
Apigenin and Hesperidin Downregulate DNA Repair Genes in MCF-7 Breast Cancer Cells and Augment Doxorubicin Toxicity

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
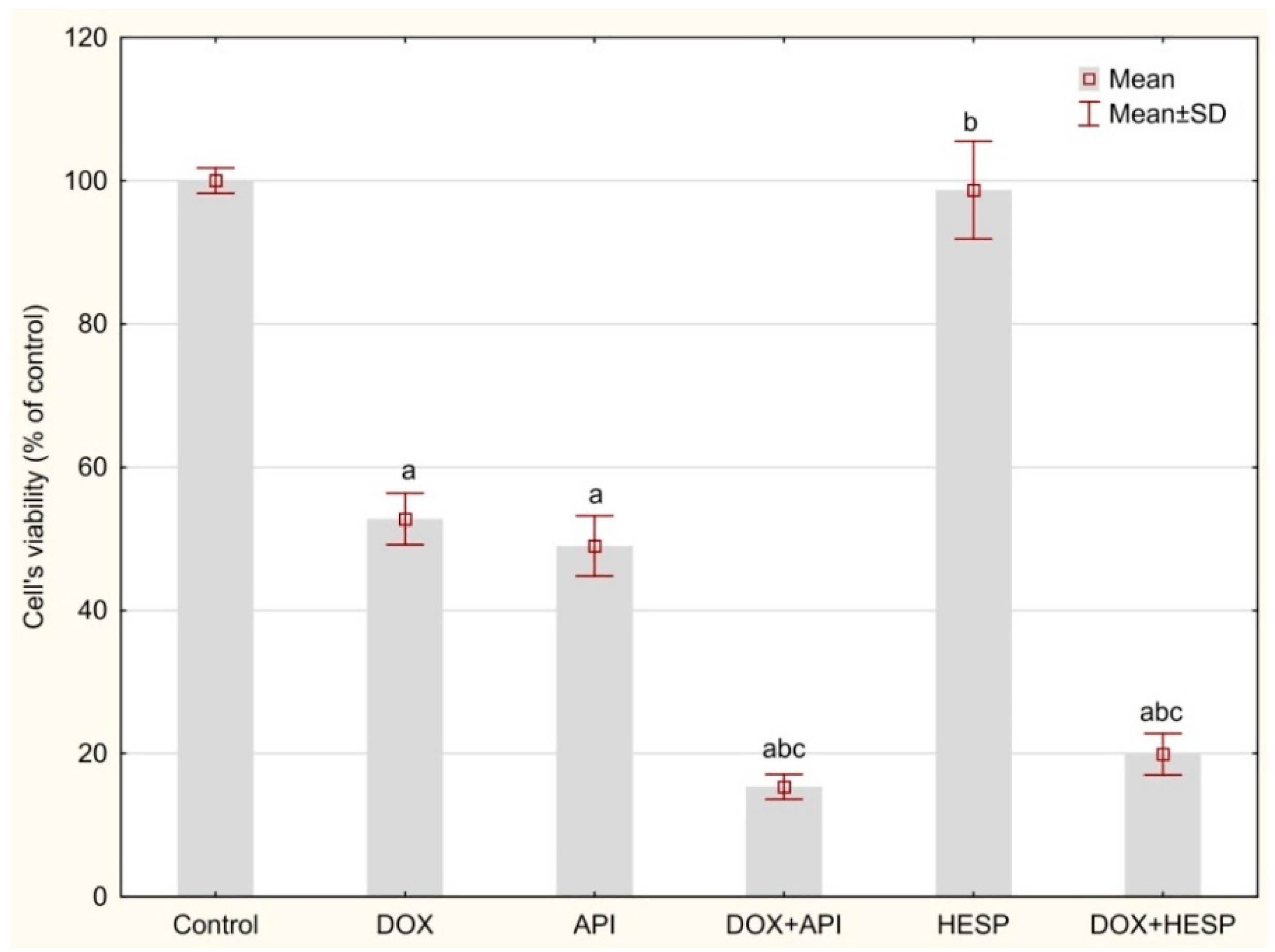
2.1. Cytotoxicity Analyses
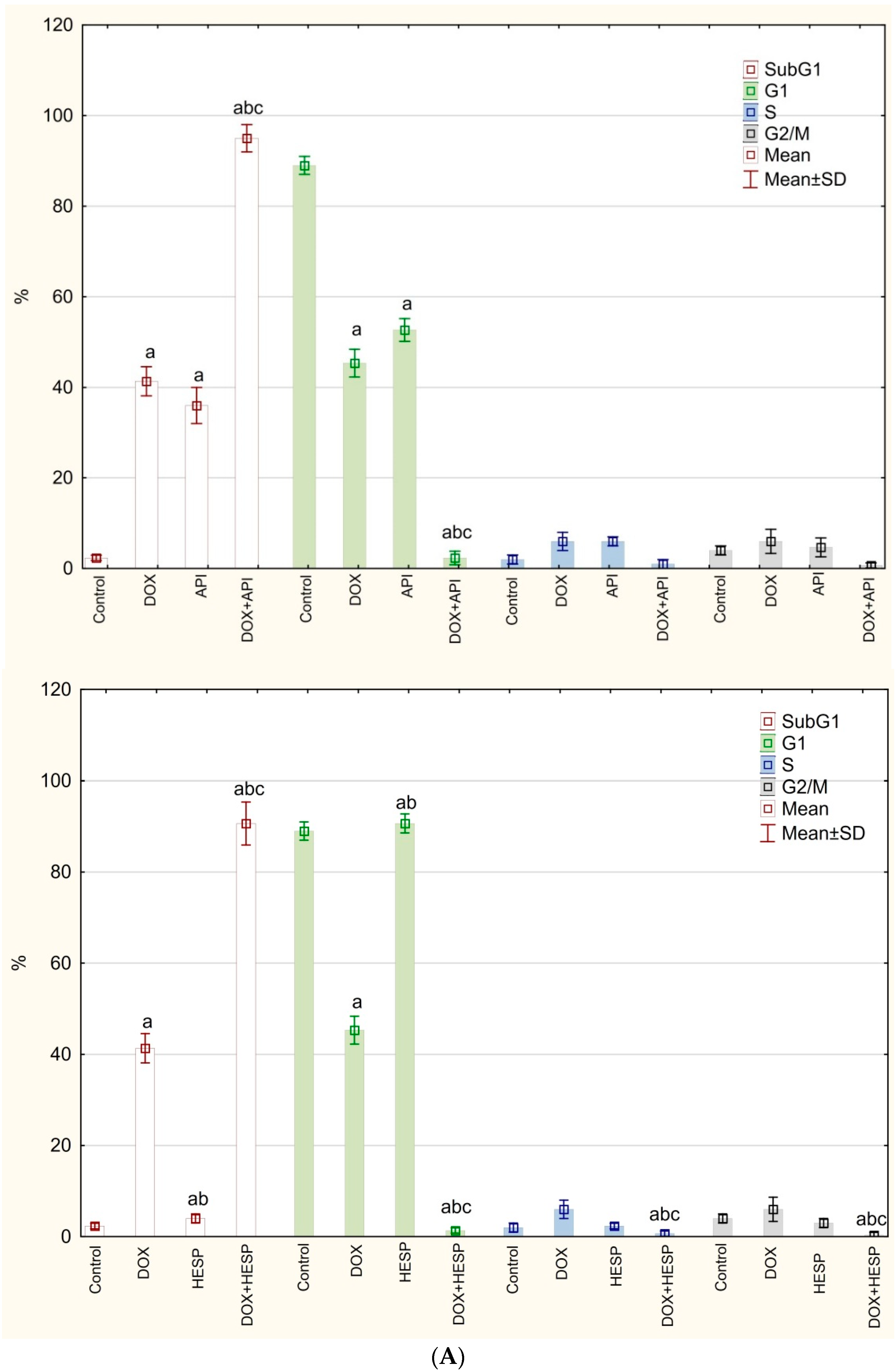
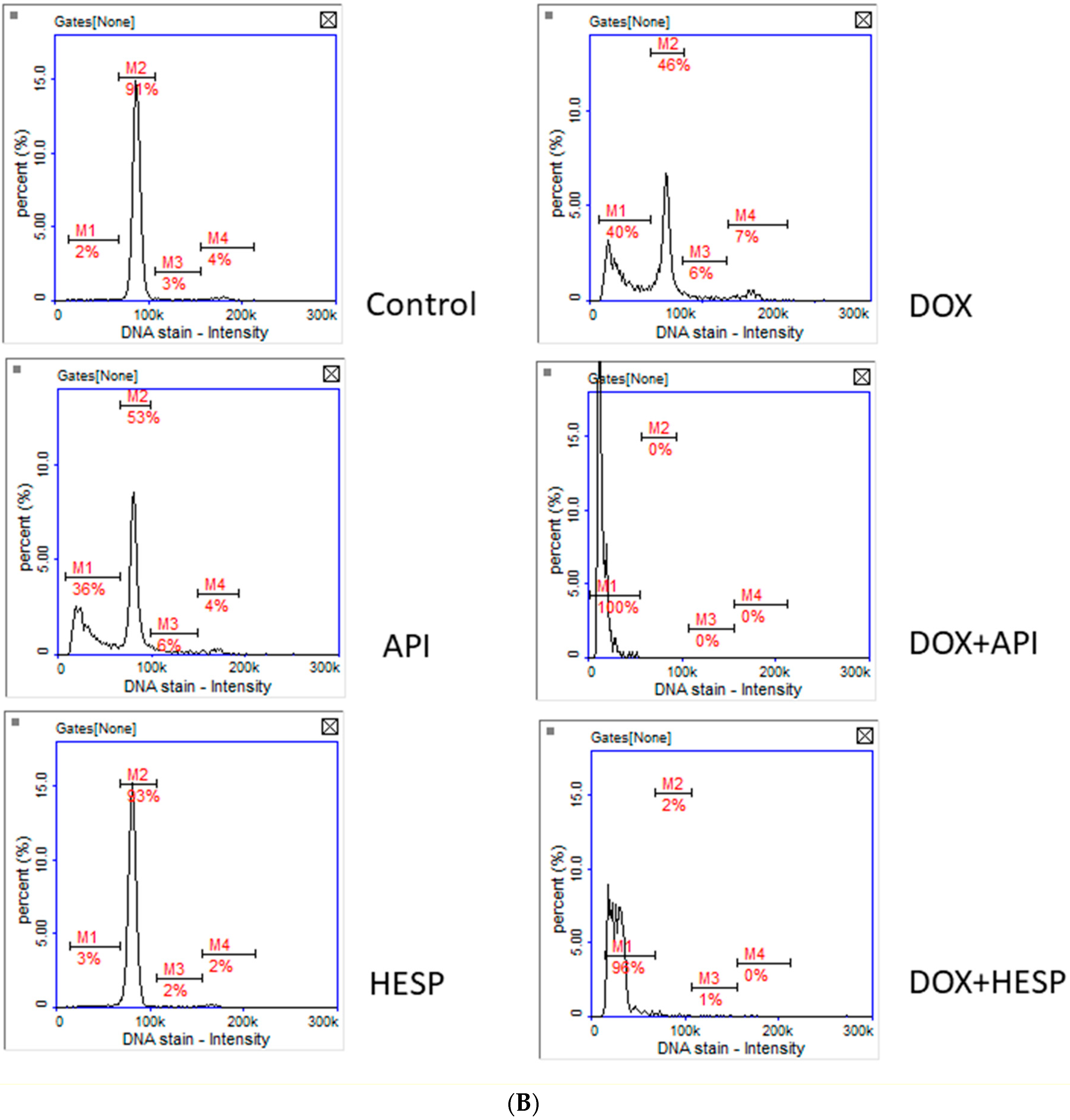
2.2. Cell Cycle Analysis
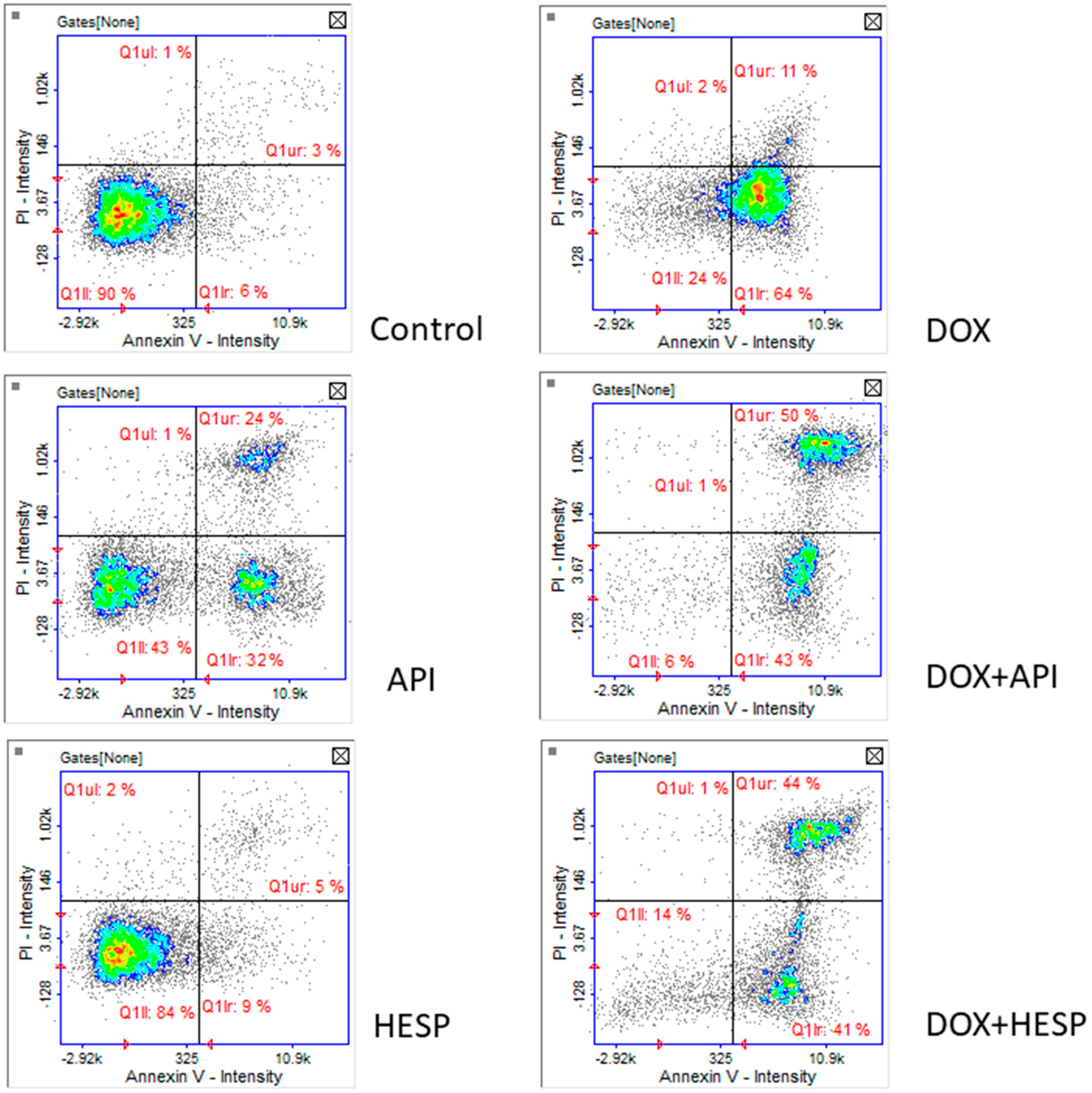
2.3. Apoptosis Detection
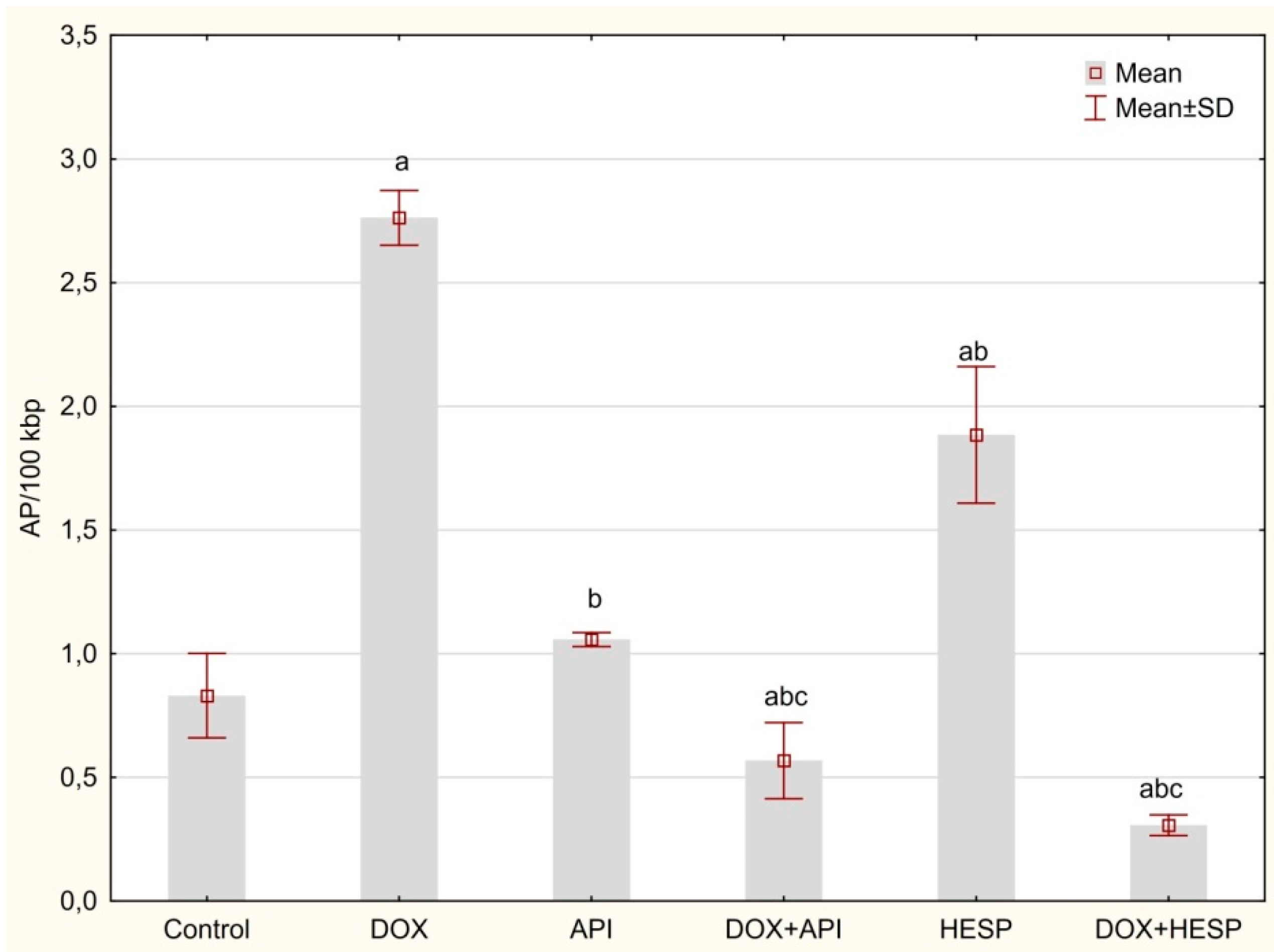
2.4. Determination of DNA Oxidative Damage
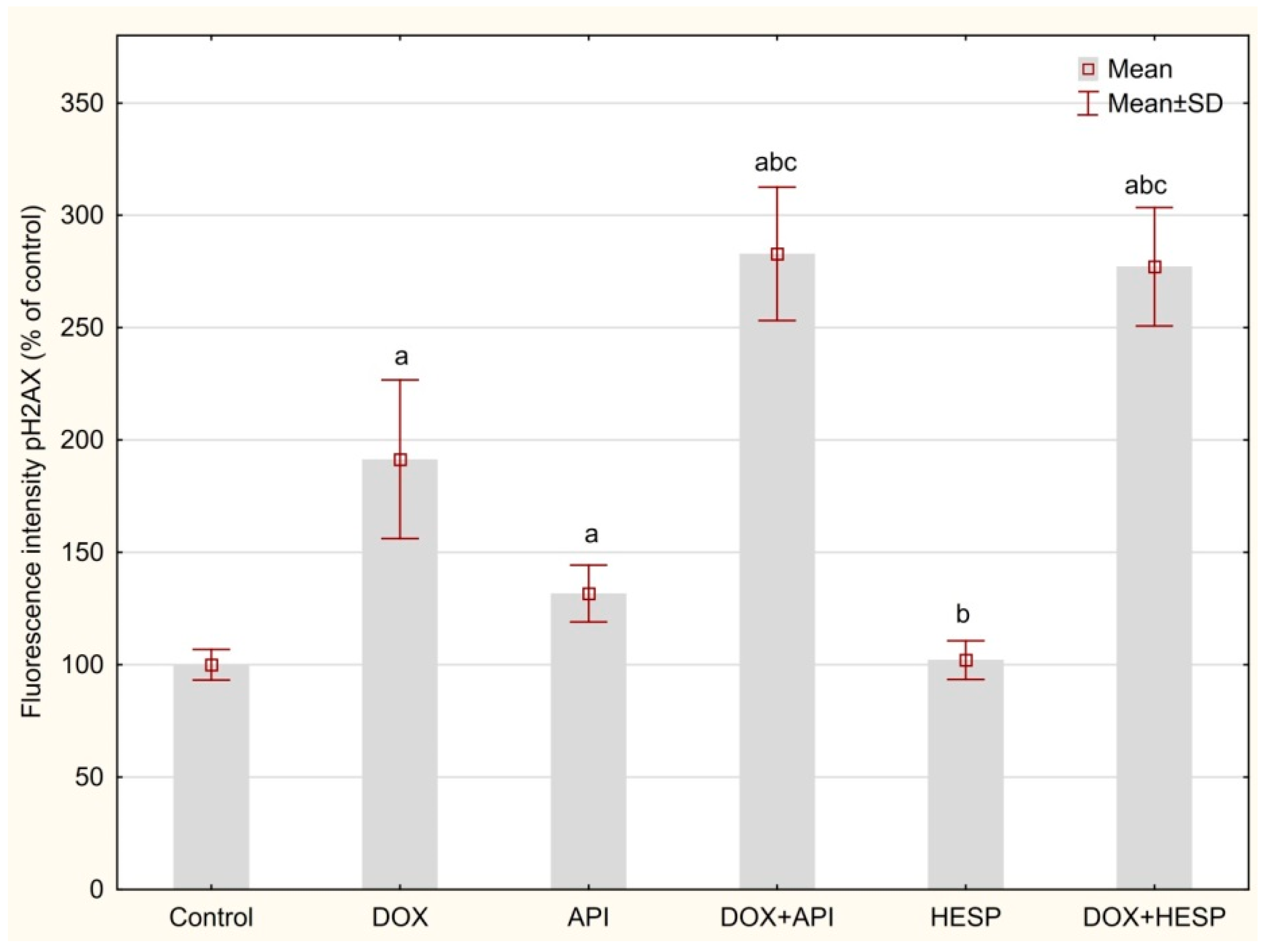
2.5. DNA Double-Strand Breaks (DSBs)
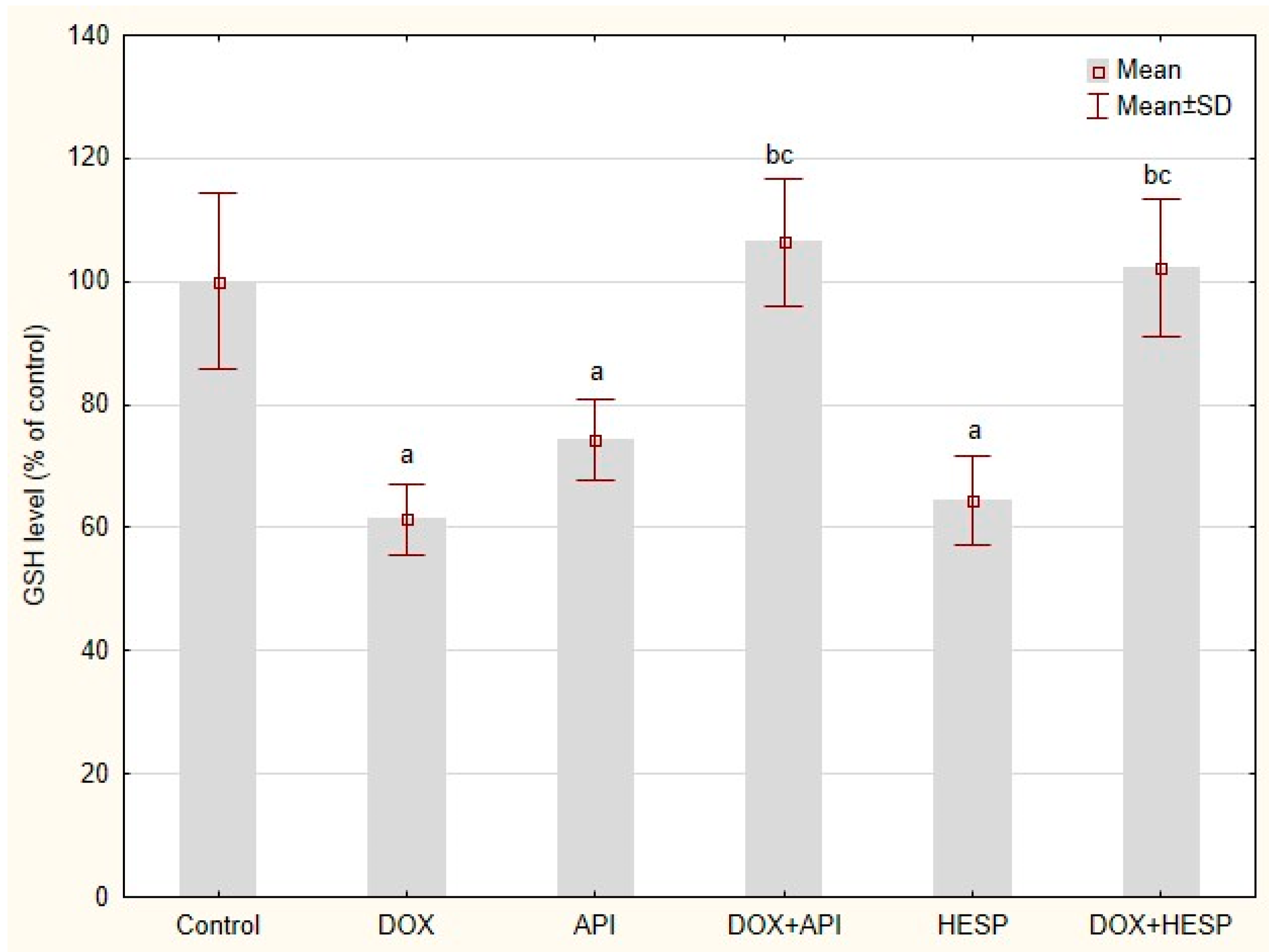
2.6. Reduced Glutathione Level
2.7. The Quantitative Real-Time PCR Analysis (qRTt-PCR)
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. MTT Assay
4.3. Assessment of Cells Morphology
4.4. Cell Cycle Analysis
4.5. Apoptosis Detection
4.6. Reduced Glutathione (GSH) Level
4.7. The Quantitative Real-Time PCR Analysis (qRT-PCR)
4.8. Determination of DNA Oxidative Damage
4.9. DNA Double-Strand Breaks (DSBs)
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wiczkowski, W.; Piskuła, M.K. Foods flavonoids. Pol. J. Food. Nutr. Sci. 2004, 54, 101–114. [Google Scholar]
- Czeczot, H. Biological activities of flavonoids—A review. Pol. J. Food Nutr. Sci. 2000, 950, 3–13. [Google Scholar]
- Muzolf, M.; Szymusiak, H.; Gliszczyńska-Swigło, A.; Rietjens, I.M.C.M.; Tyrakowska, B. pH-Dependent radical scavenging capacity of green tea catechins. J. Agric. Food Chem. 2008, 56, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Abotaleb, M.; Samuel, S.M.; Varghese, E.; Varghese, S.; Kubatka, P.; Liskova, A.; Büsselberg, D. Flavonoids in Cancer and Apoptosis. Cancers 2018, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Yen, G.C.; Duh, P.D.; Tsai, H.L.; Huang, S.L. Pro-oxidative properties of flavonoids in human lymphocytes. Biosci. Biotechnol. Biochem. 2003, 67, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Ji, P.; Liu, B.; Qiao, H.; Wang, X.; Zhou, L.; Deng, T.; Ba, Y. Apigenin enhances the cisplatin cytotoxic effect through p53-modulated apoptosis. Oncol. Lett. 2017, 3, 1024–1030. [Google Scholar] [CrossRef]
- Lamson, D.W.; Brignall, M.S. Antioxidants in cancer therapy; their actions and interactions with oncologic therapies. Altern. Med. Rev. J. Clin. Ther. 1999, 4, 304–329. [Google Scholar]
- Kikuchi, H.; Yuan, B.; Hu, X.; Okazaki, M. Chemopreventive and anticancer activity of flavonoids and its possibility for clinical use by combining with conventional chemotherapeutic agents. Am. J. Cancer. Res. 2019, 9, 1517–1535. [Google Scholar]
- Chen, M.C.; Ye, Y.Y.; Ji, G.; Liu, J.W. Hesperidin upregulates heme oxygenase-1 to attenuate hydrogen peroxide-induced cell damage in hepatic L02 cells. J. Agric. Food Chem. 2010, 58, 3330–3335. [Google Scholar] [CrossRef]
- Elavarasan, J.; Velusamy, P.; Ganesan, T.; Ramakrishnan, S.K.; Rajasekaran, D.; Periandavan, K. Hesperidin-mediated expression of Nrf2 and upregulation of antioxidant status in senescent rat heart. J. Pharm. Pharmacol. 2012, 64, 1472–1482. [Google Scholar] [CrossRef]
- Kamaraj, S.; Anandakumar, P.; Jagan, S.; Ramakrishnan, G.; Devaki, T. Hesperidin attenuates mitochondrial dysfunction during benzo(a)pyrene-induced lung carcinogenesis in mice. Fundam. Clin. Pharmacol. 2011, 25, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Parhiz, H.; Roohbakhsh, A.; Soltani, F.; Rezaee, R.; Iranshahi, M. Antioxidant and anti-inflammatory properties of the citrus flavonoids’ hesperidin and hesperetin: An updated review of their molecular mechanisms and experimental models. Phytother. Res. 2015, 29, 323–331. [Google Scholar] [CrossRef]
- Webb, M.R.; Ebeler, S.E. Comparative analysis of topoisomerase IB inhibition and DNA intercalation by flavonoids and similar compounds: Structural determinates of activity. Biochem. J. 2004, 384, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Arango, D.; Parihar, A.; Villamena, F.A.; Wang, L.; Freitas, M.A.; Grotewold, E.; Doseff, A.I. Apigenin induces DNA damage through the PKCδ-dependent activation of ATM and H2AX causing down-regulation of genes involved in cell cycle control and DNA repair. Biochem. Pharmacol. 2012, 84, 1571–1580. [Google Scholar] [CrossRef]
- Palit, S.; Kar, S.; Sharma, G.; Das, P.K. Hesperetin induces apoptosis in breast carcinoma by triggering accumulation of ROS and activation of ASK1/JNK pathway. J. Cell. Physiol. 2015, 230, 1729–1739. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, J.; Wu, D.; Wang, J.; Dong, W. Hesperetin Induces the Apoptosis of Hepatocellular Carcinoma Cells via Mitochondrial Pathway Mediated by the Increased Intracellular Reactive Oxygen Species, ATP AND Calcium. Med. Oncol. 2015, 32, 101. [Google Scholar] [CrossRef]
- Wu, D.; Zhang, J.; Wang, J.; Li, J.; Liao, F.; Dong, W. Hesperetin induces apoptosis of esophageal cancer cells via mitochondrial pathway mediated by the increased intracellular reactive oxygen species. Tumour Biol. 2016, 37, 3451–3459. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, D.; Vikash; Song, J.; Wang, J.; Yi, J.; Dong, W. Hesperetin induces the apoptosis of gastric cancer cells via activating mitochondrial pathway by increasing reactive oxygen species. Dig. Dis. Sci. 2015, 60, 2985–2995. [Google Scholar] [CrossRef]
- Wang, C.; Kurzer, M.S. Phytoestrogen concentration determines effects on DNA synthesis in human breast cancer cells. Nutr. Cancer 1997, 28, 236–247. [Google Scholar] [CrossRef]
- Wang, C.; Kurzer, M.S. Effects of phytoestrogens on DNA synthesis in MCF-7 cells in the presence of estradiol or growth factors. Nutr. Cancer 1998, 31, 90–100. [Google Scholar] [CrossRef]
- Choi, E.J.; Kim, G.H. 5-Fluorouracil combined with apigenin enhances anticancer activity through induction of apoptosis in human breast cancer MDA-MB-453 cells. Oncol. Rep. 2009, 22, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xin, Y.; Diao, Y.; Lu, C.; Fu, J.; Luo, L.; Yin, Z. Synergistic effects of apigenin and paclitaxel on apoptosis of cancer cells. PLoS ONE 2011, 6, e29169. [Google Scholar] [CrossRef] [PubMed]
- Hermawan, A.; Meiyanto, E.; Susidarti, R. Hesperidine Increases Cytotoxic Effect of Doxorubicin on MCF-7 Cells, Indones. J. Pharm. 2010, 21, 8–17. [Google Scholar] [CrossRef]
- Watanabe, N.; Hirayama, R.; Kubota, N. The chemopreventive flavonoid apigenin confers radiosensitizing effect in human tumor cells grown as monolayers and spheroids. J. Radiat. Res. 2007, 48, 45–50. [Google Scholar] [CrossRef]
- Febriansah, R.; Putri, D.D.; Sarmoko Nurulita, N.A.; Meiyanto, E.; Nugroho, A.E. Hesperidin as a preventive resistance agent in MCF-7 breastcancer cells line resistance to doxorubicin. Asian. Pac. J. Trop. Biomed. 2014, 4, 228–3391. [Google Scholar] [CrossRef]
- Desai, U.N.; Shah, K.P.; Mirza, S.H.; Panchal, D.K.; Parikh, S.K.; Rawal, R.M. Enhancement of the cytotoxic effects of cytarabine in synergism withhesperidine and silibinin in acute myeloid leukemia: An in-vitroapproach. J. Cancer Res. Ther. 2015, 11, 352–792. [Google Scholar] [CrossRef]
- Khamis, A.A.A.; Ali, E.M.M.; El-Moneim, M.A.A.; Abd-Alhaseeb, M.M.; El-Magd, M.A.; Salim, E.I. Hesperidin, piperine and bee venom synergisti-cally potentiate the anticancer effect of tamoxifen against breast cancer cells. Biomed. Pharmacother. 2018, 105, 1335–1343. [Google Scholar] [CrossRef]
- Medhat, A.M.; Azab, K.S.; Said, M.M.; El Fatih, N.M.; El Bakary, N.M. Antitumor and radiosensitizing synergistic effects of apigenin and cryptotanshinone against solid Ehrlich carcinoma in female mice. Tumour Biol. 2017, 39, 1010428317728480. [Google Scholar] [CrossRef]
- Abdel-Rafei, M.K.; Hassan, A.A.B. Radiosensitizing Efficacy of Diosmin- Hesperidin Complex Against Ehrlich Solid Carcinoma in Mice, A Potential Role of Histone Deacetylase and Pro-angiogenic Chaperones Targeting. Int. J. Cancer. Clin. Res. 2017, 13, 59–70. [Google Scholar] [CrossRef][Green Version]
- Souza, R.P.; Bonfim-Mendonça, P.S.; Gimenes, F.; Ratti, B.A.; Kaplum, V.; Bruschi, M.L.; Nakamura, C.V.; Silva, S.O.; Maria-Engler, S.S.; Consolaro, M.E. Oxidative Stress Triggered by Apigenin Induces Apoptosis in a Comprehensive Panel of Human Cervical Cancer-Derived Cell Lines. Oxid Med Cell Longev. 2017, 2017, 1512745. [Google Scholar] [CrossRef]
- Vrhovac Madunić, I.; Madunić, J.; Antunović, M.; Paradžik, M.; Garaj-Vrhovac, V.; Breljak, D.; Marijanović, I.; Gajski, G. Apigenin, a dietary flavonoid, induces apoptosis, DNA damage, and oxidative stress in human breast cancer MCF-7 and MDA MB-231 cells. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Sheng, X.; Xu, X.; Yu, C.; Lu, H. Hesperidin induces apoptosis and G0/G1 arrest in human non-small cell lung cancer A549 cells. Int. J. Mol. Med. 2018, 41, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Singal, P.K.; Li, T.; Kumar, D.; Danelisen, I.; Iliskovic, N. Adriamycin-induced heart failure: Mechanism and modulation. Mol. Cell. Biochem. 2000, 207, 77–86. [Google Scholar] [CrossRef]
- Segredo, M.P.; Salvadori, D.M.; Rocha, N.S.; Moretto, F.C.; Correa, C.R.; Camargo, E.A.; de Almeida, D.C.; Silva Reis, R.A.; Murbach Freire, C.M.; Braz, M.G.; et al. Oxidative stress on cardiotoxicity after treatment with single and multiple doses of doxorubicin. Hum. Exp. Toxicol. 2014, 33, 748–760. [Google Scholar] [CrossRef]
- Mahbub, A.; Le Maitre, C.; Haywood-Small, S.; Cross, N.; Jordan-Mahy, N. Polyphenols act synergisticallywith doxorubicin and etoposide in leukaemia cell lines. Cell Death Discov. 2015, 1, 15043. [Google Scholar] [CrossRef]
- Gao, A.M.; Ke, Z.P.; Wang, J.N.; Yang, J.Y.; Chen, S.Y.; Chen, Y. Apigenin sensitizes doxorubicin-resistant hepatocellular carcinoma BEL-7402/ADM cells to doxorubicin via inhibiting PI3K/Akt/Nrf2 pathway. Carcinogenesis 2013, 34, 1806–1814. [Google Scholar] [CrossRef]
- Gao, A.M.; Zhang, X.Y.; Ke, Z.P. Apigenin sensitizes BEL-7402/ADM cells to doxorubicin through inhibiting miR-101/Nrf2 pathway. Oncotarget 2017, 8, 82085–82091. [Google Scholar] [CrossRef]
- Korga, A.; Ostrowska, M.; Jozefczyk, A.; Iwan, M.; Wójcik, R.; Zgórka, G.; Herbet, M.; Vilarrubla, G.G.; Dudka, J. Apigenin and hesperidin augment the toxic effect of doxorubicin against HepG2 cells. BMC Pharmacol. Toxicol. 2019, 20, 22. [Google Scholar] [CrossRef]
- Kim, B.R.; Jeon, Y.K.; Nam, M.J. A mechanism of apigenin-induced apoptosis is potentially related to anti-angiogenesis and anti-migration in human hepatocellular carcinoma cells. Food Chem. Toxicol. 2011, 49, 1626–1632. [Google Scholar] [CrossRef]
- Vargo, M.A.; Voss, O.H.; Poustka, F.; Cardounel, A.J.; Grotewold, E.; Doseff, A.I. Apigenin-induced-apoptosis is mediated by the activation of PKCdelta and caspases in leukemia cells. Bioche. Pharmacol. 2006, 72, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Heim, K.E.; Tagliaferro, A.R.; Bobilya, D.J. Flavonoid antioxidants: Chemistry, metabolism and structure-activity relationships. J. Nutr. Biochem. 2002, 13, 572–584. [Google Scholar] [CrossRef]
- Silva, J.P.; Gomes, A.C.; Coutinho, O.P. Oxidative DNA damage protection and repair by polyphenolic compounds in PC12 cells. Eur. J. Pharmacol. 2008, 601, 50–60. [Google Scholar] [CrossRef]
- Das, S.; Das, J.; Paul, A.; Samadder, A.; Khuda-Bukhsh, A.R. Apigenin, a Bioactive Flavonoid from Lycopodium clavatum, Stimulates Nucleotide Excision Repair Genes to Protect Skin Keratinocytes from Ultraviolet B-Induced Reactive Oxygen Species and DNA Damage. J. Acupunct. Meridian. Stud. 2013, 6, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.C.; Young, M.R.; Cmarik, J.; Colburn, N.H. Activator protein 1 (AP-1)- and nuclear factor kappaB (NF-kappaB)-dependent transcriptional events in carcinogenesis. Free Radic. Biol. Med. 2000, 28, 1338–1348. [Google Scholar] [CrossRef]
- Zare, M.F.R.; Rakhshan, K.; Aboutaleb, N.; Nikbakht, F.; Naderi, N.; Bakhshesh, M.; Azizi, Y. Apigenin attenuates doxorubicin induced cardiotoxicity via reducing oxidative stress and apoptosis in male rats. Life Sci. 2019, 232, 116623. [Google Scholar] [CrossRef]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef]
- Terao, J. Dietary flavonoids as antioxidants in vivo: Conjugated metabolites of (−)-epicatechin and quercetin participate in antioxidative defense in blood plasma. J. Med. Investig. JMI 1999, 46, 159–168. [Google Scholar]
- Andueza, A.; García-Garzón, A.; Ruiz de Galarreta, M.; Ansorena, E.; Iraburu, M.J.; López-Zabalza, M.J.; Martínez-Irujo, J.J. Oxidation pathways underlying the pro-oxidant effects of apigenin. Free Radic. Biol. Med. 2015, 87, 169–180. [Google Scholar] [CrossRef]
- Constantin, R.P.; do Nascimento, G.S.; Constantin, R.P.; Salgueiro, C.L.; Bracht, A.; Ishii-Iwamoto, E.L.; Yamamoto, N.S.; Constantin, J. Citrus flavanones affect hepatic fatty acid oxidation in rats by acting as prooxidant agents. Biomed. Res. Int. 2013, 342973. [Google Scholar] [CrossRef]
- Rusak, G.; Piantanida, I.; Masić, L.; Kapuralin, K.; Durgo, K.; Kopjar, N. Spectrophotometric analysis of flavonoid-DNA interactions and DNA damaging/protecting and cytotoxic potential of flavonoids in human peripheral blood lymphocytes. Chem. Biol. Interact. 2010, 188, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Beckerman, R.; Prives, C. Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2010, 2, a000935. [Google Scholar] [CrossRef]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Horejsí, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Lukas, C.; Bartek, J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011, 13, 1161–1169. [Google Scholar] [CrossRef]
- Kemp, M.G.; Lindsey-Boltz, L.A.; Sancar, A. The DNA damage response kinases DNA-dependent protein kinase (DNA-PK) and ataxia telangiectasia mutated (ATM) are stimulated by bulky adduct-containing DNA. J. Biol. Chem. 2011, 286, 19237–19246. [Google Scholar] [CrossRef]
- Liu, Q.; Guntuku, S.; Cui, X.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes. Dev. 2000, 14, 1448–1459. [Google Scholar]
- Pabla, N.; Huang, S.; Mi, Q.-S.; Daniel, R.; Dong, Z. ATR-Chk2 signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis. J. Biol. Chem. 2008, 283, 6572–6583. [Google Scholar] [CrossRef]
- Jänicke, R.U. MCF-7 breast carcinoma cells do not express caspase-3. Breast Cancer Res. Treat 2009, 117, 219–221. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; He, M.; Li, L.; Liang, Z.; Zou, Z.; Tao, A. Cell-in-Cell Death Is Not Restricted by Caspase-3 Deficiency in MCF-7 Cells. J. Breast Cancer 2016, 19, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Kwan, Y.P.; Saito, T.; Ibrahim, D.; Al-Hassan, F.M.; Ein Oon, C.; Chen, Y.; Jothy, S.L.; Kanwar, J.R.; Sasidharan, S. Evaluation of the cytotoxicity, cell-cycle arrest, and apoptotic induction by Euphorbia hirta in MCF-7 breast cancer cells. Pharm. Biol. 2016, 54, 1223–1236. [Google Scholar] [CrossRef] [PubMed]
- Antonini, E.; Iori, R.; Ninfali, P.; Scarpa, E.S. A Combination of Moringin and Avenanthramide 2f Inhibits the Proliferation of Hep3B Liver Cancer Cells Inducing Intrinsic and Extrinsic Apoptosis. Nutr. Cancer 2018, 70, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- Puccini, J.; Dorstyn, L.; Kumar, S. Caspase-2 as a tumour suppressor. Cell Death Differ. 2013, 20, 1133–1139. [Google Scholar] [CrossRef]
- Yang, F.; Kemp, C.J.; Henikoff, S. Anthracyclines induce double-strand DNA breaks at active gene promoters. Mutat. Res. 2015, 773, 9–15. [Google Scholar] [CrossRef]
- Korga, A.; Dudka, J.; Burdan, F.; Sliwinska, J.; Mandziuk, S.; Dawidek-Pietryka, K. The redox imbalance and the reduction of contractile protein content in rat hearts administered with L-thyroxine and Doxorubicin. Oxid. Med. Cell Longev. 2012, 681367. [Google Scholar] [CrossRef]
- Korga, A.; Ostrowska, M.; Iwan, M.; Herbet, M.; Dudka, J. Inhibition of glycolysis disrupts cellular antioxidant defense and sensitizes HepG2 cells to doxorubicin treatment. FEBS Open Biol. 2019, 9, 959–972. [Google Scholar] [CrossRef]
- O’Connor, M. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef]
- Minchom, A.; Aversa, C.; Lopez, J. Dancing with the DNA damage response: Next-generation anti-cancer therapeutic strategies. Ther. Adv. Med. Oncol. 2018, 10, 1–18. [Google Scholar] [CrossRef]
- McHowat, J.; Swift, L.M.; Arutunyan, A.; Sarvazyan, N. Clinical concentrations of doxorubicin inhibit activity of myocardial membrane-associated, calcium-independent phospholipase A2. Cancer Res. 2001, 61, 4024–4029. [Google Scholar] [PubMed]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | DOX (1 µM) | ||||
|---|---|---|---|---|---|
| 100.04 ± 1.78 | 52.8 ± 3.59 | ||||
| API (µM) | |||||
| 10 | 25 | 50 | 100 | 200 | |
| 95.67 ± 7.45 b | 79.42 ± 6.11 ab | 49.02 ± 4.23 a | 33.07 ± 1.90 ab | 12.77 ± 2.13 ab | |
| DOX + API | |||||
| 49.04 ± 2.74 a | 44.57 ± 9.02 ac | 15.35 ± 2.35 abc | 12.89 ± 3.07 abc | 5.64 ± 1.15 abc | |
| HESP (µM) | |||||
| 10 | 25 | 50 | 100 | 200 | |
| 99.23 ± 7.73 b | 101 ± 2.27 b | 98.70 ± 6.82 b | 75.886.67 ab | 68.99 ± 3.57 ab | |
| DOX + HESP | |||||
| 50.08 ± 4.86 ac | 57.99 ± 2.31 ac | 19.93 ± 2.80 abc | 18.69 ± 4.55 abc | 25.02 ± 4.54 abc | |
| Table 1 | Control | DOX | API | DOX + API | HESP | DOX + HESP | Scale (RQ) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PARP1 | 1.000 | 0.023 | 1.057 | 0.057 | 0.816 ab | 0.080 | 0.476 abc | 0.020 | 0.391 ab | 0.013 | 0.268 abc | 0.008 | > 2.00 | |
| ERCC1 | 1.001 | 0.043 | 0.329 a | 0.043 | 0.487 ab | 0.027 | 0.208 abc | 0.013 | 0.578 ab | 0.019 | 0.126 abc | 0.007 | 1.51–1.99 | |
| ATM | 1.051 | 0.428 | 0.234 a | 0.043 | 0.622 ab | 0.036 | 0.584 ab | 0.014 | 0.715 ab | 0.025 | 0.162 ac | 0.048 | 1.11–1.50 | |
| MSH2 | 1.002 | 0.085 | 0.369 a | 0.017 | 0.235 ab | 0.014 | 0.299 ab | 0.007 | 0.765 ab | 0.024 | 0.254 abc | 0.033 | 0.91–1.10 | |
| OGG1 | 1.000 | 0.031 | 0.785 a | 0.042 | 0.766 a | 0.042 | 0.425 abc | 0.048 | 0.335 ab | 0.008 | 0.177 abc | 0.016 | 0.61–0.90 | |
| MGMT | 1.001 | 0.043 | 0.513 a | 0.028 | 0.484 a | 0.027 | 0.261 abc | 0.031 | 0.664 ab | 0.024 | 0.221 abc | 0.012 | 0.21–0.6 | |
| XPC | 1.001 | 0.041 | 2.108 a | 0.222 | 0.384 ab | 0.016 | 0.145 abc | 0.007 | 0.534 ab | 0.023 | 1.190 abc | 0.051 | < 0.21 | |
| MLH1 | 1.000 | 0.026 | 0.799 a | 0.032 | 0.637 ab | 0.043 | 0.387 abc | 0.029 | 0.517 ab | 0.008 | 0.357 abc | 0.010 | ||
| Target | Forward | Reverse |
|---|---|---|
| PARP1 | CCCCACGACTTTGGGATGAA | AGACTGTAGGCCACCTCGAT |
| ERCC1 | CTCGGAGTTTTGTGGGGGAC | CACTGGCGTCTACGTTCTCA |
| ATM | GCCGCGGTTGATACTACTTTG | GCAGCAGGGTGACAATAAACA |
| MSH2 | CAGGAGGTGAGGAGGTTTCG | CCGTGCGCCGTATAGAAGTC |
| MLH1 | GCACCGGGATCAGGAAAGAA | GCCTCACCTCGAAAGCCATA |
| XPC | GCGAAGTGGAATTTGCCCAG | TTGGCCTTGGATTTCTGGCT |
| MGMT | ACCGTTTGCGACTTGGTACT | TGCTCACAACCAGACAGCTC |
| OGG1 | CCTGTGGGGACCTTATGCTG | TGTGAATCCCCTCTCCCGAT |
| 18SRNA | GAAACTGCGAATGGCTCATTAAA | CACAGTTATCCAAGTGGGAGAGG |
| BACT | AGAGCTACGAGCTGCCTGAC | AGCACTGTGTTGGCGTACAG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korga-Plewko, A.; Michalczyk, M.; Adamczuk, G.; Humeniuk, E.; Ostrowska-Lesko, M.; Jozefczyk, A.; Iwan, M.; Wojcik, M.; Dudka, J. Apigenin and Hesperidin Downregulate DNA Repair Genes in MCF-7 Breast Cancer Cells and Augment Doxorubicin Toxicity. Molecules 2020, 25, 4421. https://doi.org/10.3390/molecules25194421
Korga-Plewko A, Michalczyk M, Adamczuk G, Humeniuk E, Ostrowska-Lesko M, Jozefczyk A, Iwan M, Wojcik M, Dudka J. Apigenin and Hesperidin Downregulate DNA Repair Genes in MCF-7 Breast Cancer Cells and Augment Doxorubicin Toxicity. Molecules. 2020; 25(19):4421. https://doi.org/10.3390/molecules25194421
Chicago/Turabian StyleKorga-Plewko, Agnieszka, Monika Michalczyk, Grzegorz Adamczuk, Ewelina Humeniuk, Marta Ostrowska-Lesko, Aleksandra Jozefczyk, Magdalena Iwan, Marta Wojcik, and Jaroslaw Dudka. 2020. "Apigenin and Hesperidin Downregulate DNA Repair Genes in MCF-7 Breast Cancer Cells and Augment Doxorubicin Toxicity" Molecules 25, no. 19: 4421. https://doi.org/10.3390/molecules25194421
APA StyleKorga-Plewko, A., Michalczyk, M., Adamczuk, G., Humeniuk, E., Ostrowska-Lesko, M., Jozefczyk, A., Iwan, M., Wojcik, M., & Dudka, J. (2020). Apigenin and Hesperidin Downregulate DNA Repair Genes in MCF-7 Breast Cancer Cells and Augment Doxorubicin Toxicity. Molecules, 25(19), 4421. https://doi.org/10.3390/molecules25194421