Potential Pharmacokinetic Drug–Drug Interaction Between Harmine, a Cholinesterase Inhibitor, and Memantine, a Non-Competitive N-Methyl-d-Aspartate Receptor Antagonist



Abstract
1. Introduction
2. Results
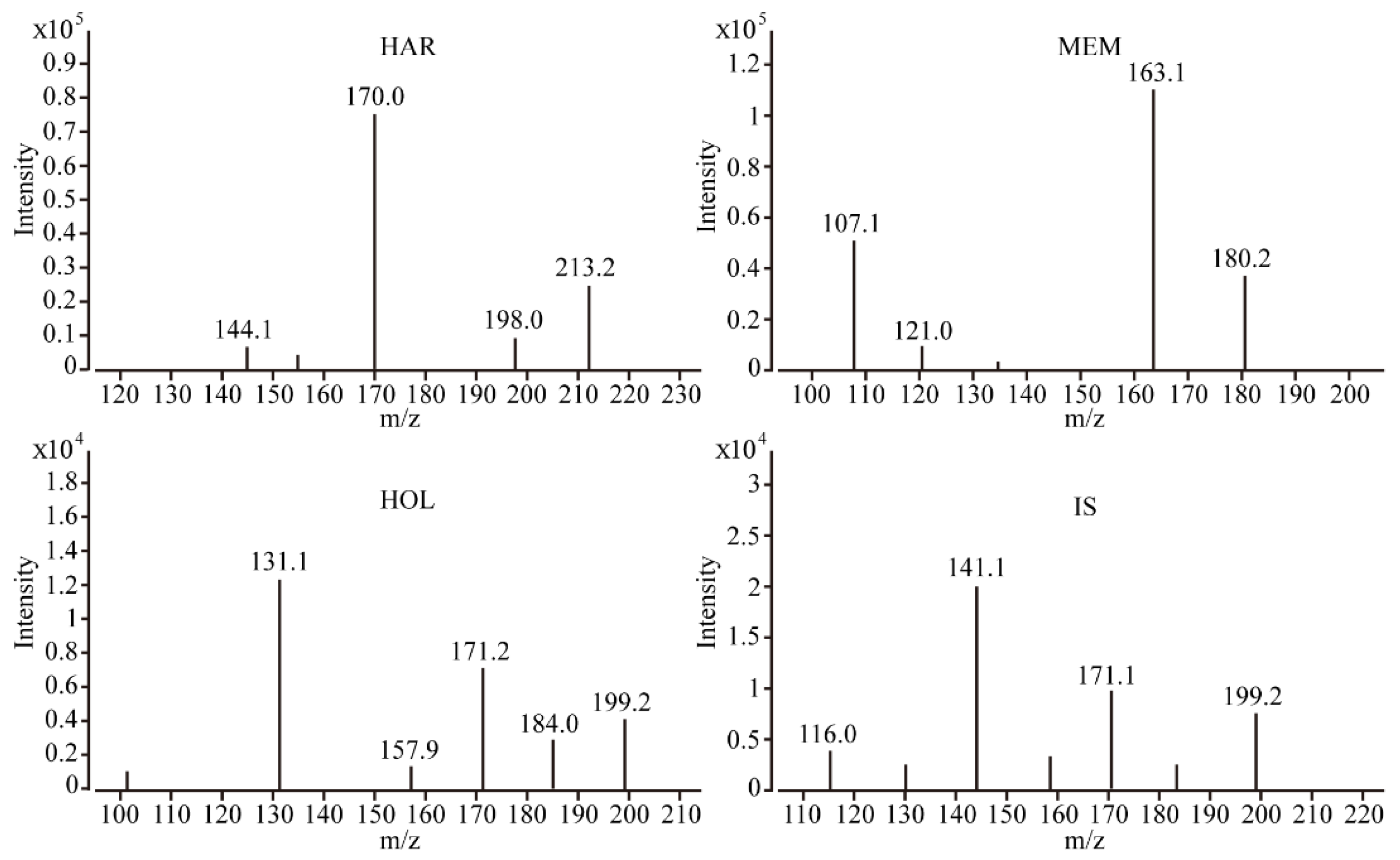
2.1. Optimization of UPLC-MS/MS Conditions
2.2. Optimization of the Pretreatment Procedure
2.3. Method Validation
2.3.1. Selectivity and Carry-Over
2.3.2. Linearity, the Low Limit of Detection (LLOD) and LLOQ
2.3.3. Within-Run and Between-Run Precision
2.3.4. Matrix Effect and Extraction Recovery
2.3.5. Stability
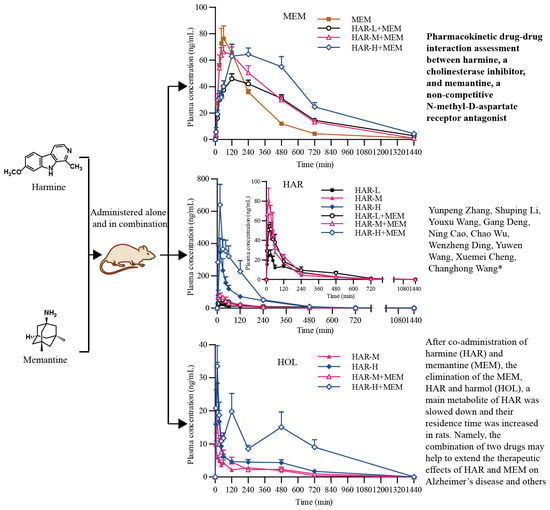
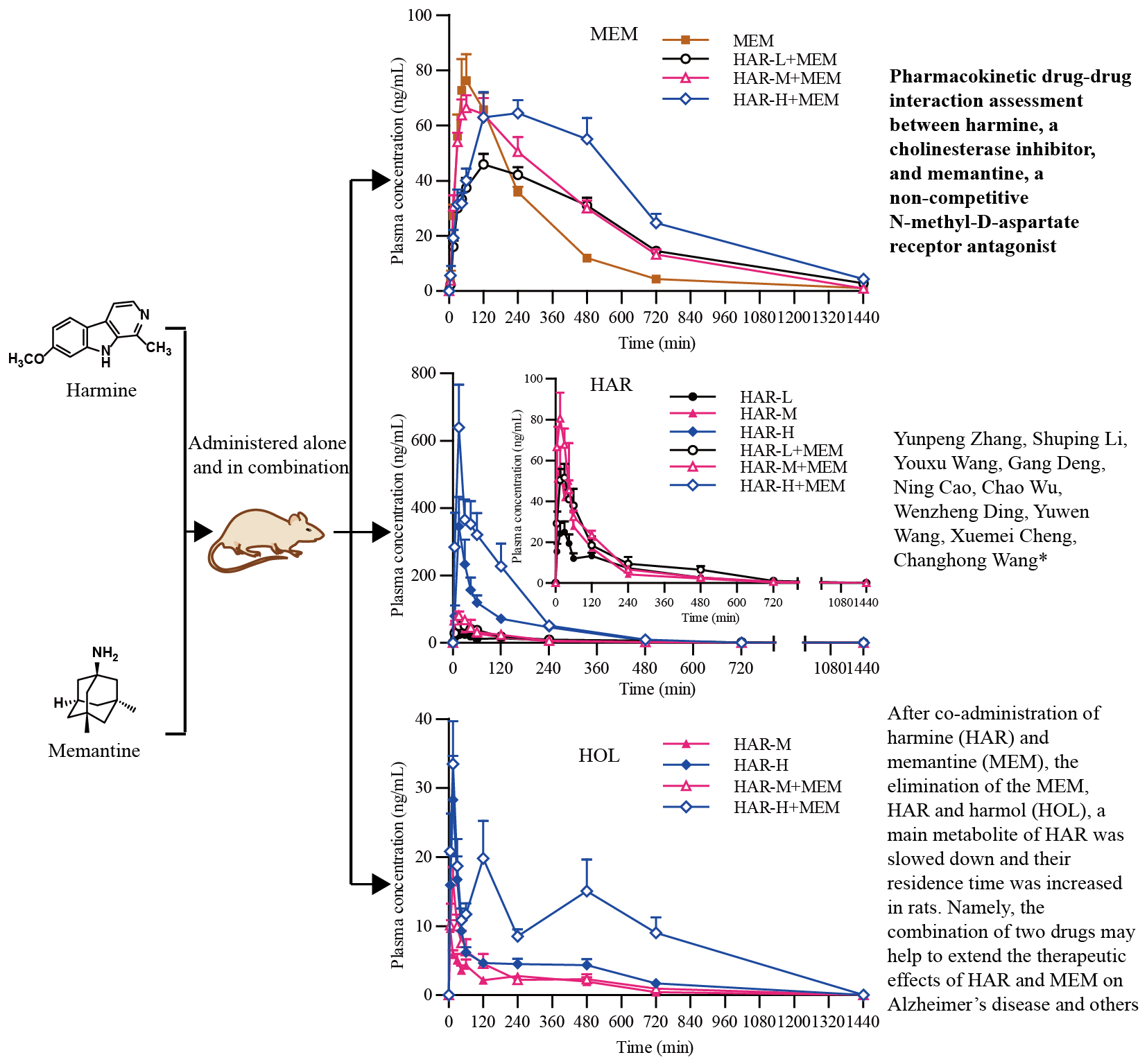
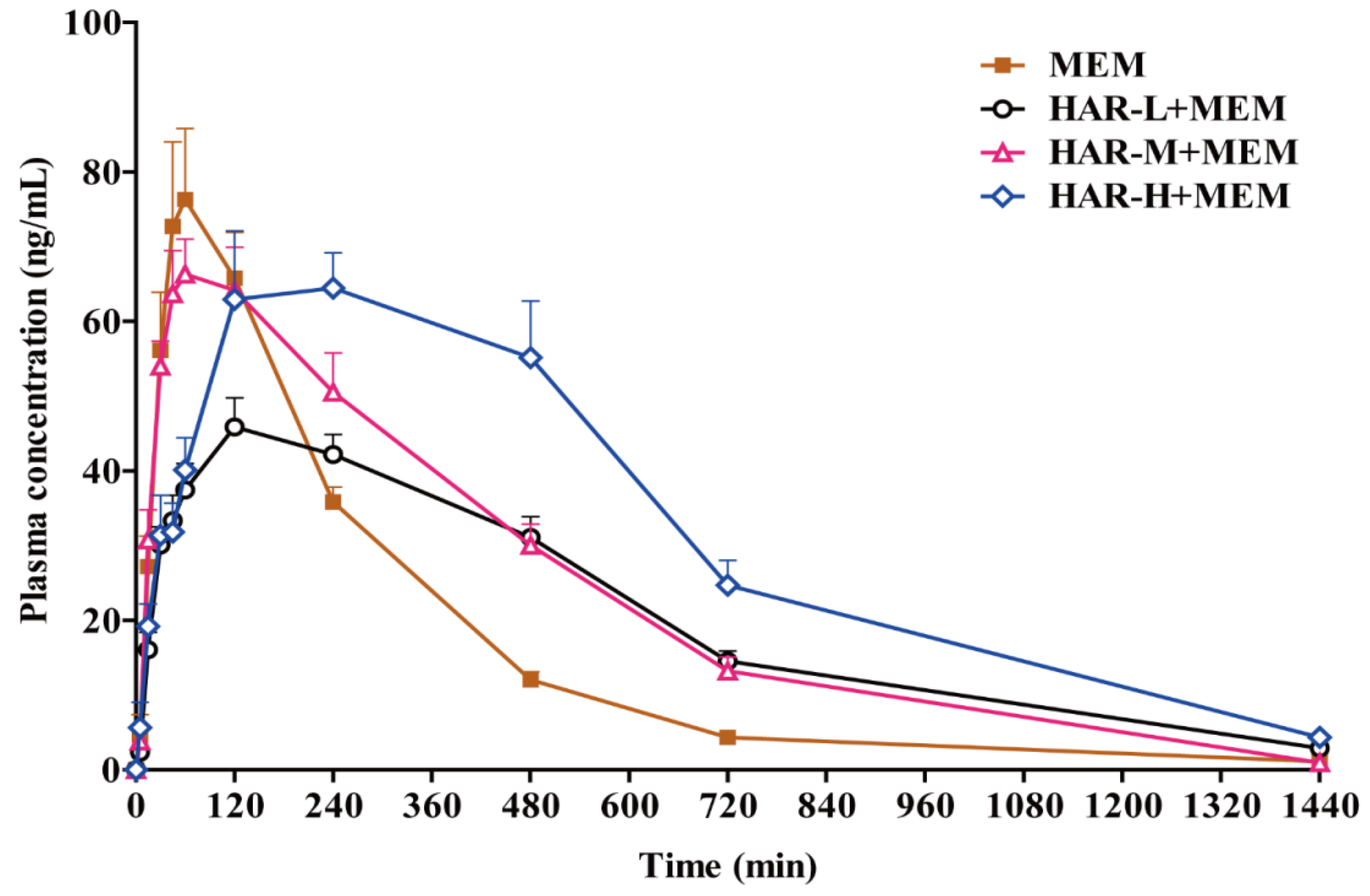
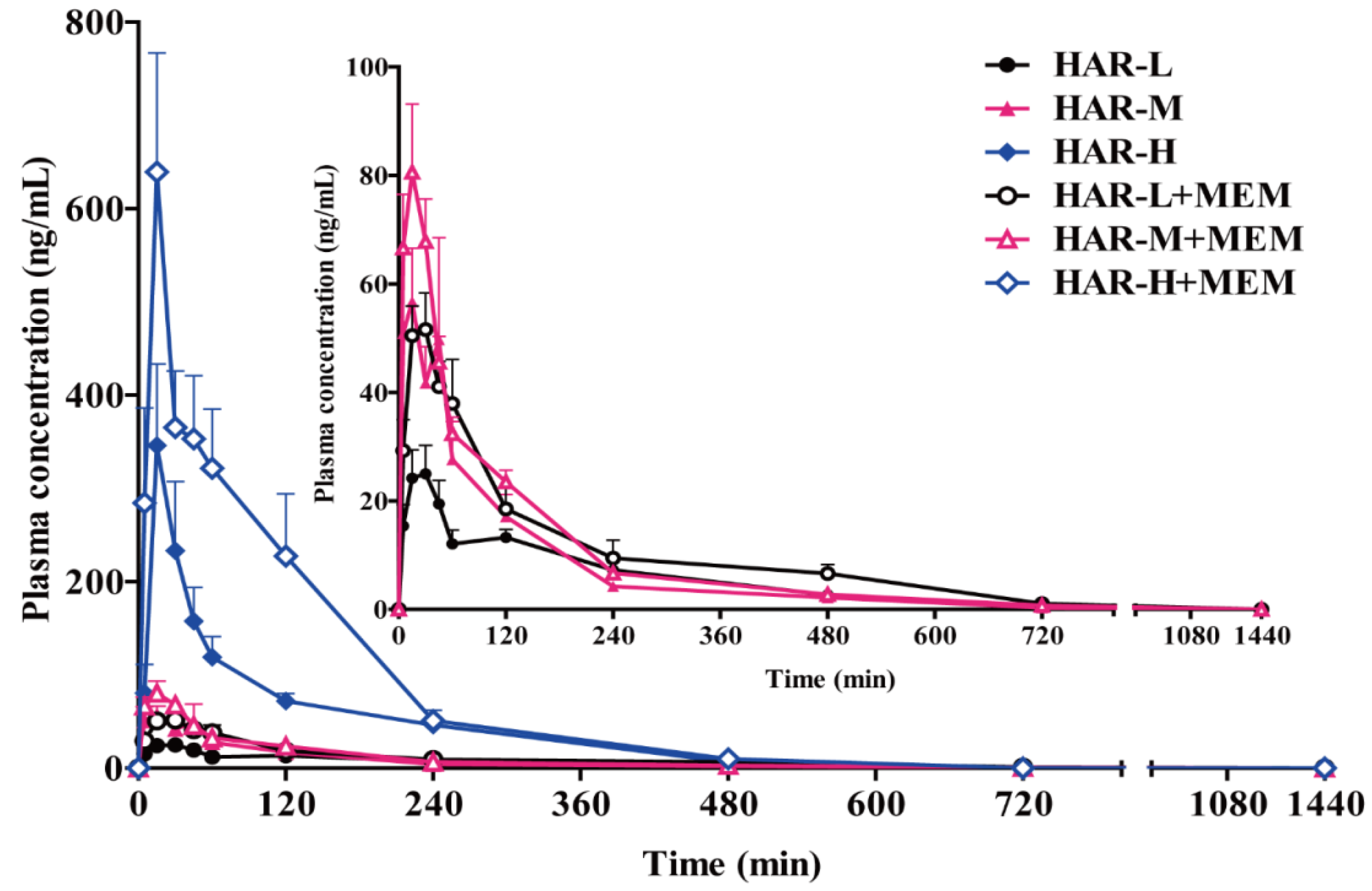
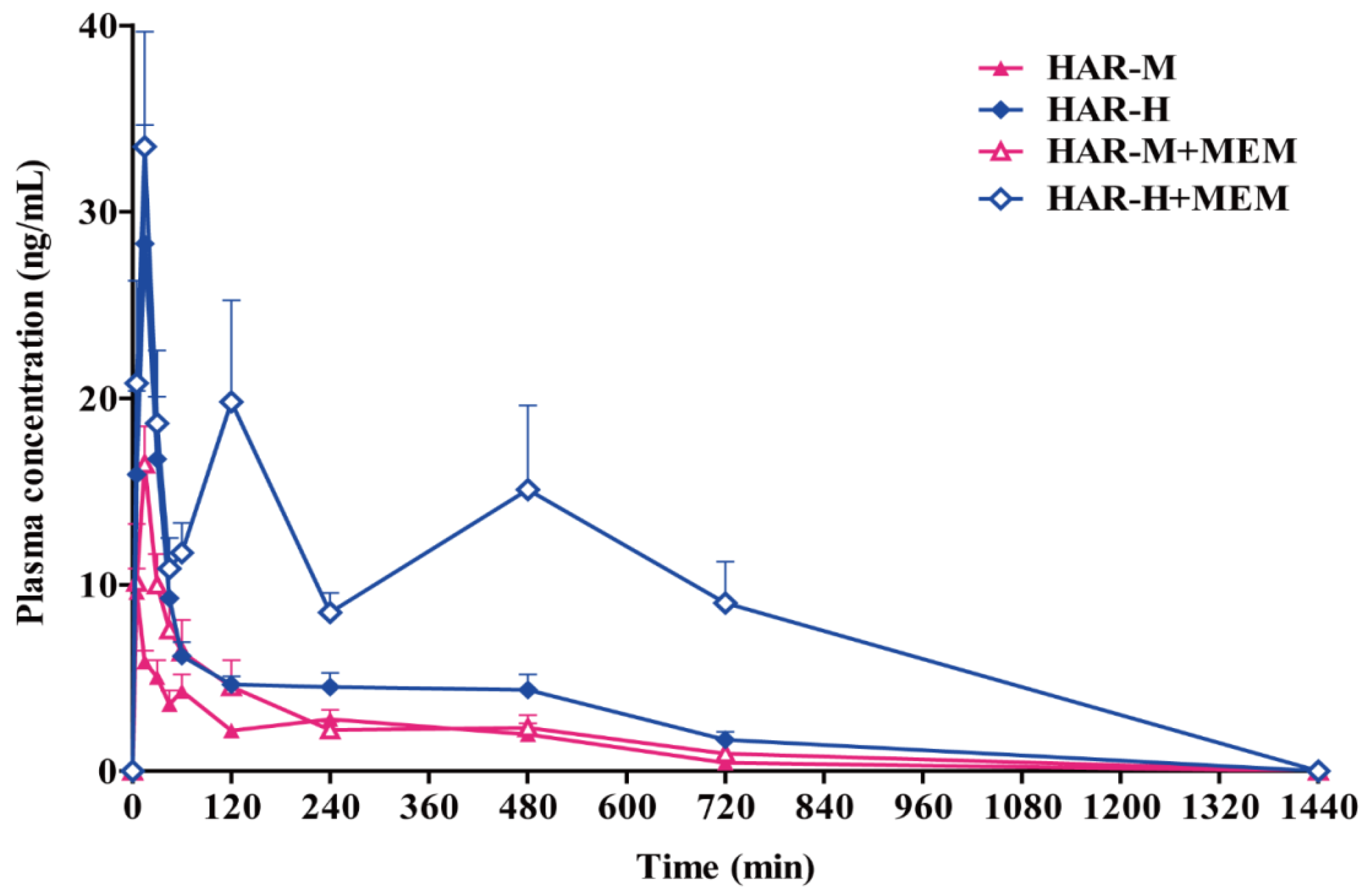
2.4. Pharmacokinetics Studies
3. Discussion
4. Materials and Methods
4.1. Reagents and Materials
4.2. Animals and Ethics Statement
4.3. Instruments and Conditions
4.3.1. Liquid Chromatography
4.3.2. Mass Spectrometric Conditions
4.4. Preparation of Standard Solutions, Calibration Standards and Quality Control Samples
4.5. Sample Preparation
4.6. Method Validation
4.6.1. Selectivity and Carry-Over
4.6.2. Linearity, LLOD, and LLOQ
4.6.3. Within-Run and Between-Run Precision
4.6.4. Matrix Effect and Extraction Recovery
4.6.5. Stability
4.7. Pharmacokinetic Study
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Allen, J.R.F.; Holmstedt, B.R. The simple β-carboline alkaloids. Phytochemistry 1980, 19, 1573–1582. [Google Scholar] [CrossRef]
- Li, S.P.; Cheng, X.M.; Wang, C.H. A review on traditional uses, phytochemistry, pharmacology, pharmacokinetics and toxicology of the genus Peganum. J. Ethnopharmacol. 2017, 203, 127–162. [Google Scholar] [CrossRef] [PubMed]
- Ida, A.; Luigi, A.; Lucia, C.; Vincenzo, D.F.; Florinda, F.; Filomena, N.; Lucèia, F.S.; Maroua, K. Chemical composition, antibacterial and phytotoxic activities of peganum harmala seed essential oils from five different localities in northern africa. Molecules 2016, 21, 1235–1248. [Google Scholar]
- Herraiz, T.; González, D.; Ancín-Azpilicueta, C.; Arán, V.J.; Guillén, H. Beta-carboline alkaloids in peganum harmala and inhibition of human monoamine oxidase (MAO). Food. Chem. Toxicol. 2010, 48, 839–845. [Google Scholar] [CrossRef]
- Jiang, B.; Li, S.P.; Liu, W.; Yang, Y.D.; Chen, W.X.; He, D.D.; Cheng, X.M.; Wang, Z.T.; Chen, W.S.; Wang, C.H. Inhibitive activities detection of monoamine oxidases (MAO) A and B inhibitors in human liver MAO incubations by UPLC-ESI-MS/MS. J. Pharm. Biomed. Anal. 2015, 115, 283–291. [Google Scholar] [CrossRef]
- Mennenga, S.E.; Gerson, J.E.; Dunckley, T.; Bimontenelson, H.A. Harmine treatment enhances short-term memory in old rats: Dissociation of cognition and the ability to perform the procedural requirements of maze testing. Physiol. Behav. 2015, 138, 260–265. [Google Scholar] [CrossRef] [PubMed]
- He, D.D.; Wu, H.; Wei, Y.; Liu, W.; Huang, F.; Shi, H.L.; Zhang, B.B.; Wu, X.J.; Wang, C.H. Effects of harmine, an acetylcholinesterase inhibitor, on spatial learning and memory of APP/PS1 transgenic mice and scopolamine-induced memory impairment mice. Eur. J. Pharmacol. 2015, 768, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Li, S.P.; Wang, Y.W.; Qi, S.L.; Zhang, Y.P.; Deng, G.; Ding, W.Z.; Ma, C.; Lin, Q.Y.; Guan, H.D.; Liu, W.; et al. Analogous β-carboline alkaloids harmaline and harmine ameliorate scopolamine-induced cognition dysfunction by attenuating acetylcholinesterase activity, oxidative stress, and inflammation in mice. Front. Pharmacol. 2018, 9, 346–361. [Google Scholar] [CrossRef]
- Collins, M.A.; Neafsey, E.J. Beta-carboline analogs of N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): Endogenous factors underlying parkinsonism? Neurosci. Lett. 1985, 55, 179–184. [Google Scholar] [CrossRef]
- Smeyne, R.J.; Lewis, V.J. The MPTP model of parkinson’s disease. Brain. Res. Mol. Brain. Res. 2005, 134, 57–66. [Google Scholar] [CrossRef]
- Serra, P.A.; Pluchino, S.; Marchetti, B.; Desole, M.S.; Miele, E. The MPTP mouse model: Cues on DA release and neural stem cell restorative role. Parkinsonism. Relat. Disord. 2008, 14, S189–S193. [Google Scholar] [CrossRef]
- Azizi, E.; Akbari-Javar, H.; Salehi, M.H.; Jalali, N. Evaluation of toxic alkaloids of seeds of peganum harmala L. as an abortion agent in pregnant rat. Pathophysiology 1998, 5, 94. [Google Scholar] [CrossRef]
- Berdai, M.A.; Labib, S.; Harandou, M. Peganum harmala L. intoxication in a pregnant woman. Vopr. Neirokhir. 2014, 2014, 12–19. [Google Scholar]
- Mahmoudian, M.; Jalipour, H.; Dardashti, P.S. Toxicity of peganum harmala: Review and a case report. Iran. J. Pharmacol. Ther. 2002, 1, 1–4. [Google Scholar]
- Llinás, R.; Volkind, R.A. The olivo-cerebellar system: Functional properties as revealed by harmaline-induced tremor. Exp. Brain. Res. 1973, 18, 69–87. [Google Scholar] [CrossRef]
- Headley, P.M.; Lodge, D.; Duggan, A.W. Drug-induced rhythmical activity in the inferior olivary complex of the rat. Brain. Res. 1976, 101, 461–478. [Google Scholar] [CrossRef]
- Ledoux, M.S.; Lorden, J.F. Abnormal spontaneous and harmaline-stimulated purkinje cell activity in the awake genetically dystonic rat. Exp. Brain. Res. 2002, 145, 457–467. [Google Scholar] [CrossRef]
- De, M.C.; Lamarre, Y. Rhythmic activity induced by harmaline in the olivo-cerebello-bulbar system of the cat. Brain. Res. 1973, 53, 81–95. [Google Scholar]
- Riedel, C.J.; Muraszko, K.M.; Youle, R.J. Diphtheria toxin mutant selectively kills cerebellar Purkinje neurons. Proc. Nati. Acad. Sci. USA. 1990, 87, 5051–5055. [Google Scholar] [CrossRef]
- Rossi, P.I.; Musante, I.; Summa, M.; Pittaluga, A.; Emionite, L.; Ikehata, M.; Rastaldi, M.P.; Ravazzolo, R.; Puliti, A. Compensatory molecular and functional mechanisms in nervous system of the grm1(crv4) mouse lacking the mglu1 receptor: A model for motor coordination deficits. Cereb. Cortex. 2013, 23, 2179–2189. [Google Scholar] [CrossRef][Green Version]
- Francis, P.T.; Ramírez, M.J.; Lai, M.K. Neurochemical basis for symptomatic treatment of alzheimer’s disease. Neuropharmacology 2010, 59, 221–229. [Google Scholar] [CrossRef]
- Sonkusare, S.K.; Kaul, C.L.; Ramarao, P. Dementia of alzheimer’s disease and other neurodegenerative disorders--memantine, a new hope. Pharmacol. Res. 2005, 51, 1–17. [Google Scholar] [CrossRef]
- Parsons, C.G.; Stöffler, A.; Danysz, W. Memantine: A NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system-too little activation is bad, too much is even worse. Neuropharmacology 2007, 53, 699–723. [Google Scholar] [CrossRef]
- Johnson, J.W.; Kotermanski, S.E. Mechanism of action of memantine. Curr. Opin. Pharmacol. 2006, 6, 61–67. [Google Scholar] [CrossRef]
- Parsons, C.G.; Danysz, W.; Quack, G. Memantine is a clinically well tolerated N-methyl-d-aspartate (NMDA) receptor antagonist--a review of preclinical data. Neuropharmacology 1999, 38, 735–767. [Google Scholar] [CrossRef]
- Doraiswamy, P.M. Non-cholinergic strategies for treating and preventing alzheimer’s disease. CNS Drugs 2002, 16, 811–824. [Google Scholar] [CrossRef]
- Molinuevo, J.L.; Lladó, A.; Rami, L. Memantine: Targeting glutamate excitotoxicity in alzheimer’s disease and other dementias. Am. J. Alzheimers Dis. Other Demen. 2005, 20, 77–85. [Google Scholar] [CrossRef]
- Lange, K.W.; Kornhuber, J.; Riederer, P. Dopamine/glutamate interactions in parkinson’s disease. Neurosci. Biobehav. Rev. 1997, 21, 393–400. [Google Scholar] [CrossRef]
- Areosa, S.A.; Sherriff, F.; Mcshane, R. Memantine for dementia. Cochrane. Database. Syst. Rev. 2006, 5, CD003154. [Google Scholar]
- Tian, Z.H.; Pang, H.H.; Zhang, Q.; Du, S.Y.; Lu, Y.; Zhang, L.; Bai, J.; Li, P.Y.; Li, D.Q.; Zhao, M.D. Effect of aspirin on the pharmacokinetics and absorption of panax notoginseng saponins. J. Chromatogr. B Analyt. Technol. Biomed Life Sci. 2018, 1074–1075, 25–33. [Google Scholar] [CrossRef]
- Liu, M.; Yuan, J.; Hu, W.J.; Ke, C.Q.; Zhang, Y.F.; Ye, Y.; Zhong, D.F.; Zhao, G.R.; Yao, S.; Liu, J. Pretreatment with broad-spectrum antibiotics alters the pharmacokinetics of major constituents of shaoyao-gancao decoction in rats after oral administration. Acta Pharmacol. Sin. 2019, 40, 288–296. [Google Scholar] [CrossRef]
- Li, S.P.; Zhang, Y.P.; Deng, G.; Wang, Y.W.; Qi, S.L.; Cheng, X.M.; Ma, Y.M.; Xie, Y.; Wang, C.H. Exposure characteristics of the analogous β-carboline alkaloids harmaline and harmine based on the efflux transporter of multidrug resistance protein 2. Front. Pharmacol. 2017, 8, 541–559. [Google Scholar] [CrossRef]
- Beconi, M.G.; Howland, D.; Park, L.; Lyons, K.; Giuliano, J.; Dominguez, C.; Munoz-Sanjuan, I.; Pacifici, R. Pharmacokinetics of memantine in rats and mice. PLoS. Curr. 2012, 3, RRN1291. [Google Scholar] [CrossRef]
- Zhao, T.; Zheng, S.S.; Zhang, B.F.; Li, Y.Y.; Bligh, S.W.; Wang, C.H.; Wang, Z.T. Metabolic pathways of the psychotropic-carboline alkaloids, harmaline and harmine, by liquid chromatography/mass spectrometry and NMR spectroscopy. Food Chem. 2012, 134, 1096–1105. [Google Scholar] [CrossRef]
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.Q. Global prevalence of dementia: A delphi consensus study. Lancet 2005, 366, 2112–2117. [Google Scholar] [CrossRef]
- Terry, A.V.; Buccafusco, J.J. The cholinergic hypothesis of age and alzheimer’s disease-related cognitive deficits: Recent challenges and their implications for novel drug development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef]
- Riederer, P.; Hoyer, S. From benefit to damage. glutamate and advanced glycation end products in alzheimer brain. J. Neural Transm. 2006, 113, 1671–1677. [Google Scholar] [CrossRef]
- Jann, D.M.W.; Shirley, K.L.; Small, G.W. Clinical pharmacokinetics and pharmacodynamics of cholinesterase inhibitors. Clin. Pharmacokinet. 2002, 41, 719–739. [Google Scholar] [CrossRef]
- Noetzli, M.; Eap, C.B. Pharmacodynamic, pharmacokinetic and pharmacogenetic aspects of drugs used in the treatment of alzheimer’s disease. Clin. Pharmacokinet. 2013, 52, 225–241. [Google Scholar] [CrossRef]
- Kornhuber, J.; Kennepohl, E.M.; Bleich, S.; Wiltfang, J.; Kraus, T.; Reulbach, U.; Meineke, I. Memantine pharmacotherapy: A naturalistic study using a population pharmacokinetic approach. Clin. Pharmacokinet. 2007, 46, 599–612. [Google Scholar] [CrossRef]
- Busch, A.E.; Karbach, U.; Miska, D.; Gorboulev, V.; Akhoundova, A.; Volk, C.; Arndt, P.; Ulzheimer, J.C.; Sonders, M.S.; Baumann, C. Human neurons express the polyspecific cation transporter hOCT2, which translocates monoamine neurotransmitters, amantadine, and memantine. Mol. Pharmacol. 1998, 54, 342–352. [Google Scholar] [CrossRef]
- Li, S.P.; Teng, L.; Liu, W.; Cheng, X.M.; Jiang, B.; Wang, Z.T.; Wang, C.H. Pharmacokinetic study of harmane and its 10 metabolites in rat after intravenous and oral administration by UPLC-ESI-MS/MS. Pharm. Biol. 2016, 54, 1768–1781. [Google Scholar] [CrossRef]
- Yu, A.M.; Idle, J.R.; Krausz, K.W.; Küpfer, A.; Gonzalez, F.J. Contribution of individual cytochrome P450 isozymes to the O-demethylation of the psychotropic beta-carboline alkaloids harmaline and harmine. J. Pharmacol. Exp. Ther. 2003, 305, 315–322. [Google Scholar] [CrossRef]
- Zhao, T.; He, Y.Q.; Wang, J.; Ding, K.M.; Wang, C.H.; Wang, Z.T. Inhibition of human cytochrome P450 enzymes 3A4 and 2D6 by β-carboline alkaloids, harmine derivatives. Phytother. Res. 2011, 25, 1671–1677. [Google Scholar] [CrossRef]
- Dovertelova, G.; Zendulka, O.; Noskova, K.; Jurica, J.; Pes, O.; Dusek, J.; Carazo, A.; Zapletalova, L.; Hiavacova, N.; Pavek, P. Effect of endocannabinoid oleamide on rat and human liver cytochrome P450 enzymes in in vitro and in vivo models. Drug Metab Dispos. 2018, 46, 913–923. [Google Scholar] [CrossRef]
- Aarons, D.H.; Rossi, G.V.; Orzechowski, R.F. Cardiovascular actions of three harmala alkaloids: Harmine, harmaline, and harmalol. J. Pharm. Sci. 2010, 66, 1244–1248. [Google Scholar] [CrossRef]
- Fortunato, J.J.; Réus, G.Z.; Kirsch, T.R.; Stringari, R.B.; Fries, G.R.; Kapczinski, F.; Hallak, J.E.; Zuardi, A.W.; Crippa, J.A.; Quevedo, J. Chronic administration of harmine elicits antidepressant-like effects and increases BDNF levels in rat hippocampus. J. Neural Transm. 2010, 117, 1131–1137. [Google Scholar] [CrossRef]
- Li, S.W.; Wang, A.Q.; Gu, F.; Wang, Z.H.; Tian, C.Q.; Qian, Z.Y.; Tang, L.Q.; Gu, Y.Q. Novel harmine derivatives for tumor targeted therapy. Oncotarget 2015, 6, 8988–9001. [Google Scholar] [CrossRef][Green Version]
- Toutain, P.L.; Ferran, A.; Bousquet, M.A. Species differences in pharmacokinetics and pharmacodynamics. Handb. Exp. Pharmacol. 2010, 199, 19–48. [Google Scholar]
- Liu, D.; Zheng, X.; Tang, Y.; Zi, J.; Nan, Y.; Wang, S.; Xiao, C.; Zhu, J.; Chen, C. Metabolism of tanshinol borneol ester in rat and human liver microsomes. Drug Metab. Dispos. 2010, 38, 1464–1470. [Google Scholar] [CrossRef]
- Li, S.P.; Teng, L.; Liu, W.; Cheng, X.M.; Jiang, B.; Wang, Z.T.; Wang, C.H. Interspecies metabolism diversity of harmaline and harmine in in vitro 11 mammalian liver microsomes. Drug Test. Anal. 2017, 9, 754–768. [Google Scholar] [CrossRef]
- Su, T.; Ding, X. Regulation of the cytochrome P450 2A genes. Toxicol. Appl. Pharmacol. 2004, 199, 285–294. [Google Scholar] [CrossRef]
- Blume, N.; Leonard, J.; Xu, Z.J.; Watanabe, O.; Remotti, H.; Fishman, J. Characterization of Cyp2d22, a novel cytochrome P450 expressed in mouse mammary cells. Arch. Biochem. Biophys. 2000, 381, 191–204. [Google Scholar] [CrossRef]
- Monshouwer, M.; Vant, K.G.A.; Nijmeijer, S.M.; Witkamp, R.F.; Van, M.A.S. Characterization of cytochrome P450 isoenzymes in primary cultures of pig hepatocytes. Toxicol. In Vitro 1998, 12, 715–723. [Google Scholar] [CrossRef]
- Chu, X.; Bleasby, K.; Evers, R. Species differences in drug transporters and implications for translating preclinical findings to humans. Expert. Opin. Drug Metab. Toxicol. 2013, 9, 237–252. [Google Scholar] [CrossRef]
- Yang, Y.D.; Cheng, X.M.; Liu, W.; Chou, G.X.; Wang, Z.T.; Wang, C.H. Potent AChE and BChE inhibitors isolated from seeds of peganum harmala linn by a bioassay-guided fractionation. J. Ethnopharmacol. 2015, 168, 279–286. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds harmine, memantine, harmol and tacrine are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacokinetics Parameters | MEM | HAR-L + MEM | HAR-M + MEM | HAR-H + MEM |
|---|---|---|---|---|
| Ke (/h) | 0.261 ± 0.0282 | 0.191 ± 0.0508 ** | 0.215 ± 0.0728 | 0.203 ± 0.0723 |
| Kd (/h) | 0.548 ± 0.669 | 0.320 ± 0.0934 | 0.488 ± 0.262 | 0.485 ± 0.202 |
| Ka (/h) | 1.73 ± 0.848 | 0.595 ± 0.315 ** | 1.08 ± 1.05 | 0.481 ± 0.203 ** |
| T1/2e (h) | 2.69 ± 0.306 | 3.96 ± 1.44 * | 3.62 ± 1.42 | 4.27 ± 3.03 |
| T1/2d (h) | 3.08 ± 2.37 | 2.42 ± 1.06 | 1.67 ± 0.560 | 1.61 ± 0.529 |
| T1/2a (h) | 0.482 ± 0.202 | 1.37 ± 0.468 *** | 0.945 ± 0.398 ** | 1.61 ± 0.459 *** bb |
| Cmax (ng/mL) | 84.8 ± 25.8 | 50.5 ± 7.59 ** | 74.9 ± 15.0b | 74.3 ± 26.6 |
| Tmax (h) | 1.31 ± 0.579 | 3.13 ± 2.23 | 1.56 ± 1.11 | 4.00 ± 1.85 ** b |
| AUC(0-t) (ng*h/mL) | 349 ± 46.2 | 392 ± 55.2 | 472 ± 105 * | 603 ± 149 ** b |
| AUC(0-∞)(ng*h/mL) | 367 ± 4.87 | 477 ± 75.1 ** | 547 ± 142 ** | 781 ± 285 ** b |
| MRT (h) | 4.04 ± 0.808 | 7.45 ± 1.51 *** | 6.25 ± 1.25 *** | 8.50 ± 3.74 ** |
| Vd (L/kg) | 53.8 ± 10.6 | 60.1 ± 19.3 | 48.4 ± 16.4 | 38.3 ± 13.1 * |
| CL/F (L/h/kg) | 13.8 ± 1.50 | 10.7 ± 1.79 ** | 9.70 ± 2.57 ** | 7.10 ± 2.29 *** b |
| Pharmacokinetics Parameters | HAR-L | HAR-M | HAR-H | HAR-L + MEM | HAR-M + MEM | HAR-H + MEM |
|---|---|---|---|---|---|---|
| Ke (/h) | 0.428 ± 0.226 | 0.477 ± 0.277 | 0.419 ± 0.271 | 0.312 ± 0.115 | 0.174 ± 0.0543 * | 0.456 ± 0.407 b |
| Kd (/h) | 1.62 ± 2.43 | 2.06 ± 2.79 | 1.11 ± 1.11 | 1.40 ± 1.28 | 0.743 ± 0.741 | 0.921 ± 0.559 |
| Ka (/h) | 1.66 ± 2.27 | 2.69 ± 2.60 | 2.80 ± 2.74 | 1.91 ± 2.46 | 1.52 ± 1.08 | 2.93 ± 2.06 |
| T1/2e (h) | 2.05 ± 1.11 | 2.28 ± 1.86 | 2.35 ± 1.44 | 2.89 ± 2.26 | 4.33 ± 1.31 * | 3.21 ± 3.17 |
| T1/2d (h) | 2.14 ± 2.57 | 0.960 ± 0.734 | 1.36 ± 1.37 | 1.22 ± 1.24 | 2.33 ± 2.71 | 1.40 ± 1.74 |
| T1/2a (h) | 1.29 ± 1.25 | 0.529 ± 0.478 | 0.605 ± 0.546 | 1.57 ± 2.28 | 1.18 ± 1.22 | 0.357 ± 0.228 |
| Cmax (ng/mL) | 31.4 ± 16.3 | 77.0 ± 53.8 | 368 ± 253 aaa | 64.5 ± 14.7 *** | 86.6 ± 31.0 | 755 ± 298 * bbb |
| Tmax (h) | 0.500 ± 0.267 | 0.365 ± 0.271 | 0.344 ± 0.186 | 0.500 ± 0.267 | 0.240 ± 0.129 | 0.448 ± 0.630 |
| AUC(0-t) (ng*h/mL) × 10 | 7.49 ± 3.63 | 10.0 ± 4.48 | 50.2 ± 18.0 aaa | 14.1 ± 5.67 * | 13.9 ± 4.06 | 106 ± 54.0 * bbb |
| AUC(0-∞)(ng*h/mL) × 10 | 8.31 ± 3.89 | 10.9 ± 4.79 | 58.7 ± 27.5 aaa | 15.8 ± 6.54 * | 15.3 ± 4.31 | 111 ± 50.8 * bbb |
| MRT (h) | 3.36 ± 1.38 | 2.83 ± 1.41 | 3.17 ± 1.48 | 4.02 ± 1.96 | 3.28 ± 1.02 | 2.88 ± 1.93 |
| Vd (L/kg) × 10 | 80.3 ± 39.2 | 136 ± 105 | 51.4 ± 33.1 | 59.0 ± 35.1 | 172 ± 61.6 | 60.3 ± 89.6 |
| CL/F (L/h/kg) | 323 ± 206 | 431 ± 178 | 163 ± 70.4 | 158 ± 90.6 | 278 ± 68.5 * | 91.2 ± 50.8 * bbb |
| Pharmacokinetics Parameters | HAR-M | HAR-H | HAR-M + MEM | HAR-H +MEM |
|---|---|---|---|---|
| Ke (/h) | 0.400 ± 0.133 | 0.226 ± 0.0896 aa | 0.312 ± 0.271 | 0.0899 ± 0.0912 ** b |
| Kd(/h) | 0.433 ± 0.152 | 0.840 ± 0.948 | 0.966 ± 1.59 | 0.971 ± 1.10 |
| Ka (/h) | 0.808 ± 1.28 | 1.17 ± 1.96 | 1.98 ± 1.72 | 0.864 ± 1.00 |
| T1/2e (h) | 1.94 ± 0.712 | 3.57 ± 1.50 a | 3.53 ± 2.19 | 17.2 ± 14.7 * b |
| T1/2d (h) | 1.89 ± 1.07 | 2.02 ± 1.72 | 2.05 ± 1.61 | 3.48 ± 4.35 |
| T1/2a (h) | 2.22 ± 1.67 | 2.02 ± 1.52 | 2.78 ± 4.59 | 2.15 ± 2.05 |
| Cmax (ng/mL) | 10.4 ± 2.13 | 32.4 ± 16.5 aa | 18.0 ± 5.95 * | 40.4 ± 10.9 bbb |
| Tmax (h) | 0.198 ± 0.324 | 0.271 ± 0.159 | 0.229 ± 0.0589 | 2.39 ± 3.52 |
| AUC(0-t) (ng*h/mL) | 27.1 ± 10.8 | 57.8 ± 19.3 aaa | 35.5 ± 15.3 | 158 ± 59.9 ** bbb |
| AUC(0-∞)(ng*h/mL) | 29.6 ± 11.3 | 69.7 ± 23.1 aaa | 43.1 ± 21.9 | 327 ± 140 *** bbb |
| MRT (h) | 4.44 ± 1.78 | 6.04 ± 2.12 | 5.52 ± 2.55 | 23.5 ± 19.3 * b |
| Vd (L/kg) × 10 | 463 ± 308 | 616 ± 225 | 499 ± 312 | 633 ± 462 |
| CL/F (L/h/kg) × 10 | 186 ± 171 | 130 ± 56.7 | 107 ± 34.1 | 27.8 ± 9.14 *** bbb |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Li, S.; Wang, Y.; Deng, G.; Cao, N.; Wu, C.; Ding, W.; Wang, Y.; Cheng, X.; Wang, C. Potential Pharmacokinetic Drug–Drug Interaction Between Harmine, a Cholinesterase Inhibitor, and Memantine, a Non-Competitive N-Methyl-d-Aspartate Receptor Antagonist. Molecules 2019, 24, 1430. https://doi.org/10.3390/molecules24071430
Zhang Y, Li S, Wang Y, Deng G, Cao N, Wu C, Ding W, Wang Y, Cheng X, Wang C. Potential Pharmacokinetic Drug–Drug Interaction Between Harmine, a Cholinesterase Inhibitor, and Memantine, a Non-Competitive N-Methyl-d-Aspartate Receptor Antagonist. Molecules. 2019; 24(7):1430. https://doi.org/10.3390/molecules24071430
Chicago/Turabian StyleZhang, Yunpeng, Shuping Li, Youxu Wang, Gang Deng, Ning Cao, Chao Wu, Wenzheng Ding, Yuwen Wang, Xuemei Cheng, and Changhong Wang. 2019. "Potential Pharmacokinetic Drug–Drug Interaction Between Harmine, a Cholinesterase Inhibitor, and Memantine, a Non-Competitive N-Methyl-d-Aspartate Receptor Antagonist" Molecules 24, no. 7: 1430. https://doi.org/10.3390/molecules24071430
APA StyleZhang, Y., Li, S., Wang, Y., Deng, G., Cao, N., Wu, C., Ding, W., Wang, Y., Cheng, X., & Wang, C. (2019). Potential Pharmacokinetic Drug–Drug Interaction Between Harmine, a Cholinesterase Inhibitor, and Memantine, a Non-Competitive N-Methyl-d-Aspartate Receptor Antagonist. Molecules, 24(7), 1430. https://doi.org/10.3390/molecules24071430