Lactosylated Albumin Nanoparticles: Potential Drug Nanovehicles with Selective Targeting Toward an In Vitro Model of Hepatocellular Carcinoma

 , , , ,
, , , ,
Abstract
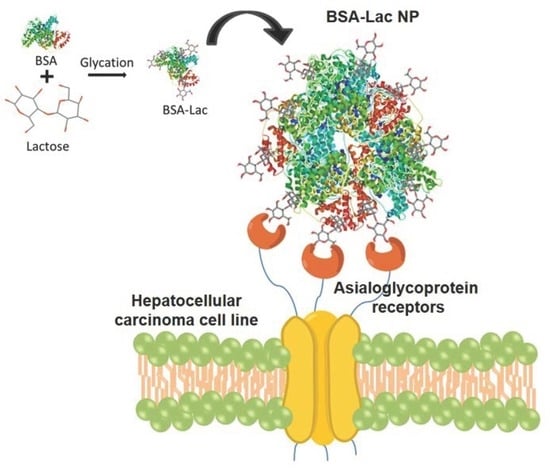
1. Introduction
2. Results and Discussion
2.1. Characterization of the BSA-Lac
2.1.1. Biorecognition Assays of BSA-Lac with RCA
2.1.2. Electrophoresis SDS-PAGE
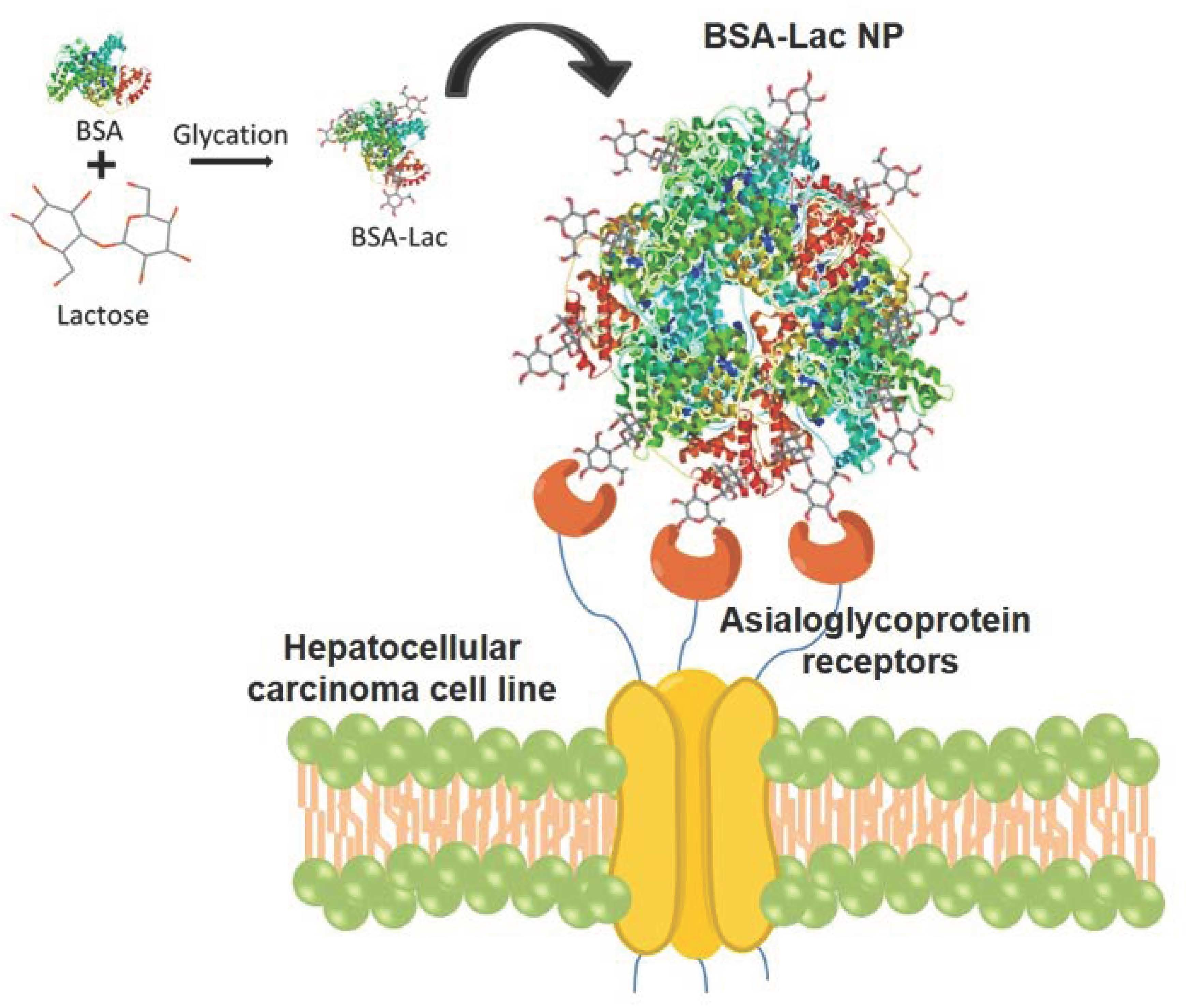
2.1.3. FT-IR
2.2. Characterization of Nanoparticles
2.2.1. Particle Size and Zeta Potential
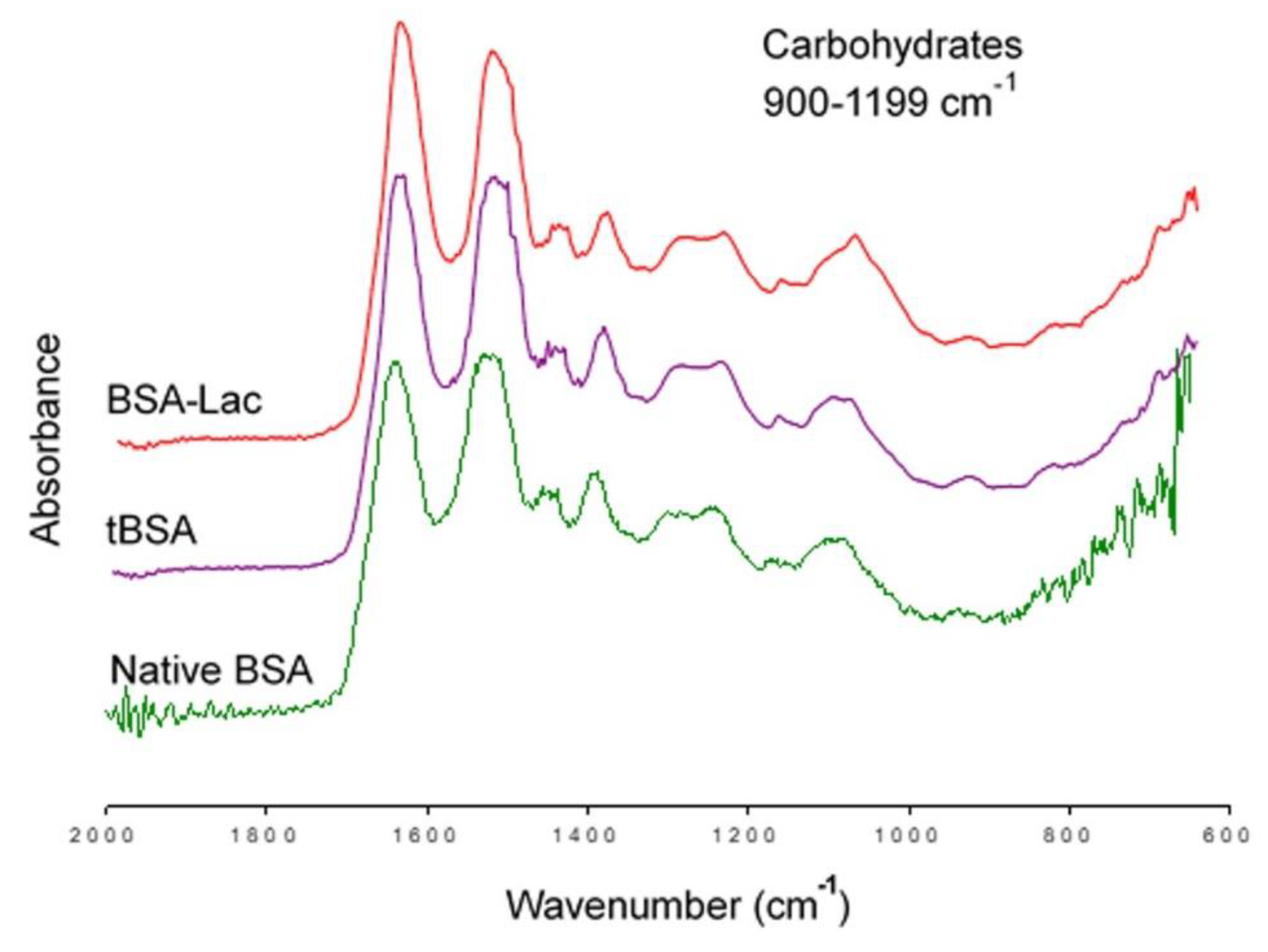
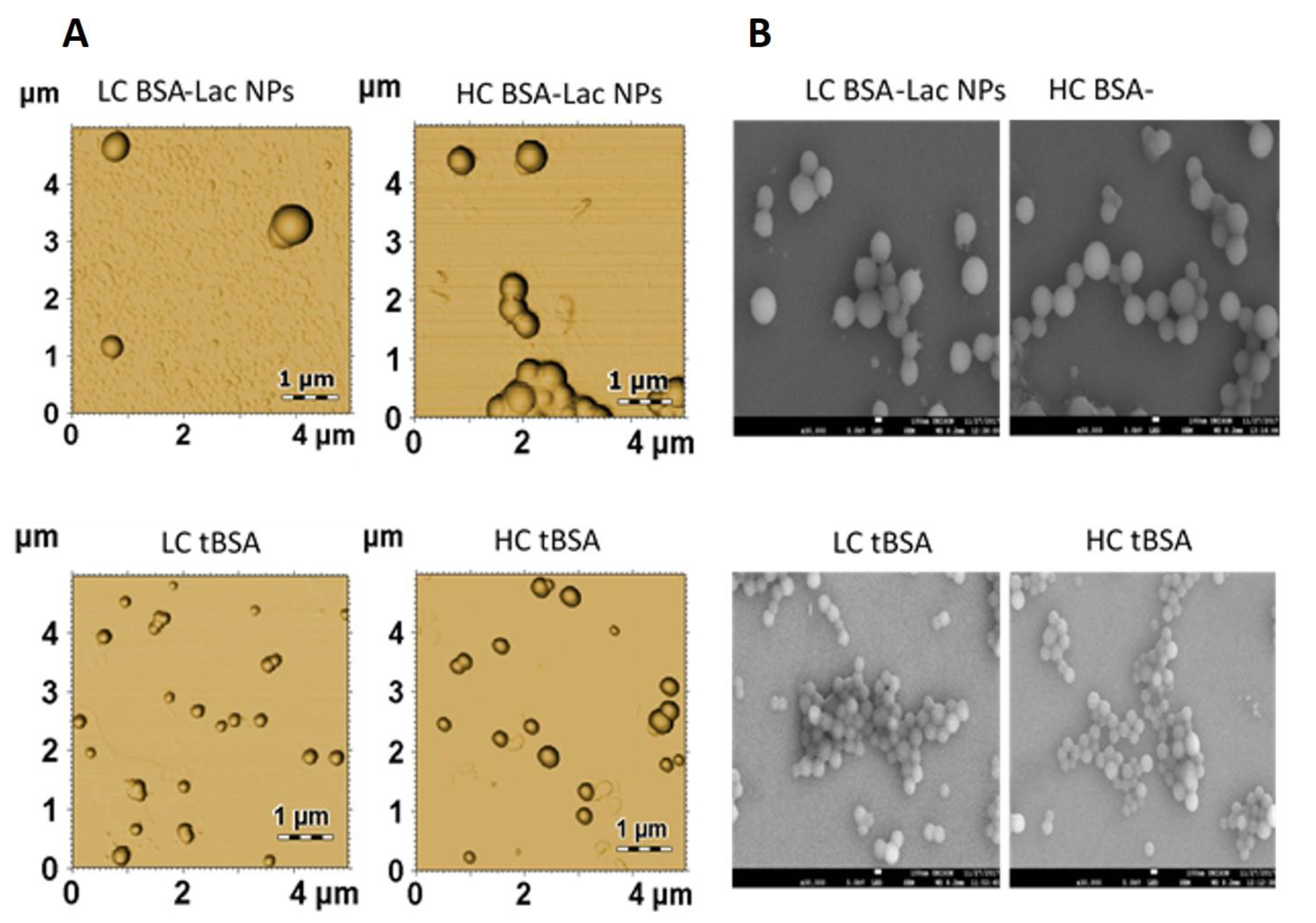
2.2.2. Nanoparticle Morphology
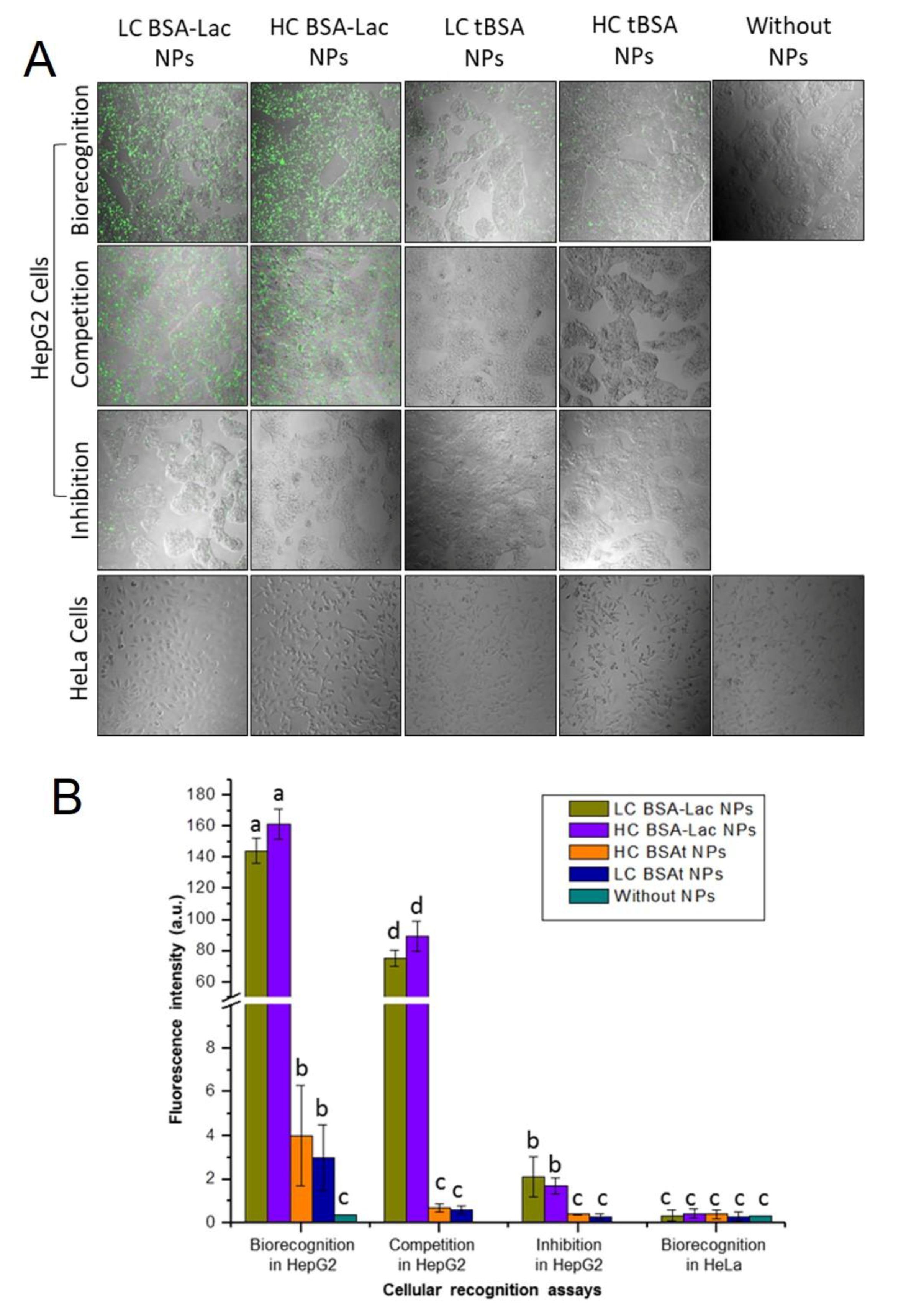
2.3. Evaluation of Specific Cellular Recognition
3. Materials and Methods
3.1. Material
3.2. Lactosylation of Albumin
3.3. Characterization of BSA-Lac
3.3.1. Enzyme-linked Lectin Recognition Assays (ELLA)
3.3.2. SDS-PAGE Electrophoresis
3.3.3. Lectin-blotting Assay
3.3.4. Fourier-transform Infrared Spectroscopy
3.4. Synthesis of BSA-Lac Nanoparticles (BSA-Lac NPs)
3.5. Characterization of BSA-Lac NPs
3.5.1. Particle Size and Zeta Potential
3.5.2. Nanoparticle Morphology
3.6. Cellular Uptake Evaluation and Specificity of Recognition
3.7. Statistical Analysis
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| HCC | hepatocellular carcinoma |
| BSA | bovine serum albumin |
| BSA-Lac | bovine serum albumin conjugated with lactose |
| tBSA | bovine serum albumin treated with heat |
| HC | high-crosslinker |
| LC | low-crosslinker |
| ASGPR | asialoglycoprotein receptor |
| EPR | effect of permeability and retention |
| FDA | food and drug administration |
| NPs | nanoparticles |
| RCA | Ricinus communis agglutinin |
| ELLAs | assay with enzyme-linked lectins |
| FT-IR | Fourier transform infrared spectroscopy |
References
- Li, M.; Zhang, W.; Wang, B.; Gao, Y.; Song, Z.; Zheng, Q.C. Ligand-based targeted therapy: A novel strategy for hepatocellular carcinoma. Int. J. Nanomed. 2016, 11, 5645–5669. [Google Scholar] [CrossRef]
- Dutta, R.; Mahato, R.I. Recent advances in hepatocellular carcinoma therapy. Pharmacol Ther. 2017, 173, 106–117. [Google Scholar] [CrossRef]
- Liu, X.; Han, M.; Xu, J.; Geng, S.; Zhang, Y.; Ye, X.; Gou, J.; Yin, T.; He, H.; Tang, X. Asialoglycoprotein receptor-targeted liposomes loaded with a norcantharimide derivative for hepatocyte-selective targeting. Int. J. Pharm. 2017, 520, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Li, N.M.; Gao, P.; Yang, S.; Ning, Q.; Huang, W.; Li, Z.P.; Ye, P.J.; Xiang, L.; He, D.X.; et al. In vitro and in vivo evaluation of macromolecular prodrug GC-FUA based nanoparticle for hepatocellular carcinoma chemotherapy. Drug Deliv. 2017, 24, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, R.; Gramantieri, L.; Bolondi, L. Hepatocellular carcinoma: Epidemiology and clinical aspects. Mol. Aspects Med. 2008, 29, 130–143. [Google Scholar] [CrossRef]
- Thao, L.Q.; Lee, C.; Kim, B.; Lee, S.; Kim, T.H.; Kim, J.O.; Lee, E.S.; Oh, K.T.; Choi, H.G.; Yoo, S.D.; et al. Doxorubicin and paclitaxel co-bound lactosylated albumin nanoparticles having targetability to hepatocellular carcinoma. Colloids Surf. B 2017, 152, 183–191. [Google Scholar] [CrossRef]
- Mohamed, N.K.; Hamad, M.A.; Hafez, M.Z.; Wooley, K.L.; Elsabahy, M. Nanomedicine in management of hepatocellular carcinoma: Challenges and opportunities. Int. J. Cancer 2017, 140, 1475–1484. [Google Scholar] [CrossRef]
- Quan, G.; Pan, X.; Wang, Z.; Wu, Q.; Li, G.; Dian, L.; Chen, B.; Wu, C. Lactosaminated mesoporous silica nanoparticles for asialoglycoprotein receptor targeted anticancer drug delivery. J. Nanobio. 2015, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Prabhakar, U.; Maeda, H.; Jain, R.K.; Sevick-Muraca, E.M.; Zamboni, W.; Farokhzad, O.C.; Barry, S.T.; Gabizon, A.; Grodzinski, P.; Blakey, D.C. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013, 2013, 2412–2417. [Google Scholar] [CrossRef]
- Elzoghby, A.O.; Samy, W.M.; Elgindy, N.A. Albumin-based nanoparticles as potential controlled release drug delivery systems. J. Control Release 2012, 157, 168–182. [Google Scholar] [CrossRef]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable controlled-release polymers and polymeric nanoparticles: Mechanisms of controlling drug release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef]
- Xu, R.; Fisher, M.; Juliano, R.L. Targeted albumin-based nanoparticles for delivery of amphipathic drugs. Bioconjug Chem. 2011, 22, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Doppalapudi, S.; Jain, A.; Domb, A.J.; Khan, W. Biodegradable polymers for targeted delivery of anti-cancer drugs. Expert Opin. Drug Deliv. 2016, 13, 891–909. [Google Scholar] [CrossRef]
- Sarabia-Sainz, A.I.; Ramos-Clamont Montfort, G.; Winzerling, J.; Vázquez-Moreno, L. Bacterial recognition of thermal glycation products derived from porcine serum albumin with lactose. Acta Biochim. Pol. 2011, 58, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Gallegos-Tabanico, A.; Sarabia-Sainz, J.A.; Sarabia-Sainz, H.M.; Torres, R.C.; Guzman-Partida, A.M.; Ramos-Clamont Montfort, G.; Silva-Campa, E.; Burgara-Estrella, A.J.; Angulo-Molina, A.; Acosta-Elias, M.; et al. Molecular Recognition of Glyconanoparticles by RCA and E. coli K88; Designing Transports for Targeted Therapy. Acta Biochim. Pol. 2017, 64, 671–677. [Google Scholar] [CrossRef]
- D’souza, A.A.; Devarajan, P.V. Asialoglycoprotein receptor mediated hepatocyte targeting-Strategies and applications. J. Control Release 2015, 203, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Ledesma-Osuna, A.I.; Ramos-Clamont, G.; Vázquez-Moreno, L. Characterization of bovine serum albumin glycated with glucose, galactose and lactose. Acta Biochim. Pol. 2008, 55, 491–497. [Google Scholar]
- Güler, G.; Vorob’ev, M.M.; Vogel, V.; Mäntele, W. Proteolytically-induced changes of secondary structural protein conformation of bovine serum albumin monitored by Fourier transform infrared (FT-IR) and UV-circular dichroism spectroscopy. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2016, 161, 8–18. [Google Scholar] [CrossRef]
- Jian, W.; He, J.; Sun, Y.; Pang, J. Comparative studies on physicochemical properties of bovine serum albumin-glucose and bovine serum albumin-mannose conjugates formed via Maillard reaction. LWT–Food Sci Technol. 2016, 69, 358–364. [Google Scholar] [CrossRef]
- López-Pablos, A.L.; Leyva-Porras, C.C.; Silva-Cázares, M.B.; Longoria-Rodríguez, F.E.; Pérez-García, S.A.; Vértiz-Hernández, Á.A.; Saavedra-Leos, M.Z. Preparation and characterization of high purity anhydrous β-Lactose from α-Lactose monohydrate at mild temperature. Int. J. Polym. Sci. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Rubio-Ruiz, M.E.; Díaz-Díaz, E.; Cárdenas-León, M.; Argüelles-Medina, R.; Sánchez-Canales, P.; Larrea-Gallo, F.; Guarner-Lans, V. Glycation does not modify bovine serum albumin (BSA)-induced reduction of rat aortic relaxation: The response to glycated and nonglycated BSA is lost in metabolic syndrome. Glycobiology 2008, 18, 517–525. [Google Scholar] [CrossRef]
- Jun, J.Y.; Nguyen, H.H.; Chun, H.S.; Kang, B.C.; Ko, S. Preparation of size-controlled bovine serum albumin (BSA) nanoparticles by a modified desolvation method. Food Chem. 2011, 127, 1892–1898. [Google Scholar] [CrossRef]
- Schäfer, V.; Briesen, V.H.; Rubsamen-Waigmann, H.; Steffan, A.M.; Royer, C.; Kreuter, J. Phagocytosis and degradation of human serum albumin microspheres and nanoparticles in human macrophages. J. Microencapsul. 1994, 11, 261–269. [Google Scholar] [CrossRef]
- Patil, A.N.; Shinkar, D.M.; Saudagar, R.B. Review on Nanoparticle Drug Delivery System. Int. J. Pharma Chem. Res. 2017, 3, 114–119. [Google Scholar]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano–bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef]
- Peñaloza, J.P.; Márquez-Miranda, V.; Cabaña-Brunod, M.; Reyes-Ramírez, R.; Llancalahuen, F.M.; Maldonado-Biermann, F.; Velásquez, L.A.; Fuentes, J.A.; González-Nilo, F.D.; Otero, C. Intracellular trafficking and cellular uptake mechanism of PHBV nanoparticles for targeted delivery in epithelial cell lines. J. Nanobiotechnol. 2017, 15, 1–15. [Google Scholar]
- Schwartz, A.L.; Fridovich, S.E.; Knowles, B.B.; Lodish, H.F. Characterization of the asialoglycoprotein receptor in a continuous hepatoma line. J. Biol. Chem. 1981, 256, 8878–8881. [Google Scholar]
- Zhang, L.; Tian, Y.; Wen, Z.; Zhang, F.; Qi, Y.; Huang, W.; Wang, Y. Asialoglycoprotein receptor facilitates infection of PLC/PRF/5 cells by HEV through interaction with ORF2. J. Med. Virol. 2016, 88, 2186–2195. [Google Scholar] [CrossRef]
- Spiess, M.; Lodish, H.F. Sequence of a second human asialoglycoprotein receptor: Conservation of two receptor genes during evolution. Proc. Natl. Acad. Sci. USA 1985, 82, 6465–6469. [Google Scholar] [CrossRef]
- Hu, X.L.; Zang, Y.; Li, J.; Chen, G.R.; James, T.D.; He, X.P.; Tian, H. Targeted multimodal theranostics via biorecognition controlled aggregation of metallic nanoparticle composites. Chem. Sci. 2016, 7, 4004–4008. [Google Scholar] [CrossRef]
- Larsen, M.T.; Kuhlmann, M.; Hvam, M.L.; Howard, K.A. Albumin-based drug delivery: Harnessing nature to cure disease. Mol. Cell Ther. 2016, 4, 3. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680. [Google Scholar] [CrossRef]
- Lagarda-Diaz, I.; Guzman-Partida, A.M.; Urbano-Hernandez, G.; Ortega-Nieblas, M.M.; Robles-Burgueño, M.R.; Winzerling, J.; Vazquez-Moreno, L. Insecticidal action of PF2 lectin from Olneya tesota (Palo Fierro) against Zabrotes subfasciatus larvae and midgut glycoconjugate binding. J. Agr. Food Chem. 2008, 57, 689–694. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NPs | ||||
|---|---|---|---|---|
| LC BSA-Lac | HC BSA-Lac | LC tBSA | HC tBSA | |
| Size (nm) | 560 ± 18.0 | 539 ± 9.0 | 241 ± 2.5 | 246 ± 4.4 |
| Zeta potential (mv) | −26 ± 0.1 | −24 ± 0.4 | −30 ± 0.9 | −27 ± 0.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teran-Saavedra, N.G.; Sarabia-Sainz, J.A.-i.; Silva-Campa, E.; Burgara-Estrella, A.J.; Guzmán-Partida, A.M.; Ramos-Clamont Montfort, G.; Pedroza-Montero, M.; Vazquez-Moreno, L. Lactosylated Albumin Nanoparticles: Potential Drug Nanovehicles with Selective Targeting Toward an In Vitro Model of Hepatocellular Carcinoma. Molecules 2019, 24, 1382. https://doi.org/10.3390/molecules24071382
Teran-Saavedra NG, Sarabia-Sainz JA-i, Silva-Campa E, Burgara-Estrella AJ, Guzmán-Partida AM, Ramos-Clamont Montfort G, Pedroza-Montero M, Vazquez-Moreno L. Lactosylated Albumin Nanoparticles: Potential Drug Nanovehicles with Selective Targeting Toward an In Vitro Model of Hepatocellular Carcinoma. Molecules. 2019; 24(7):1382. https://doi.org/10.3390/molecules24071382
Chicago/Turabian StyleTeran-Saavedra, Nayelli Guadalupe, Jose Andre-i Sarabia-Sainz, Erika Silva-Campa, Alexel J. Burgara-Estrella, Ana María Guzmán-Partida, Gabriela Ramos-Clamont Montfort, Martín Pedroza-Montero, and Luz Vazquez-Moreno. 2019. "Lactosylated Albumin Nanoparticles: Potential Drug Nanovehicles with Selective Targeting Toward an In Vitro Model of Hepatocellular Carcinoma" Molecules 24, no. 7: 1382. https://doi.org/10.3390/molecules24071382
APA StyleTeran-Saavedra, N. G., Sarabia-Sainz, J. A.-i., Silva-Campa, E., Burgara-Estrella, A. J., Guzmán-Partida, A. M., Ramos-Clamont Montfort, G., Pedroza-Montero, M., & Vazquez-Moreno, L. (2019). Lactosylated Albumin Nanoparticles: Potential Drug Nanovehicles with Selective Targeting Toward an In Vitro Model of Hepatocellular Carcinoma. Molecules, 24(7), 1382. https://doi.org/10.3390/molecules24071382