total phenolic content |
| pNPG | p-nitrophenyl-α-d-glucopyranoside |
| BPC | base peak chromatogram |
| ESI | electrospray ionization source |
| ANOVA | one-way analysis of variance |
References
- Leroux-Stewart, J.; Rabasa-Lhoret, R.; Chiasson, J. α-Glucosidase inhibitors. Ther. Diabetes Mellit. Relat. Disorders. 2014, 416. [Google Scholar]
- Kumar, V.; Prakash, O.; Kumar, S.; Narwal, S. α-glucosidase inhibitors from plants: A natural approach to treat diabetes. Pharmacogn Rev. 2011, 5, 19–29. [Google Scholar] [CrossRef]
- Razavi-Nematollahi, L.; Ismail-Beigi, F. Adverse effects of glycemia-lowering medications in type 2 diabetes. Curr. Diabetes Rep. 2019, 19, 132. [Google Scholar] [CrossRef]
- Zhang, L.; Tu, Z.; Yuan, T.; Wang, H.; Xie, X.; Fu, Z. Antioxidants and α-glucosidase inhibitors from Ipomoea batatas leaves identified by bioassay-guided approach and structure-activity relationships. Food Chem. 2016, 208, 61–67. [Google Scholar] [CrossRef]
- Zhang, L.; Tu, Z.; Yuan, T.; Ma, H.; Niesen, D.B.; Wang, H.; Seeram, N.P. New Gallotannin and other Phytochemicals from Sycamore Maple (Acer pseudoplatanus) Leaves. Nat. Prod. Commun. 2015, 10, 1977–1980. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, L.; Ye, Y.; Zhu, M.; Li, J.; Tu, Z.; Yang, S.; Liao, H. Phytochemical profiles and screening of α-glucosidase inhibitors of four Acer species leaves with ultra-filtration combined with UPLC-QTOF-MS/MS. Ind. Crops Prod. 2019, 129, 156–168. [Google Scholar] [CrossRef]
- Wang, S.; Xie, X.; Zhang, L.; Hu, Y.; Wang, H.; Tu, Z. Inhibition mechanism of α-glucosidase inhibitors screened from Artemisia selengensis Turcz root. Ind. Crops Prod. 2020, 143, 111941. [Google Scholar] [CrossRef]
- Belayneh, Y.M.; Birhanu, Z.; Birru, E.M.; Getenet, G. Evaluation of in vivo antidiabetic, antidyslipidemic, and in vitro antioxidant activities of hydromethanolic root extract of Datura stramonium L.(Solanaceae). J. Exp. Med. 2019, 11, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Casanova, L.M.; De Silva, D.; Sola-Penna, M.; Camargo, L.M.D.; Celestrini, D.D.; Tinoco, L.W.; Costa, S.S. Identification of chicoric acid as a hypoglycemic agent from Ocimum gratissimum leaf extract in a biomonitoring in vivo study. Fitoterapia 2014, 93, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Tabaraki, R.; Nateghi, A. Optimization of ultrasonic-assisted extraction of natural antioxidants from rice bran using response surface methodology. Ultrason. Sonochem. 2011, 18, 1279–1286. [Google Scholar] [CrossRef]
- Camel, V. Microwave-assisted solvent extraction of environmental samples. TrAC Trends Anal. Chem. 2000, 19, 229–248. [Google Scholar] [CrossRef]
- Yolmeh, M.; Najafi, M.B.H.; Farhoosh, R. Optimisation of ultrasound-assisted extraction of natural pigment from annatto seeds by response surface methodology (RSM). Food Chem. 2014, 155, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Chemat, F.; Rombaut, N.; Sicaire, A.; Meullemiestre, A.; Fabiano-Tixier, A.; Abert-Vian, M. Ultrasound assisted extraction of food and natural products. Mechanisms, techniques, combinations, protocols and applications. A review. Ultrason. Sonochem. 2017, 34, 540–560. [Google Scholar] [CrossRef]
- Savic Gajic, I.; Savic, I.; Boskov, I.; Žerajić, S.; Markovic, I.; Gajic, D. Optimization of ultrasound-assisted extraction of phenolic compounds from Black Locust (Robiniae Pseudoacaciae) flowers and comparison with conventional methods. Antioxidants 2019, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Ilaiyaraja, N.; Likhith, K.R.; Babu, G.R.S.; Khanum, F. Optimisation of extraction of bioactive compounds from Feronia limonia (wood apple) fruit using response surface methodology (RSM). Food Chem. 2015, 173, 348–534. [Google Scholar] [CrossRef]
- Benchikh, Y.; Zaoui, A.; Derbal, R.; Bey, M.B.; Louaileche, H. Optimisation of extraction conditions of phenolic compounds and antioxidant activity of Ruta chalepensis L. using response surface methodology. J. Food Meas. Charact. 2019, 13, 883–891. [Google Scholar] [CrossRef]
- Zerajić, S.A.; Savić-Gajić, I.M.; Savić, I.M.; Nikolić, G.S. The optimization of ultrasound-assisted extraction of total flavonoids from pot marigold (Calendulae officinalis L.) flowers. Adv. Technol. 2019, 8, 10–18. [Google Scholar] [CrossRef]
- Yang, B.; Zhang, M.; Weng, H.; Xu, Y.; Zeng, L. Optimization of ultrasound assisted extraction (UAE) of einsenoside eompound from Anoectochilus roxburghii (Wall.) Lindl by response surface methodology (RSM). Molecules 2020, 25, 193. [Google Scholar] [CrossRef]
- Taranhalli, A.D.; Kadam, A.M.; Karale, S.S.; Warke, Y.B. Evaluation of antidiarrhoeal and wound healing potentials of Ceratophyllum demersum Linn. whole plant in rats. Lat. Am. J. Pharm. 2011, 30, 297–303. [Google Scholar]
- Karatas, M.; Dogan, M.; Emsen, B.; Aasim, M. Determination of in vitro free radical scavenging activities of various extracts from in vitro propagated Ceratophyllum demersum L. Fresen. Environ. Bull. 2015, 24, 2946–2952. [Google Scholar]
- Malathy, R.; Amalan, S.S. Studies on the potential therapeutic effects on the aquatic macrophytes namely Cabomba aquatica, Ceratophyllum demersum and Hygrophila corymbosa. J. Chem. Pharm. Res. 2015, 7, 479–483. [Google Scholar]
- Dogan, M.; Emsen, B.; Aasim, M.; Yildirim, E. Ceratophyllum demersum L. extract as a botanical insecticide for controlling the Maize Weevil, Sitophilus zeamais Motschulsky (Coleoptera: Curculionidae). Egypt. J. Pest Control. 2017, 27, 11–15. [Google Scholar]
- Lu, X.; Qiao, Y.; Zhang, X.; Ma, B.; Qiu, M. Chemical constituents from Ceratophyllum demersum (Ceratophyllaceae). Aeta Bot. Yunnaniea. 2007, 29, 263–264. [Google Scholar]
- Bankova, V.; Ivanova, P.; Christov, R.; Popov, S.; Dimitrova-Konaklieva, S. Secondary metabolites of Ceratophyllum demersum. Hydrobiologia 1995, 316, 59–61. [Google Scholar] [CrossRef]
- Mohammadpour, H.; Sadrameli, S.M.; Eslami, F.; Asoodeh, A. Optimization of ultrasound-assisted extraction of Moringa peregrina oil with response surface methodology and comparison with soxhlet method. Ind. Crops Prod. 2019, 131, 106–116. [Google Scholar] [CrossRef]
- Tomšik, A.; Pavlić, B.; Vladić, J.; Ramić, M.; Brindza, J.; Vidović, S. Optimization of ultrasound-assisted extraction of bioactive compounds from wild garlic (Allium ursinum L.). Ultrason. Sonochem. 2016, 29, 502–511. [Google Scholar] [CrossRef]
- Belwal, T.; Huang, H.; Li, L.; Duan, Z.; Zhang, X.; Aalim, H.; Luo, Z. Optimization model for ultrasonic-assisted and scale-up extraction of anthocyanins from Pyrus communis Starkrimson fruit peel. Food Chem. 2019, 297, 124993. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Sánchez, C.; Lozano-Sánchez, J.; Rodríguez-Pérez, C.; Segura-Carretero, A.; Fernández-Gutiérrez, A. Comprehensive, untargeted, and qualitative Rp-HPLC-ESI-QTOF/MS2 metabolite profiling of green asparagus (Asparagus officinalis). J. Food Compos. Anal. 2016, 46, 78–87. [Google Scholar] [CrossRef]
- Luo, Y.; Wen, Q.; Lai, C.; Feng, Y.; Tan, T. Characterization of polymeric phenolic acids and flavonoids in Clerodendranthi Spicati Herba using ultrahigh-performance liquid chromatography coupled to quadrupole time-of-flight tandem mass spectrometry with target and nontarget data mining strategy. Rapid Commun. Mass Sp. 2019, 33, 1884–1893. [Google Scholar] [CrossRef]
- Sun, Y.; Qin, Y.; Li, H.; Peng, H.; Chen, H.; Xie, H.; Deng, Z. Rapid characterization of chemical constituents in Radix Tetrastigma, a functional herbal mixture, before and after metabolism and their antioxidant/antiproliferative activities. J. Funct. Foods. 2015, 18, 300–318. [Google Scholar] [CrossRef]
- Abu-Reidah, I.M.; Ali-Shtayeh, M.S.; Jamous, R.M.; Arráez-Román, D.; Segura-Carretero, A. Comprehensive metabolite profiling of Arum palaestinum (Araceae) leaves by using liquid chromatography-tandem mass spectrometry. Food Res. Int. 2015, 70, 74–86. [Google Scholar] [CrossRef]
- Sun, H.; Liu, J.; Zhang, A.; Zhang, Y.; Meng, X.; Han, Y.; Zhang, Y.; Wang, X. Characterization of the multiple components of Acanthopanax Senticosus stem by ultra high performance liquid chromatography with quadrupole time-of-flight tandem mass spectrometry. J. Sep. Sci. 2016, 39, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Zhu, Y.; Gao, X.; Chang, Y.; Wang, M.; Zhang, P. Qualitative and quantitative analysis of the major constituents in Chinese medicinal preparation Dan-Lou tablet by ultra high performance liquid chromatography/diode-array detector/quadrupole time-of-flight tandem mass spectrometry. J. Pharmaceut. Biomed. 2013, 80, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Tu, Z.; Zhang, L.; Liao, H. Antioxidant, metabolic enzymes inhibitory ability of Torreya grandis kernels, and phytochemical profiling identified by HPLC-QTOF-MS/MS. J. Food Biochem. 2019, 43, e13043. [Google Scholar]
- Zengin, G.; Mahomoodally, M.F.; Paksoy, M.Y.; Picot-Allain, C.; Glamocilja, J.; Sokovic, M.; Diuzheva, A.; Jeko, J.; Cziaky, Z.; Rodrigues, M.J.; et al. Phytochemical characterization and bioactivities of five Apiaceae species: Natural sources for novel ingredients. Ind. Crops Prod. 2019, 135, 107–121. [Google Scholar] [CrossRef]
- Xie, X.; Tu, Z.; Zhang, L.; Zhao, Y.; Wang, H.; Wang, Z.; Zhang, N.; Zhong, B. Antioxidant activity, α-glucosidase inhibition, and phytochemical fingerprints of Anoectochilus roxburghii formula tea residues with HPLC-QTOF-MS/MS. J. Food Biochem. 2017, 41, e12402. [Google Scholar] [CrossRef]
- Fabre, N.; Rustan, I.; Hoffmann, E.; Quetin-Leclercq, J. Determination of flavone, flavonol, and flavanone aglycones by negative ion liquid chromatography electrospray ion trap mass spectrometry. J. Am. Soc. Mass Spectr. 2001, 12, 707–715. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, M.; Tu, Z.; Zhao, Y.; Wang, H.; Li, G.; Wang, H.; Sha, X. α-Glucosidase inhibition, anti-glycation and antioxidant activities of Liquidambar formosana Hance leaf, and identification of phytochemical profile. S. Afr. J. Bot. 2017, 113, 239–247. [Google Scholar] [CrossRef]
- Kumar, S.; Singh, A.; Kumar, B. Identification and characterization of phenolics and terpenoids from ethanolic extracts of Phyllanthus species by HPLC-ESI-QTOF-MS/MS. J. Pharm. Anal. 2017, 7, 214–222. [Google Scholar] [CrossRef]
- Zhang, L.; Tu, Z.; Wang, H.; Fu, Z.; Wen, Q.; Fan, D. Metabolic profiling of antioxidants constituents in Artemisia selengensis leaves. Food Chem. 2015, 186, 123–132. [Google Scholar] [CrossRef]
- Zhu, H.; Yin, R.; Han, F.; Guan, J.; Zhang, X.; Mao, X.; Zhao, L.; Li, Q.; Hou, X.; Bi, K. Characterization of chemical constituents in Zhi-Zi-Da-Huang decoction by ultra high performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry. J. Sep. Sci. 2014, 37, 3489–3496. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Xu, T.; Qin, M.; Zhuang, X.; Fang, Y.; Zhang, Z. Phenolic characterization of young wines made from spine grape (Vitis davidii Foex) grown in Chongyi County (China). Food Res. Int. 2012, 49, 664–671. [Google Scholar] [CrossRef]
- Bao, J.; Gao, X.; Jones, A.D. Unusual negative charge-directed fragmentation: Collision-induced dissociation of cyclopentenone oxylipins in negative ion mode. Rapid Commun. Mass Sp. 2014, 28, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Ludovici, M.; Ialongo, C.; Reverberi, M.; Beccaccioli, M.; Scarpari, M.; Scala, V. Quantitative profiling of oxylipins through comprehensive LC-MS/MS analysis of Fusarium verticillioides and maize kernels. Food Addit. Contam. Part. A. 2014, 31, 2026–2033. [Google Scholar] [CrossRef] [PubMed]
- Strassburg, K.; Huijbrechts, A.M.L.; Kortekaas, K.A.; Lindeman, J.H.; Pedersen, T.L.; Dane, A.; Berger, R.; Brenkman, A.; Hankemeier, T.; Duynhoven, J.; et al. Quantitative profiling of oxylipins through comprehensive LC-MS/MS analysis: Application in cardiac surgery. Anal. Bioanal. Chem. 2012, 404, 1413–1426. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Tong, X.; Wang, J.; Zou, W.; Cao, H.; Su, W. Rapid separation and identification of multiple constituents in traditional Chinese medicine formula Shenqi Fuzheng Injection by ultra-fast liquid chromatography combined with quadrupole-time-of-flight mass spectrometry. J. Pharmaceut. Biomed. 2013, 74, 141–155. [Google Scholar] [CrossRef]
- Ammar, S.; Contreras, M.D.; Belguith-Hadrich, O.; Segura-Carretero, A.; Bouaziz, M. Assessment of the distribution of phenolic compounds and contribution to the antioxidant activity in Tunisian fig leaves, fruits, skins and pulps using mass spectrometry-based analysis. Food Funct. 2015, 6, 3663–3677. [Google Scholar] [CrossRef]
- Zhang, L.; Tu, Z.; Xie, X.; Lu, Y.; Wang, Z.; Wang, H.; Sha, X. Antihyperglycemic, antioxidant activities of two Acer palmatum cultivars, and identification of phenolics profile by UPLC-QTOF-MS/MS: New natural sources of functional constituents. Ind. Crops Prod. 2016, 89, 522–532. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Std no | A: Liquid-to-Solid Ratio (mL/g) | B: Power (W) | C: Time (min) | Response: IC50 (µg DM/mL) |
|---|---|---|---|---|
| 1 | 40:1 | 300 | 40 | 159.20 |
| 2 | 30:1 | 350 | 40 | 182.30 |
| 3 | 40:1 | 350 | 80 | 189.97 |
| 4 | 40:1 | 350 | 60 | 149.14 |
| 5 | 50:1 | 300 | 60 | 160.40 |
| 6 | 30:1 | 400 | 60 | 169.88 |
| 7 | 40:1 | 400 | 40 | 172.22 |
| 8 | 50:1 | 400 | 60 | 159.50 |
| 9 | 40:1 | 350 | 60 | 146.90 |
| 10 | 30:1 | 300 | 60 | 172.71 |
| 11 | 50:1 | 350 | 80 | 198.20 |
| 12 | 40:1 | 400 | 80 | 187.48 |
| 13 | 50:1 | 350 | 40 | 156.40 |
| 14 | 40:1 | 350 | 60 | 148.13 |
| 15 | 40:1 | 350 | 60 | 142.26 |
| 16 | 40:1 | 300 | 80 | 183.31 |
| 17 | 40:1 | 350 | 60 | 146.47 |
| Source | Sum of Squares | df | Mean Square | F Value | p-Value Prob > F | Significance |
|---|---|---|---|---|---|---|
| Model | 4764.62 | 9 | 529.40 | 37.69 | <0.0001 | ** |
| A-liquid-to-solid ratio | 203.74 | 1 | 203.74 | 14.50 | 0.0066 | ** |
| B-Power | 22.65 | 1 | 22.65 | 1.61 | 0.2447 | |
| C-Time | 986.78 | 1 | 986.78 | 70.25 | <0.0001 | ** |
| A2 | 668.94 | 1 | 668.94 | 47.62 | 0.0002 | ** |
| B2 | 174.45 | 1 | 174.45 | 12.42 | 0.0097 | ** |
| C2 | 2138.47 | 1 | 2138.47 | 152.25 | <0.0001 | ** |
| AB | 0.94 | 1 | 0.94 | 0.067 | 0.8034 | |
| AC | 291.45 | 1 | 291.45 | 20.75 | 0.0026 | ** |
| BC | 19.55 | 1 | 19.55 | 1.39 | 0.2767 | |
| Residual | 98.32 | 7 | 14.05 | |||
| Lack of Fit | 70.57 | 3 | 23.52 | 3.39 | 0.1345 | |
| Pure Error | 27.75 | 4 | 6.94 | |||
| Total | 4862.94 | 16 | ||||
| R2 = 0.9798 R2Adj = 0.9538 | ||||||
| No. | Rt (min) | Found at m/z | Expected at m/z | Error (ppm) | Molecular Formula | MS/MS | Proposed Compounds |
|---|---|---|---|---|---|---|---|
| Organic acids | |||||||
| 3 | 4.53 | 195.0514 | 195.0510 | 1.9 | C6H12O7 | - | Gluconic acid |
| 5 | 5.57 | 133.0148 | 133.0142 | 4.2 | C4H6O5 | 115.002[M − H − H2O]− | Malic acid |
| 10 | 6.75 | 191.0200 | 191.0197 | 1.4 | C6H8O7 | 111.0083[M − H − CO2 − 2H2O]− | Citric acid |
| 18 | 9.07 | 163.0404 | 163.0401 | 2.2 | C9H8O3 | 119.0487[M − H − CO2]− | p-Coumaric acid |
| 19 | 9.08 | 325.0937 | 325.0938 | 0.3 | C15H18O8 | 163.0394[M − H − hexose]−, 119.0499[M − H − hexose − CO2]− | Coumaroyl hexose |
| 20 | 9.32 | 325.0939 | 325.0942 | −0.8 | C15H18O8 | 145.0927[M − H − hexose − H2O]−, 117.0342[M − H − hexose − H2O − CO]− | Coumaroyl hexose |
| 24 | 9.69 | 325.0941 | 325.0939 | 0.7 | C15H18O8 | 145.0927[M − H − hexose − H2O]−, 117.0342[M − H − hexose − H2O − CO]− | Coumaroyl hexose |
| 49 | 14.92 | 187.0984 | 187.0982 | 1.0 | C9H16O4 | 125.0970[M − H − C2H2O2]− | Azelaic acid |
| Phenols acids and derivatives | |||||||
| 9 | 6.31 | 329.0879 | 329.0878 | 0.3 | C14H18O9 | 167.0341[M − H − glucose]−, 152.0120[M − H − C7H13O5]−, 123.0438[M − H − C7H10O7]−, 108.0210[M − H − C8H13O7]− | Vanilloyl glucoside |
| 11 | 7.81 | 341.0884 | 341.0883 | 0.4 | C15H18O9 | 179.0344[M − H − hexose]−, 161.0244[M − H − C6H12O6]−, 133.0293[M − H − C7H12O7]−, | Caffeoyl-hexose |
| 13 | 8.18 | 517.1584 | 517.1563 | 4.2 | C22H30O14 | 193.0506[ferulic acid − H]− | Feruloyl sucrose |
| 14 | 8.44 | 223.0621 | 223.0612 | 4.2 | C11H12O5 | 208.0322[M − H − CH3]−, 193.0122[M − H − CH2O]−, 165.0175[M − H − C2H2COOH]−, 135.0440[M − H − C3H4O3]−, 121.0298[M − H − C4H6O3]− | Sinapinic acid |
| 15 | 8.44 | 385.1159 | 385.1140 | 4.8 | C17H22O10 | 223.0606[M − H − glucose]−, 208.0365[M − H − C7H13O5]−, 193.0154[M − H − C8H16O5]−, 179.0714[M − H − C7H10O7]−, 164.0476[M − H − C8H13O7]−, 149.0235[M − H − C9H16O7]− | Sinapoylglucose |
| 16 | 8.66 | 341.0886 | 341.0881 | 1.1 | C15H18O9 | 179.0354[M − H − hexose]−, 135.0449[M − H − C7H10O7]− | Caffeoyl hexose |
| 23 | 9.51 | 355.1052 | 355.1035 | −0.2 | C16H20O9 | 193.0511[M − glucose]−, 178.0272[M − H − C7H13O5]−, 149.0606[M − H − C7H10O7]−, 134.0372[M − H − C8H13O7]− | Feruloyl glucose |
| 25 | 9.84 | 385.1158 | 385.1140 | 4.7 | C17H22O10 | 223.0606[M − H − glucose]−, 208.0365[M − H − C7H13O5]−, 193.0154[M − H − C8H16O5]−, 179.0714[M − H − C7H10O7]−, 164.0476[M − H − C8H13O7]−, 149.0235[M − H − C9H16O7]− | Sinapoylglucose |
| 26 | 10.28 | 183.0307 | 183.0299 | 4.3 | C8H8O5 | 124.0158[M − H − C2H3O2]− | Methyl gallate |
| 42 | 13.05 | 197.0465 | 197.0455 | 5.0 | C9H10O5 | 169.0138[M − H − C2H4]−, 125.0235[M − H − C3H4O2]−, 124.0163[M − H − C3H5O2]− | Ethyl gallate |
| 47 | 13.84 | 193.0507 | 193.0506 | 0.3 | C10H10O4 | 178.0253[M − H − CH3]−, 134.0368[M − H − CH3 − CO2]−, 133.0287[M − H − C4H4O2]− | Ferulic acid |
| Flavonoids | |||||||
| 17 | 8.85 | 577.1378 | 577.1315 | 4.7 | C30H26O12 | 289.0720[(Epi) catechin − H]− | Procyanidin dimmer |
| 21 | 9.32 | 401.1471 | 401.1453 | 4.5 | C18H26O10 | 355.1037[M − H − H2O − CO]−, 269.1040[apigenin − H]−, 223.0582[M − H − C7H14O5]−, 161.0448[M − H − C9H20O7]−, | Apigenin pentose |
| 22 | 9.41 | 289.0724 | 289.0718 | 2.3 | C15H14O6 | 245.0782[M − H − CO2]−, 137.0234[M − H − C8H8O3]−, 125.0232[M − H − C9H8O3]−, 109.0228[B-ring − H]− | (Epi)catechin |
| 27 | 10.28 | 289.0722 | 289.0718 | 1.7 | C15H14O6 | 245.0782[M − H − CO2]−, 137.0234[M − H − C8H8O3]−, 125.0232[M − H − C9H8O3]−, 109.0228[B-ring − H]− | (Epi)catechin |
| 28 | 10.50 | 463.0900 | 463.0898 | 0.5 | C21H20O12 | 463.0898[M − H]−, 301.0354[M − H − hexose]−, 300.0280[M − H − C6H11O5]− | Quercetin-3-O-hexoside |
| 30 | 10.85 | 479.0842 | 479.0831 | 2.2 | C21H20O13 | 259.0262[M − H − C8H12O7]− | Myricetin-3-O-hexoside |
| 32 | 11.12 | 609.1495 | 609.1490 | 0.5 | C27H30O16 | 301.0357[M − H − rutinose]− | Quercetin-3-O-rutinoside |
| 33 | 11.14 | 593.1549 | 593.1546 | 0.5 | C27H30O15 | 285.0411[M − H − rutinose]−, 284.0320[M − H − C12H21O9]−, 151.0027[M − H − rutinose − C8H5O]− | Kaempferol-3-O-rutinoside |
| 34 | 11.93 | 447.0960 | 447.0959 | −0.1 | C21H20O11 | 447.0963[M − H]−, 285.0419[M − H − hexose]−, 284.0336[M − H − C6H11O5]− | Luteolin-7-O-hexoside |
| 35 | 11.93 | 493.1007 | 493.0988 | 3.9 | C22H22O13 | 331.0465[M − H − hexose]−, 315.0157[M − H − C6H10O6]− | Laricitrin-3-O-hexoside |
| 36 | 12.05 | 463.0901 | 463.0898 | 0.5 | C21H20O12 | 301.0363[M − H − hexose]−, 300.0282[M − H − C6H11O5]− | Quercetin-3-O-hexoside |
| 37 | 12.23 | 577.1621 | 577.1621 | 0.0 | C27H30O14 | 269.0459[M − H − rutinose]−, 268.0375[M − H − C12H21O9]− | Apigenin-7-O-rutinoside |
| 38 | 12.38 | 579.1751 | 579.1743 | 1,4 | C27H32O14 | 271.0622[M − H − C12H20O9]−, 151.0035[M − H − C20H28O10]− | Naringin |
| 40 | 12.86 | 507.1174 | 507.1176 | −0.4 | C23H24O13 | 345.0619[M − H − hexose]−, 344.0553[M − H − C6H11O5]−, 329.0309[M − H − C6H10O6]−, 273.0416[M − H − C8H10O8]− | Syringetin-3-O-hexoside |
| 41 | 12.98 | 447.0951 | 447.0953 | −0.4 | C21H20O11 | 285.0481[M − H − hexose]−, 284.0339[M − H − C6H11O5]−, 227.0361[M − H − C8H12O7]− | Kaempferol-3-O-hexoside |
| 43 | 13.25 | 431.1000 | 431.0980 | 3.7 | C21H20O10 | 431.0983[M − H]−, 269.0463[M − H − glucose]−, 268.0388[M − H − C6H11O5]− | Apigenin-7-O-glucoside |
| 44 | 13.38 | 461.1095 | 461.1089 | 1.2 | C22H22O11 | 446.0876[M − H − CH3]−, 299.0553[M − H − hexoside]−, 298.0487[M − H − C6H11O5]−, 283.0249[M − H − C6H10O6]−, 255.0305[M − H − C8H10O7]− | Chrysoeriol-O-hexoside |
| 45 | 13.60 | 433.1157 | 433.1140 | 3.8 | C21H22O10 | 271.0622[M − H − glucose]−, 151.0029[M − H − C14H18O6], 119.0493[M − H − C13H14O9]− | Naringenin-7-O-glucoside |
| 46 | 13.84 | 477.0959 | 477.0960 | −0.1 | C21H20O11 | 285.0412[M − H − hexoside]− | Kaempferol-3-O-hexoside |
| 48 | 14.02 | 463.0918 | 463.0908 | 2.2 | C21H20O12 | 301.0363[M − H − hexoside]− | Quercetin-3-O-hexoside |
| 50 | 15.34 | 301.0367 | 301.0354 | 4.6 | C15H10O7 | 301.0363[M − H]−, 151.0027[M − H − C8H8O3]−, 149.0240[M − H − C8H10O3]− | Quercetin |
| 51 | 16.08 | 287.0565 | 287.0561 | 1.2 | C15H12O6 | 259.0611[M − H − CO]−, 177.0553[M − H − C5H4O3]−, 151.0028[M − H − C8H8O2]−, 125.0239[M − H − C9H6O3]− | Dihydrokaempferol |
| 54 | 17.56 | 285.0417 | 285.0415 | 0.8 | C15H10O6 | 285.0414[M − H]−, 175.0400[M − H − C5H2O3]−, 151.0030[M − H − C8H6O2]−, 133.0297[M − H − C7H4O4]−, | Luteolin |
| 58 | 19.82 | 269.0466 | 269.0456 | 4.0 | C15H10O5 | 269.0455[M − H]−, 151.0030[M − H − C8H6O]−, 149.0238[M − H − C7H4O2]−, 117.0338[M − H − C7H4O4]− | Apigenin |
| 59 | 20.11 | 271.0624 | 271.0612 | 4.5 | C15H12O5 | 151.0030[M − H − C8H8O]−, 119.0499[M − H − C7H4O4]− | Naringenin |
| Fatty acids | |||||||
| 55 | 18.08 | 327.2186 | 327.2183 | −1.3 | C18H32O5 | 291.1957[M − H − 2H2O]−, 229.1442[M − H − 3H2O − CO2]−, 171.1030[M − H − C8H12O3]− | Trihydroxy octadecadienoic acid |
| 56 | 18.29 | 327.2177 | 327.2181 | 4.5 | C18H32O5 | 291.1971[M − H − 2H2O]−, 229.1442[M − H − 3H2O − CO2]−, 171.1032[M − H − C8H12O3]− | Trihydroxy octadecadienoic acid |
| 57 | 19.25 | 329.2353 | 329.2351 | 0.7 | C18H34O5 | 211.1345[M − H − C6H14O2]−, 171.1029[M − H − C8H14O3]− | Trihydroxy octadecenoic acid |
| 60 | 22.09 | 309.2075 | 309.2071 | 1.2 | C18H30O4 | 291.1973[M − H − H2O]−, 265.2159[M − H − C3H8]−, 171.1018[M − H − C9H14O]− | Hydroxy octadecatrienoic acid |
| 64 | 26.29 | 309.2075 | 309.2071 | 1.2 | C18H30O4 | 291.1973[M − H − H2O]−, 185.1188[M − H − C8H12O]−, 171.1031[M − H − C9H14O]− | Hydroxy octadecatrienoic acid |
| 65 | 26.80 | 309.2077 | 309.2071 | 1.8 | C18H30O4 | 209.1554[M − H − C6H12O]−, 197.1187[M − H − C7H12O]− | 11-Hydroperoxy octadecatrienoic acid |
| 66 | 27.08 | 309.2083 | 309.2071 | 4.0 | C18H30O4 | 291.1962[M − H − H2O]−, 185.1183[M − H − C8H12O]−, 171.1028[M − H − C9H14O]− | Hydroxy octadecatrienoic acid |
| 67 | 27.27 | 311.2240 | 311.2228 | 3.8 | C18H32O4 | 293.2107[M − H − H2O]−, 185.1172[M − H − C8H14O]−, 171.1023[M − H − C9H16O]− | 9-Hydroperoxy-octadecadienoic acid |
| 68 | 27.37 | 309.2086 | 309.2071 | 4.6 | C18H30O4 | 211.1333[M − H − C6H12O]−, 197.1180[M − H − C7H11 − H2O]− | 11-Hydroperoxy octadecatrienoic acid |
| 69 | 28.35 | 311.2241 | 311.2228 | 4.1 | C18H32O4 | 293.2138[M − H − H2O]−, 185.1181[M − H − C8H14O]−, 171.1030[M − H − C9H16O]− | 9-Hydroperoxy-octadecadienoic acid |
| 70 | 28.89 | 329.2234 | 329.2333 | 0.3 | C18H34O5 | 211.1351[M − H − C6H14O2]−, 171.1025[M − H − C8H14O3]− | Trihydroxy octadecenoic acid |
| 71 | 29.27 | 311.2239 | 311.2228 | 3.6 | C18H32O4 | 293.2133[M − H − H2O]−, 185.1183[M − H − C8H14O]−, 171.1029[M − H − C9H16O]− | 9-Hydroperoxy-octadecadienoic acid |
| 72 | 30.29 | 291.1980 | 291.1966 | 5.0 | C18H28O3 | 273.1857[M − H − H2O]−, 247.2078[M − H − H2O − CO2]− | 12-Oxo-phytodienoic acid |
| 73 | 30.56 | 559.3142 | 559.3124 | 3.3 | C28H48O11 | 277.2186[M − H − C10H18O9]− | Dirhamosyl linolenic acid |
| 74 | 30.85 | 293.2135 | 293.2122 | 4.3 | C18H30O3 | 275.2031[M − H − H2O]−, 183.1390[M − H − C7H10O]−, 171.1032[M − H − C9H14]−, | Hydroxy octadecatrienoic acid |
| 75 | 31.29 | 293.2135 | 293.2122 | 4.3 | C18H30O3 | 275.2016[M − H − H2O]−, 223.1335[M − H − C5H10]−, 195.1387[M − H − C6H10O]− | Hydroxy octadecatrienoic acid |
| 76 | 32.80 | 291.1977 | 291.1966 | 4.0 | C18H28O3 | 211.1334[M − H − C6H8]−, 197.1183[M − H − C7H10]−, 185.1177[M − H − C8H10]−, | Oxo-octadecatrienoic acid |
| 77 | 33.62 | 295.2283 | 295.2279 | 1.5 | C18H32O3 | 277.2158[M − H − H2O]−, 195.1387[M − H − C6H12O]−, 171.1026[M − H − C9H16]− | 9-Hydroxy-10, 12-octadecadienoic acid Hydroxy octadecadienoic acid |
| 78 | 34.81 | 293.2135 | 293.2122 | 4.3 | C18H30O3 | 249.2215[M − H − CO2]−, 195.1385[M − H − C6H10O]−, 179.1071[M − H − C6H10O2]−, 113.0965[M − H − C11H16O2]− | Oxo-octadecadienoic acid |
| 79 | 35.48 | 293.3133 | 293.2122 | 3.6 | C18H30O3 | 185.1179[M − H − C8H12]−, 125.0961[M − H − C9H12O3]− | Oxo-octadecadienoic acid |
| 80 | 36.00 | 293.2123 | 293.2122 | 0.2 | C18H30O3 | 185.1157[M − H − C8H12]−, 125.0963[M − H − C9H12O3]− | Oxo-octadecadienoic acid |
| Others | |||||||
| 1 | 3.06 | 341.1101 | 341.1089 | 3.4 | C12H22O11 | 179.0595[M − H − C6H10O5]−, 161.0470[M − H − C6H12O6]−, 113.0229[M − H − C7H16O8]− | Sucrose |
| 2 | 3.06 | 179.0566 | 179.0561 | 2.6 | C6H12O6 | 113.0234[M − 2H2O − CH2OH]−, | Monose |
| 4 | 5.40 | 305.1598 | 305.1606 | −2.6 | C14H26O7 | 175.0250, 161.0230, 133.0296 | Unidentified |
| 6 | 5.80 | 137.0247 | 137.0244 | 2.0 | C7H6O3 | - | Protocatechualdehyde |
| 7 | 5.80 | 299.0783 | 299.0772 | 3.5 | C13H16O7 | 137.0270 | Unidentified |
| 8 | 6.31 | 305.1616 | 305.1606 | 3.4 | C14H26O7 | 289.1306, 272.1043, 247.1083, 148.0521, 134.0375 | Unidentified |
| 12 | 8.02 | 391.0828 | 391.0823 | 1.3 | C22H16O7 | 193.0513, 178.0272, 149.0605, 134.0374 | Unidentified |
| 29 | 10.85 | 177.0204 | 177.0201 | 1.8 | C9H6O4 | 177.0180[M − H]−, 149.0234[M − H − CO]−, 133.0285[M − H − CO2]−, 105.0336[M − H − C2O3]− | Dihydroxycoumarin |
| 31 | 11.12 | 431.1938 | 431.1935 | 0.6 | C20H32O10 | 385.1837, 223.1382, 205.1203, 163.1131, 119.0333, 113.0281, 101.0234 | Hydroxy-2,4,4-trimethyl-3-(3-oxobutyl)-2-cyclohexen-1-one glucoside |
| 39 | 12.46 | 723.5092 | 723.5089 | 0.4 | C41H72O10 | 677.5014, 659.4905, 550.4370, 451.3300, 433.316, 367.2732, 341.2932, 309.2213, 225.1609, 207.1497, 143.0814, 125.0709, | Unidentified |
| 52 | 16.62 | 193.0513 | 193.0508 | 2.4 | C10H10O4 | 161.0244, 133.0296 | Unidentified |
| 53 | 17.18 | 201.1145 | 201.1144 | 0.2 | C10H18O4 | 183.1026[M − H − H2O]−, 139.1128[M − H − H2O − CO2]− | Dibutyl oxalate |
| 61 | 23.00 | 307.1928 | 307.1915 | 4.4 | C18H28O4 | 235.1346[M − H − C5H12]−, 211.1343, 185.1188, 137.0966 | Dihydrocapsiate |
| 62 | 24.18 | 311.1878 | 311.1878 | −0.2 | C17H28O5 | 293.1750[M − H − H2O]−, 267.1966[M − H − CO2]− | Dihydroartemisinin ethyl ether |
| 63 | 25.37 | 305.1770 | 305.1758 | 4.0 | C18H26O4 | 249.1499, 135.0809 | Unidentified |
| Factors | Coded Symbols | Levels | ||
|---|---|---|---|---|
| − 1 | 0 | 1 | ||
| Liquid-to-solid ratio (mL/g) | X1 | 30 | 40 | 50 |
| Power (W) | X2 | 40 | 60 | 80 |
| Time (min) | X3 | 300 | 350 | 400 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Tu, Z.; Wang, H.; Zhang, L. Ultrasound-Assisted Extraction Optimization of α-Glucosidase Inhibitors from Ceratophyllum demersum L. and Identification of Phytochemical Profiling by HPLC-QTOF-MS/MS. Molecules 2020, 25, 4507. https://doi.org/10.3390/molecules25194507
Li Z, Tu Z, Wang H, Zhang L. Ultrasound-Assisted Extraction Optimization of α-Glucosidase Inhibitors from Ceratophyllum demersum L. and Identification of Phytochemical Profiling by HPLC-QTOF-MS/MS. Molecules. 2020; 25(19):4507. https://doi.org/10.3390/molecules25194507
Chicago/Turabian StyleLi, Zhen, Zongcai Tu, Hui Wang, and Lu Zhang. 2020. "Ultrasound-Assisted Extraction Optimization of α-Glucosidase Inhibitors from Ceratophyllum demersum L. and Identification of Phytochemical Profiling by HPLC-QTOF-MS/MS" Molecules 25, no. 19: 4507. https://doi.org/10.3390/molecules25194507
APA StyleLi, Z., Tu, Z., Wang, H., & Zhang, L. (2020). Ultrasound-Assisted Extraction Optimization of α-Glucosidase Inhibitors from Ceratophyllum demersum L. and Identification of Phytochemical Profiling by HPLC-QTOF-MS/MS. Molecules, 25(19), 4507. https://doi.org/10.3390/molecules25194507