
Bifunctional HDAC Therapeutics: One Drug to Rule Them All?

Abstract
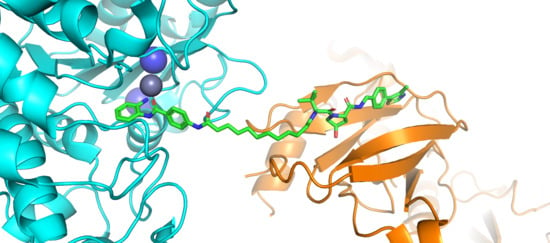
1. Introduction
2. Dual HDAC Inhibitors
3. Class-I HDAC Dual Inhibitors
3.1. Class-I HDAC/Kinase Dual Inhibitors
3.2. Class-I HDAC/Non-Kinase Dual Inhibitors
3.3. HDAC8-Selective Dual Inhibitors
4. Class-II HDAC Dual Inhibitors
4.1. JAK-Targeting HDAC6-Selective Dual Inhibitors
4.2. Other HDAC6-Selective Dual Inhibitors
5. HDAC Complex-Selective Dual Inhibitors
6. HDAC Proteolysis-Targeting Chimeras (PROTACs)
7. Conclusions and Future Outlook
Funding
Conflicts of Interest
References
- Mai, A.; Massa, S.; Rotili, D.; Cerbara, I.; Valente, S.; Pezzi, R.; Simeoni, S.; Ragno, R. Histone deacetylation in epigenetics: An attractive target for anticancer therapy. Med. Res. Rev. 2005, 25, 261–309. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef]
- Gregoretti, I.; Lee, Y.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Disc. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef]
- Aldana-Masangkay, G.I.; Sakamoto, K.M. The role of HDAC6 in cancer. J. Biomed. Biotechnol. 2011, 2011, 875824. [Google Scholar] [CrossRef]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2008, 277, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Kilgore, M.; Miller, C.A.; Fass, D.M.; Hennig, K.M.; Haggarty, S.J.; Sweatt, J.D.; Rumbaugh, G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2010, 35, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.A.; Coppola, G.; Desplats, P.A.; Tang, B.; Soragni, E.; Burnett, R.; Gao, F.; Fitzgerald, K.M.; Borok, J.F.; Herman, D.; et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 15564–15569. [Google Scholar] [CrossRef] [PubMed]
- Sumner, C.J.; Huynh, T.N.; Markowitz, J.A.; Perhac, J.S.; Hill, B.; Coovert, D.D.; Schussler, K.; Chen, X.; Jarecki, J.; Burghes, A.H.M.; et al. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann. Neurol. 2003, 54, 647–654. [Google Scholar] [CrossRef]
- Jenssen, K.; Burnett, R.; Soragni, E.; Herman, D.; Perlman, S.L.; Gottesfeld, J.M. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat. Chem. Biol. 2006, 2, 551–558. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, X.; Wu, Y.; Wiest, O. Inhibition and mechanism of HDAC8 revisited. J. Am. Chem. Soc. 2014, 136, 11636–11643. [Google Scholar] [CrossRef]
- Gantt, S.M.L.; Decroos, C.; Lee, M.S.; Gullett, L.E.; Bowman, C.M.; Christianson, D.W.; Fierke, C.A. General base–general acid catalysis in human histone Deacetylase 8. Biochemistry 2016, 55, 820–832. [Google Scholar] [CrossRef]
- Roche, J.; Bertrand, P. Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef]
- Dokmanovic, M.; Marks, P.A. Prospects: Histone deacetylase inhibitors. J. Cell. Biochem. 2005, 96, 293–304. [Google Scholar] [CrossRef]
- Lane, A.A.; Chabner, B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef]
- McKinsey, T.A. Isoform-selective HDAC inhibitors: Closing in on translational medicine for the heart. J. Mol. Cell. Cardiol. 2010, 51, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval summary: Vorinostat for treatment of advanced primary cutaneous T-Cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- VanderMolen, K.M.; McCulloch, W.; Pearce, C.J.; Oberlies, N.H. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): A natural product recently approved for cutaneous T-cell lymphoma. J. Antibiot. 2011, 64, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Laubach, J.P.; Moreau, P.; San-Miguel, J.F.; Richardson, P.G. Panobinostat for the treatment of multiple myeloma. Clin. Cancer Res. 2015, 21, 4767–4773. [Google Scholar] [CrossRef] [PubMed]
- Poole, R. Belinostat: First global approval. Drugs 2014, 74, 1543–1554. [Google Scholar] [CrossRef]
- Lu, X.; Ning, Z.; Li, Z.; Cao, H.; Wang, X. Development of chidamide for peripheral T-cell lymphoma, the first orphan drug approved in China. Intractable Rare Dis. Res. 2016, 5, 185–191. [Google Scholar] [CrossRef]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty years of HDAC inhibitors: 2020 insight and hindsight. J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109, 31–39. [Google Scholar] [CrossRef]
- Vansteenkiste, J.; Van Cutsem, E.; Dumez, H.; Chen, C.; Ricker, J.; Randolph, S.; Schöffski, P. Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Investig. New Drugs 2008, 26, 483–488. [Google Scholar] [CrossRef]
- Schneider, B.; Kalemkerian, G.; Bradley, D.; Smith, D.; Egorin, M.; Daignault, S.; Dunn, R.; Hussain, M. Phase I study of vorinostat (suberoylanilide hydroxamic acid, NSC 701852) in combination with docetaxel in patients with advanced and relapsed solid malignancies. Investig. New Drugs 2012, 30, 249–257. [Google Scholar] [CrossRef]
- Shah, M.H.; Binkley, P.; Chan, K.; Xiao, J.; Arbogast, D.; Collamore, M.; Farra, Y.; Young, D.; Grever, M. Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin. Cancer Res. 2006, 12, 3997–4003. [Google Scholar] [CrossRef] [PubMed]
- DeAngelo, D.J.; Mesa, R.A.; Fiskus, W.; Tefferi, A.; Paley, C.; Wadleigh, M.; Ritchie, E.K.; Snyder, D.S.; Begna, K.; Ganguly, S.; et al. Phase II trial of panobinostat, an oral pan-deacetylase inhibitor in patients with primary myelofibrosis, post-essential thrombocythaemia, and post-polycythaemia vera myelofibrosis. Br. J. Haematol. 2013, 162, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, R.P.; Rankin, C.; Hoff, P.M.G.; Gold, P.J.; Billingsley, K.G.; Chapman, R.A.; Wong, L.; Ward, J.H.; Blanke, C.D. Phase II trial of depsipeptide (NSC-630176) in previously treated colorectal cancer patients with advanced disease: A southwest oncology group study (S0336). Investig. New Drugs 2009, 27, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Luu, T.H.; Morgan, R.J.; Leong, L.; Lim, D.; McNamara, M.; Portnow, J.; Frankel, P.; Smith, D.D.; Doroshow, J.H.; Gandara, D.R.; et al. A Phase II Trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: A California cancer consortium study. Clin. Cancer Res. 2008, 14, 7138–7142. [Google Scholar] [CrossRef]
- Traynor, A.M.; Dubey, S.; Eickhoff, J.C.; Kolesar, J.M.; Schell, K.; Huie, M.S.; Groteluschen, D.L.; Marcotte, S.M.; Hallahan, C.M.; Weeks, H.R.; et al. Vorinostat (NSC# 701852) in patients with relapsed non-small cell lung cancer: A wisconsin oncology network phase II study. J. Thorac. Oncol. 2009, 4, 522–526. [Google Scholar] [CrossRef]
- Woyach, J.A.; Kloos, R.T.; Ringel, M.D.; Arbogast, D.; Collamore, M.; Zwiebel, J.A.; Grever, M.; Villalona-Calero, M.; Shah, M.H. Lack of therapeutic effect of the histone deacetylase inhibitor vorinostat in patients with metastatic radioiodine-refractory thyroid carcinoma. J. Clin. Endocrinol. Metab. 2009, 94, 164–170. [Google Scholar] [CrossRef]
- Stadler, W.M.; Margolin, K.; Ferber, S.; McCulloch, W.; Thompson, J.A. A phase II study of depsipeptide in refractory metastatic renal cell cancer. Clin. Genitourin. Cancer 2006, 5, 57–60. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Infante, J.R.; Spigel, D.R.; Arrowsmith, E.R.; Boccia, R.V.; Burris, H.A. A phase II trial of panobinostat, a histone deacetylase inhibitor, in the treatment of patients with refractory metastatic renal cell carcinoma. Cancer Investig. 2011, 29, 451–455. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R. Hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Zhou, H.; Tong, Z.; McLeod, J.F. “Cocktail” approaches and strategies in drug development: Valuable tool or flawed science? J. Clin. Pharmacol. 2004, 44, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Lehár, J.; Krueger, A.S.; Avery, W.; Heilbut, A.M.; Johansen, L.M.; Price, E.R.; Rickles, R.J.; Short III, G.F.; Staunton, J.E.; Jin, X.; et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat. Biotechnol. 2009, 27, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Peters, J. Polypharmacology—Foe or friend? J. Med. Chem. 2013, 56, 8955–8971. [Google Scholar] [CrossRef] [PubMed]
- León, R.; Garcia, A.G.; Marco-Contelles, J. Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease. Med. Res. Rev. 2013, 33, 139–189. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.M.S.; Dhar, K.L. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef]
- Fu, R.; Sun, Y.; Sheng, W.; Liao, D. Designing multi-targeted agents: An emerging anticancer drug discovery paradigm. Eur. J. Med. Chem. 2017, 136, 195–211. [Google Scholar] [CrossRef]
- Zimmermann, G.R.; Lehár, J.; Keith, C.T. Multi-target therapeutics: When the whole is greater than the sum of the parts. Drug Discov. Today 2007, 12, 34–42. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Morphy, R.; Kay, C.; Rankovic, Z. From magic bullets to designed multiple ligands. Drug Discov. Today 2004, 9, 641–651. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- Chuang, D.; Leng, Y.; Marinova, Z.; Kim, H.; Chiu, C. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009, 32, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Zwinderman, M.R.H.; de Weerd, S.; Dekker, F.J. Targeting HDAC complexes in asthma and COPD. Epigenomes 2019, 3, 19. [Google Scholar] [CrossRef]
- Christensen, D.P.; Dahllöf, M.; Lundh, M.; Rasmussen, D.N.; Nielsen, M.D.; Billestrup, N.; Grunnet, L.G.; Mandrup-Poulsen, T. Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol. Med. 2011, 17, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Thurn, K.T.; Thomas, S.; Moore, A.; Munster, P.N. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future Oncol. 2011, 7, 263–283. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination therapy with histone deacetylase inhibitors (HDACi) for the Treatment of cancer: Achieving the full therapeutic potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef]
- de Lera, A.R.; Ganesan, A. Epigenetic polypharmacology: From combination therapy to multitargeted drugs. Clin. Epigenetics 2016, 8, 105. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.N.; Thurn, K.T.; Thomas, S.; Raha, P.; Lacevic, M.; Miller, A.; Melisko, M.; Ismail-Khan, R.; Rugo, H.; Moasser, M.; et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br. J. Cancer 2011, 104, 1828–1835. [Google Scholar] [CrossRef]
- Hesham, H.M.; Lasheen, D.S.; Abouzid, K.A.M. Chimeric HDAC inhibitors: Comprehensive review on the HDAC-based strategies developed to combat cancer. Med. Res. Rev. 2018, 38, 2058–2109. [Google Scholar] [CrossRef]
- Luan, Y.; Li, J.; Bernatchez, J.A.; Li, R. Kinase and histone deacetylase hybrid inhibitors for cancer therapy. J. Med. Chem. 2019, 62, 3171–3183. [Google Scholar] [CrossRef]
- Cai, X.; Zhai, H.; Wang, J.; Forrester, J.; Qu, H.; Yin, L.; Lai, C.; Bao, R.; Qian, C. Discovery of 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J. Med. Chem. 2010, 53, 2000–2009. [Google Scholar] [CrossRef]
- Qian, C.; Lai, C.; Bao, R.; Wang, D.; Wang, J.; Xu, G.; Atoyan, R.; Qu, H.; Yin, L.; Samson, M.; et al. Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and phosphatidylinositol 3-kinase signaling. Clin. Cancer Res. 2012, 18, 4104–4113. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; LoRusso, P.M.; Papadopoulos, K.P.; Patnaik, A.; Beeram, M.; Smith, L.S.; Rasco, D.W.; Mays, T.A.; Chambers, G.; Ma, A.; et al. Phase I first-in-human study of CUDC-101, a multitargeted inhibitor of HDACs, EGFR, and HER2 in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 5032–5040. [Google Scholar] [CrossRef] [PubMed]
- Galloway, T.J.; Wirth, L.J.; Colevas, A.D.; Gilbert, J.; Bauman, J.E.; Saba, N.F.; Raben, D.; Mehra, R.; Ma, A.W.; Atoyan, R.; et al. A phase I study of CUDC-101, a multitarget inhibitor of HDACs, EGFR, and HER2, in combination with chemoradiation in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 1566–1573. [Google Scholar] [CrossRef] [PubMed]
- Oki, Y.; Kelly, K.R.; Flinn, I.; Patel, M.R.; Gharavi, R.; Ma, A.; Parker, J.; Hafeez, A.; Tuck, D.; Younes, A. CUDC-907 in relapsed/refractory diffuse large B-cell lymphoma, including patients with MYC-alterations: Results from an expanded phase I trial. Haematologica 2017, 102, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Beckers, T.; Burkhardt, C.; Wieland, H.; Gimmnich, P.; Ciossek, T.; Maier, T.; Sanders, K. Distinct pharmacological properties of second generation HDAC inhibitors with the benzamide or hydroxamate head group. Int. J. Cancer 2007, 121, 1138–1148. [Google Scholar] [CrossRef]
- Wang, D.; Wiest, O.; Helquist, P.; Lan-Hargest, H.; Wiech, N.L. On the function of the 14 Å long internal cavity of histone deacetylase-like protein: implications for the design of histone deacetylase inhibitors. J. Med. Chem. 2004, 47, 3409–3417. [Google Scholar] [CrossRef]
- Bressi, J.C.; Jennings, A.J.; Skene, R.; Wu, Y.; Melkus, R.; Jong, R.D.; O’Connell, S.; Grimshaw, C.E.; Navre, M.; Gangloff, A.R. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg. Med. Chem. Lett. 2010, 20, 3142–3145. [Google Scholar] [CrossRef]
- Moradei, O.M.; Mallais, T.C.; Frechette, S.; Paquin, I.; Tessier, P.E.; Leit, S.M.; Fournel, M.; Bonfils, C.; Trachy-Bourget, M.; Liu, J.; et al. Novel aminophenyl benzamide-type histone deacetylase inhibitors with enhanced potency and selectivity. J. Med. Chem. 2007, 50, 5543–5546. [Google Scholar] [CrossRef]
- Witter, D.J.; Harrington, P.; Wilson, K.J.; Chenard, M.; Fleming, J.C.; Haines, B.; Kral, A.M.; Secrist, J.P.; Miller, T.A. Optimization of biaryl selective HDAC1&2 inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 2008, 18, 726–731. [Google Scholar] [CrossRef]
- Methot, J.L.; Chakravarty, P.K.; Chenard, M.; Close, J.; Cruz, J.C.; Dahlberg, W.K.; Fleming, J.; Hamblett, C.L.; Hamill, J.E.; Harrington, P.; et al. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 2008, 18, 973–978. [Google Scholar] [CrossRef]
- Adhikari, N.; Amin, S.A.; Trivedi, P.; Jha, T.; Ghosh, B. HDAC3 is a potential validated target for cancer: An overview on the benzamide-based selective HDAC3 inhibitors through comparative SAR/QSAR/QAAR approaches. Eur. J. Med. Chem. 2018, 157, 1127–1142. [Google Scholar] [CrossRef] [PubMed]
- Fuller, N.O.; Pirone, A.; Lynch, B.A.; Hewitt, M.C.; Quinton, M.S.; McKee, T.D.; Ivarsson, M. CoREST complex-selective histone deacetylase inhibitors show prosynaptic effects and an improved safety profile to enable treatment of synaptopathies. ACS Chem. Neurosci. 2019, 10, 1729–1743. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Nimmanapalli, R.; Fuino, L.; Stobaugh, C.; Richon, V.; Bhalla, K. Cotreatment with the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) enhances imatinib-induced apoptosis of Bcr-Abl-positive human acute leukemia cells. Blood 2003, 101, 3236–3239. [Google Scholar] [CrossRef]
- Nimmanapalli, R.; Fuino, L.; Bali, P.; Gasparetto, M.; Glozak, M.; Tao, J.; Moscinski, L.; Smith, C.; Wu, J.; Jove, R.; et al. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of Bcr-Abl and induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous Leukemia-Blast crisis cells. Cancer Res. 2003, 63, 5126–5135. [Google Scholar] [PubMed]
- Zhang, W.; Peyton, M.; Xie, Y.; Soh, J.; Minna, J.D.; Gazdar, A.F.; Frenkel, E.P. Histone deacetylase inhibitor romidepsin enhances anti-tumor effect of erlotinib in non-small cell lung cancer (NSCLC) cell lines. J. Thorac. Oncol. 2009, 4, 161–166. [Google Scholar] [CrossRef]
- Zhang, Q.; Jiang, S.; Zhang, S.; Ma, X. Histone deacetylase inhibitor trichostatin a enhances antitumor effects of docetaxel or erlotinib in A549 cell line. Asian Pac. J. Cancer Prev. 2012, 13, 3471–3476. [Google Scholar] [CrossRef]
- Mahboobi, S.; Dove, S.; Sellmer, A.; Winkler, M.; Eichhorn, E.; Pongratz, H.; Ciossek, T.; Baer, T.; Maier, T.; Beckers, T. Design of chimeric histone deacetylase- and tyrosine kinase-inhibitors: A series of imatinib hybrides as potent inhibitors of wild-type and mutant BCR-ABL, PDGF-Rβ, and histone deacetylases. J. Med. Chem. 2009, 52, 2265–2279. [Google Scholar] [CrossRef]
- Beckers, T.; Mahboobi, S.; Sellmer, A.; Winkler, M.; Eichhorn, E.; Pongratz, H.; Maier, T.; Ciossek, T.; Baer, T.; Kelter, G.; et al. Chimerically designed HDAC- and tyrosine kinase inhibitors. A series of erlotinib hybrids as dual-selective inhibitors of EGFR, HER2 and histone deacetylases. Med. Chem. Commun. 2012, 3, 829–835. [Google Scholar] [CrossRef]
- Zang, J.; Liang, X.; Huang, Y.; Jia, Y.; Li, X.; Xu, W.; Chou, C.J.; Zhang, Y. Discovery of novel pazopanib-based HDAC and VEGFR dual inhibitors targeting cancer epigenetics and angiogenesis simultaneously. J. Med. Chem. 2018, 61, 5304–5322. [Google Scholar] [CrossRef]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A.; et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2010, 28, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- van der Graaf, W.T.A.; Blay, J.; Chawla, S.P.; Kim, D.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Rajendra, R.; Jones, R.L.; Pollack, S.M. Targeted treatment for advanced soft tissue sarcoma: Profile of pazopanib. OncoTargets Ther. 2013, 6, 217–222. [Google Scholar] [CrossRef][Green Version]
- Tavallai, S.; Hamed, H.A.; Grant, S.; Poklepovic, A.; Dent, P. Pazopanib and HDAC inhibitors interact to kill sarcoma cells. Cancer Biol. Ther. 2014, 15, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Roberts, J.L.; Sander, C.; Lee, J.; Kirkwood, J.M.; Poklepovic, A.; Dent, P. The HDAC inhibitor AR42 interacts with pazopanib to kill trametinib/dabrafenib-resistant melanoma cells in vitro and in vivo. Oncotarget 2017, 8, 16367–16386. [Google Scholar] [CrossRef]
- Lu, D.; Yan, J.; Wang, L.; Liu, H.; Zeng, L.; Zhang, M.; Duan, W.; Ji, Y.; Cao, J.; Geng, M.; et al. Design, synthesis, and biological evaluation of the first c-Met/HDAC inhibitors based on pyridazinone derivatives. ACS Med. Chem. Lett. 2017, 8, 830–834. [Google Scholar] [CrossRef]
- Xing, W.; Ai, J.; Jin, S.; Shi, Z.; Peng, X.; Wang, L.; Ji, Y.; Lu, D.; Liu, Y.; Geng, M.; et al. Enhancing the cellular anti-proliferation activity of pyridazinones as c-met inhibitors using docking analysis. Eur. J. Med. Chem. 2015, 95, 302–312. [Google Scholar] [CrossRef]
- Geng, A.; Cui, H.; Zhang, L.; Chen, X.; Li, H.; Lu, T.; Zhu, Y. Discovery of novel phenoxybenzamide analogues as Raf/HDAC dual inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 1605–1608. [Google Scholar] [CrossRef]
- Lachenmayer, A.; Toffanin, S.; Cabellos, L.; Alsinet, C.; Hoshida, Y.; Villanueva, A.; Minguez, B.; Tsai, H.; Ward, S.C.; Thung, S.; et al. Combination therapy for hepatocellular carcinoma: Additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. J. Hepatol. 2012, 56, 1343–1350. [Google Scholar] [CrossRef]
- Cheng, C.; Yun, F.; Ullah, S.; Yuan, Q. Discovery of novel cyclin-dependent kinase (CDK) and histone deacetylase (HDAC) dual inhibitors with potent in vitro and in vivo anticancer activity. Eur. J. Med. Chem. 2020, 189, 112073. [Google Scholar] [CrossRef]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Zuco, V.; De Cesare, M.; Cincinelli, R.; Nannei, R.; Pisano, C.; Zaffaroni, N.; Zunino, F. Synergistic antitumor effects of novel HDAC inhibitors and paclitaxel in vitro and in vivo. PLoS ONE 2011, 6, e29085. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, J.; Tong, L.; Luo, Y.; Su, M.; Zang, Y.; Li, J.; Lu, W.; Chen, Y. The discovery of colchicine-SAHA hybrids as a new class of antitumor agents. Bioorg. Med. Chem. 2013, 21, 3240–3244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kong, Y.; Zhang, J.; Su, M.; Zhou, Y.; Zang, Y.; Li, J.; Chen, Y.; Fang, Y.; Zhang, X.; et al. Design, synthesis and biological evaluation of colchicine derivatives as novel tubulin and histone deacetylase dual inhibitors. Eur. J. Med. Chem. 2015, 95, 127–135. [Google Scholar] [CrossRef]
- Sampath, D.; Zabka, T.S.; Misner, D.L.; O’Brien, T.; Dragovich, P.S. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol. Ther. 2015, 151, 16–31. [Google Scholar] [CrossRef]
- von Heideman, A.; Berglund, Å.; Larsson, R.; Nygren, P. Safety and efficacy of NAD depleting cancer drugs: Results of a phase I clinical trial of CHS 828 and overview of published data. Cancer Chemother. Pharmacol. 2010, 65, 1165–1172. [Google Scholar] [CrossRef]
- Zheng, X.; Bauer, P.; Baumeister, T.; Buckmelter, A.J.; Caligiuri, M.; Clodfelter, K.H.; Han, B.; Ho, Y.; Kley, N.; Lin, J.; et al. Structure-based identification of ureas as novel nicotinamide phosphoribosyltransferase (nampt) inhibitors. J. Med. Chem. 2013, 56, 4921–4937. [Google Scholar] [CrossRef]
- Dong, G.; Chen, W.; Wang, X.; Yang, X.; Xu, T.; Wang, P.; Zhang, W.; Rao, Y.; Miao, C.; Sheng, C. Small molecule inhibitors simultaneously targeting cancer metabolism and epigenetics: Discovery of novel nicotinamide phosphoribosyltransferase (NAMPT) and histone deacetylase (HDAC) dual inhibitors. J. Med. Chem. 2017, 60, 7965–7983. [Google Scholar] [CrossRef]
- Colombano, G.; Travelli, C.; Galli, U.; Caldarelli, A.; Chini, M.G.; Canonico, P.L.; Sorba, G.; Bifulco, G.; Tron, G.C.; Genazzani, A.A. A novel potent nicotinamide phosphoribosyltransferase inhibitor synthesized via click chemistry. J. Med. Chem. 2010, 53, 616–623. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, S.; Wu, S.; Dong, G.; Sheng, C. Evodiamine-inspired dual inhibitors of histone deacetylase 1 (HDAC1) and topoisomerase 2 (TOP2) with potent antitumor activity. Acta Pharm. Sin. B 2020, 10, 1294–1308. [Google Scholar] [CrossRef]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Blake, M.; Baek, J.H.; Kohlhagen, G.; Pommier, Y.; Carrier, F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs. Cancer Res. 2003, 63, 7291–7300. [Google Scholar] [PubMed]
- Guerrant, W.; Patil, V.; Canzoneri, J.C.; Oyelere, A.K. Dual targeting of histone deacetylase and topoisomerase II with novel bifunctional inhibitors. J. Med. Chem. 2012, 55, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Guerrant, W.; Patil, V.; Canzoneri, J.C.; Yao, L.; Hood, R.; Oyelere, A.K. Dual-acting histone deacetylase-topoisomerase I inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 3283–3287. [Google Scholar] [CrossRef]
- Chen, J.; Li, D.; Li, W.; Yin, J.; Zhang, Y.; Yuan, Z.; Gao, C.; Liu, F.; Jiang, Y. Design, synthesis and anticancer evaluation of acridine hydroxamic acid derivatives as dual Topo and HDAC inhibitors. Bioorg. Med. Chem. 2018, 26, 3958–3966. [Google Scholar] [CrossRef]
- Irwin, M.H.; Moos, W.H.; Faller, D.V.; Steliou, K.; Pinkert, C.A. Epigenetic treatment of neurodegenerative disorders: Alzheimer and parkinson diseases. Drug Dev. Res. 2016, 77, 109–123. [Google Scholar] [CrossRef]
- García-Osta, A.; Cuadrado-Tejedor, M.; García-Barroso, C.; Oyarzábal, J.; Franco, R. Phosphodiesterases as therapeutic targets for Alzheimer’s disease. ACS Chem. Neurosci. 2012, 3, 832–844. [Google Scholar] [CrossRef]
- Puzzo, D.; Staniszewski, A.; Deng, S.X.; Privitera, L.; Leznik, E.; Liu, S.; Zhang, H.; Feng, Y.; Palmeri, A.; Landry, D.W.; et al. Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-β load in an Alzheimer’s disease mouse model. J. Neurosci. 2009, 29, 8075–8086. [Google Scholar] [CrossRef]
- Rabal, O.; Sánchez-Arias, J.A.; Cuadrado-Tejedor, M.; de Miguel, I.; Pérez-González, M.; García-Barroso, C.; Ugarte, A.; Estella-Hermoso de Mendoza, A.; Sáez, E.; Espelosin, M.; et al. Discovery of in vivo chemical probes for treating Alzheimer’s disease: Dual phosphodiesterase 5 (PDE5) and class I histone deacetylase selective inhibitors. ACS Chem. Neurosci. 2019, 10, 1765–1782. [Google Scholar] [CrossRef]
- Chou, C.J.; Herman, D.; Gottesfeld, J.M. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J. Biol. Chem. 2008, 283, 35402–35409. [Google Scholar] [CrossRef]
- Lauffer, B.E.L.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.; Chen, Z.; Fredrickson, T.; Zhu, Y.; Kirkpatrick, R.; Zhang, G.; Johanson, K.; Sung, C.; Liu, R.; Winkler, J. Cloning and characterization of a novel human class I histone deacetylase that functions as a transcription repressor. J. Biol. Chem. 2000, 275, 15254–15264. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Rezai-Zadeh, N.; Seto, E. Negative regulation of histone deacetylase 8 activity by cyclic AMP-dependent protein kinase A. Mol. Cell Biol. 2004, 24, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Conway, S.J. Small molecules as tools to study the chemical epigenetics of lysine acetylation. Curr. Opin. Chem. Biol. 2018, 45, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Nagaiah, G.; Remick, S.C. Combretastatin A4 phosphate: A novel vascular disrupting agent. Future Oncol. 2010, 6, 1219–1228. [Google Scholar] [CrossRef]
- Yeung, S.J.; She, M.; Yang, H.; Pan, J.; Sun, L.; Chaplin, D. Combination chemotherapy including combretastatin A4 phosphate and paclitaxel is effective against anaplastic thyroid cancer in a nude mouse xenograft model. J. Clin. Endocrinol. Metab. 2007, 92, 2902–2909. [Google Scholar] [CrossRef]
- Nathan, P.; Zweifel, M.; Padhani, A.R.; Koh, D.; Ng, M.; Collins, D.J.; Harris, A.; Carden, C.; Smythe, J.; Fisher, N.; et al. Phase I trial of combretastatin A4 phosphate (CA4P) in combination with bevacizumab in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 3428–3439. [Google Scholar] [CrossRef]
- Lamaa, D.; Lin, H.; Zig, L.; Bauvais, C.; Bollot, G.; Bignon, J.; Levaique, H.; Pamlard, O.; Dubois, J.; Ouaissi, M.; et al. Design and synthesis of tubulin and histone deacetylase inhibitor based on iso-combretastatin A-4. J. Med. Chem. 2018, 61, 6574–6591. [Google Scholar] [CrossRef]
- Halder, A.K.; Mallick, S.; Shikha, D.; Saha, A.; Saha, K.D.; Jha, T. Design of dual MMP-2/HDAC-8 inhibitors by pharmacophore mapping, molecular docking, synthesis and biological activity. RSC Adv. 2015, 5, 72373–72386. [Google Scholar] [CrossRef]
- Amin, S.A.; Adhikari, N.; Jha, T. Is dual inhibition of metalloenzymes HDAC-8 and MMP-2 a potential pharmacological target to combat hematological malignancies? Pharmacol. Res. 2017, 122, 8–19. [Google Scholar] [CrossRef]
- Somiari, S.B.; Somiari, R.I.; Heckman, C.M.; Olsen, C.H.; Jordan, R.M.; Russell, S.J.; Shriver, C.D. Circulating MMP2 and MMP9 in breast cancer—Potential role in classification of patients into low risk, high risk, benign disease and breast cancer categories. Int. J. Cancer 2006, 119, 1403–1411. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chang, H.; Chiang, L.; Hung, W. Histone deacetylase inhibitor up-regulates RECK to inhibit MMP-2 activation and cancer cell invasion. Cancer Res. 2003, 63, 3069–3072. [Google Scholar] [PubMed]
- Agrawal, A.; Romero-Perez, D.; Jacobsen, J.; Villarreal, F.; Cohen, S. Zinc-binding groups modulate selective inhibition of MMPs. ChemMedChem 2008, 3, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Halder, A.K.; Saha, A.; Jha, T. Exploring QSAR and pharmacophore mapping of structurally diverse selective matrix metalloproteinase-2 inhibitors. J. Pharm. Pharmacol. 2013, 65, 1541–1554. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Likos, J.J.; Zhu, L.; Woodward, H.; Munie, G.; McDonald, J.J.; Stevens, A.M.; Howard, C.P.; De Crescenzo, G.A.; Welsch, D.; et al. Solution structure and backbone dynamics of the catalytic domain of matrix metalloproteinase-2 complexed with a hydroxamic acid inhibitor. Biochim. Biophys. Acta Proteins Proteom. 2002, 1598, 10–23. [Google Scholar] [CrossRef]
- Whitehead, L.; Dobler, M.R.; Radetich, B.; Zhu, Y.; Atadja, P.W.; Claiborne, T.; Grob, J.E.; McRiner, A.; Pancost, M.R.; Patnaik, A.; et al. Human HDAC isoform selectivity achieved via exploitation of the acetate release channel with structurally unique small molecule inhibitors. Bioorg. Med. Chem. 2011, 19, 4626–4634. [Google Scholar] [CrossRef] [PubMed]
- Osko, J.D.; Christianson, D.W. Structural basis of catalysis and inhibition of HDAC6 CD1, the enigmatic catalytic domain of histone deacetylase 6. Biochemistry 2019, 58, 4912–4924. [Google Scholar] [CrossRef] [PubMed]
- Porter, N.J.; Osko, J.D.; Diedrich, D.; Kurz, T.; Hooker, J.M.; Hansen, F.K.; Christianson, D.W. Histone deacetylase 6-selective inhibitors and the influence of capping groups on hydroxamate-Zinc denticity. J. Med. Chem. 2018, 61, 8054–8060. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [PubMed]
- Kalin, J.H.; Bergman, J.A. Development and therapeutic implications of selective histone deacetylase 6 inhibitors. J. Med. Chem. 2013, 56, 6297–6313. [Google Scholar] [CrossRef] [PubMed]
- Rabal, O.; Sánchez-Arias, J.A.; Cuadrado-Tejedor, M.; de Miguel, I.; Pérez-González, M.; García-Barroso, C.; Ugarte, A.; Estella-Hermoso de Mendoza, A.; Sáez, E.; Espelosin, M.; et al. Design, synthesis, biological evaluation and in vivo testing of dual phosphodiesterase 5 (PDE5) and histone deacetylase 6 (HDAC6)-selective inhibitors for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018, 150, 506–524. [Google Scholar] [CrossRef] [PubMed]
- Hai, Y.; Christianson, D.W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Keusch, J.J.; Wang, L.; Saito, M.; Hess, D.; Wang, X.; Melancon, B.J.; Helquist, P.; Gut, H.; Matthias, P. Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol. 2016, 12, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Kiu, H.; Nicholson, S.E. Biology and significance of the JAK/STAT signalling pathways. Growth Factors 2012, 30, 88–106. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Mustafa, N.; Tan, E.C.; Poulsen, A.; Singh, P.; Duong-Thi, M.; Lee, J.X.T.; Ramanujulu, P.M.; Chng, W.J.; Yen, J.J.Y.; et al. Design and synthesis of ligand efficient dual inhibitors of janus kinase (JAK) and histone deacetylase (HDAC) based on ruxolitinib and vorinostat. J. Med. Chem. 2017, 60, 8336–8357. [Google Scholar] [CrossRef]
- O’Shea, J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef]
- Kontzias, A.; Kotlyar, A.; Laurence, A.; Changelian, P.; O’Shea, J.J. Jakinibs: A new class of kinase inhibitors in cancer and autoimmune disease. Curr. Opin. Pharmacol. 2012, 12, 464–470. [Google Scholar] [CrossRef]
- Marotta, L.L.C.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24– stem cell–like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Hoffman, R. Ruxolitinib: The First FDA Approved therapy for the treatment of myelofibrosis. Clin. Cancer Res. 2012, 18, 3008–3014. [Google Scholar] [CrossRef]
- Amaru Calzada, A.; Todoerti, K.; Donadoni, L.; Pellicioli, A.; Tuana, G.; Gatta, R.; Neri, A.; Finazzi, G.; Mantovani, R.; Rambaldi, A.; et al. The HDAC inhibitor Givinostat modulates the hematopoietic transcription factors NFE2 and C-MYB in JAK2V617F myeloproliferative neoplasm cells. Exp. Hematol. 2012, 40, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Evrot, E.; Ebel, N.; Romanet, V.; Roelli, C.; Andraos, R.; Qian, Z.; Dölemeyer, A.; Dammassa, E.; Sterker, D.; Cozens, R.; et al. JAK1/2 and pan-deacetylase inhibitor combination therapy yields improved efficacy in preclinical mouse models of JAK2V617F-driven disease. Clin. Cancer Res. 2013, 19, 6230–6241. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.G.; Mustafa, N.; Tan, E.C.; Poulsen, A.; Ramanujulu, P.M.; Chng, W.J.; Yen, J.J.Y.; Dymock, B.W. Design and synthesis of janus kinase 2 (JAK2) and histone deacetlyase (HDAC) bispecific inhibitors based on pacritinib and evidence of dual pathway inhibition in hematological cell lines. J. Med. Chem. 2016, 59, 8233–8262. [Google Scholar] [CrossRef] [PubMed]
- Chu-Farseeva, Y.; Mustafa, N.; Poulsen, A.; Tan, E.C.; Yen, J.J.Y.; Chng, W.J.; Dymock, B.W. Design and synthesis of potent dual inhibitors of JAK2 and HDAC based on fusing the pharmacophores of XL019 and vorinostat. Eur. J. Med. Chem. 2018, 158, 593–619. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Zang, J.; Li, X.; Tang, S.; Huang, M.; Geng, M.; Chou, C.J.; Li, C.; Cao, Y.; Xu, W.; et al. Discovery of novel janus kinase (JAK) and histone deacetylase (HDAC) dual inhibitors for the treatment of hematological malignancies. J. Med. Chem. 2019, 62, 3898–3923. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Dong, G.; Li, H.; Liu, N.; Zhang, W.; Sheng, C. Discovery of janus kinase 2 (JAK2) and histone deacetylase (HDAC) dual inhibitors as a novel strategy for the combinational treatment of leukemia and invasive fungal infections. J. Med. Chem. 2018, 61, 6056–6074. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Huang, H.; HuangFu, W.; Wu, Y.; Nepali, K.; Lai, M.; Su, C.; Sung, T.; Chen, Y.; Pan, S.; et al. 1-Aroylindoline-hydroxamic acids as anticancer agents, inhibitors of HSP90 and HDAC. Eur. J. Med. Chem. 2018, 150, 667–677. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Dong, G.; Wu, S.; Fang, K.; Miao, Z.; Wang, W.; Sheng, C. Small molecules simultaneously inhibiting p53-murine double minute 2 (MDM2) interaction and histone deacetylases (HDACs): Discovery of novel multitargeting antitumor agents. J. Med. Chem. 2018, 61, 7245–7260. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. p53 regulation by ubiquitin. FEBS Lett. 2011, 585, 2803–2809. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Li, Y.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Palani, C.; Beck, J.; Sonnemann, J. Histone deacetylase inhibitors enhance the anticancer activity of nutlin-3 and induce p53 hyperacetylation and downregulation of MDM2 and MDM4 gene expression. Investig. New Drugs 2012, 30, 25–36. [Google Scholar] [CrossRef] [PubMed]
- McCormack, E.; Haaland, I.; Venås, G.; Forthun, R.B.; Huseby, S.; Gausdal, G.; Knappskog, S.; Micklem, D.R.; Lorens, J.B.; Bruserud, Ø.; et al. Synergistic induction of p53 mediated apoptosis by valproic acid and nutlin-3 in acute myeloid leukemia. Leukemia 2012, 26, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Millard, C.J.; Watson, P.J.; Fairall, L.; Schwabe, J.W.R. Targeting class I histone deacetylases in a “complex” environment. Trends Pharmacol. Sci. 2017, 38, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Guenther, M.G.; Lane, W.S.; Fischle, W.; Verdin, E.; Lazar, M.A.; Shiekhattar, R. A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes Dev. 2000, 14, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Perissi, V.; Staszewski, L.M.; Yang, W.; Krones, A.; Glass, C.K.; Rosenfeld, M.G.; Seto, E. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc. Natl. Acad. Sci. USA 2000, 97, 7202–7207. [Google Scholar] [CrossRef]
- You, A.; Tong, J.K.; Grozinger, C.M.; Schreiber, S.L. CoREST is an integral component of the CoREST-human histone deacetylase complex. Proc. Natl. Acad. Sci. USA 2001, 98, 1454–1458. [Google Scholar] [CrossRef]
- Yang, M.; Gocke, C.B.; Luo, X.; Borek, D.; Tomchick, D.R.; Machius, M.; Otwinowski, Z.; Yu, H. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol. Cell 2006, 23, 377–387. [Google Scholar] [CrossRef]
- Bantscheff, M.; Hopf, C.; Savitski, M.M.; Dittmann, A.; Grandi, P.; Michon, A.; Schlegl, J.; Abraham, Y.; Becher, I.; Bergamini, G.; et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011, 29, 255–265. [Google Scholar] [CrossRef]
- Itoh, T.; Fairall, L.; Muskett, F.W.; Milano, C.P.; Watson, P.J.; Arnaudo, N.; Saleh, A.; Millard, C.J.; El-Mezgueldi, M.; Martino, F.; et al. Structural and functional characterization of a cell cycle associated HDAC1/2 complex reveals the structural basis for complex assembly and nucleosome targeting. Nucleic Acids Res. 2015, 43, 2033–2044. [Google Scholar] [CrossRef]
- Wade, P.A.; Jones, P.L.; Vermaak, D.; Wolffe, A.P. A multiple subunit Mi-2 histone deacetylase from Xenopus laevis cofractionates with an associated Snf2 superfamily ATPase. Curr. Biol. 1998, 8, 843–848. [Google Scholar] [CrossRef]
- Zhang, Y.; LeRoy, G.; Seelig, H.; Lane, W.S.; Reinberg, D. The dermatomyositis-specific autoantigen Mi2 is a component of a complex containing histone deacetylase and nucleosome remodeling activities. Cell 1998, 95, 279–289. [Google Scholar] [CrossRef]
- Laherty, C.D.; Yang, W.; Sun, J.; Davie, J.R.; Seto, E.; Eisenman, R.N. Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell 1997, 89, 349–356. [Google Scholar] [CrossRef]
- Hassig, C.A.; Fleischer, T.C.; Billin, A.N.; Schreiber, S.L.; Ayer, D.E. Histone deacetylase activity is required for full transcriptional repression by mSin3A. Cell 1997, 89, 341–347. [Google Scholar] [CrossRef]
- Ding, Z.; Gillespie, L.L.; Paterno, G.D. Human MI-ER1 alpha and beta function as transcriptional repressors by recruitment of histone deacetylase 1 to their conserved ELM2 domain. Mol. Cell. Biol. 2003, 23, 250–258. [Google Scholar] [CrossRef][Green Version]
- Derwish, R.; Paterno, G.D.; Gillespie, L.L. Differential HDAC1 and 2 recruitment by members of the MIER family. PLoS ONE 2017, 12, e0169338. [Google Scholar] [CrossRef] [PubMed]
- Plaster, N.; Sonntag, C.; Schilling, T.F.; Hammerschmidt, M. REREa/Atrophin-2 interacts with histone deacetylase and Fgf8 signaling to regulate multiple processes of zebrafish development. Dev. Dyn. 2007, 236, 1891–1904. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.W.; Cowley, S. The physiological roles of histone deacetylase (HDAC) 1 and 2: Complex co-stars with multiple leading parts. Biochem. Soc. Trans. 2013, 41, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Beyer, J.N.; Raniszewski, N.R.; Burslem, G.M. Epigenetic chemical biology: Opportunities beyond the advances and opportunities in epigenetic chemical biology. ChemBioChem 2020. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Wynder, C.; Cooch, N.; Shiekhattar, R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 2005, 437, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Lakowski, B.; Roelens, I.; Jacob, S. CoREST-like complexes regulate chromatin modification and neuronal gene expression. J. Mol. Neurosci. 2006, 29, 227–239. [Google Scholar] [CrossRef]
- Lee, M.G.; Wynder, C.; Bochar, D.A.; Hakimi, M.; Cooch, N.; Shiekhattar, R. Functional interplay between histone demethylase and deacetylase enzymes. Mol. Cell. Biol. 2006, 26, 6395–6402. [Google Scholar] [CrossRef]
- Song, Y.; Dagil, L.; Fairall, L.; Robertson, N.; Wu, M.; Ragan, T.J.; Savva, C.G.; Saleh, A.; Morone, N.; Kunze, M.B.A.; et al. Mechanism of crosstalk between the LSD1 demethylase and HDAC1 deacetylase in the CoREST complex. Cell Rep. 2020, 30, 2699–2711. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Matson, C.; Lan, F.; Iwase, S.; Baba, T.; Shi, Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol. Cell 2005, 19, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.M.; Manton, C.A.; Bhat, K.P.; Tsai, W.; Aldape, K.; Barton, M.C.; Chandra, J. Inhibition of LSD1 sensitizes glioblastoma cells to histone deacetylase inhibitors. Neuro Oncol. 2011, 13, 894–903. [Google Scholar] [CrossRef]
- Huang, Y.; Huang, Y.; Vasilatos, S.; Vasilatos, S.; Boric, L.; Boric, L.; Shaw, P.; Shaw, P.; Davidson, N.; Davidson, N. Inhibitors of histone demethylation and histone deacetylation cooperate in regulating gene expression and inhibiting growth in human breast cancer cells. Breast Cancer Res. Treat. 2012, 131, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Sharma, S.; Shah, B.; Portier, B.P.; Devaraj, S.G.T.; Liu, K.; Iyer, S.P.; Bearss, D.; Bhalla, K.N. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia 2014, 28, 2155–2164. [Google Scholar] [CrossRef]
- Prusevich, P.; Kalin, J.H.; Ming, S.A.; Basso, M.; Givens, J.; Li, X.; Hu, J.; Taylor, M.S.; Cieniewicz, A.M.; Hsiao, P.; et al. A Selective phenelzine analogue inhibitor of histone demethylase LSD1. ACS Chem. Biol. 2014, 9, 1284–1293. [Google Scholar] [CrossRef]
- Duan, Y.; Ma, Y.; Qin, W.; Ding, L.; Zheng, Y.; Zhu, Y.; Zhai, X.; Yang, J.; Ma, C.; Guan, Y. Design and synthesis of tranylcypromine derivatives as novel LSD1/HDACs dual inhibitors for cancer treatment. Eur. J. Med. Chem. 2017, 140, 392–402. [Google Scholar] [CrossRef]
- Kalin, J.H.; Wu, M.; Gomez, A.V.; Song, Y.; Das, J.; Hayward, D.; Adejola, N.; Wu, M.; Panova, I.; Chung, H.J.; et al. Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nature Commun. 2018, 9, 53. [Google Scholar] [CrossRef]
- Zhijun, H.; Shusheng, W.; Han, M.; Jianping, L.; Li-sen, Q.; Dechun, L. Pre-clinical characterization of 4SC-202, a novel class I HDAC inhibitor, against colorectal cancer cells. Tumor Biol. 2016, 37, 10257–10267. [Google Scholar] [CrossRef]
- Inui, K.; Zhao, Z.; Yuan, J.; Jayaprakash, S.; Le, L.T.M.; Drakulic, S.; Sander, B.; Golas, M.M. Stepwise assembly of functional C-terminal REST/NRSF transcriptional repressor complexes as a drug target. Protein Sci. 2017, 26, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.M.; Zylla, J.S.; Bhattacharya, S.; Calar, K.; Hartman, T.W.; Bhardwaj, R.D.; Miskimins, W.K.; de la Puente, P.; Gnimpieba, E.Z.; Messerli, S.M. Analysis of dual class i histone deacetylase and lysine demethylase inhibitor domatinostat (4SC-202) on growth and cellular and genomic landscape of atypical teratoid/rhabdoid. Cancers 2020, 12, 756. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed]
- Nalawansha, D.A.; Crews, C.M. PROTACs: An emerging therapeutic modality in precision medicine. Cell Chem. Biol. 2020, 27, 998–1014. [Google Scholar] [CrossRef] [PubMed]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Mares, A.; Smith, I.E.D.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.; Crew, A.; Coleman, K.; et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef]
- Burslem, G.M.; Crews, C.M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef]
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Oláh, J.; Ovádi, J.; Sippl, W.; Jung, M. Chemically induced degradation of sirtuin 2 (Sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals). J. Med. Chem. 2018, 61, 482–491. [Google Scholar] [CrossRef]
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Gerhardt, S.; Ovádi, J.; Sippl, W.; Einsle, O.; Jung, M. Structure-based development of an affinity probe for Sirtuin 2. Angew. Chem. Int. Ed. 2016, 55, 2252–2256. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Song, Y.; Xie, H.; Wu, Y.; Wu, H.; Leisten, E.D.; Tang, W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg. Med. Chem. Lett. 2018, 28, 2493–2497. [Google Scholar] [CrossRef]
- Yang, H.; Lv, W.; He, M.; Deng, H.; Li, H.; Wu, W.; Rao, Y. Plasticity in designing PROTACs for selective and potent degradation of HDAC6. Chem. Commun. 2019, 55, 14848–14851. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Lv, W.; Su, S.; Wu, W.; Rao, Y. Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell 2019, 10, 606–609. [Google Scholar] [CrossRef]
- Wu, H.; Yang, K.; Zhang, Z.; Leisten, E.D.; Li, Z.; Xie, H.; Liu, J.; Smith, K.A.; Novakova, Z.; Barinka, C.; et al. Development of multifunctional histone deacetylase 6 degraders with potent antimyeloma activity. J. Med. Chem. 2019, 62, 7042–7057. [Google Scholar] [CrossRef]
- Yang, K.; Wu, H.; Zhang, Z.; Leisten, E.D.; Nie, X.; Liu, B.; Wen, Z.; Zhang, J.; Cunningham, M.D.; Tang, W. Development of selective histone deacetylase 6 (HDAC6) degraders recruiting von hippel-lindau (VHL) E3 ubiquitin ligase. ACS Med. Chem. Lett. 2020, 11, 575–581. [Google Scholar] [CrossRef]
- Smalley, J.P.; Adams, G.E.; Millard, C.J.; Song, Y.; Norris, J.K.S.; Schwabe, J.W.R.; Cowley, S.M.; Hodgkinson, J.T. PROTAC-mediated degradation of class I histone deacetylase enzymes in corepressor complexes. Chem. Commun. 2020, 56, 4476–4479. [Google Scholar] [CrossRef]
- Galdeano, C.; Gadd, M.S.; Soares, P.; Scaffidi, S.; Van Molle, I.; Birced, I.; Hewitt, S.; Dias, D.M.; Ciulli, A. Structure-guided design and optimization of small molecules targeting the protein-protein interaction between the von Hippel–Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J. Med. Chem. 2014, 57, 8657–8663. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, J.; Zhao, L.Y.; Chen, X.; Zheng, G.; Zhang, X.; Liao, D. Discovery of histone deacetylase 3 (HDAC3)-specific PROTACs. Chem. Commun. 2020, 56, 9866–9869. [Google Scholar] [CrossRef]
- Wang, Y.; Stowe, R.; Pinello, C.; Tian, G.; Madoux, F.; Li, D.; Zhao, L.; Li, J.; Wang, Y.; Wang, Y.; et al. Identification of histone deacetylase inhibitors with benzoylhydrazide scaffold that selectively inhibit class I histone deacetylases. Chem. Biol. 2015, 22, 273–284. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hybrid | IC50 (nM) | |||||||
|---|---|---|---|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3 | HDAC6 | HDAC8 | CDK1 | CDK2 | CDK4,6,7 | |
| 7 | 240 | 0.24 | >1000 | >1000 | >1000 | 12.58 | 0.56 | >1000 |
| 8 | 6.4 | 0.25 | 45 | >1000 | >1000 | 8.63 | 0.30 | >1000 |
| HDAC Class HDAC Isoform | I | IIa | IIb | IV | ||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 8 | 4,5,7,9 | 6 | 10 | 11 | |
| IC50 (nM) or % inhibition | 222 | 49 | 2170 | 740 | <20% at 10 µM | 2.1 | 80 | 930 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smalley, J.P.; Cowley, S.M.; Hodgkinson, J.T. Bifunctional HDAC Therapeutics: One Drug to Rule Them All? Molecules 2020, 25, 4394. https://doi.org/10.3390/molecules25194394
Smalley JP, Cowley SM, Hodgkinson JT. Bifunctional HDAC Therapeutics: One Drug to Rule Them All? Molecules. 2020; 25(19):4394. https://doi.org/10.3390/molecules25194394
Chicago/Turabian StyleSmalley, Joshua P., Shaun M. Cowley, and James T. Hodgkinson. 2020. "Bifunctional HDAC Therapeutics: One Drug to Rule Them All?" Molecules 25, no. 19: 4394. https://doi.org/10.3390/molecules25194394
APA StyleSmalley, J. P., Cowley, S. M., & Hodgkinson, J. T. (2020). Bifunctional HDAC Therapeutics: One Drug to Rule Them All? Molecules, 25(19), 4394. https://doi.org/10.3390/molecules25194394