
Design, Synthesis, and In Vitro Biological Activities of a Bio-Oxidizable Prodrug to Deliver Both ChEs and DYRK1A Inhibitors for AD Therapy


Abstract
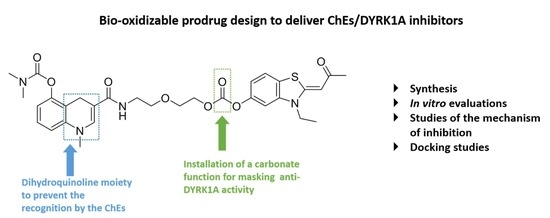
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. In Vitro Activity Evaluation
2.3. Docking
3. Conclusions
4. Experimental Section
4.1. General Information
4.2. Chemistry
4.3. Biological Evaluation
4.3.1. hAChE and hBuChE Inhibition Assays
4.3.2. Propidium Competition Assays
4.3.3. DYRK1A Inhibition Assay
4.3.4. PAMPA-BBB Permeability Assay
4.4. Docking Simulation
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Available online: http://www.who.int (accessed on 8 February 2019).
- Scheltens, P.; Blennow, K.; Breteler, M.M.; De Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef]
- Anderson, R.M.; Hadjichrysanthou, C.; Evans, S.; Wong, M.M. Why do so many clinical trials of therapies for Alzheimer’s disease fail? Lancet 2017, 390, 2327–2329. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, B.; Bernhardt, T.; Moeller, H.J.; Heuser, I.; Frölich, L. Combination therapy in Alzheimer’s disease: A review of current evidence. CNS Drugs 2004, 18, 827–844. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Rosini, M.; Andrisano, V.; Bartolini, M.; Minarini, A.; Tumiatti, V.; Melchiorre, C. MTDL design strategy in the context of Alzheimer’s disease: From lipocrine to memoquin and beyond. Curr. Pharm. Des. 2009, 15, 601–613. [Google Scholar] [CrossRef]
- Ferris, C.F.; Kulkarni, P.; Yee, J.R.; Nedelman, M.; de Jong, I.E.M. The Serotonin Receptor 6 Antagonist Idalopirdine and Acetylcholinesterase Inhibitor Donepezil Have Synergistic Effects on Brain Activity—A Functional MRI Study in the Awake Rat. Front. Pharmacol. 2017, 8, 279. [Google Scholar] [CrossRef] [PubMed]
- Freret, T.; Bouet, V.; Quiedeville, A.; Nee, G.; Dallemagne, P.; Rochais, C.; Boulouard, M. Synergistic effect of acetylcholinesterase inhibition (donepezil) and 5-HT4 receptor activation (RS67333) on object recognition in mice. Behav. Brain Res. 2012, 230, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Green, K.D.; Fosso, M.Y.; Garneau-Tsodikova, S. Multifunctional Donepezil Analogues as Cholinesterase and BACE1 Inhibitors. Molecules 2018, 23, 3252. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.J.; Gong, D.M.; Jiang, Y.R.; Guo, D.; Zhu, Y.; Deng, Y.C. Multipotent AChE and BACE-1 inhibitors for the treatment of Alzheimer’s disease: Design, synthesis and bio-analysis of 7-amino-1,4-dihydro2h-isoquilin-3-one derivates. Eur. J. Med. Chem. 2017, 138, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, P.; Cariati, L.; Desiderio, D.; Sgammato, R.; Lamberti, A.; Arcone, R.; Salerno, R.; Nardi, M.; Masullo, M.; Oliverio, M. Design, synthesis, and evaluation of donepezil-like compounds as AChE and BACE-1 inhibitors. ACS Med. Chem. Lett. 2016, 7, 470–475. [Google Scholar] [CrossRef]
- Bautista-Aguilera, Ó.M.; Hagenow, S.; Palomino-Antolin, A.; Farre-Alins, V.; Ismaili, L.; Joffrin, P.-L.; Jimeno, M.L.; Soukup, O.; Janočková, J.; Kalinowsky, L.; et al. Multitarget-Directed Ligands Combining Cholinesterase and Monoamine Oxidase Inhibition with Histamine H3R Antagonism for Neurodegenerative Diseases. Angew. Chem. Int. Ed. 2017, 56, 12765–12769. [Google Scholar] [CrossRef]
- Sagi, Y.; Drigues, N.; Youdim, M.B. The neurochemical and behavioral effects of the novel cholinesterase-monoamine oxidase inhibitor, ladostigil, in response to L-dopa and L-tryptophan, in rats. Br. J. Pharmacol. 2005, 146, 553–560. [Google Scholar] [CrossRef]
- Lecoutey, C.; Hedou, D.; Freret, T.; Giannoni, P.; Gaven, F.; Since, M.; Bouet, V.; Ballandonne, C.; Corvaisier, S.; Malzert Freon, A.; et al. Design of donecopride, a dual serotonin subtype 4 receptor agonist/acetylcholinesterase inhibitor with potential interest for Alzheimer’s disease treatment. Proc. Natl. Acad. Sci. USA 2014, 111, E3825–E3830. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.; Xu, Y.; Liu, W.; Gong, Q.; Wang, W.; Qiu, X.; Zhu, J.; Mao, F.; Zhang, H.; et al. Novel Vilazodone-Tacrine Hybrids as Potential Multitarget-Directed Ligands for the Treatment of Alzheimer’s Disease Accompanied with Depression: Design, Synthesis, and Biological Evaluation. ACS Chem. Neurosci. 2017, 8, 2708–2721. [Google Scholar] [CrossRef]
- Więckowska, A.; Kołaczkowski, M.; Bucki, A.; Godyń, J.; Marcinkowska, M.; Więckowski, K.; Paula Zaręba, P.; Siwek, A.; Kazek, G.; Głuch-Lutwin, M.; et al. Novel multi-target-directed ligands for Alzheimer’s disease: Combining cholinesterase inhibitors and 5-HT 6 receptor antagonists. Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2016, 124, 63–81. [Google Scholar] [CrossRef]
- Jiang, X.Y.; Chen, T.-K.; Zhou, J.-T.; He, S.-Y.; Yang, H.-Y.; Chen, Y.; Qu, W.; Feng, F.; Sun, H.-P. Dual GSK-3β/AChE Inhibitors as a New Strategy for Multitargeting Anti-Alzheimer’s Disease Drug Discovery. ACS Med. Chem. Lett. 2018, 9, 171–176. [Google Scholar] [CrossRef]
- Oukoloff, K.; Coquelle, N.; Bartolini, M.; Naldi, M.; Le Guevel, R.; Bach, S.; Josselin, B.; Ruchaud, S.; Catto, M.; Pisani, L.; et al. Design, biological evaluation and Xray crystallography of nanomolar multifunctional ligands targeting simultaneously acetylcholinesterase and glycogen synthase kinase-3. Eur. J. Med. Chem. 2019, 106, 553–565. [Google Scholar] [CrossRef]
- Agatonovic-Kustrin, S.; Kettle, C.; Morton, D.W. A molecular approach in drug development for Alzheimer’s disease. Biomed. Pharmacother. 2018, 106, 553–565. [Google Scholar] [CrossRef]
- Agis-Torres, A.; Sollhuber, M.; Fernandez, M.; Sanchez-Montero, J.M. Multi-Target-Directed Ligands and other Therapeutic Strategies in the Search of a Real Solution for Alzheimer’s Disease. Curr. Neuropharmacol. 2014, 12, 2–36. [Google Scholar] [CrossRef]
- Kochi, A.; Eckroat, T.J.; Green, K.D.; Mayhoub, A.S.; Lim, M.H.; Garneau-Tsodikova, S. A novel hybrid of 6-chlorotacrine and metal-amyloid-β modulator for inhibition of acetylcholine and metal-induced amyloid-β aggregation. Chem. Sci. 2013, 4, 4137–4145. [Google Scholar] [CrossRef]
- Lalut, J.; Santoni, G.; Karila, D.; Lecoutey, C.; Davis, A.; Nachon, F.; Silman, I.; Sussman, J.; Weik, M.; Maurice, T.; et al. Novel multitarget-directed ligands targeting acetylcholinesterase and σ1 receptors as lead compounds for treatment of Alzheimer’s disease: Synthesis, evaluation, and structural characterization of their complexes with acetylcholinesterase. Eur. J. Med. Chem. 2019, 162, 234–248. [Google Scholar] [CrossRef]
- Estrada Valencia, M.; Herrera-Arozamena, C.; de Andres, L.; Perez, C.; Morales-Garcia, J.A.; Perez-Castillo, A.; Ramos, E.; Romero, A.; Vina, D.; Yanez, M.; et al. Neurogenic and neuroprotective donepezil-flavonoid hybrids with sigma-1 affinity and inhibition of key enzymes in Alzheimer’s disease. Eur. J. Med. Chem. 2018, 156, 534–553. [Google Scholar] [CrossRef]
- Mao, F.; Wang, H.; Ni, W.; Zheng, X.; Wang, M.; Bao, K.; Ling, D.; Li, X.; Xu, Y.; Zhang, H.; et al. Design, Synthesis, and Biological Evaluation of Orally Available First-Generation Dual-Target Selective Inhibitors of Acetylcholinesterase (AChE) and Phosphodiesterase 5 (PDE5) for the Treatment of Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 328–345. [Google Scholar] [CrossRef]
- Branca, C.; Shaw, D.M.; Belfiore, R.; Gokhale, V.; Shaw, A.Y.; Foley, C.; Smith, B.; Hulme, C.; Dunckley, T.; Meechoovet, B.; et al. Dyrk1 inhibition improves Alzheimer’s disease-like pathology. Aging Cell. 2017, 16, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Medda, F.; Gokhale, V.; Dunckley, T.; Hulme, C. Recent advances in the design, synthesis, and biological evaluation of selective DYRK1A inhibitors: A new avenue for a disease modifying treatment of Alzheimer’s? ACS Chem. Neurosci. 2012, 3, 857–872. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Fruit, C.; Hérault, Y.; Meijer, L.; Besson, T. Dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) inhibitors: A survey of recent patent literature. Expert Opin. Ther. Pat. 2017, 27, 1183–1199. [Google Scholar] [CrossRef]
- Stotani, S.; Giordanetto, F.; Medda, F. DYRK1A inhibition as potential treatment for Alzheimer’s disease. Future Med. Chem. 2016, 8, 681–696. [Google Scholar] [CrossRef]
- Jarhad, D.B.; Mashelkar, K.K.; Kim, H.-R.; Noh, M.; Jeong, L.S. Dual-Specificity Tyrosine Phosphorylation-Regulated Kinase 1A (DYRK1A) Inhibitors as Potential Therapeutics. J. Med. Chem. 2018, 61, 9791–9810. [Google Scholar] [CrossRef]
- Pathak, A.; Rohilla, A.; Gupta, T.; Akhtar, M.J.; Haider, M.R.; Sharma, K.; Haider, K.; ShaharYar, M. DYRK1A kinase inhibition with emphasis on neurodegeneration: A comprehensive evolution story-cum-perspective. Eur. J. Med. Chem. 2018, 158, 559–592. [Google Scholar] [CrossRef]
- Ferrer, I.; Barrachina, M.; Puig, B.; Martinez de Lagran, M.; Marti, E.; Avila, J.; Dierssen, M. Constitutive Dyrk1A is abnormally expressed in Alzheimer disease, Down syndrome, Pick disease, and related transgenic models. Neurobiol. Dis. 2005, 20, 392–400. [Google Scholar] [CrossRef]
- Azorsa, D.O.; Robeson, R.H.; Frost, D.; Meec hoovet, B.; Brautigam, G.R.; Dickey, C.; Beaudry, C.; Basu, G.D.; Holz, D.R.; Hernandez, J.A.; et al. High-content siRNA screening of the kinome identifies kinases involved in Alzheimer’s disease-related tau hyperphosphorylation. BMC Genom. 2010, 11, 25. [Google Scholar] [CrossRef]
- Coutadeur, S.; Benyamine, H.; Delalonde, L.; de Oliveira, C.; Leblond, B.; Foucourt, A.; Besson, T.; Casagrande, A.S.; Taverne, T.; Girard, A.; et al. A novel DYRK1A (dual specificity tyrosine phosphorylation-regulated kinase 1A) inhibitor for the treatment of Alzheimer’s disease: Effect on Tau and amyloid pathologies in vitro. J. Neurochem. 2015, 133, 440–451. [Google Scholar] [CrossRef]
- Kimura, R.; Kamino, K.; Yamamoto, M.; Nuripa, A.; Kida, T.; Kazui, H.; Hashimoto, R.; Tanaka, T.; Kudo, T.; Yamagata, H.; et al. The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between beta-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 2007, 16, 15–23. [Google Scholar] [CrossRef]
- Marsais, F.; Bohn, P.; Levacher, V.; Le Fur, N. Preparation of Dihydro Pyridines, Quinolines, and Isoquinolines as Anti-Alzheimer Agents. PCT International Applications WO 2006103120, 5 October 2006. [Google Scholar]
- Marsais, F.; Levacher, V.; Papamicael, C.; Bohn, P.; Peauger, L.; Gembus, V.; Le Fur, N.; Dumartin-Lepine, M.-L. Oxidisable pyridine derivatives, their preparation and use as anti-alzheimer agents. PCT International Applications WO 2014114742, 31 July 2014. [Google Scholar]
- Bohn, P.; Le Fur, N.; Hagues, G.; Costentin, J.; Torquet, N.; Papamicael, C.; Marsais, F.; Levacher, V. Rational design of central selective acetylcholinesterase inhibitors by means of a “bio-oxidisable prodrug” strategy. Org. Biomol. Chem. 2009, 7, 2612–2618. [Google Scholar] [CrossRef]
- Peauger, L.; Azzouz, R.; Gembus, V.; Tintas, M.-L.; Sopkova-de Oliveira Santos, J.; Bohn, P.; Papamicael, C.; Levacher, V. Donepezil-based central acetylcholinesterase inhibitors by means of a “bio-oxidizable” prodrug strategy: Design, synthesis, and in vitro biological evaluation. J. Med. Chem. 2017, 60, 5909–5926. [Google Scholar] [CrossRef]
- Azzouz, R.; Peauger, L.; Gembus, V.; Tintas, M.-L.; Sopkova-de Oliveira Santos, J.; Papamicael, C.; Levacher, V. Novel donepezil-like N-benzylpyridinium salt derivatives as AChE inhibitors and their corresponding dihydropyridine “bio-oxidizable” prodrugs: Synthesis, biological evaluation and structure-activity relationship. Eur. J. Med. Chem. 2018, 145, 165–190. [Google Scholar] [CrossRef]
- Tintas, M.-L.; Gembus, V.; Alix, F.; Barre, A.; Coadou, G.; Truong, L.; Sebban, M.; Papamicael, C.; Oulyadi, H.; Levacher, V. Rational design of carbamate-based dual binding site and central AChE inhibitors by a “biooxidisable” prodrug approach: Synthesis, in vitro evaluation and docking studies. Eur. J. Med. Chem. 2018, 155, 171–182. [Google Scholar] [CrossRef]
- Alix, F.; Gembus, V.; Coquet, L.; Hubert-Roux, M.; Chan, P.; Truong, L.; Sebban, M.; Coadou, G.; Oulyadi, H.; Papamicael, C.; et al. Dihydroquinoline Carbamate DQS1-02 as a Prodrug of a Potent Acetylcholinesterase Inhibitor for Alzheimer’s Disease Therapy: Multigram-Scale Synthesis, Mechanism Investigations, in Vitro Safety Pharmacology, and Preliminary in Vivo Toxicology Profile. ACS Omega 2018, 3, 18387–18397. [Google Scholar] [CrossRef]
- Ogawa, Y.; Nonaka, Y.; Goto, T.; Ohnishi, E.; Hiramatsu, T.; Kii, I.; Yoshida, M.; Ikura, T.; Onogi, H.; Shibuya, H.; et al. Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1A. Nat. Commun. 2010, 1, 86. [Google Scholar] [CrossRef]
- Muraki, M.; Ohkawara, B.; Hosoya, T.; Onogi, H.; Koizumi, J.; Koizumi, T.; Sumi, K.; Yomoda, J.; Murray, M.V.; Kimura, H.; et al. Manipulation of Alternative Splicing by a Newly Developed Inhibitor of Clks. J. Biol. Chem. 2004, 279, 24246–24254. [Google Scholar] [CrossRef] [PubMed]
- Barre, A.; Tintas, M.-L.; Alix, F.; Gembus, V.; Papamicael, C.; Levacher, V. Palladium-Catalyzed Carbonylation of (Hetero)Aryl, Alkenyl and Allyl Halides by Means of N-Hydroxysuccinimidyl Formate as CO Surrogate. J. Org. Chem. 2015, 80, 6537–6544. [Google Scholar] [CrossRef] [PubMed]
- Greig, N.H.; Utsuki, T.; Ingram, D.K.; Wang, Y.; Pepeu, G.; Scali, C.; Yu, Q.S.; Mamczarz, J.; Holloway, H.W.; Giordano, T.; et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer beta-amyloid peptide in rodent. Proc. Natl. Acad. Sci. USA 2005, 102, 17213–17218. [Google Scholar] [CrossRef]
- Nordberg, A.; Ballard, C.; Bullock, R.; Darreh-Shori, T.; Somogyi, M. A Review of Butyrylcholinesterase as a Therapeutic Target in the Treatment of Alzheimer’s Disease. Prim. Care Companion CNS Disord. 2013, 15. [Google Scholar] [CrossRef]
- Johnson, G.; Moore, S.W. The peripheral anionic site of acetylcholinesterase: Structure, functions and potential role in rational drug design. Curr. Pharm. Des. 2006, 12, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Hosie, L.; Sutton, L.D.; Quinn, D.M. p-Nitrophenyl and Cholesteryl-N-Alkyl Carbamates as Inhibitors of Cholesterol Esterase. J. Biol. Chem. 1987, 262, 260–264, CODEN: JBCHA3. [Google Scholar] [PubMed]
- Feaster, S.R.; Lee, K.; Baker, N.; Hui, D.Y.; Quinn, D.M. Molecular Recognition by Cholesterol Esterase of Active Site Ligands: Structure-Reactivity Effects for Inhibition by Aryl Carbamates and Subsequent Carbamyl enzyme Turnover. Biochemistry 1996, 35, 16723–16734. [Google Scholar] [CrossRef] [PubMed]
- GraphPad Prism Version 8.0.1; GraphPad Software: San Diego, CA, USA, 2018.
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- PyMol 0.99rc6; DeLano Scientific: San Carlo, CA, USA, 2006.
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Marvin Sketch 17.29.0. ChemAxon Ltd. 1998–2017. Available online: http://www.chemaxon.com (accessed on 31 March 2019).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A Programming Language for Software Integration and Development. J. Mol. Graph. Mod. 1999, 17, 57–61. [Google Scholar]
Sample Availability: Not available. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 hAChE (nM) 1 | IC50 hBuChE (nM) 1 | DYRK1a Inhibition (%) 1,2 | Propidium Iodide Displacement Inhibition (%) 3 |
|---|---|---|---|---|
| 2a2 | 37.1 ± 0.1 | 1440.2 ± 286 | 0 | - |
| 14 | 81.4 ± 0.4 | 482 ± 21 | 5 | 26 ± 1 |
| 15 | >10,000 | 6400 ± 714 | 6 | - |
| 4 | 1023.1 ± 72.1 | 961 ± 22 | 8 | 4 ± 2 |
| 16 | >10,000 | >10,000 | 7 | - |
| Indy | >10,000 | >10,000 | 92 | - |
| Donepezil | 15.3 ± 1.7 | - | - | 18 ± 1 |
| Tacrine | - | 17.1 ± 0.1 | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barré, A.; Azzouz, R.; Gembus, V.; Papamicaël, C.; Levacher, V. Design, Synthesis, and In Vitro Biological Activities of a Bio-Oxidizable Prodrug to Deliver Both ChEs and DYRK1A Inhibitors for AD Therapy. Molecules 2019, 24, 1264. https://doi.org/10.3390/molecules24071264
Barré A, Azzouz R, Gembus V, Papamicaël C, Levacher V. Design, Synthesis, and In Vitro Biological Activities of a Bio-Oxidizable Prodrug to Deliver Both ChEs and DYRK1A Inhibitors for AD Therapy. Molecules. 2019; 24(7):1264. https://doi.org/10.3390/molecules24071264
Chicago/Turabian StyleBarré, Anaïs, Rabah Azzouz, Vincent Gembus, Cyril Papamicaël, and Vincent Levacher. 2019. "Design, Synthesis, and In Vitro Biological Activities of a Bio-Oxidizable Prodrug to Deliver Both ChEs and DYRK1A Inhibitors for AD Therapy" Molecules 24, no. 7: 1264. https://doi.org/10.3390/molecules24071264
APA StyleBarré, A., Azzouz, R., Gembus, V., Papamicaël, C., & Levacher, V. (2019). Design, Synthesis, and In Vitro Biological Activities of a Bio-Oxidizable Prodrug to Deliver Both ChEs and DYRK1A Inhibitors for AD Therapy. Molecules, 24(7), 1264. https://doi.org/10.3390/molecules24071264