
Mapping Magnetic Properties and Relaxation in Vanadium(IV) Complexes with Lanthanides by Electron Paramagnetic Resonance

 , , ,
, , ,  ,
,  , and
, and
Abstract
:1. Introduction
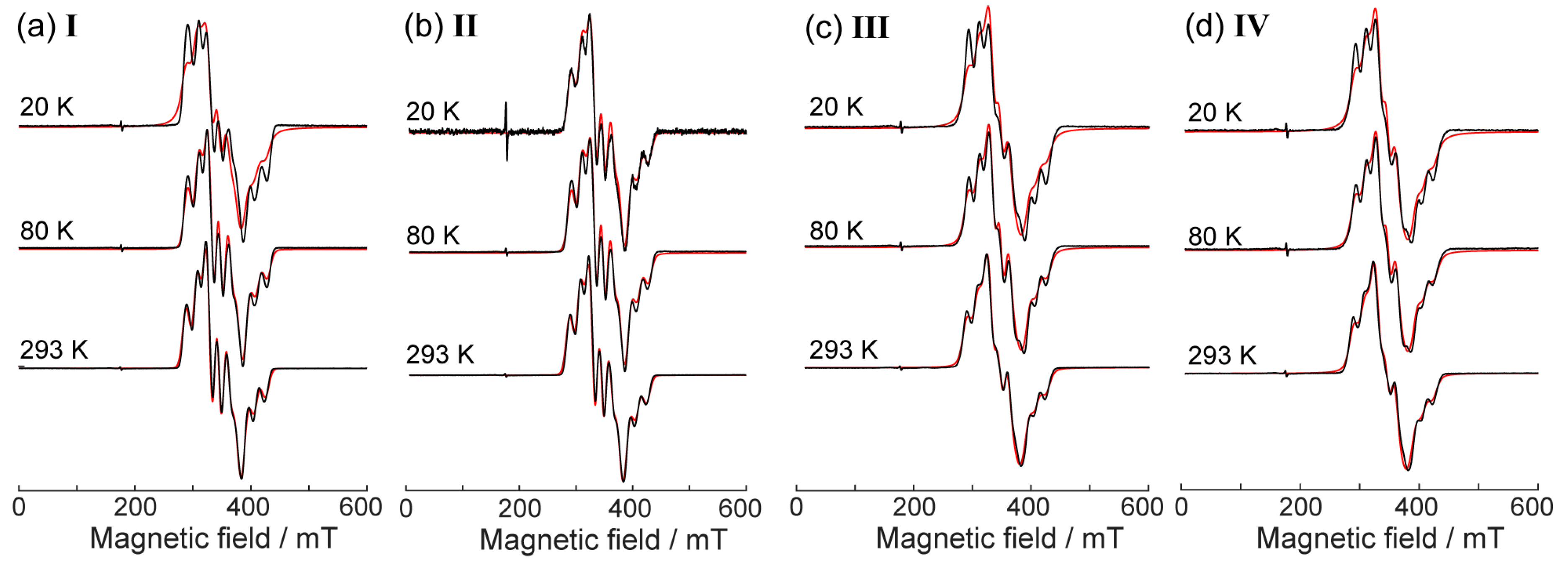
2. Results and Discussion
2.1. Synthesis
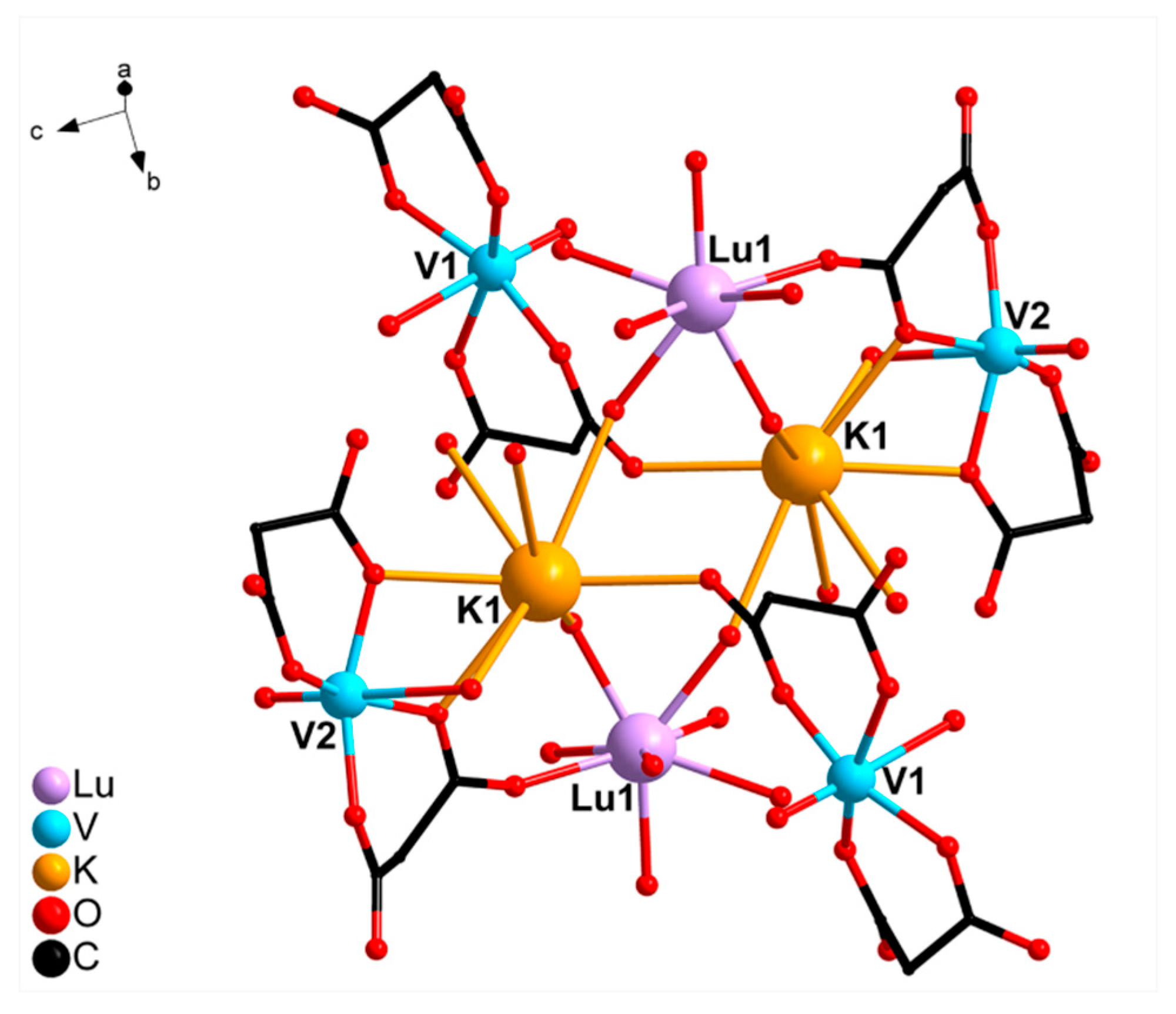
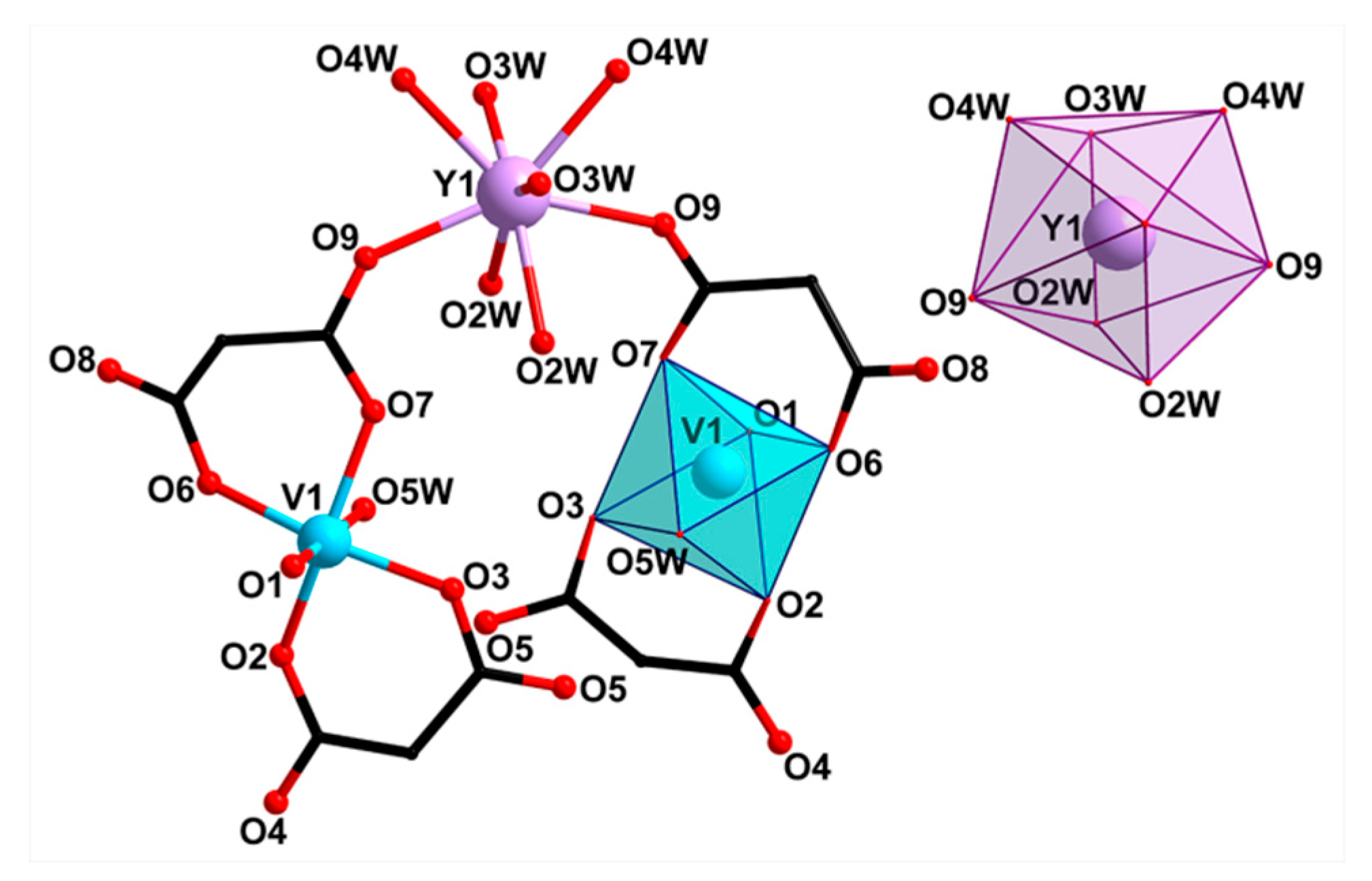
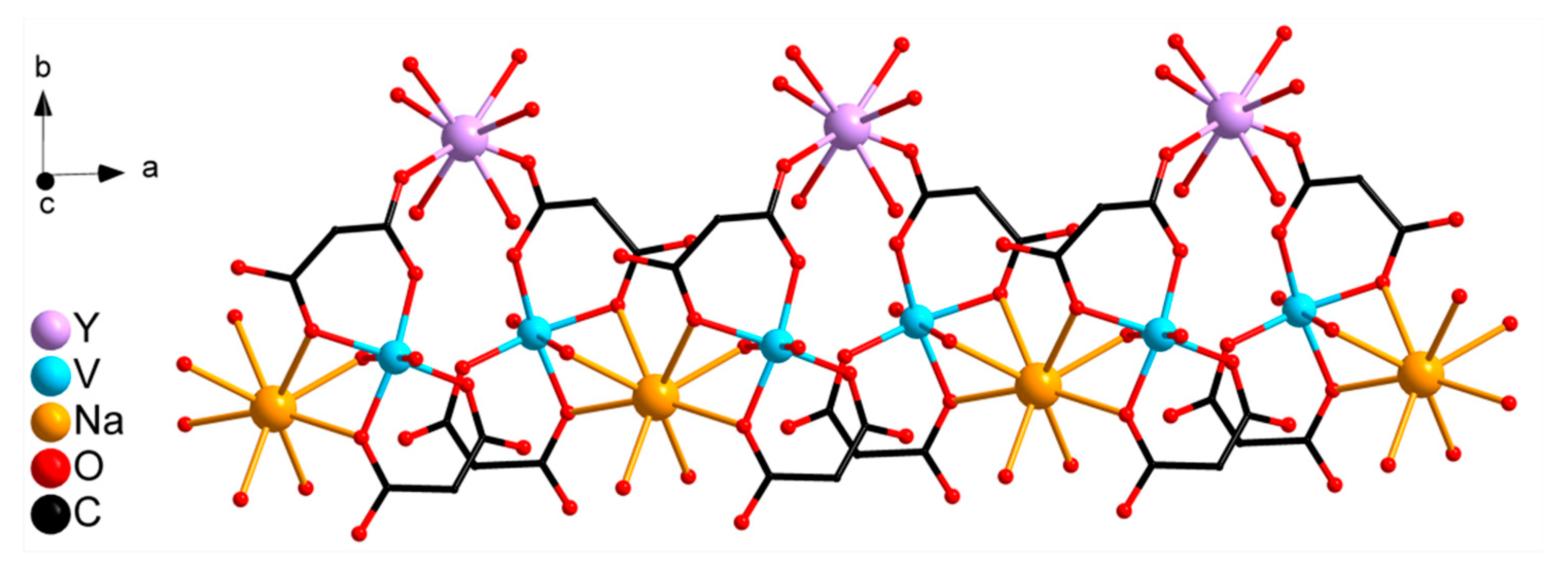
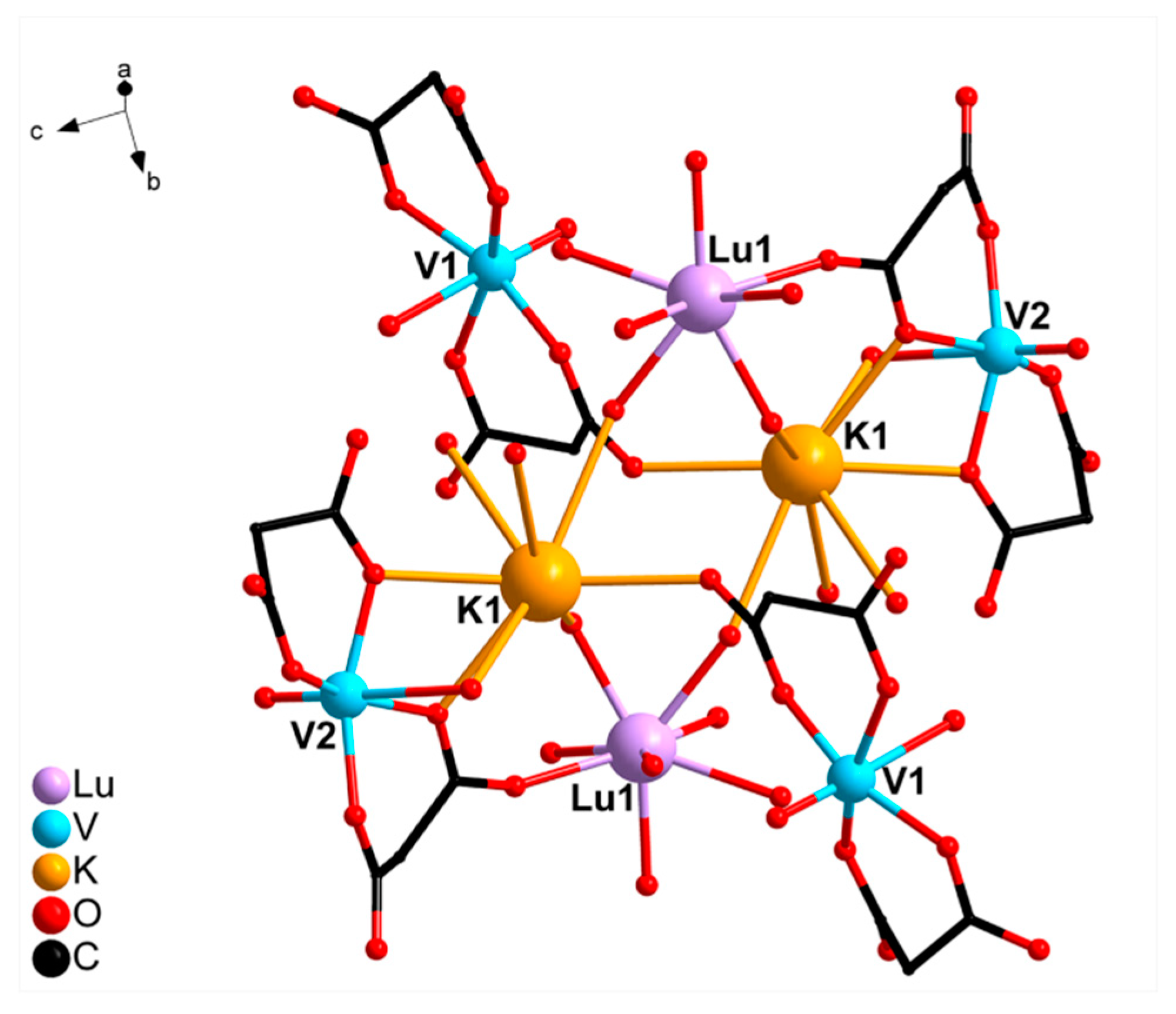
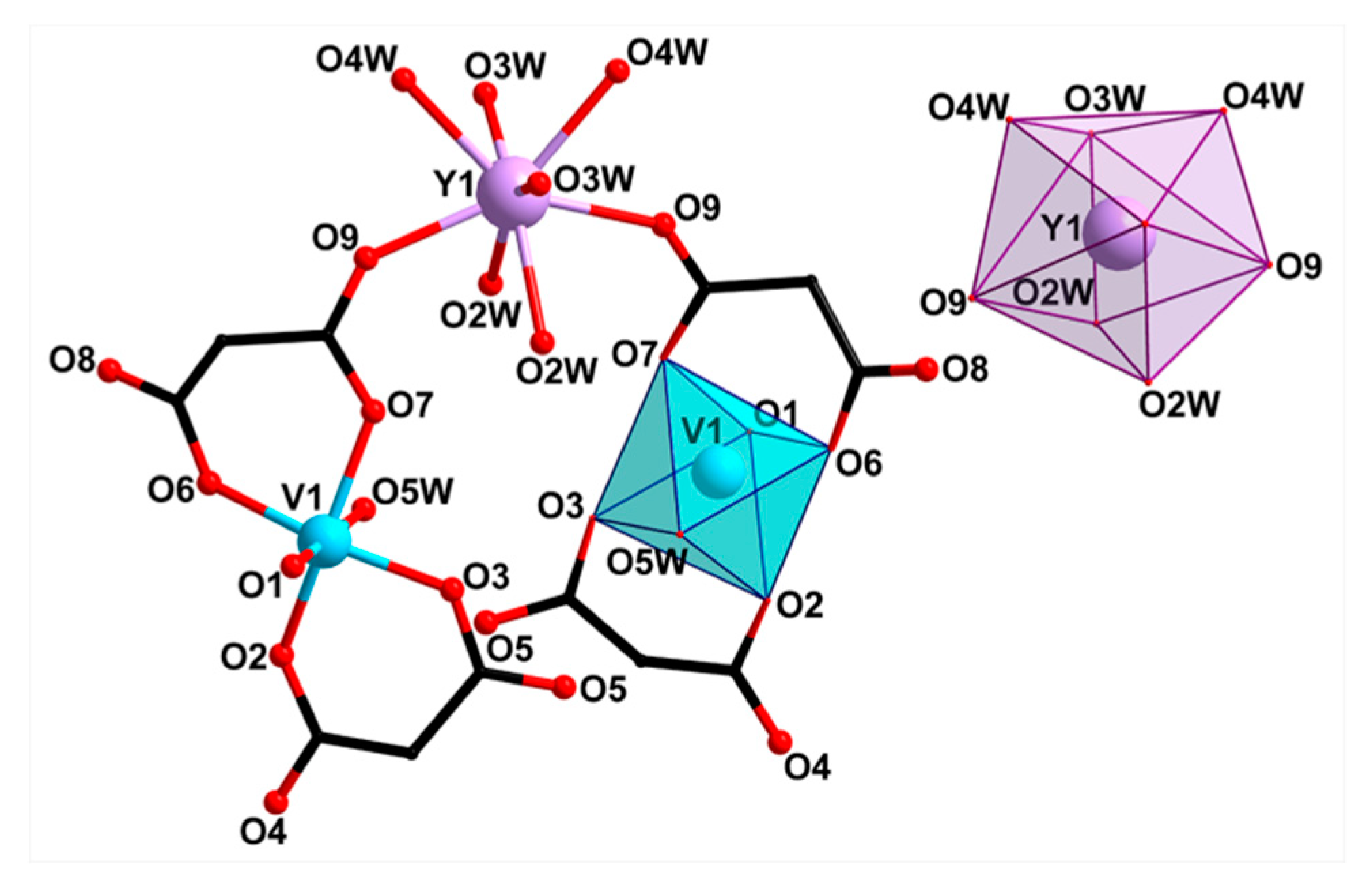
2.2. Crystal Structure Description
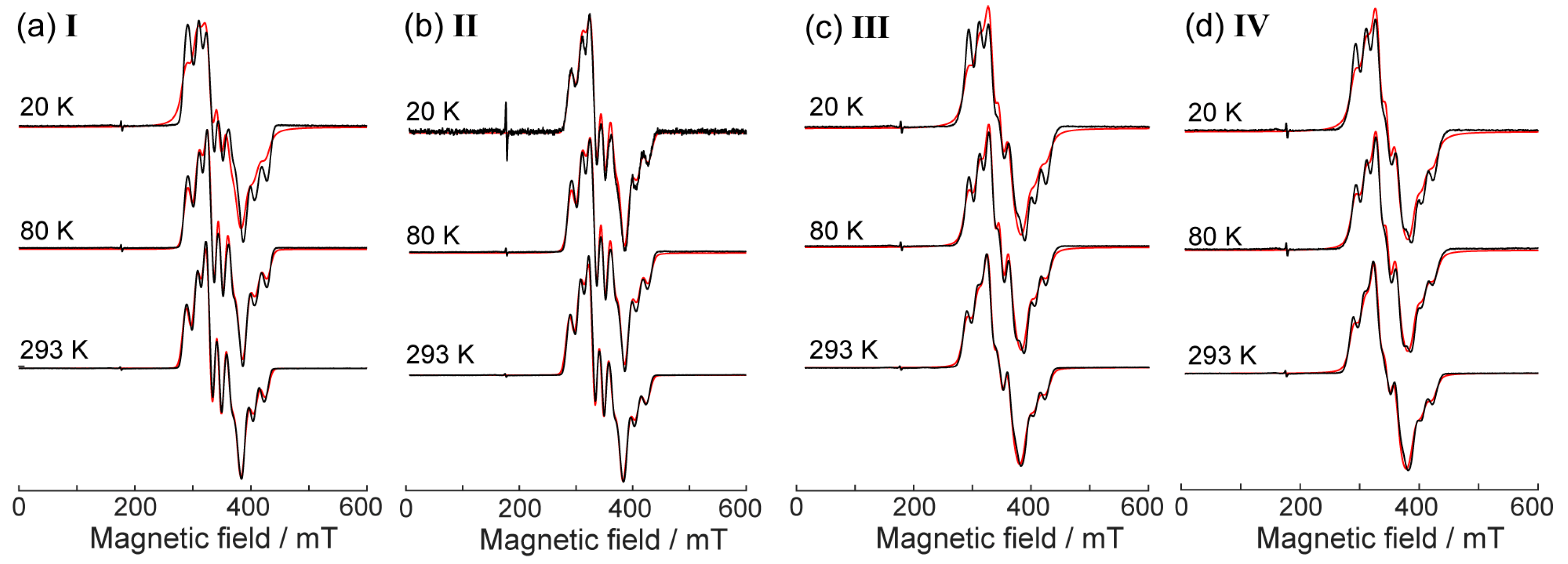
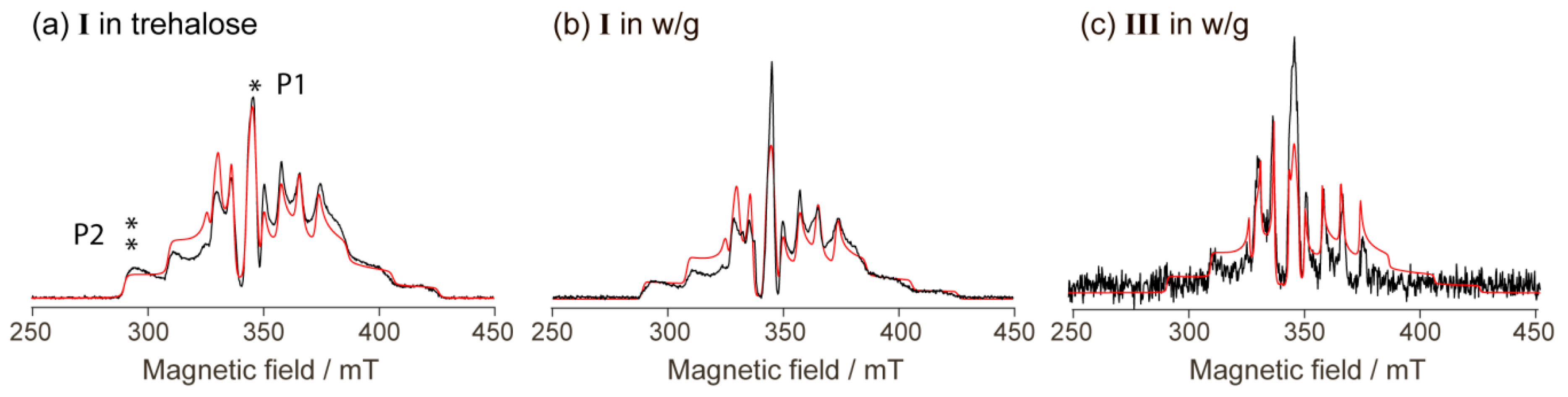
2.3. CW EPR Study of Solid Phases
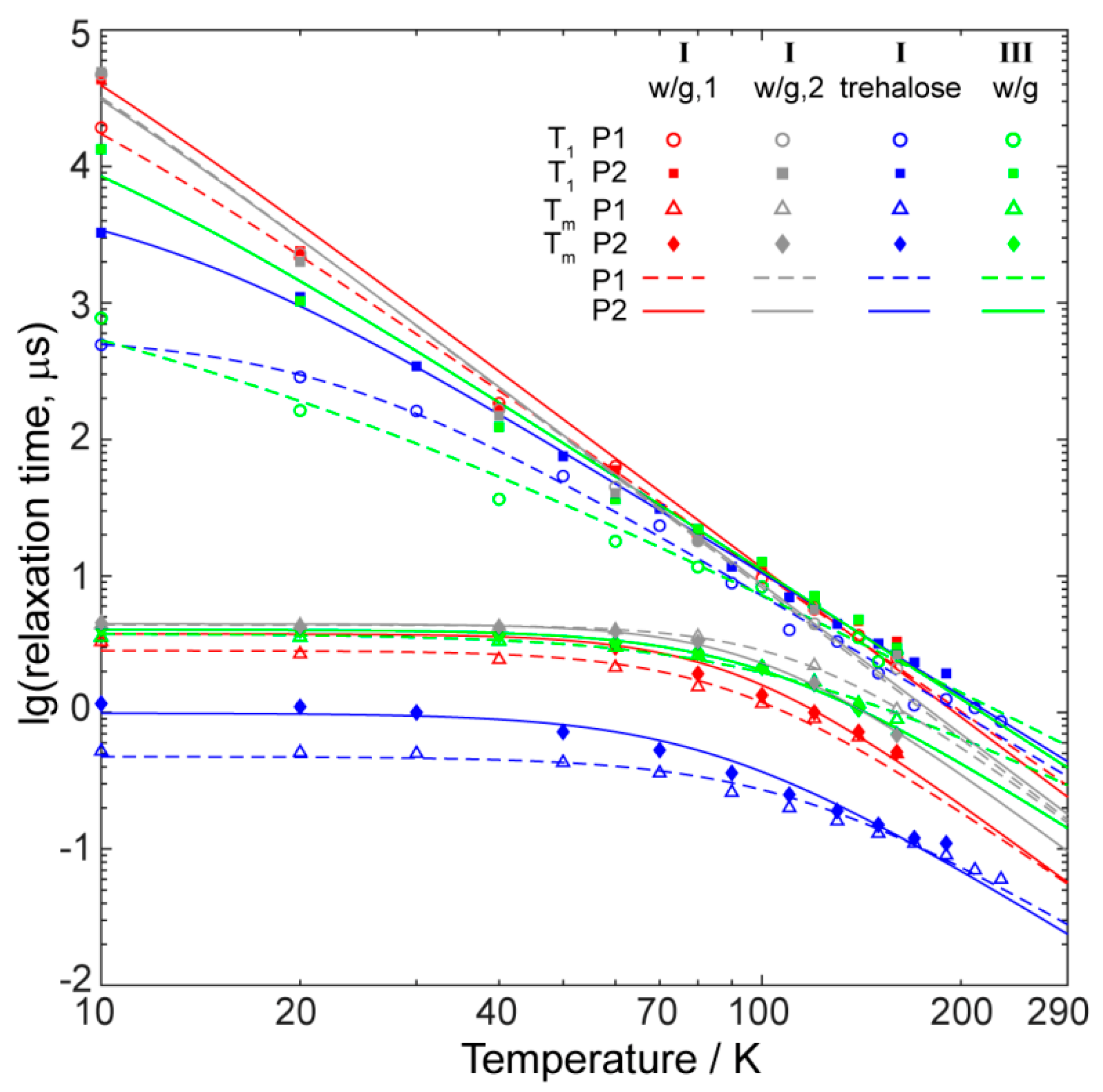
2.4. Relaxation Properties of Complexes in Frozen Solutions
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Troiani, F.; Affronte, M. Molecular spins for quantum information technologies. Chem. Soc. Rev. 2011, 40, 3119–3129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aromi, G.; Aguila, D.; Gamez, P.; Luis, F.; Roubeau, O. Design of magnetic coordination complexes for quantum computing. Chem. Soc. Rev. 2012, 41, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Zadrozny, J.M.; Niklas, J.; Poluektov, O.G.; Freedman, D.E. Multiple quantum coherences from hyperfine transitions in a vanadium(IV) complex. J. Am. Chem. Soc. 2014, 136, 15841–15844. [Google Scholar] [CrossRef] [PubMed]
- Zadrozny, J.M.; Niklas, J.; Poluektov, O.G.; Freedman, D.E. Millisecond Coherence Time in a Tunable Molecular Electronic Spin Qubit. ACS Cent. Sci. 2015, 1, 488–492. [Google Scholar] [CrossRef]
- Atzori, M.; Tesi, L.; Morra, E.; Chiesa, M.; Sorace, L.; Sessoli, R. Room-Temperature Quantum Coherence and Rabi Oscillations in Vanadyl Phthalocyanine: Toward Multifunctional Molecular Spin Qubits. J. Am. Chem. Soc. 2016, 138, 2154–2157. [Google Scholar] [CrossRef]
- Atzori, M.; Morra, E.; Tesi, L.; Albino, A.; Chiesa, M.; Sorace, L.; Sessoli, R. Quantum Coherence Times Enhancement in Vanadium(IV)-based Potential Molecular Qubits: The Key Role of the Vanadyl Moiety. J. Am. Chem. Soc. 2016, 138, 11234–11244. [Google Scholar] [CrossRef]
- Yu, C.J.; Graham, M.J.; Zadrozny, J.M.; Niklas, J.; Krzyaniak, M.D.; Wasielewski, M.R.; Poluektov, O.G.; Freedman, D.E. Long Coherence Times in Nuclear Spin-Free Vanadyl Qubits. J. Am. Chem. Soc. 2016, 138, 14678–14685. [Google Scholar] [CrossRef]
- Atzori, M.; Tesi, L.; Benci, S.; Lunghi, A.; Righini, R.; Taschin, A.; Torre, R.; Sorace, L.; Sessoli, R. Spin Dynamics and Low Energy Vibrations: Insights from Vanadyl-Based Potential Molecular Qubits. J. Am. Chem. Soc. 2017, 139, 4338–4341. [Google Scholar] [CrossRef] [Green Version]
- Graham, M.J.; Krzyaniak, M.D.; Wasielewski, M.R.; Freedman, D.E. Probing Nuclear Spin Effects on Electronic Spin Coherence via EPR Measurements of Vanadium(IV) Complexes. Inorg. Chem. 2017, 56, 8106–8113. [Google Scholar] [CrossRef]
- Atzori, M.; Chiesa, A.; Morra, E.; Chiesa, M.; Sorace, L.; Carretta, S.; Sessoli, R. A two-qubit molecular architecture for electron mediated nuclear quantum simulation. Chem. Sci. 2018, 9, 6183–6192. [Google Scholar] [CrossRef] [Green Version]
- Yamabayashi, T.; Atzori, M.; Tesi, L.; Cosquer, G.; Santanni, F.; Boulon, M.E.; Morra, E.; Benci, S.; Torre, R.; Chiesa, M.; et al. Scaling Up Electronic Spin Qubits into a Three-Dimensional Metal Organic Framework. J. Am. Chem. Soc. 2018, 140, 12090–12101. [Google Scholar] [CrossRef] [PubMed]
- Atzori, M.; Sessoli, R. The Second Quantum Revolution: Role and Challenges of Molecular Chemistry. J. Am. Chem. Soc. 2019, 141, 11339–11352. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Ngendahimana, T.; Eaton, G.R.; Eaton, S.S.; Zadrozny, J.M. Counterion influence on dynamic spin properties in a V(IV) complex. Chem. Sci. 2019, 10, 548–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, C.E.; Lin, C.-Y.; Johnson, S.H.; van Tol, J.; Zadrozny, J.M. Nuclear-spin-pattern control of electron-spin dynamics in a series of V(IV) complexes. Chem. Sci. 2019, 10, 8447–8454. [Google Scholar] [CrossRef]
- Fataftah, M.S.; Krzyaniak, M.D.; Vlaisavljevich, B.; Wasielewski, M.R.; Zadrozny, J.M.; Freedman, D.E. Metal-ligand covalency enables room temperature molecular qubit candidates. Chem. Sci. 2019, 10, 6707–6714. [Google Scholar] [CrossRef] [Green Version]
- Shevelev, G.Y.; Krumkacheva, O.A.; Lomzov, A.A.; Kuzhelev, A.A.; Rogozhnikova, O.Y.; Trukhin, D.V.; Troitskaya, T.I.; Tormyshev, V.M.; Fedin, M.V.; Pyshnyi, D.V.; et al. Physiological-Temperature Distance Measurement in Nucleic Acid Using Triarylmethyl-Based Spin Labels and Pulsed Dipolar EPR Spectroscopy. J. Am. Chem. Soc. 2014, 136, 9874–9877. [Google Scholar] [CrossRef]
- Kuzhelev, A.A.; Shevelev, G.Y.; Krumkacheva, O.A.; Tormyshev, V.M.; Pyshnyi, D.V.; Fedin, M.V.; Bagryanskaya, E.G. Saccharides as Prospective Immobilizers of Nucleic Acids for Room-Temperature Structural EPR Studies. J. Phys. Chem. Lett. 2016, 7, 2544–2548. [Google Scholar] [CrossRef] [Green Version]
- Krumkacheva, O.; Bagryanskaya, E. EPR-based distance measurements at ambient temperature. J. Magn. Reson. 2017, 280, 117–126. [Google Scholar] [CrossRef]
- Bazhina, E.S.; Aleksandrov, G.G.; Kiskin, M.A.; Korlyukov, A.A.; Efimov, N.N.; Bogomyakov, A.S.; Starikova, A.A.; Mironov, V.S.; Ugolkova, E.A.; Minin, V.V.; et al. The First Series of Heterometallic LnIII-VIV Complexes Based on Substituted Malonic Acid Anions: Synthesis, Structure and Magnetic Properties. Eur. J. Inorg. Chem. 2018, 5075–5090. [Google Scholar] [CrossRef]
- Casanova, D.; Llunell, M.; Alemany, P.; Alvarez, S. The rich stereochemistry of eight-vertex polyhedra: A continuous shape measures study. Chem. Eur. J. 2005, 11, 1479–1494. [Google Scholar] [CrossRef]
- Bernhardt, E.; Willner, H.; Kornath, A.; Breidung, J.; Bühl, M.; Jonas, V.; Thiel, W. D3d Ground-State Structure of V(CO)6: A Combined Matrix Isolation and ab Initio Study of the Jahn-Teller Effect. J. Phys. Chem. A 2003, 107, 859–868. [Google Scholar] [CrossRef]
- Rubinson, K.A. The Electron Paramagnetic Resonance and Optical Spectra of Vanadium Hexacarbonyl. J. Am. Chem. Soc. 1976, 98, 5188–5191. [Google Scholar] [CrossRef]
- Bratt, S.W.; Kassyk, A.; Perutz, R.N.; Symons, M.C.R. Electron Paramagnetic Resonance Spectra and Molecular Structure of Vanadium Hexacarbonyl. J. Am. Chem. Soc. 1982, 104, 490–494. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- SMART (Control) and SAINT (Integration) Software, Version 5.0; Bruker AXS, Inc.: Madison, WI, USA, 1997.
- Sheldrick, G.M. SADABS, Program for Scaling and Correction of Area Detector Data; Göttingen University: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXT – Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H.J. OLEX2: A complete structure solution, refinement and analysis program. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
Sample Availability: Not available. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | [g⊥; g‖] | [A⊥; A‖]/MHz |
|---|---|---|
| I | [1.979; 1.938] | [187; 529] |
| II | [1.978; 1.937] | [189; 525] |
| III | [1.977; 1.941] | [157; 520] |
| IV | [1.977; 1.943] | [151; 512] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurganskii, I.V.; Bazhina, E.S.; Korlyukov, A.A.; Babeshkin, K.A.; Efimov, N.N.; Kiskin, M.A.; Veber, S.L.; Sidorov, A.A.; Eremenko, I.L.; Fedin, M.V. Mapping Magnetic Properties and Relaxation in Vanadium(IV) Complexes with Lanthanides by Electron Paramagnetic Resonance. Molecules 2019, 24, 4582. https://doi.org/10.3390/molecules24244582
Kurganskii IV, Bazhina ES, Korlyukov AA, Babeshkin KA, Efimov NN, Kiskin MA, Veber SL, Sidorov AA, Eremenko IL, Fedin MV. Mapping Magnetic Properties and Relaxation in Vanadium(IV) Complexes with Lanthanides by Electron Paramagnetic Resonance. Molecules. 2019; 24(24):4582. https://doi.org/10.3390/molecules24244582
Chicago/Turabian StyleKurganskii, Ivan V., Evgeniya S. Bazhina, Alexander A. Korlyukov, Konstantin A. Babeshkin, Nikolay N. Efimov, Mikhail A. Kiskin, Sergey L. Veber, Alexey A. Sidorov, Igor L. Eremenko, and Matvey V. Fedin. 2019. "Mapping Magnetic Properties and Relaxation in Vanadium(IV) Complexes with Lanthanides by Electron Paramagnetic Resonance" Molecules 24, no. 24: 4582. https://doi.org/10.3390/molecules24244582
APA StyleKurganskii, I. V., Bazhina, E. S., Korlyukov, A. A., Babeshkin, K. A., Efimov, N. N., Kiskin, M. A., Veber, S. L., Sidorov, A. A., Eremenko, I. L., & Fedin, M. V. (2019). Mapping Magnetic Properties and Relaxation in Vanadium(IV) Complexes with Lanthanides by Electron Paramagnetic Resonance. Molecules, 24(24), 4582. https://doi.org/10.3390/molecules24244582