
Methyltransferase Inhibitors: Competing with, or Exploiting the Bound Cofactor


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
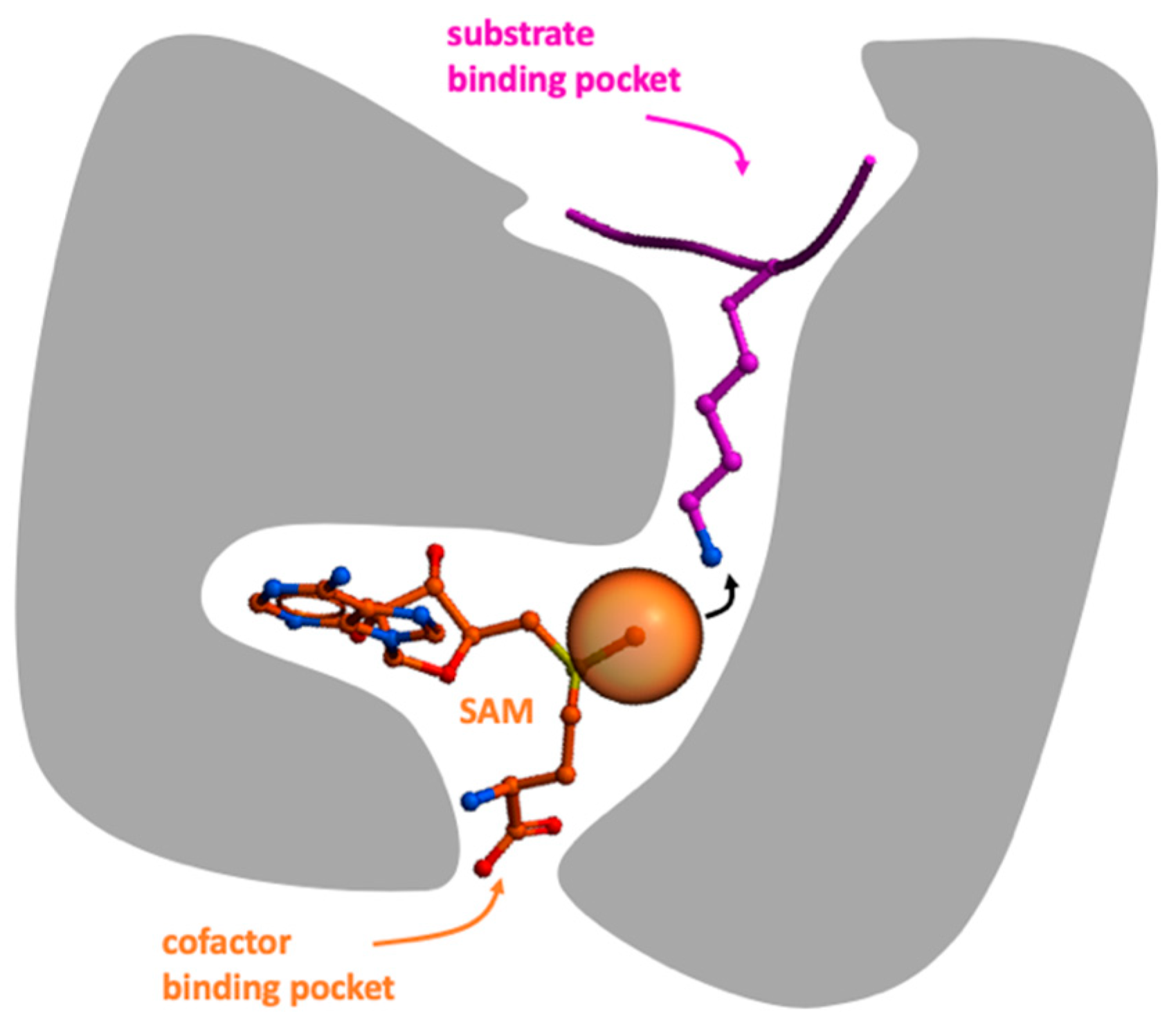
1. Introduction
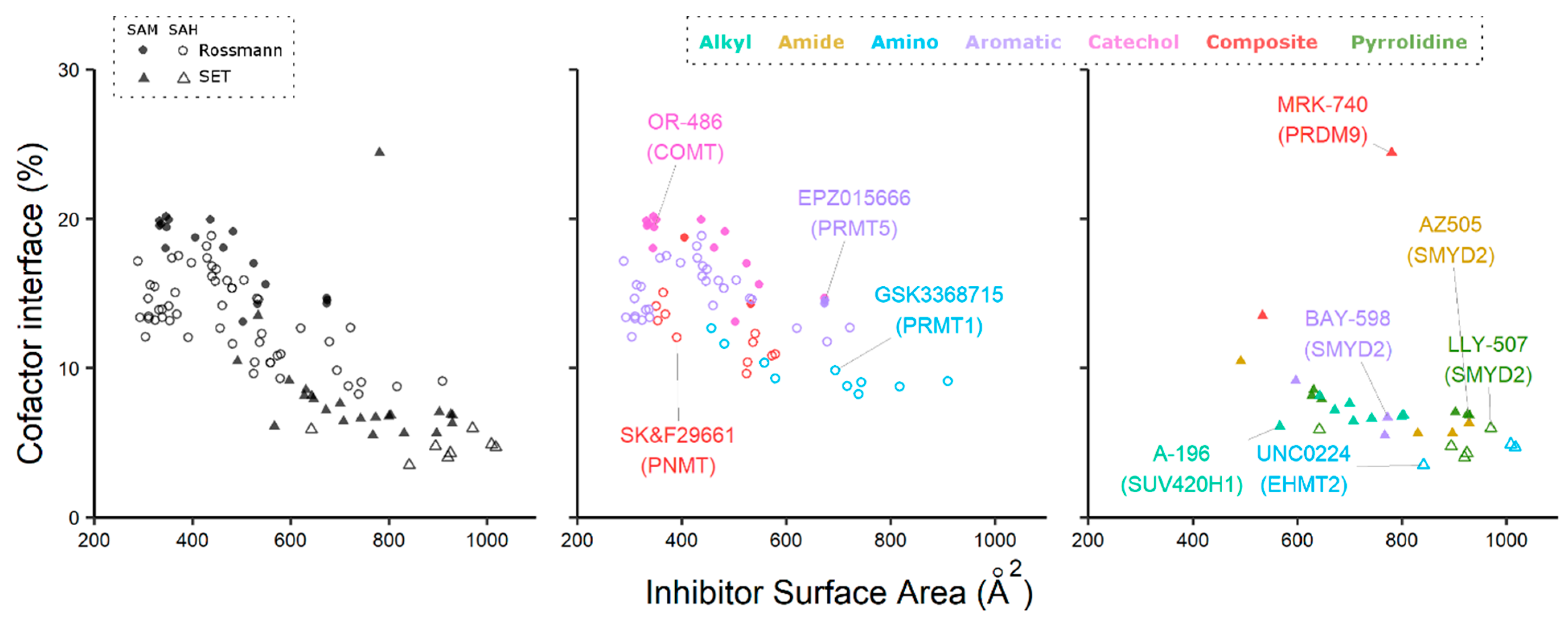
2. Cofactor Competitors
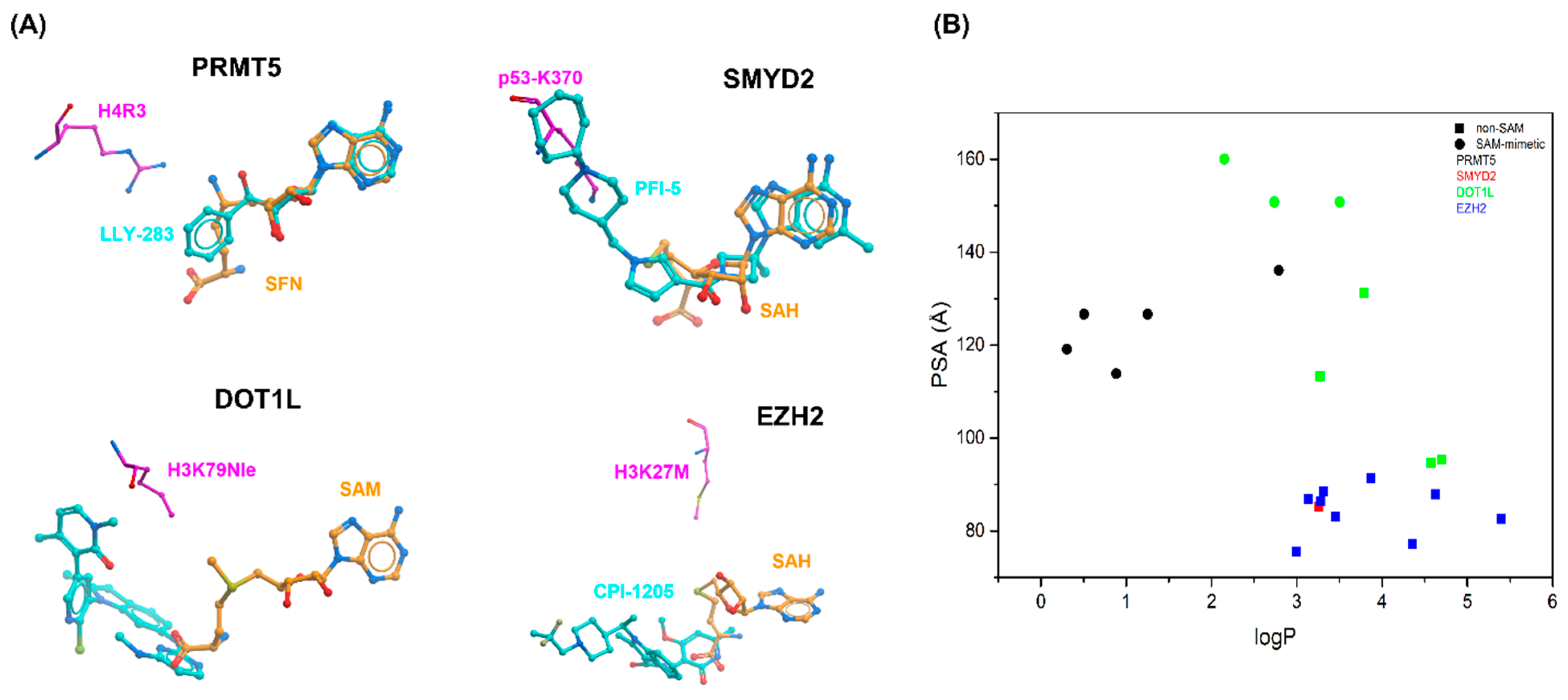
2.1. PRMT5
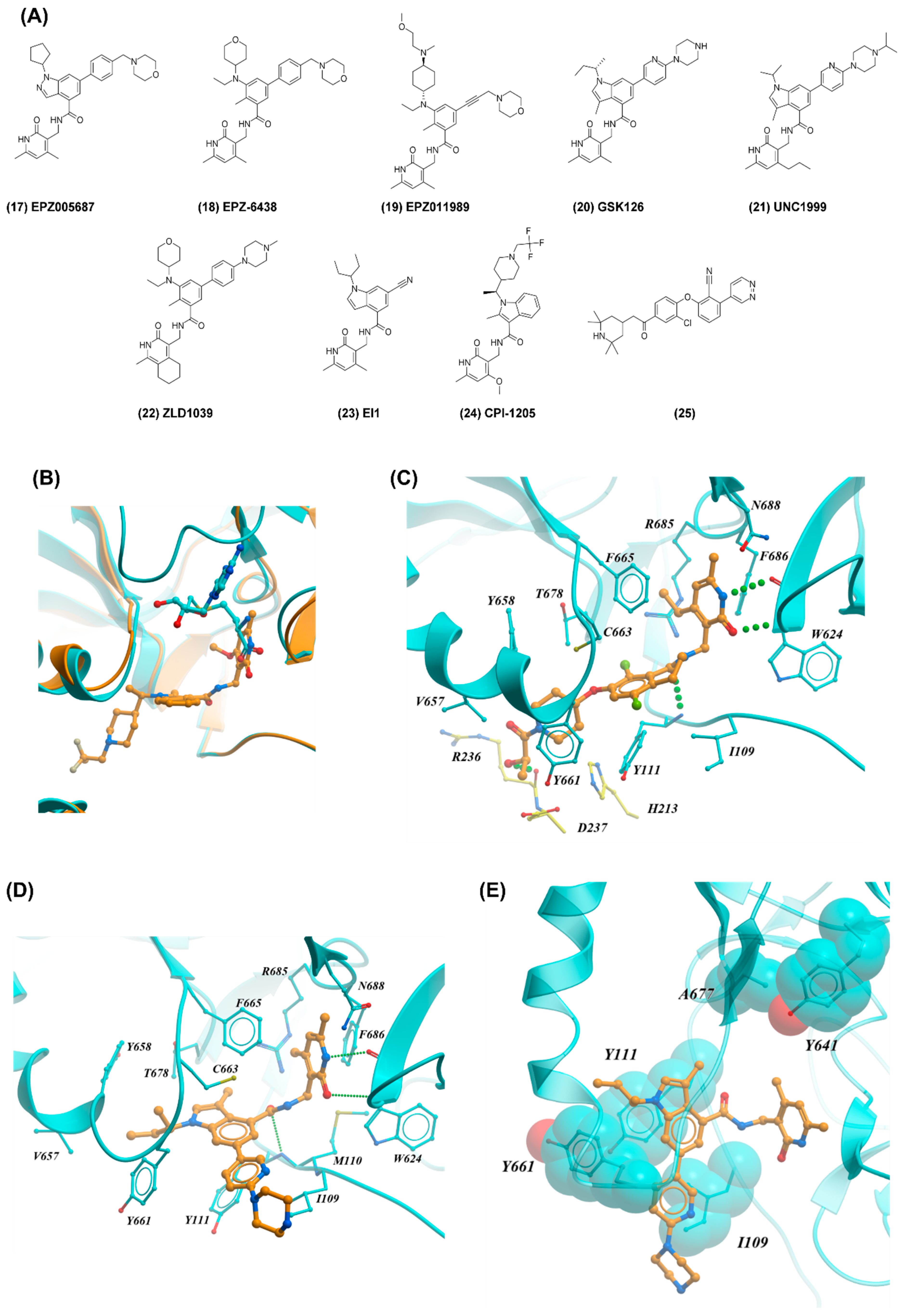
2.2. SMYD2
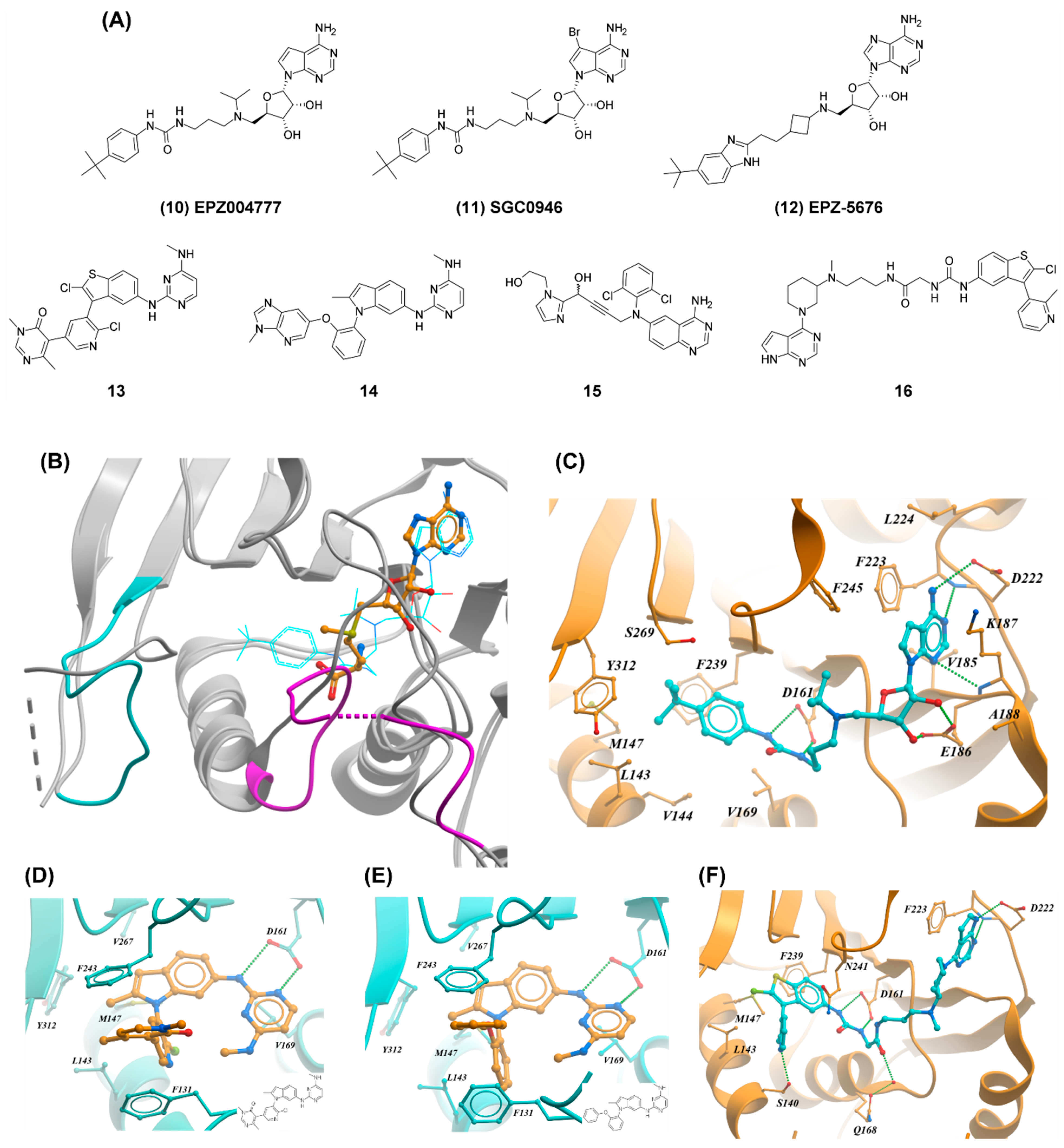
2.3. DOT1L
2.4. EZH2
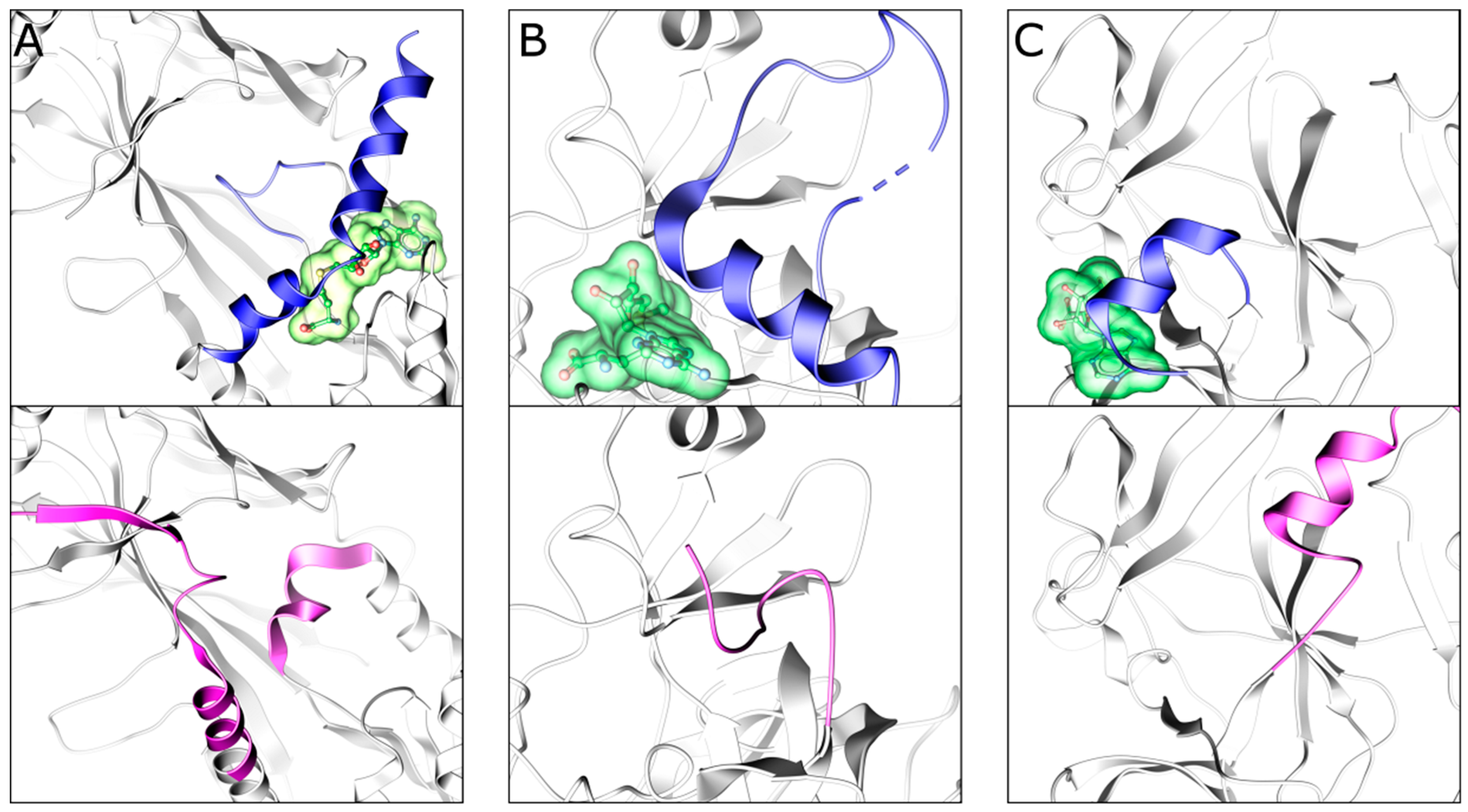
3. Exploiting the Bound Cofactor
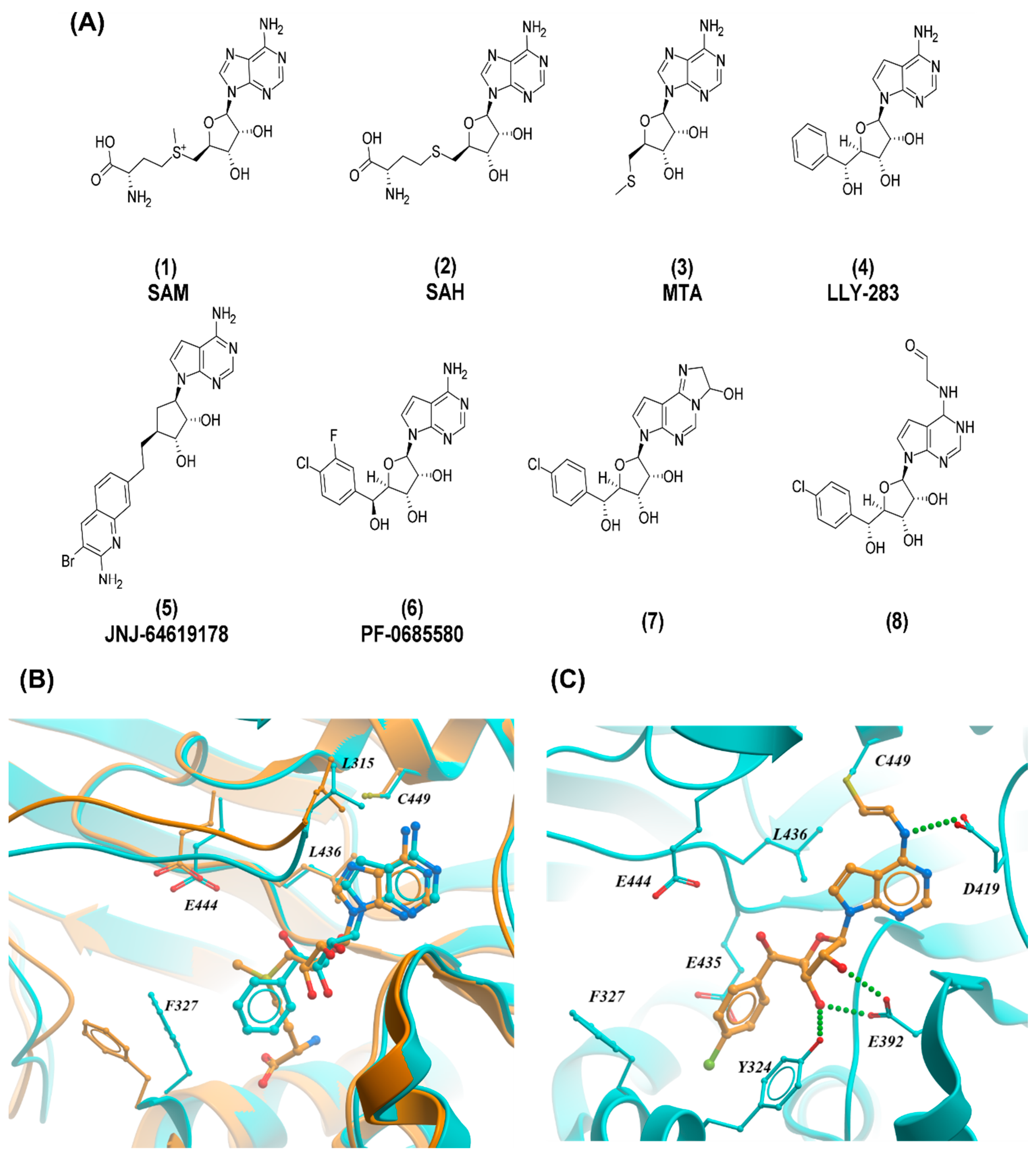
3.1. PRMT5
3.2. Type I PRMTs
3.3. EHMT1/2 (G9a/GLP)
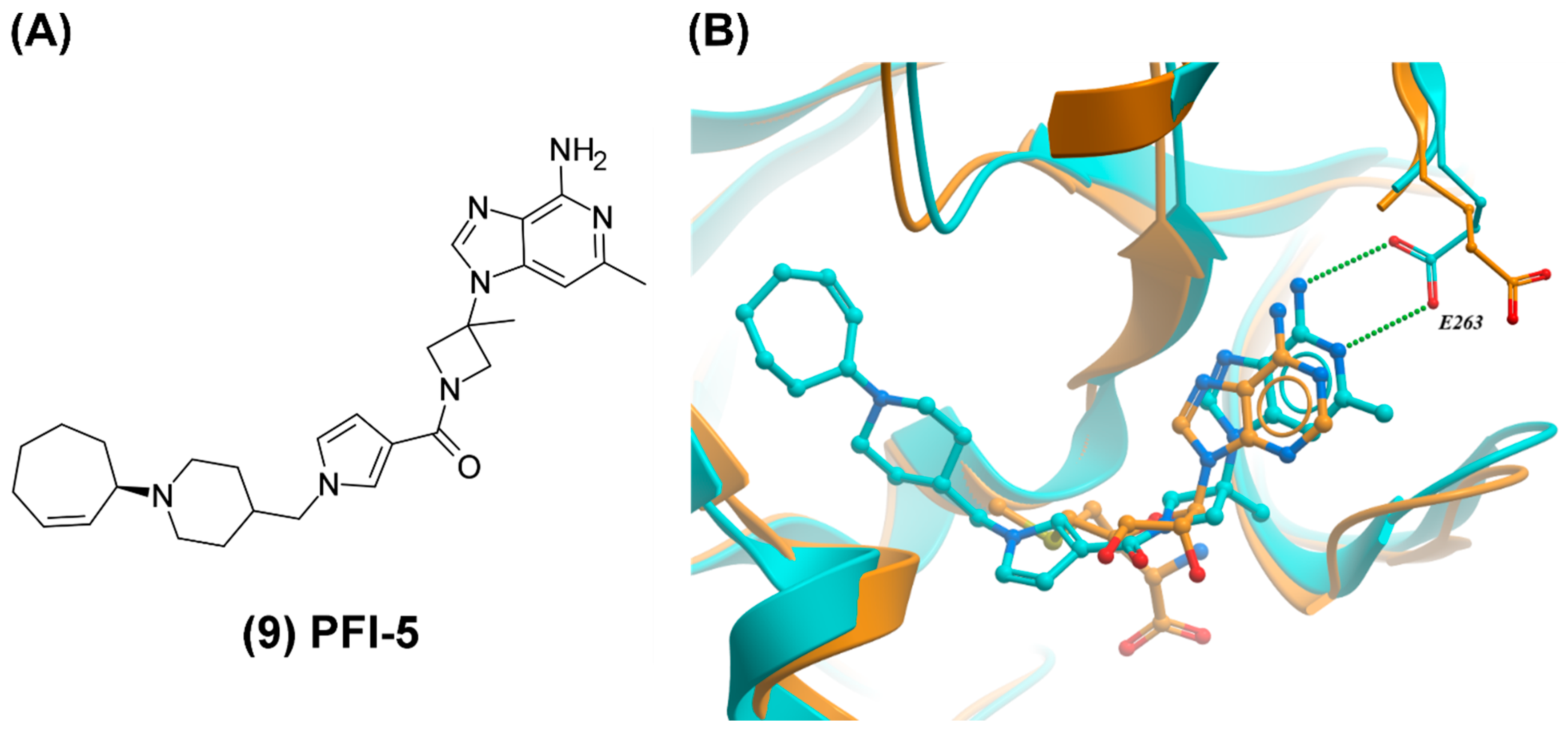
3.4. SETD7, SMYD2, and SUV420H1/2
3.5. PRDM9
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schapira, M. Chemical Inhibition of Protein Methyltransferases. Cell Chem. Biol. 2016, 23, 1067–1076. [Google Scholar] [CrossRef]
- Copeland, R.A.; Solomon, M.E.; Richon, V.M. Protein methyltransferases as a target class for drug discovery. Nat. Rev. Drug Discov. 2009, 8, 724–732. [Google Scholar] [CrossRef]
- Copeland, R.A.; Moyer, M.P.; Richon, V.M. Targeting genetic alterations in protein methyltransferases for personalized cancer therapeutics. Oncogene 2013, 32, 939–946. [Google Scholar] [CrossRef]
- Dilworth, D.; Barsyte-Lovejoy, D. Targeting protein methylation: From chemical tools to precision medicines. Cell. Mol. Life Sci. 2019, 76, 2967–2985. [Google Scholar] [CrossRef]
- Kim, K.H.; Roberts, C.W.M. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Guccione, E.; Richard, S. The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 642–657. [Google Scholar] [CrossRef]
- Scheer, S.; Ackloo, S.; Medina, T.S.; Schapira, M.; Li, F.; Ward, J.A.; Lewis, A.M.; Northrop, J.P.; Richardson, P.L.; Kaniskan, H.Ü.; et al. A chemical biology toolbox to study protein methyltransferases and epigenetic signaling. Nat. Commun. 2019, 10, 19. [Google Scholar] [CrossRef]
- Mirabella, F.; Wu, P.; Wardell, C.P.; Kaiser, M.F.; Walker, B.A.; Johnson, D.C.; Morgan, G.J. MMSET is the key molecular target in t(4;14) myeloma. Blood Cancer J. 2013, 3, e114. [Google Scholar] [CrossRef]
- Jaffe, J.D.; Wang, Y.; Chan, H.M.; Zhang, J.; Huether, R.; Kryukov, G.V.; Bhang, H.C.; Taylor, J.E.; Hu, M.; Englund, N.P.; et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 1386–1391. [Google Scholar] [CrossRef]
- Ceol, C.J.; Houvras, Y.; Jane-Valbuena, J.; Bilodeau, S.; Orlando, D.A.; Battisti, V.; Fritsch, L.; Lin, W.M.; Hollmann, T.J.; Ferré, F.; et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 2011, 471, 513–517. [Google Scholar] [CrossRef]
- Rodriguez-Paredes, M.; Martinez de Paz, A.; Simó-Riudalbas, L.; Sayols, S.; Moutinho, C.; Moran, S.; Villanueva, A.; Vázquez-Cedeira, M.; Lazo, P.A.; Carneiro, F.; et al. Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene 2014, 33, 2807–2813. [Google Scholar] [CrossRef]
- Campagna-Slater, V.; Mok, M.W.; Nguyen, K.T.; Feher, M.; Najmanovich, R.; Schapira, M. Structural Chemistry of the Histone Methyltransferases Cofactor Binding Site. J. Chem. Inf. Model. 2011, 51, 612–623. [Google Scholar] [CrossRef]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef]
- Knutson, S.K.; Wigle, T.J.; Warholic, N.M.; Sneeringer, C.J.; Allain, C.J.; Klaus, C.R.; Sacks, J.D.; Raimondi, A.; Majer, C.R.; Song, J.; et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 2012, 8, 890–896. [Google Scholar] [CrossRef]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A.; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef]
- Chan-Penebre, E.; Kuplast, K.G.; Majer, C.R.; Boriack-Sjodin, P.A.; Wigle, T.J.; Johnston, L.D.; Rioux, N.; Munchhof, M.J.; Jin, L.; Jacques, S.L.; et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432–437. [Google Scholar] [CrossRef]
- Bonday, Z.Q.; Cortez, G.S.; Grogan, M.J.; Antonysamy, S.; Weichert, K.; Bocchinfuso, W.P.; Li, F.; Kennedy, S.; Li, B.; Mader, M.M.; et al. LLY-283, a Potent and Selective Inhibitor of Arginine Methyltransferase 5, PRMT5, with Antitumor Activity. ACS Med. Chem. Lett. 2018, 9, 612–617. [Google Scholar] [CrossRef]
- Taylor, A.P.; Swewczyk, M.; Kennedy, S.; Trush, V.V.; Wu, H.; Zeng, H.; Dong, A.; Ferreira de Freitas, R.; Tatlock, J.; Kumpf, R.A.; et al. Selective, Small-Molecule Co-Factor Binding Site Inhibition of a Su(var)3-9, Enhancer of Zeste, Trithorax Domain Containing Lysine Methyltransferase. J. Med. Chem. 2019, 62, 7669–7683. [Google Scholar] [CrossRef]
- Horiuchi, K.Y.; Eason, M.M.; Ferry, J.J.; Planck, J.L.; Walsh, C.P.; Smith, R.F.; Howitz, K.T.; Ma, H. Assay development for histone methyltransferases. Assay Drug Dev. Techn. 2013, 11, 227–236. [Google Scholar] [CrossRef]
- Richon, V.M.; Johnston, D.; Sneeringer, C.J.; Jin, L.; Majer, C.R.; Elliston, K.; Jerva, L.F.; Scott, M.P.; Copeland, R.A. Chemogenetic Analysis of Human Protein Methyltransferases. Chem. Biol. Drug Des. 2011, 78, 199–210. [Google Scholar] [CrossRef]
- Copeland, R.A. Protein methyltransferase inhibitors as precision cancer therapeutics: A decade of discovery. Philos. Tr. Royal Soc. B. 2018, 373, 20170080. [Google Scholar] [CrossRef]
- Fong, J.Y.; Pignata, L.; Goy, P.-A.; Kawabata, K.C.; Lee, S.C.-W.; Koh, C.M.; Musiani, D.; Massignani, E.; Kotini, A.G.; Penson, A.; et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell 2019, 36, 194–209.e9. [Google Scholar] [CrossRef]
- Hamard, P.-J.; Santiago, G.E.; Liu, F.; Karl, D.L.; Martinez, C.; Man, N.; Mookhtiar, A.K.; Duffort, S.; Greenblatt, S.; Verdun, R.E.; et al. PRMT5 Regulates DNA Repair by Controlling the Alternative Splicing of Histone-Modifying Enzymes. Cell Rep. 2018, 24, 2643–2657. [Google Scholar] [CrossRef]
- Tan, D.Q.; Li, Y.; Yang, C.; Li, J.; Tan, S.H.; Chin, D.W.L.; Nakamura-Ishizu, A.; Yang, H.; Suda, T. PRMT5 Modulates Splicing for Genome Integrity and Preserves Proteostasis of Hematopoietic Stem Cells. Cell Rep. 2019, 26, 2316–2328. [Google Scholar] [CrossRef]
- Zhu, F.; Rui, L. PRMT5 in gene regulation and hematologic malignancies. Genes & Diseases 2019, 6, 247–257. [Google Scholar]
- Zhu, F.; Guo, H.; Bates, P.D.; Zhang, S.; Zhang, H.; Nomie, K.J.; Li, Y.; Lu, L.; Seibold, K.R.; Wang, F.; et al. PRMT5 is upregulated by B-cell receptor signaling and forms a positive-feedback loop with PI3K/AKT in lymphoma cells. Leukemia 2019, 33, 2898–2911. [Google Scholar] [CrossRef]
- Braun, C.J.; Stanciu, M.; Boutz, P.L.; Patterson, J.C.; Calligaris, D.; Higuchi, F.; Neupane, R.; Fenoglio, S.; Cahill, D.P.; Wakimoto, H.; et al. Coordinated Splicing of Regulatory Detained Introns within Oncogenic Transcripts Creates an Exploitable Vulnerability in Malignant Glioma. Cancer Cell 2017, 32, 411–426. [Google Scholar] [CrossRef]
- Srour, N.; Mersaoui, S.Y.; Richard, S. M-TAP Dance: Targeting PRMT1 and PRMT5 Family Members to Push Cancer Cells Over the Edge. Cancer Cell 2019, 36, 3–5. [Google Scholar] [CrossRef]
- Antonysamy, S.; Bonday, Z.; Campbell, R.M.; Doyle, B.; Druzina, Z.; Gheyi, T.; Han, B.; Jungheim, L.N.; Qian, Y.; Rauch, C.; et al. Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. USA 2012, 109, 17960–17965. [Google Scholar] [CrossRef]
- Smil, D.; Eram, M.S.; Li, F.; Kennedy, S.; Szewczyk, M.M.; Brown, P.J.; Barsyte-Lovejoy, D.; Arrowsmith, C.H.; Vedadi, M.; Schapira, M. Discovery of a Dual PRMT5–PRMT7 Inhibitor. ACS Med. Chem. Lett. 2015, 6, 408–412. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Wilson, F.H.; Ruth, J.R.; Paulk, J.; Tsherniak, A.; Marlow, S.E.; Vazquez, F.; Weir, B.A.; Fitzgerald, M.E.; Tanaka, M.; et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 2016, 351, 1214–1218. [Google Scholar] [CrossRef] [PubMed]
- Marjon, K.; Cameron, M.J.; Quang, P.; Clasquin, M.F.; Mandley, E.; Kunii, K.; McVay, M.; Choe, S.; Kernytsky, A.; Gross, S.; et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep. 2016, 15, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Mavrakis, K.J.; McDonald, E.R.; Schlabach, M.R.; Billy, E.; Hoffman, G.R.; deWeck, A.; Ruddy, D.A.; Venkatesan, K.; Yu, J.; McAllister, G.; et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 2016, 351, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Brehmer, D.; Wu, T.; Mannens, G.; Beke, L.; Vinken, P.; Gaffney, D.; Sun, W.; Pande, V.; Thuring, J.-W.; Millar, H.; et al. Abstract DDT02-04: A novel PRMT5 inhibitor with potent in vitro and in vivo activity in preclinical lung cancer models. Cancer Res. 2017, 77, DDT02–DDT04. [Google Scholar]
- Lin, H.; Luengo, J.I. Nucleoside protein arginine methyltransferase 5 (PRMT5) inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 1264–1269. [Google Scholar] [CrossRef]
- Luengo, J.; Vaddi, K. Selective Inhibitors of Protein Arginine Methyltransferase 5 (PRMT5). WO2017218802A1, 21 December 2017. [Google Scholar]
- Lin, H.; Wang, M.; Zhang, Y.W.; Tong, S.; Leal, R.A.; Shetty, R.; Vaddi, K.; Luengo, J.I. Discovery of Potent and Selective Covalent Protein Arginine Methyltransferase 5 (PRMT5) Inhibitors. ACS Med. Chem. Lett. 2019, 10, 1033–1038. [Google Scholar] [CrossRef]
- Alinari, L.; Mahasenan, K.V.; Yan, F.; Karkhanis, V.; Chung, J.-H.; Smith, E.M.; Quinion, C.; Smith, P.L.; Kim, L.; Patton, J.T.; et al. Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood 2015, 125, 2530–2543. [Google Scholar] [CrossRef]
- Mao, R.; Shao, J.; Zhu, K.; Zhang, Y.; Ding, H.; Zhang, C.; Shi, Z.; Jiang, H.; Sun, D.; Duan, W.; et al. Potent, Selective, and Cell Active Protein Arginine Methyltransferase 5 (PRMT5) Inhibitor Developed by Structure-Based Virtual Screening and Hit Optimization. J. Med. Chem. 2017, 60, 6289–6304. [Google Scholar] [CrossRef]
- Zhu, K.; Jiang, C.; Tao, H.; Liu, J.; Zhang, H.; Luo, C. Identification of a novel selective small-molecule inhibitor of protein arginine methyltransferase 5 (PRMT5) by virtual screening, resynthesis and biological evaluations. Bioorg. Med. Chem. Lett. 2018, 28, 1476–1483. [Google Scholar] [CrossRef]
- Brown, M.A.; Sims, R.J.; Gottlieb, P.D.; Tucker, P.W. Identification and characterization of Smyd2: A split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol. Cancer 2006, 5, 26. [Google Scholar] [CrossRef]
- Huang, J.; Perez-Burgos, L.; Placek, B.J.; Sengupta, R.; Richter, M.; Dorsey, J.A.; Kubicek, S.; Opravil, S.; Jenuwein, T.; Berger, S.L. Repression of p53 activity by Smyd2-mediated methylation. Nature 2006, 444, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Saddic, L.A.; West, L.E.; Aslanian, A.; Yates, J.R.; Rubin, S.M.; Gozani, O.; Sage, J. Methylation of the Retinoblastoma Tumor Suppressor by SMYD2. J. Biol. Chem. 2010, 285, 37733–37740. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, T.; Andresen, C.; Unger, A.; Just, S.; Rottbauer, W.; Linke, W.A. Lysine methyltransferase Smyd2 regulates Hsp90-mediated protection of the sarcomeric titin springs and cardiac function. Biochim. Biophys. Acta 2013, 1833, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tanaka, K.; Yan, J.; Li, J.; Peng, D.; Jiang, Y.; Yang, Z.; Barton, M.C.; Wen, H.; Shi, X. Regulation of estrogen receptor α by histone methyltransferase SMYD2-mediated protein methylation. Proc. Natl. Acad. Sci. USA 2013, 110, 17284–17289. [Google Scholar] [CrossRef]
- Eggert, E.; Hillig, R.C.; Koehr, S.; Stöckigt, D.; Weiske, J.; Barak, N.; Mowat, J.; Brumby, T.; Christ, C.D.; ter Laak, A.; et al. Discovery and Characterization of a Highly Potent and Selective Aminopyrazoline-Based in Vivo Probe (BAY-598) for the Protein Lysine Methyltransferase SMYD2. J. Med. Chem. 2016, 59, 4578–4600. [Google Scholar] [CrossRef]
- Ferguson, A.D.; Larsen, N.A.; Howard, T.; Pollard, H.; Green, I.; Grande, C.; Cheung, T.; Garcia-Arenas, R.; Cowen, S.; Wu, J.; et al. Structural basis of substrate methylation and inhibition of SMYD2. Structure 2011, 19, 1262–1273. [Google Scholar] [CrossRef]
- Nguyen, H.; Allali-Hassani, A.; Antonysamy, S.; Chang, S.; Chen, L.H.; Curtis, C.; Emtage, S.; Fan, L.; Gheyi, T.; Li, F.; et al. LLY-507, a Cell-Active, Potent and Selective Inhibitor of Protein Lysine Methyltransferase SMYD2. J. Biol. Chem. 2015, 290, 13641–13653. [Google Scholar] [CrossRef]
- Thomenius, M.J.; Totman, J.; Harvey, D.; Mitchell, L.H.; Riera, T.V.; Cosmopoulos, K.; Grassian, A.R.; Klaus, C.; Foley, M.; Admirand, E.A.; et al. Small molecule inhibitors and CRISPR/Cas9 mutagenesis demonstrate that SMYD2 and SMYD3 activity are dispensable for autonomous cancer cell proliferation. PLoS ONE 2018, 13, e0197372. [Google Scholar] [CrossRef]
- McLean, C.M.; Karemaker, I.D.; Leeuwen, F. van The emerging roles of DOT1L in leukemia and normal development. Leukemia 2014, 28, 2131–2138. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Zhang, Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011, 25, 1345–1358. [Google Scholar] [CrossRef]
- Steger, D.J.; Lefterova, M.I.; Ying, L.; Stonestrom, A.J.; Schupp, M.; Zhuo, D.; Vakoc, A.L.; Kim, J.-E.; Chen, J.; Lazar, M.A.; et al. DOT1L/KMT4 Recruitment and H3K79 Methylation Are Ubiquitously Coupled with Gene Transcription in Mammalian Cells. Mol. Cell. Biol. 2008, 28, 2825–2839. [Google Scholar] [CrossRef] [PubMed]
- Tatum, D.; Li, S. Evidence That the Histone Methyltransferase Dot1 Mediates Global Genomic Repair by Methylating Histone H3 on Lysine 79. J. Biol. Chem. 2011, 286, 17530–17535. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhu, H.; Stauffer, F.; Caravatti, G.; Vollmer, S.; Machauer, R.; Holzer, P.; Möbitz, H.; Scheufler, C.; Klumpp, M.; et al. Discovery of Novel Dot1L Inhibitors through a Structure-Based Fragmentation Approach. ACS Med. Chem. Lett. 2016, 7, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chory, E.J.; Wernimont, A.K.; Tempel, W.; Scopton, A.; Federation, A.; Marineau, J.J.; Qi, J.; Barsyte-Lovejoy, D.; Yi, J.; et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 2012, 3, 1288. [Google Scholar] [CrossRef]
- Basavapathruni, A.; Jin, L.; Daigle, S.R.; Majer, C.R.A.; Therkelsen, C.A.; Wigle, T.J.; Kuntz, K.W.; Chesworth, R.; Pollock, R.M.; Scott, M.P.; et al. Conformational adaptation drives potent, selective and durable inhibition of the human protein methyltransferase DOT1L. Chem. Biol. Drug Des. 2012, 80, 971–980. [Google Scholar] [CrossRef]
- Möbitz, H.; Machauer, R.; Holzer, P.; Vaupel, A.; Stauffer, F.; Ragot, C.; Caravatti, G.; Scheufler, C.; Fernandez, C.; Hommel, U.; et al. Discovery of Potent, Selective, and Structurally Novel Dot1L Inhibitors by a Fragment Linking Approach. ACS Med. Chem. Lett. 2017, 8, 338–343. [Google Scholar] [CrossRef]
- Scheufler, C.; Möbitz, H.; Gaul, C.; Ragot, C.; Be, C.; Fernández, C.; Beyer, K.S.; Tiedt, R.; Stauffer, F. Optimization of a Fragment-Based Screening Hit toward Potent DOT1L Inhibitors Interacting in an Induced Binding Pocket. ACS Med. Chem. Lett. 2016, 7, 730–734. [Google Scholar] [CrossRef]
- Jiao, L.; Liu, X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 2015, 350, aac4383. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Comet, I.; Riising, E.M.; Leblanc, B.; Helin, K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 2016, 16, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Warholic, N.M.; Wigle, T.J.; Klaus, C.R.; Allain, C.J.; Raimondi, A.; Scott, M.P.; Chesworth, R.; Moyer, M.P.; Copeland, R.A.; et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2013, 110, 7922–7927. [Google Scholar] [CrossRef] [PubMed]
- Konze, K.D.; Ma, A.; Li, F.; Barsyte-Lovejoy, D.; Parton, T.; MacNevin, C.J.; Liu, F.; Gao, C.; Huang, X.-P.; Kuznetsova, E.; et al. An Orally Bioavailable Chemical Probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013, 8, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Chan, H.; Teng, L.; Li, L.; Chuai, S.; Zhang, R.; Zeng, J.; Li, M.; Fan, H.; Lin, Y.; et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl. Acad. Sci. USA 2012, 109, 21360–21365. [Google Scholar] [CrossRef]
- Song, X.; Gao, T.; Wang, N.; Feng, Q.; You, X.; Ye, T.; Lei, Q.; Zhu, Y.; Xiong, M.; Xia, Y.; et al. Selective inhibition of EZH2 by ZLD1039 blocks H3K27methylation and leads to potent anti-tumor activity in breast cancer. Sci. Rep. 2016, 6, 1–15. [Google Scholar]
- Vaswani, R.G.; Gehling, V.S.; Dakin, L.A.; Cook, A.S.; Nasveschuk, C.G.; Duplessis, M.; Iyer, P.; Balasubramanian, S.; Zhao, F.; Good, A.C.; et al. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a Potent and Selective Inhibitor of Histone Methyltransferase EZH2, Suitable for Phase I Clinical Trials for B-Cell Lymphomas. J. Med. Chem. 2016, 59, 9928–9941. [Google Scholar]
- Nasveschuk, C.G.; Gagnon, A.; Garapaty-Rao, S.; Balasubramanian, S.; Campbell, R.; Lee, C.; Zhao, F.; Bergeron, L.; Cummings, R.; Trojer, P.; et al. Discovery and Optimization of Tetramethylpiperidinyl Benzamides as Inhibitors of EZH2. ACS Med. Chem. Lett. 2014, 5, 378–383. [Google Scholar] [CrossRef]
- Gibaja, V.; Shen, F.; Harari, J.; Korn, J.; Ruddy, D.; Saenz-Vash, V.; Zhai, H.; Rejtar, T.; Paris, C.G.; Yu, Z.; et al. Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene 2016, 35, 558–566. [Google Scholar] [CrossRef]
- He, Y.; Selvaraju, S.; Curtin, M.L.; Jakob, C.G.; Zhu, H.; Comess, K.M.; Shaw, B.; The, J.; Lima-Fernandes, E.; Szewczyk, M.M.; et al. The EED protein–protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat. Chem. Biol. 2017, 13, 389–395. [Google Scholar] [CrossRef]
- Qi, W.; Zhao, K.; Gu, J.; Huang, Y.; Wang, Y.; Zhang, H.; Zhang, M.; Zhang, J.; Yu, Z.; Li, L.; et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat. Chem. Biol. 2017, 13, 381–388. [Google Scholar] [CrossRef]
- Schapira, M.; Ferreira de Freitas, R. Structural biology and chemistry of protein arginine methyltransferases. Medchemcomm 2014, 5, 1779–1788. [Google Scholar] [CrossRef] [PubMed]
- Troffer-Charlier, N.; Cura, V.; Hassenboehler, P.; Moras, D.; Cavarelli, J. Functional insights from structures of coactivator-associated arginine methyltransferase 1 domains. EMBO J. 2007, 26, 4391–4401. [Google Scholar] [CrossRef] [PubMed]
- Schubert, H.L.; Blumenthal, R.M.; Cheng, X. Many paths to methyltransfer: A chronicle of convergence. Trends Biochem. Sci. 2003, 28, 329–335. [Google Scholar] [CrossRef]
- Kwon, T.; Chang, J.H.; Kwak, E.; Lee, C.W.; Joachimiak, A.; Kim, Y.C.; Lee, J.; Cho, Y. Mechanism of histone lysine methyl transfer revealed by the structure of SET7/9-AdoMet. EMBO J. 2003, 22, 292–303. [Google Scholar] [CrossRef]
- Xiao, B.; Jing, C.; Wilson, J.R.; Walker, P.A.; Vasisht, N.; Kelly, G.; Howell, S.; Taylor, I.A.; Blackburn, G.M.; Gamblin, S.J. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature 2003, 421, 652–656. [Google Scholar] [CrossRef]
- Wu, H.; Mathioudakis, N.; Diagouraga, B.; Dong, A.; Dombrovski, L.; Baudat, F.; Cusack, S.; de Massy, B.; Kadlec, J. Molecular basis for the regulation of the H3K4 methyltransferase activity of PRDM9. Cell Rep. 2013, 5, 13–20. [Google Scholar] [CrossRef]
- Fedoriw, A.; Rajapurkar, S.R.; O’Brien, S.; Gerhart, S.V.; Mitchell, L.H.; Adams, N.D.; Rioux, N.; Lingaraj, T.; Ribich, S.A.; Pappalardi, M.B.; et al. Anti-tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 2019, 36, 100–114. [Google Scholar] [CrossRef]
- Eram, M.S.; Shen, Y.; Szewczyk, M.; Wu, H.; Senisterra, G.; Li, F.; Butler, K.V.; Kaniskan, H.Ü.; Speed, B.A.; Dela Seña, C.; et al. A Potent, Selective, and Cell-Active Inhibitor of Human Type I Protein Arginine Methyltransferases. ACS Chem. Biol. 2016, 11, 772–781. [Google Scholar] [CrossRef]
- Nakayama, K.; Szewczyk, M.M.; Dela Sena, C.; Wu, H.; Dong, A.; Zeng, H.; Li, F.; de Freitas, R.F.; Eram, M.S.; Schapira, M.; et al. TP-064, a potent and selective small molecule inhibitor of PRMT4 for multiple myeloma. Oncotarget 2018, 9, 18480–18493. [Google Scholar] [CrossRef]
- Chang, Y.; Ganesh, T.; Horton, J.R.; Spannhoff, A.; Liu, J.; Sun, A.; Zhang, X.; Bedford, M.T.; Shinkai, Y.; Snyder, J.P.; et al. Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases. J. Mol. Biol. 2010, 400, 1–7. [Google Scholar] [CrossRef]
- Liu, F.; Chen, X.; Allali-Hassani, A.; Quinn, A.M.; Wasney, G.A.; Dong, A.; Barsyte, D.; Kozieradzki, I.; Senisterra, G.; Chau, I.; et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a. J. Med. Chem. 2009, 52, 7950–7953. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Min, J.; Lunin, V.V.; Antoshenko, T.; Dombrovski, L.; Zeng, H.; Allali-Hassani, A.; Campagna-Slater, V.; Vedadi, M.; Arrowsmith, C.H.; et al. Structural biology of human H3K9 methyltransferases. PLoS ONE 2010, 5, e8570. [Google Scholar] [CrossRef] [PubMed]
- Barsyte-Lovejoy, D.; Li, F.; Oudhoff, M.J.; Tatlock, J.H.; Dong, A.; Zeng, H.; Wu, H.; Freeman, S.A.; Schapira, M.; Senisterra, G.A.; et al. (R)-PFI-2 is a potent and selective inhibitor of SETD7 methyltransferase activity in cells. Proc. Natl. Acad. Sci. USA 2014, 111, 12853–12858. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, S.; Dirk, L.M.A.; Yesselman, J.D.; Nimtz, J.S.; Adhikari, U.; Mehl, R.A.; Scheiner, S.; Houtz, R.L.; Al-Hashimi, H.M.; Trievel, R.C. Conservation and functional importance of carbon-oxygen hydrogen bonding in AdoMet-dependent methyltransferases. J. Am. Chem. Soc. 2013, 135, 15536–15548. [Google Scholar] [CrossRef]
- Mitchell, L.H.; Boriack-Sjodin, P.A.; Smith, S.; Thomenius, M.; Rioux, N.; Munchhof, M.; Mills, J.E.; Klaus, C.; Totman, J.; Riera, T.V.; et al. Novel Oxindole Sulfonamides and Sulfamides: EPZ031686, the First Orally Bioavailable Small Molecule SMYD3 Inhibitor. ACS Med. Chem. Lett. 2016, 7, 134–138. [Google Scholar] [CrossRef]
- Sweis, R.F.; Wang, Z.; Algire, M.; Arrowsmith, C.H.; Brown, P.J.; Chiang, G.G.; Guo, J.; Jakob, C.G.; Kennedy, S.; Li, F.; et al. Discovery of A-893, A New Cell-Active Benzoxazinone Inhibitor of Lysine Methyltransferase SMYD2. ACS Med. Chem. Lett. 2015, 6, 695–700. [Google Scholar] [CrossRef]
- Bromberg, K.D.; Mitchell, T.R.H.; Upadhyay, A.K.; Jakob, C.G.; Jhala, M.A.; Comess, K.M.; Lasko, L.M.; Li, C.; Tuzon, C.T.; Dai, Y.; et al. The SUV4-20 inhibitor A-196 verifies a role for epigenetics in genomic integrity. Nat. Chem. Biol. 2017, 13, 317–324. [Google Scholar] [CrossRef]
- Huang, C.; Liew, S.S.; Lin, G.R.; Poulsen, A.; Ang, M.J.Y.; Chia, B.C.S.; Chew, S.Y.; Kwek, Z.P.; Wee, J.L.K.; Ong, E.H.; et al. Discovery of Irreversible Inhibitors Targeting Histone Methyltransferase, SMYD3. ACS Med. Chem. Lett. 2019, 10, 978–984. [Google Scholar] [CrossRef]
- Schapira, M. Structural Chemistry of Human SET Domain Protein Methyltransferases. Curr. Chem. Genomics 2011, 5, 85–94. [Google Scholar] [CrossRef]
- Allali-Hassani, A.; Szewczyk, M.; Ivanochko, D.; Organ, S.; Bok, J.; Sook Yuin Ho, J.; Gay, F.; Li, F.; Blazer, L.; Eram, M.S.; et al. Discovery of a Chemical Probe for PRDM9. Nat. Commun. 2019, in press. [Google Scholar]
- Kaniskan, H.Ü.; Eram, M.S.; Zhao, K.; Szewczyk, M.M.; Yang, X.; Schmidt, K.; Luo, X.; Xiao, S.; Dai, M.; He, F.; et al. Discovery of Potent and Selective Allosteric Inhibitors of Protein Arginine Methyltransferase 3 (PRMT3). J. Med. Chem. 2018, 61, 1204–1217. [Google Scholar] [CrossRef] [PubMed]
- Grebien, F.; Vedadi, M.; Getlik, M.; Giambruno, R.; Grover, A.; Avellino, R.; Skucha, A.; Vittori, S.; Kuznetsova, E.; Smil, D.; et al. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPα N-terminal leukemia. Nat. Chem. Biol. 2015, 11, 571–578. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira de Freitas, R.; Ivanochko, D.; Schapira, M. Methyltransferase Inhibitors: Competing with, or Exploiting the Bound Cofactor. Molecules 2019, 24, 4492. https://doi.org/10.3390/molecules24244492
Ferreira de Freitas R, Ivanochko D, Schapira M. Methyltransferase Inhibitors: Competing with, or Exploiting the Bound Cofactor. Molecules. 2019; 24(24):4492. https://doi.org/10.3390/molecules24244492
Chicago/Turabian StyleFerreira de Freitas, Renato, Danton Ivanochko, and Matthieu Schapira. 2019. "Methyltransferase Inhibitors: Competing with, or Exploiting the Bound Cofactor" Molecules 24, no. 24: 4492. https://doi.org/10.3390/molecules24244492
APA StyleFerreira de Freitas, R., Ivanochko, D., & Schapira, M. (2019). Methyltransferase Inhibitors: Competing with, or Exploiting the Bound Cofactor. Molecules, 24(24), 4492. https://doi.org/10.3390/molecules24244492