
SAR by Space: Enriching Hit Sets from the Chemical Space

 , , ,
, , ,
Abstract
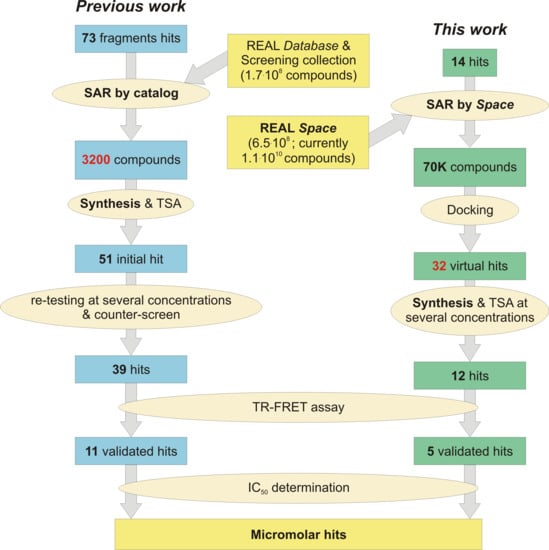
1. Introduction
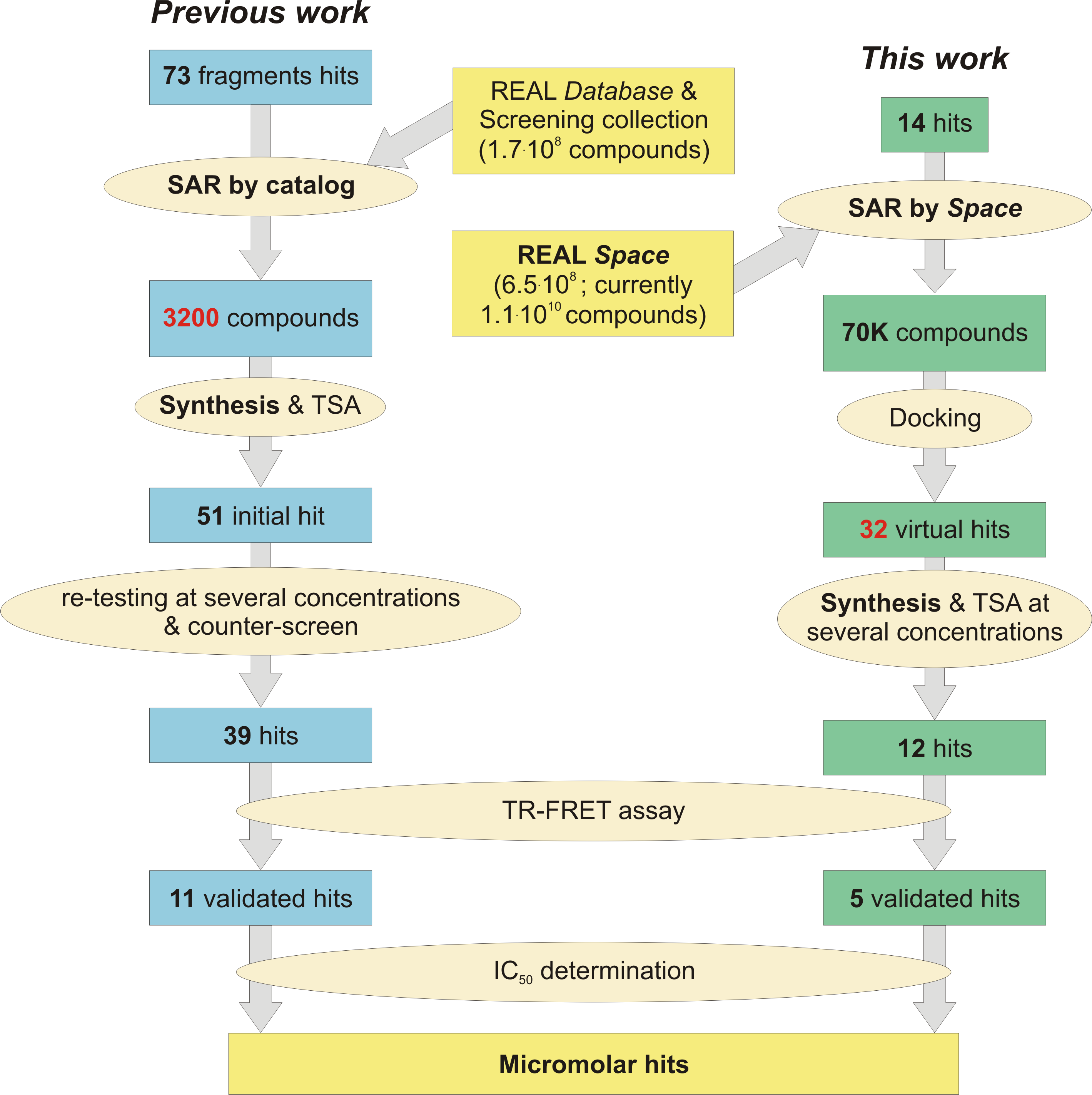
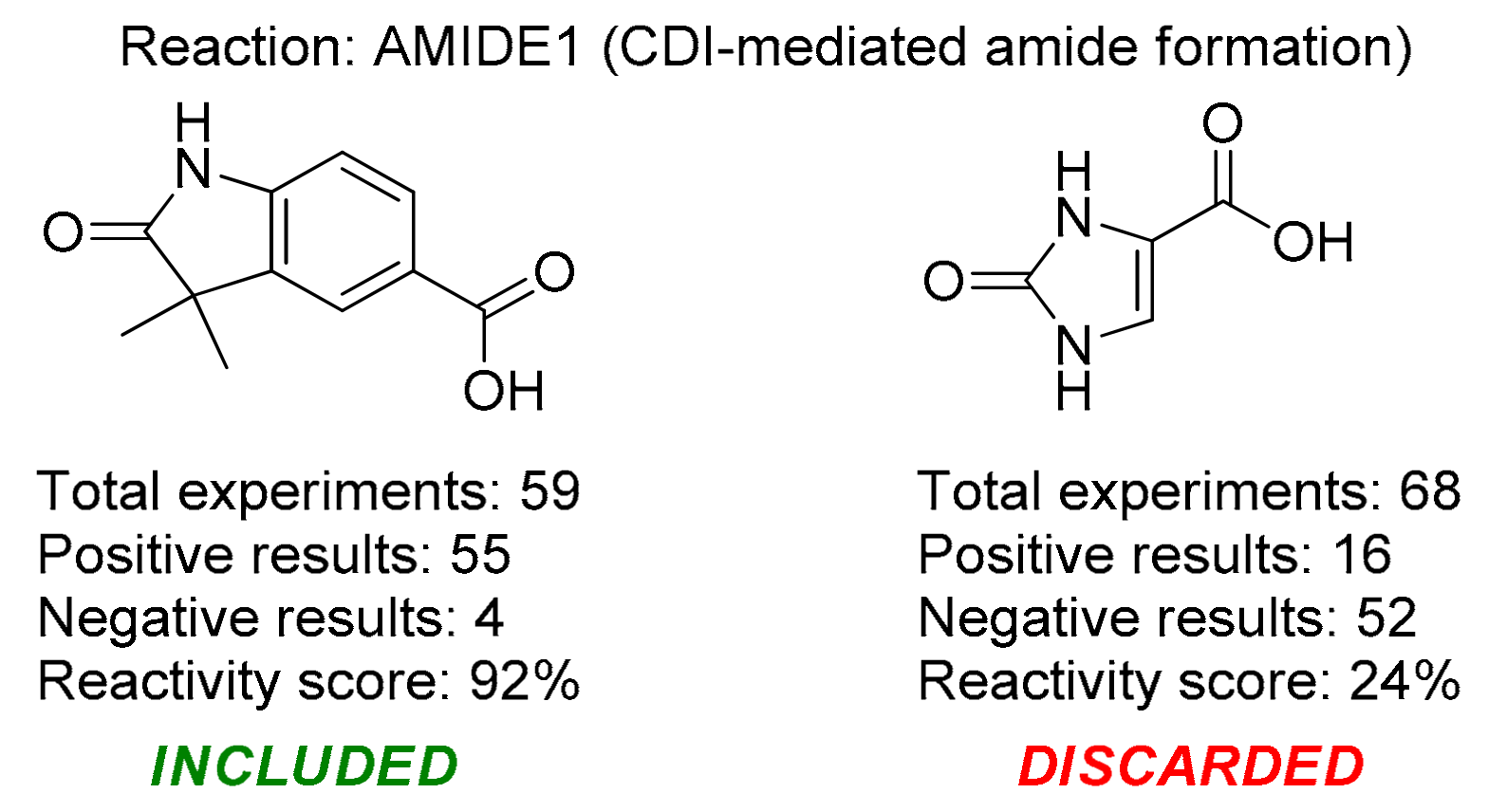
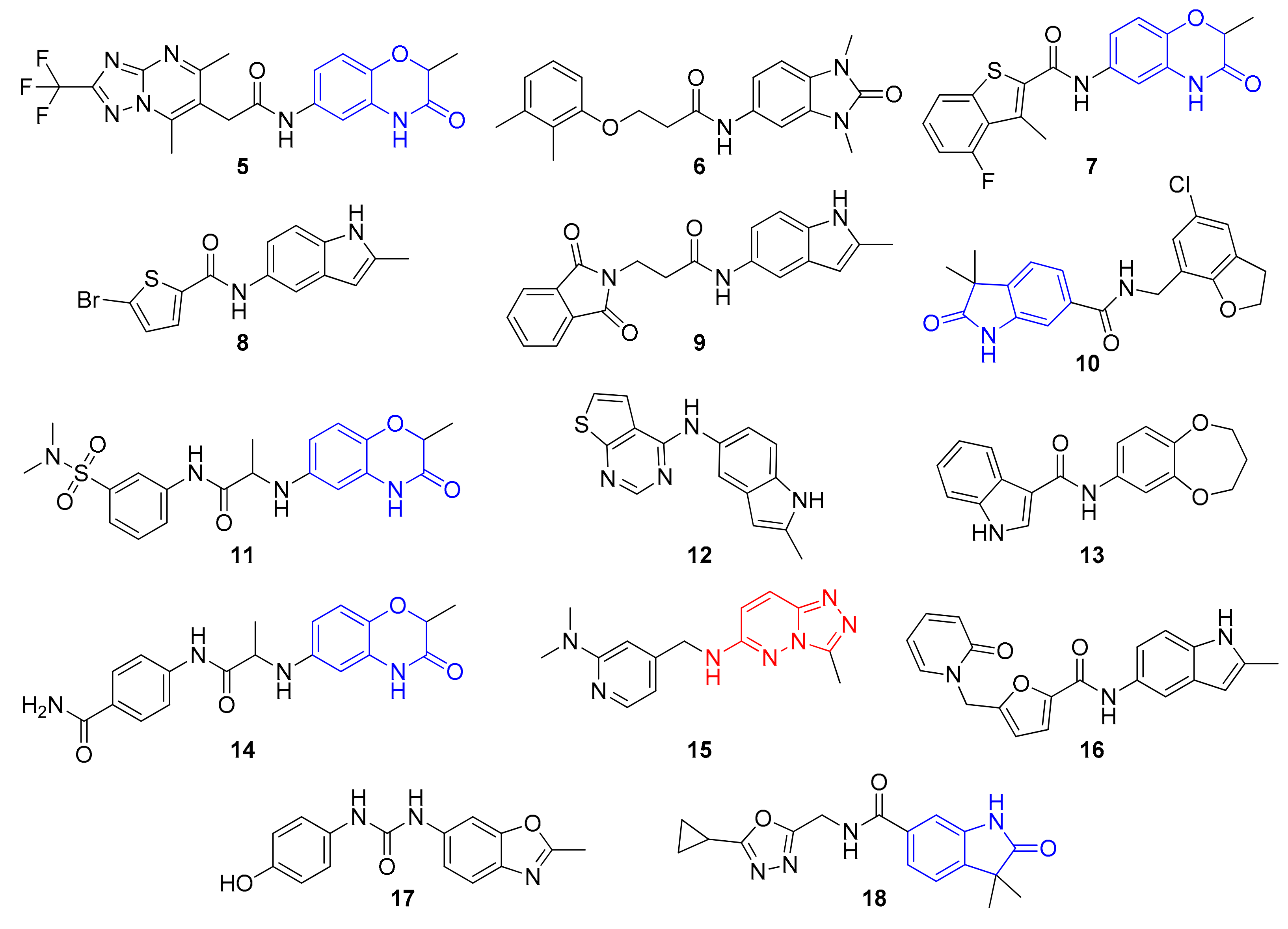
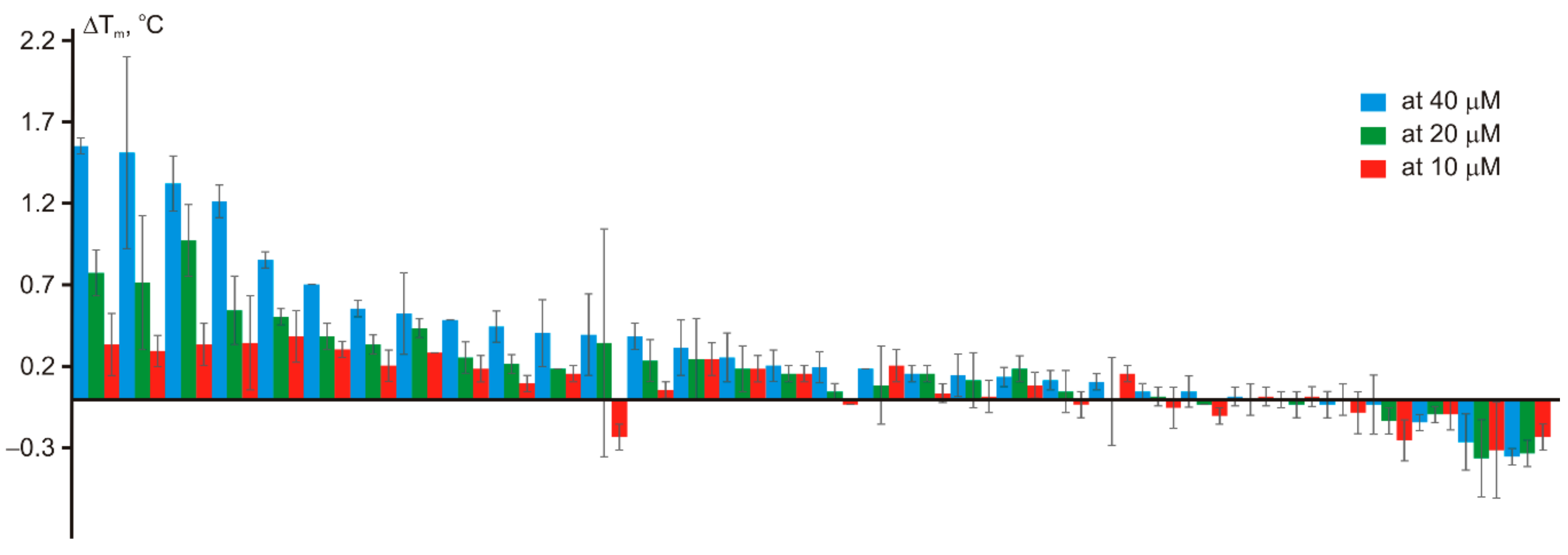
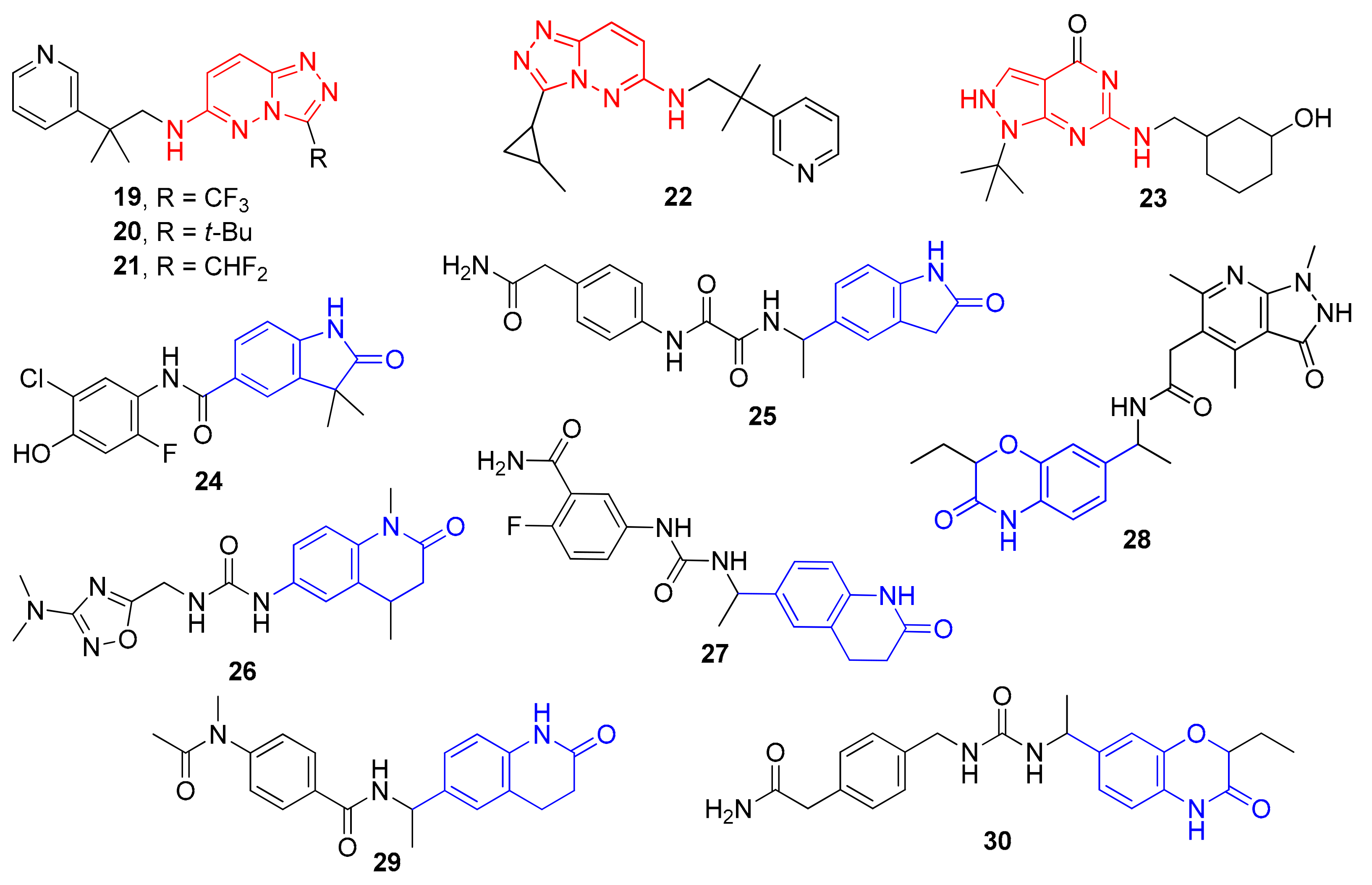
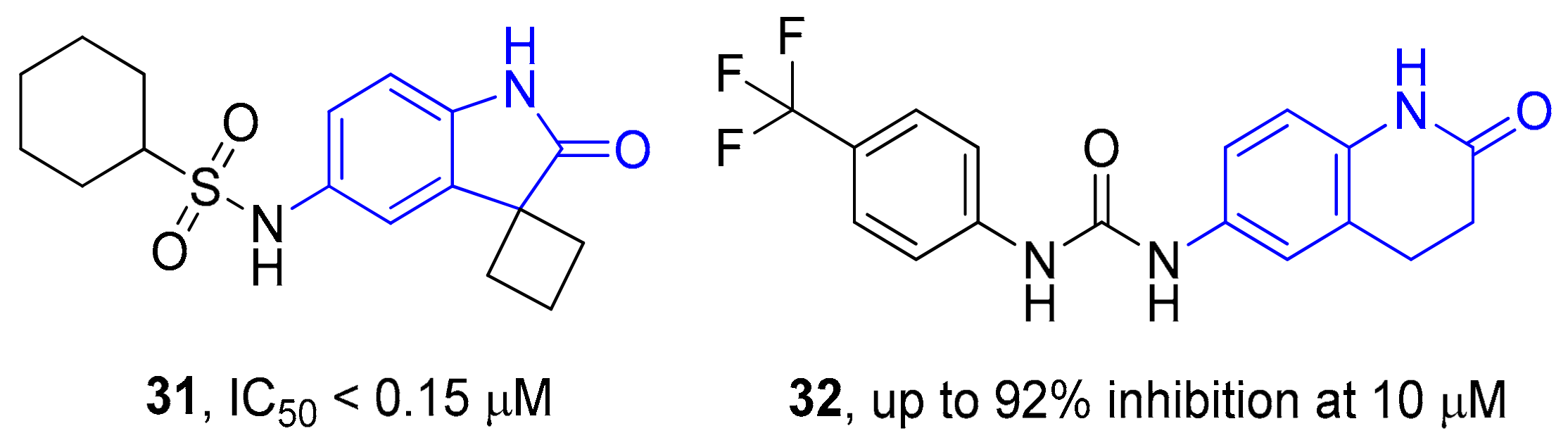
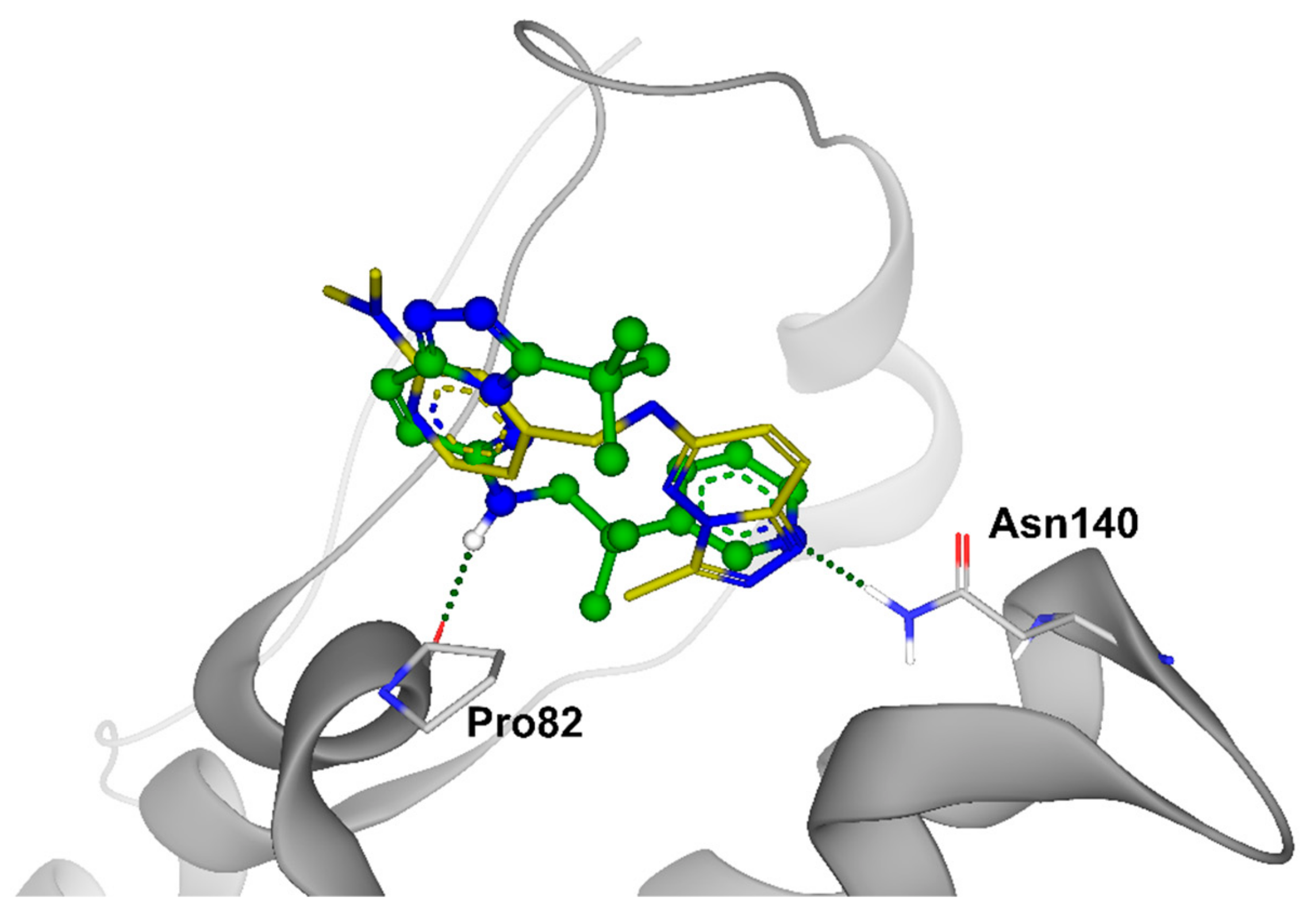
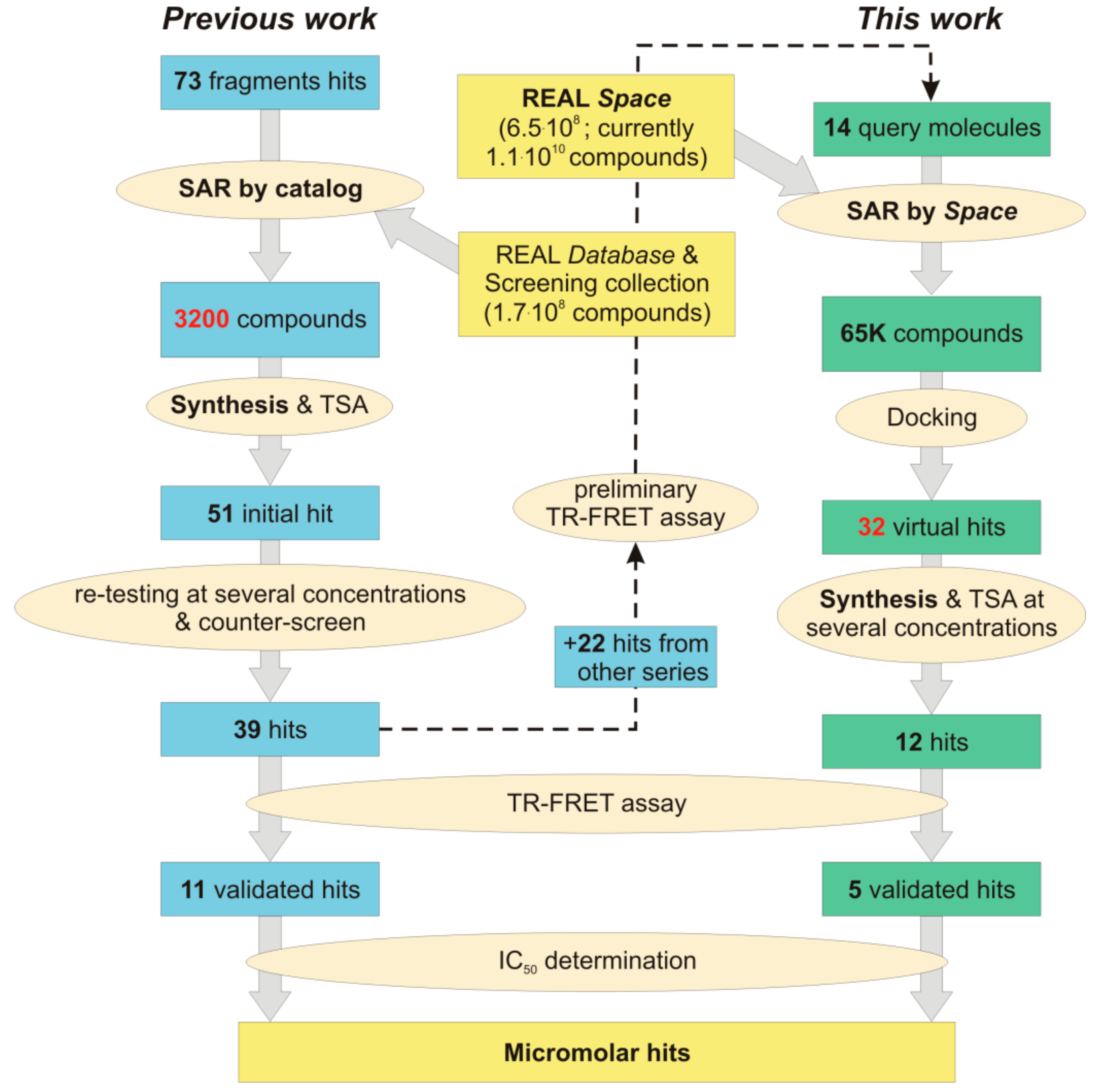
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Thermal Shift Assays
3.3. TR-FRET Assays and IC50 Measurements
3.4. Molecular Docking Studies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bohacek, R.S.; McMartin, C.; Guida, W.C. The art and practice of structure-based drug design: A molecular modeling perspective. Med. Res. Rev. 1996, 16, 3–50. [Google Scholar] [CrossRef]
- Walters, W.P. Virtual chemical libraries. J. Med. Chem. 2019, 62, 1116–1124. [Google Scholar] [CrossRef]
- Hoffmann, T.; Gastreich, M. The next level in chemical space navigation: Going far beyond enumerable compound libraries. Drug Discov. Today 2019, 24, 1148–1156. [Google Scholar] [CrossRef] [PubMed]
- Reymond, J.-L. The chemical space project. Acc. Chem. Res. 2015, 48, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Lucas, X.; Grüning, B.A.; Bleher, S.; Günther, S. The purchasable chemical space: A detailed picture. J. Chem. Inf. Model. 2015, 55, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Knehans, T.; Klingler, F.-M.; Kraut, H.; Saller, H.; Herrmann, A.; Rippmann, F.; Eiblmaier, J.; Lemmen, C.; Krier, M. Merck AcceSSible InVentory (MASSIV): In silico synthesis guided by chemical transforms obtained through bootstrapping reaction databases. In Proceedings of the Abstracts of Papers, 254th ACS National Meeting & Exposition, Washington, DC, USA; 2017. [Google Scholar]
- Hu, Q.; Peng, Z.; Sutton, S.C.; Na, J.; Kostrowicki, J.; Yang, B.; Thacher, T.; Kong, X.; Mattaparti, S.; Zhou, J.Z.; et al. Pfizer global virtual library (PGVL): A chemistry design tool powered by experimentally validated parallel synthesis information. ACS Comb. Sci. 2012, 14, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Hartenfeller, M.; Zettl, H.; Walter, M.; Rupp, M.; Reisen, F.; Proschak, E.; Weggen, S.; Stark, H.; Schneider, G. DOGS: Reaction-driven de novo design of bioactive compounds. PLoS Comput. Biol. 2012, 8, 1–12. [Google Scholar] [CrossRef]
- Chevillard, F.; Stotani, S.; Karawajczyk, A.; Hristeva, S.; Pardon, E.; Steyaert, J.; Tzalis, D.; Kolb, P. Interrogating dense ligand chemical space with a forward-synthetic library. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 11496–11501. [Google Scholar] [CrossRef]
- Shivanyuk, A.; Ryabukhin, S.V.; Bogolubsky, A.V.; Tolmachev, A. Enamine real database: Making chemical diversity real. Chem. Today 2007, 25, 58–59. [Google Scholar]
- Enamine REAL Space and REAL Database. Available online: https://enamine.net/library-synthesis/real-compounds (accessed on 7 September 2018).
- Lyu, J.; Irwin, J.J.; Roth, B.L.; Shoichet, B.K.; Levit, A.; Wang, S.; Tolmachova, K.; Singh, I.; Tolmachev, A.A.; Che, T.; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [Google Scholar] [CrossRef]
- Nicolaou, C.A.; Watson, I.A.; Hu, H.; Wang, J. The proximal lilly collection: Mapping, exploring and exploiting feasible chemical space. J. Chem. Inf. Model. 2016, 56, 1253–1266. [Google Scholar] [CrossRef]
- Borysko, P.; Moroz, Y.S.; Vasylchenko, O.V.; Hurmach, V.V.; Starodubtseva, A.; Stefanishena, N.; Nesteruk, K.; Zozulya, S.; Kondratov, I.S.; Grygorenko, O.O. Straightforward hit identification approach in fragment-based discovery of bromodomain-containing protein 4 (BRD4) inhibitors. Bioorganic Med. Chem. 2018, 26, 3399–3405. [Google Scholar] [CrossRef]
- Rarey, M.; Stahl, M. Similarity searching in large combinatorial chemistry spaces. J. Comput. Aided. Mol. Des. 2001, 15, 497–520. [Google Scholar] [CrossRef]
- Cochran, A.G.; Conery, A.R.; Sims, R.J. Bromodomains: A new target class for drug development. Nat. Rev. Drug Discov. 2019, 18, 609–628. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, P.; Chen, H.; Wold, E.A.; Tian, B.; Brasier, A.R.; Zhou, J. Drug discovery targeting bromodomain-containing protein 4. J. Med. Chem. 2017, 60, 4533–4558. [Google Scholar] [CrossRef]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef]
- Ali, I.; Conrad, R.J.; Verdin, E.; Ott, M. Lysine acetylation goes global: From epigenetics to metabolism and therapeutics. Chem. Rev. 2018, 118, 1216–1252. [Google Scholar] [CrossRef]
- Duan, Y.; Guan, Y.; Qin, W.; Zhai, X.; Yu, B.; Liu, H. Targeting Brd4 for cancer therapy: Inhibitors and degraders. Medchemcomm 2018, 9, 1779–1802. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Bradbury, R.H.; Callis, R.; Carr, G.R.; Chen, H.; Clark, E.; Feron, L.; Glossop, S.; Graham, M.A.; Hattersley, M.; Jones, C.; et al. Optimization of a series of bivalent triazolopyridazine based bromodomain and extraterminal inhibitors: The discovery of (3 R )-4-[2-[4-[1-(3-Methoxy-[1,2,4]triazolo[4,3-b]pyridazin-6-yl)-4-piperidyl]phenoxy]ethyl]-1,3-dimethyl-piperazin-2. J. Med. Chem. 2016, 59, 7801–7817. [Google Scholar] [CrossRef]
- Rarey, M.; Dixon, J.S. Feature trees: A new molecular similarity measure based on tree matching. J. Comput. Aided. Mol. Des. 1998, 12, 471–490. [Google Scholar] [CrossRef]
- BioSolveIT GmbH SeeSAR version 7.2. Available online: https://www.biosolveit.de/SeeSAR/ (accessed on 10 January 2018).
- Schärfer, C.; Schulz-Gasch, T.; Hert, J.; Heinzerling, L.; Schulz, B.; Inhester, T.; Stahl, M.; Rarey, M. CONFECT: Conformations from an expert collection of torsion patterns. ChemMedChem 2013, 8, 1690–1700. [Google Scholar] [CrossRef]
- Ember, S.W.J.; Zhu, J.-Y.; Olesen, S.H.; Martin, M.P.; Becker, A.; Berndt, N.; Georg, G.I.; Schönbrunn, E. Acetyl-lysine binding site of bromodomain-containing protein 4 (BRD4) interacts with diverse kinase inhibitors. ACS Chem. Biol. 2014, 9, 1160–1171. [Google Scholar] [CrossRef]
- Romero, F.A.; Taylor, A.M.; Crawford, T.D.; Tsui, V.; Côté, A.; Magnuson, S. Disrupting acetyl-lysine recognition: progress in the development of bromodomain inhibitors. J. Med. Chem. 2016, 59, 1271–1298. [Google Scholar] [CrossRef]
- Liu, Z.; Tian, B.; Chen, H.; Wang, P.; Brasier, A.R.; Zhou, J. Discovery of potent and selective BRD4 inhibitors capable of blocking TLR3-induced acute airway inflammation. Eur. J. Med. Chem. 2018, 151, 450–461. [Google Scholar] [CrossRef]
- UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [CrossRef]
- Lo, M.-C.; Aulabaugh, A.; Jin, G.; Cowling, R.; Bard, J.; Malamas, M.; Ellestad, G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 2004, 332, 153–159. [Google Scholar] [CrossRef]
- Matulis, D.; James, K.; Kranz, F.; Raymond Salemme, A.; Todd, M.J. Thermodynamic stability of carbonic anhydrase: Measurements of binding affinity and stoichiometry using thermofluor. Biochemistry 2005, 44, 5258–5266. [Google Scholar] [CrossRef]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef]
- BRD4 (BD1+BD2) TR-FRET Assay Kit Data Sheet. Available online: http://bpsbioscience.com/media/wysiwyg/BRDs/32612_150629.pdf (accessed on 18 January 2018).
- KNIME AG KNIME version 3.5.3. Available online: https://www.knime.com/downloads/download-knime (accessed on 12 December 2017).
- Urbaczek, S.; Kolodzik, A.; Fischer, J.R.; Lippert, T.; Heuser, S.; Groth, I.; Schulz-Gasch, T.; Rarey, M. NAOMI: On the almost trivial task of reading molecules from different file formats. J. Chem. Inf. Model. 2011, 51, 3199–3207. [Google Scholar] [CrossRef]
- Bietz, S.; Urbaczek, S.; Schulz, B.; Rarey, M. Protoss: A holistic approach to predict tautomers and protonation states in protein-ligand complexes. J. Cheminform. 2014, 6, 1–12. [Google Scholar] [CrossRef]
- BioSolveIT GmbH LeadIT version 2.32. Available online: https://www.biosolveit.de/LeadIT/ (accessed on 27 August 2017).
- Reulecke, I.; Lange, G.; Albrecht, J.; Klein, R.; Rarey, M. Towards an integrated description of hydrogen bonding and dehydration: Decreasing false positives in virtual screening with the HYDE scoring function. ChemMedChem 2008, 3, 885–897. [Google Scholar] [CrossRef]
- Schneider, N.; Hindle, S.; Lange, G.; Klein, R.; Albrecht, J.; Briem, H.; Beyer, K.; Claußen, H.; Gastreich, M.; Lemmen, C.; et al. Substantial improvements in large-scale redocking and screening using the novel HYDE scoring function. J. Comput. Aided. Mol. Des. 2012, 26. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 19–30 are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Query in the REAL Space | Hit | Similarity | ΔTm (°C) at | IC50 (μM) | |||
|---|---|---|---|---|---|---|---|---|
| Tanimoto | FTrees | 40 μM | 20 μM | 10 μM | ||||
| 1 |  |  | 0.456 | 0.956 | 0.71 ± 0.01 | 0.39 ± 0.08 | 0.31 ± 0.05 | 10.7 |
| 2 |  |  | 0.277 | 0.920 | 1.52 ± 0.59 | 0.72 ± 0.41 | 0.30 ± 0.10 | 26.4 |
| 3 |  |  | 0.323 | 0.933 | 1.33 ± 0.17 | 0.98 ± 0.22 | 0.34 ± 0.13 | 44.6 |
| 4 |  | 0.333 | 0.953 | 1.56 ± 0.05 | 0.78 ± 0.14 | 0.34 ± 0.19 | 68.2 | |
| 5 |  |  | 0.356 | 0.932 | 0.53 ± 0.25 | 0.44 ± 0.06 | 0.29 ± 0.01 | 141 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klingler, F.-M.; Gastreich, M.; Grygorenko, O.O.; Savych, O.; Borysko, P.; Griniukova, A.; Gubina, K.E.; Lemmen, C.; Moroz, Y.S. SAR by Space: Enriching Hit Sets from the Chemical Space. Molecules 2019, 24, 3096. https://doi.org/10.3390/molecules24173096
Klingler F-M, Gastreich M, Grygorenko OO, Savych O, Borysko P, Griniukova A, Gubina KE, Lemmen C, Moroz YS. SAR by Space: Enriching Hit Sets from the Chemical Space. Molecules. 2019; 24(17):3096. https://doi.org/10.3390/molecules24173096
Chicago/Turabian StyleKlingler, Franca-Maria, Marcus Gastreich, Oleksandr O. Grygorenko, Olena Savych, Petro Borysko, Anastasia Griniukova, Kateryna E. Gubina, Christian Lemmen, and Yurii S. Moroz. 2019. "SAR by Space: Enriching Hit Sets from the Chemical Space" Molecules 24, no. 17: 3096. https://doi.org/10.3390/molecules24173096
APA StyleKlingler, F.-M., Gastreich, M., Grygorenko, O. O., Savych, O., Borysko, P., Griniukova, A., Gubina, K. E., Lemmen, C., & Moroz, Y. S. (2019). SAR by Space: Enriching Hit Sets from the Chemical Space. Molecules, 24(17), 3096. https://doi.org/10.3390/molecules24173096