Position Impact of Hydroxy Groups on Spectral, Acid–Base Profiles and DNA Interactions of Several Monohydroxy Flavanones

 ,
,  ,
, 
Abstract
1. Introduction
2. Results and Discussion
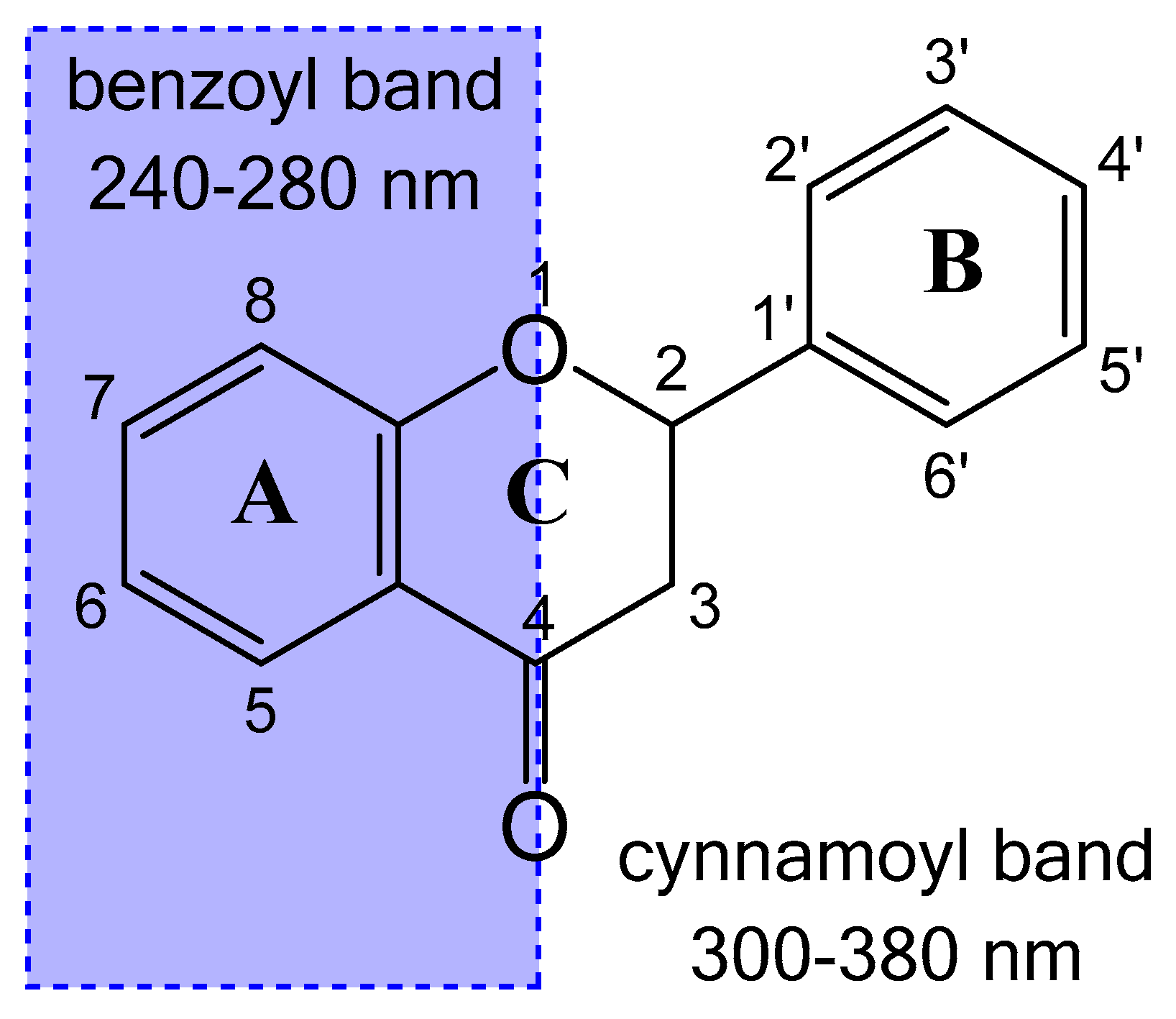
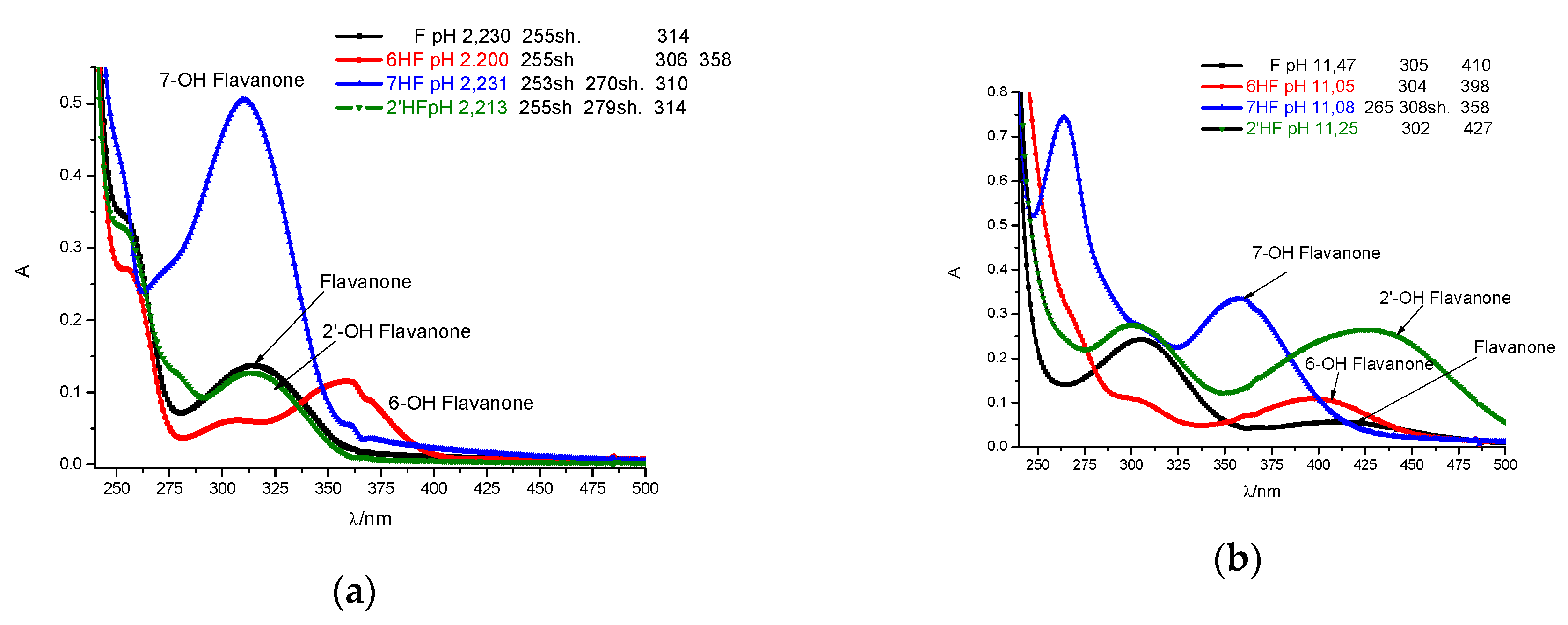
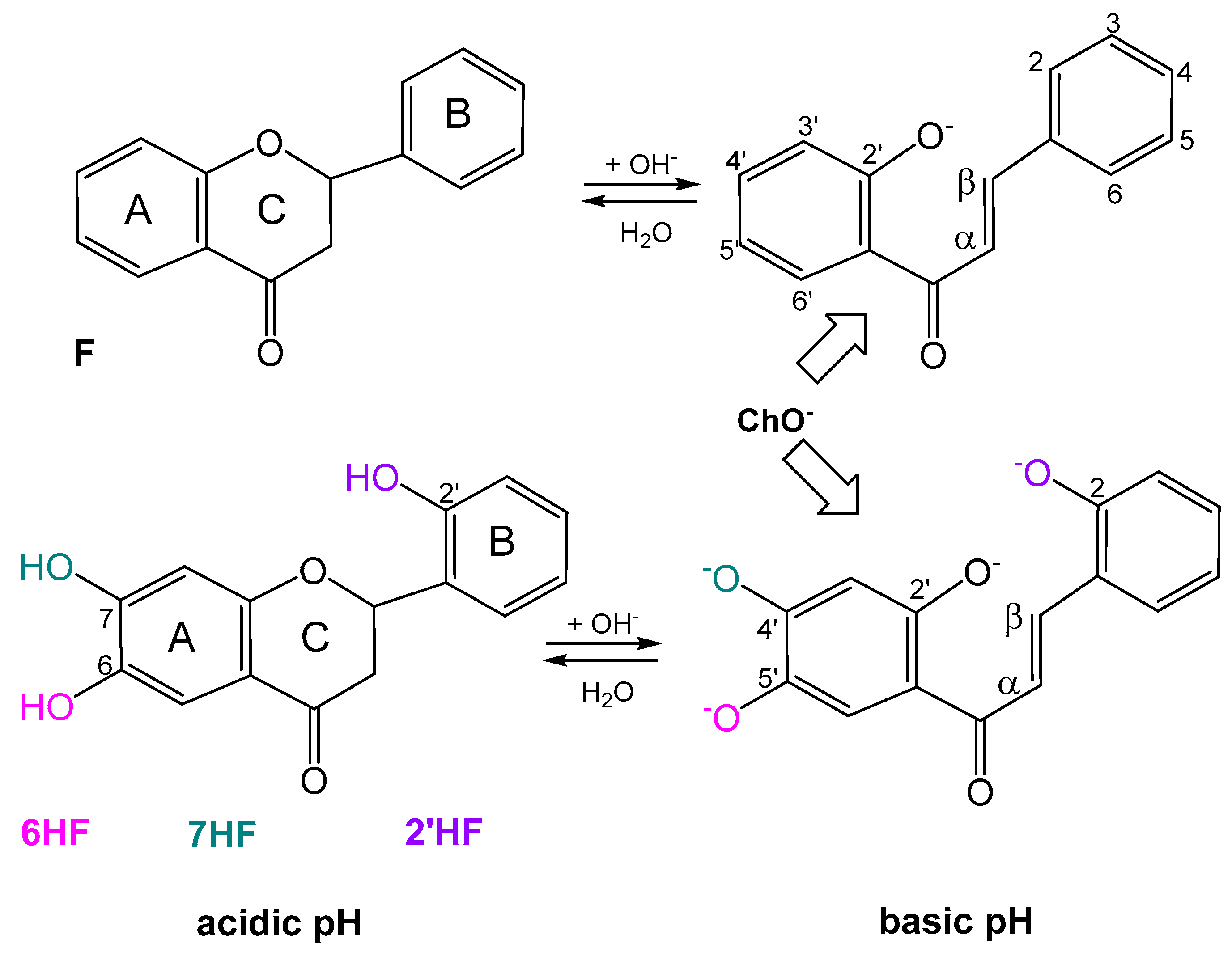
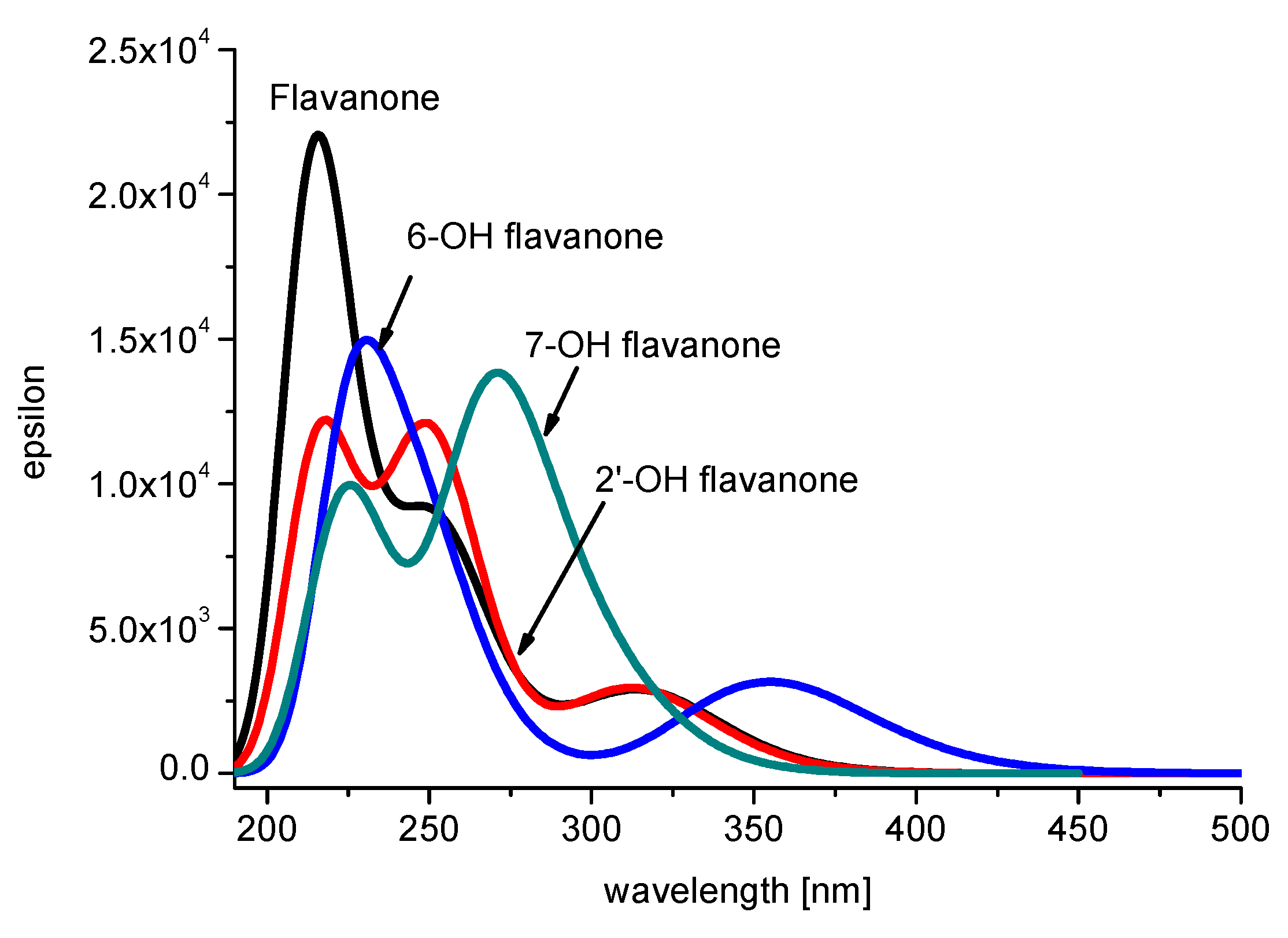
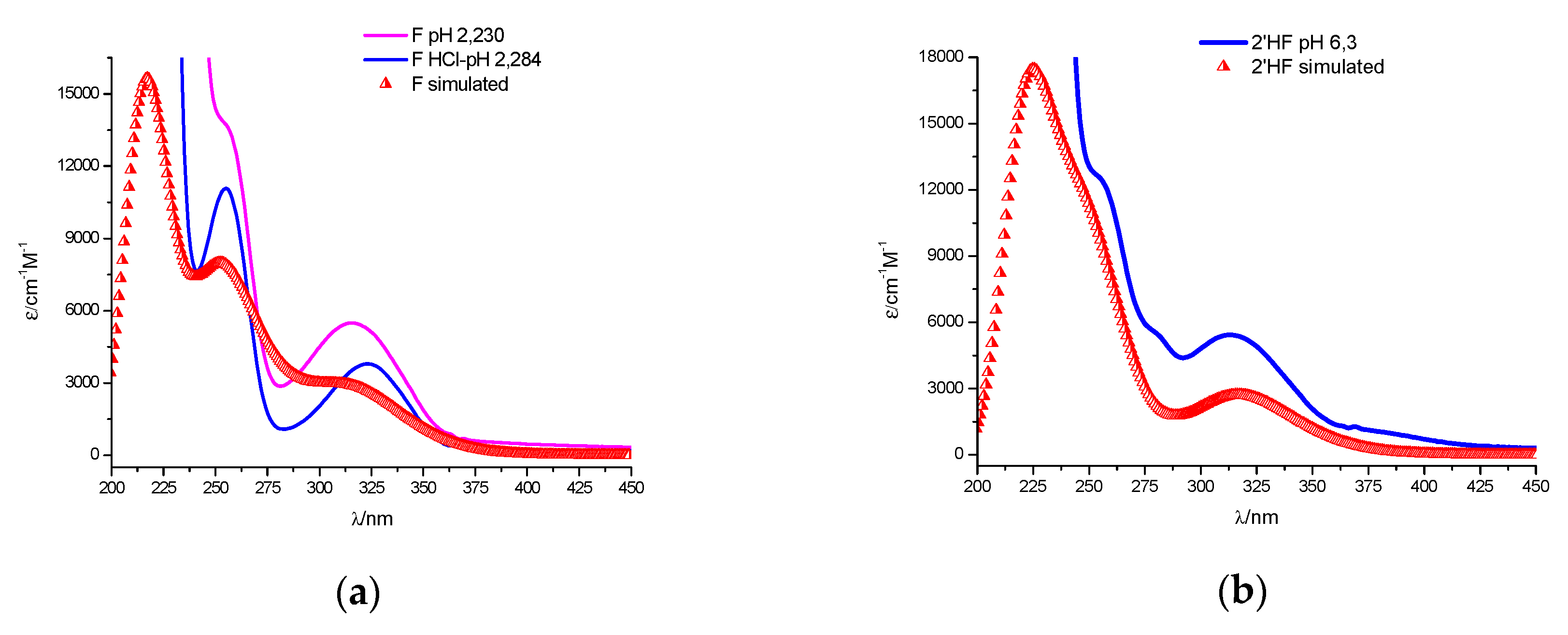
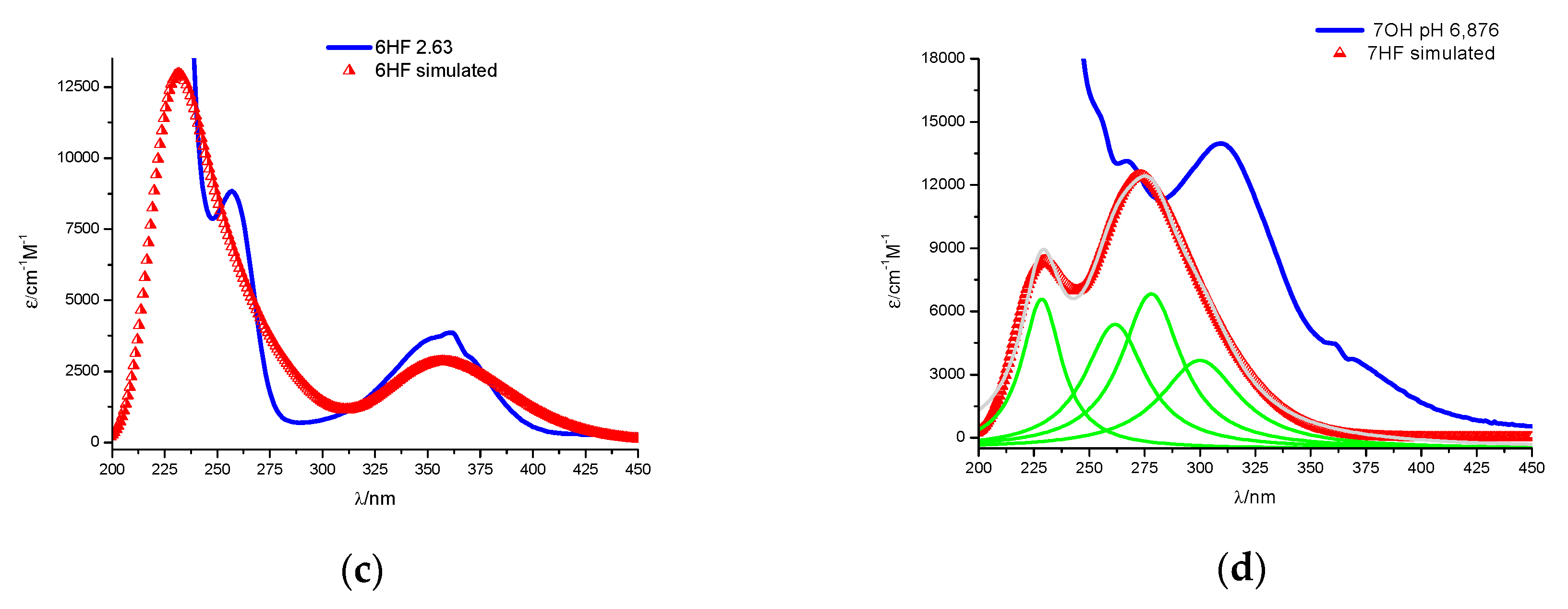
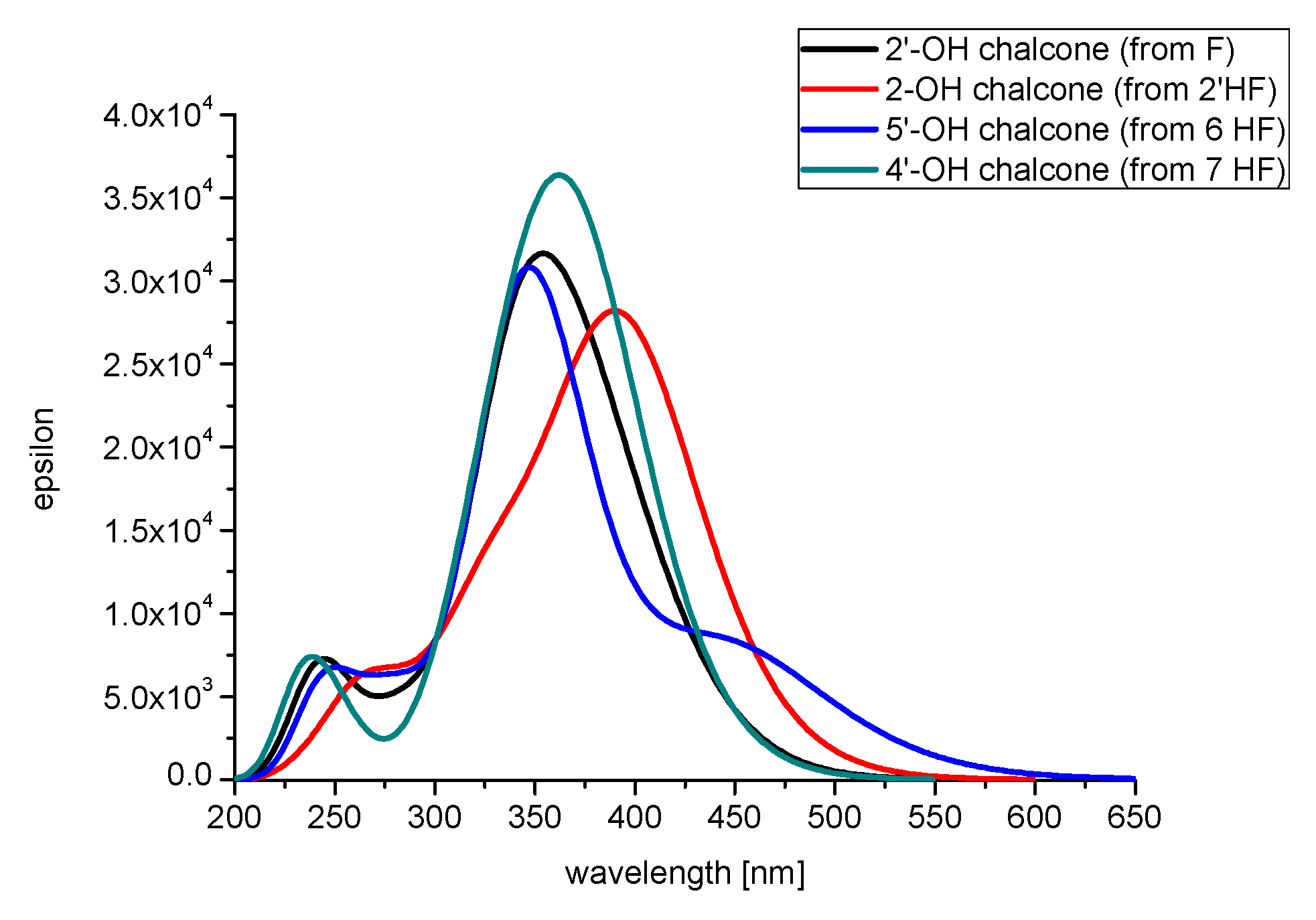
2.1. Spectral Profiles of Flavanones
2.2. Theoretical Calculations
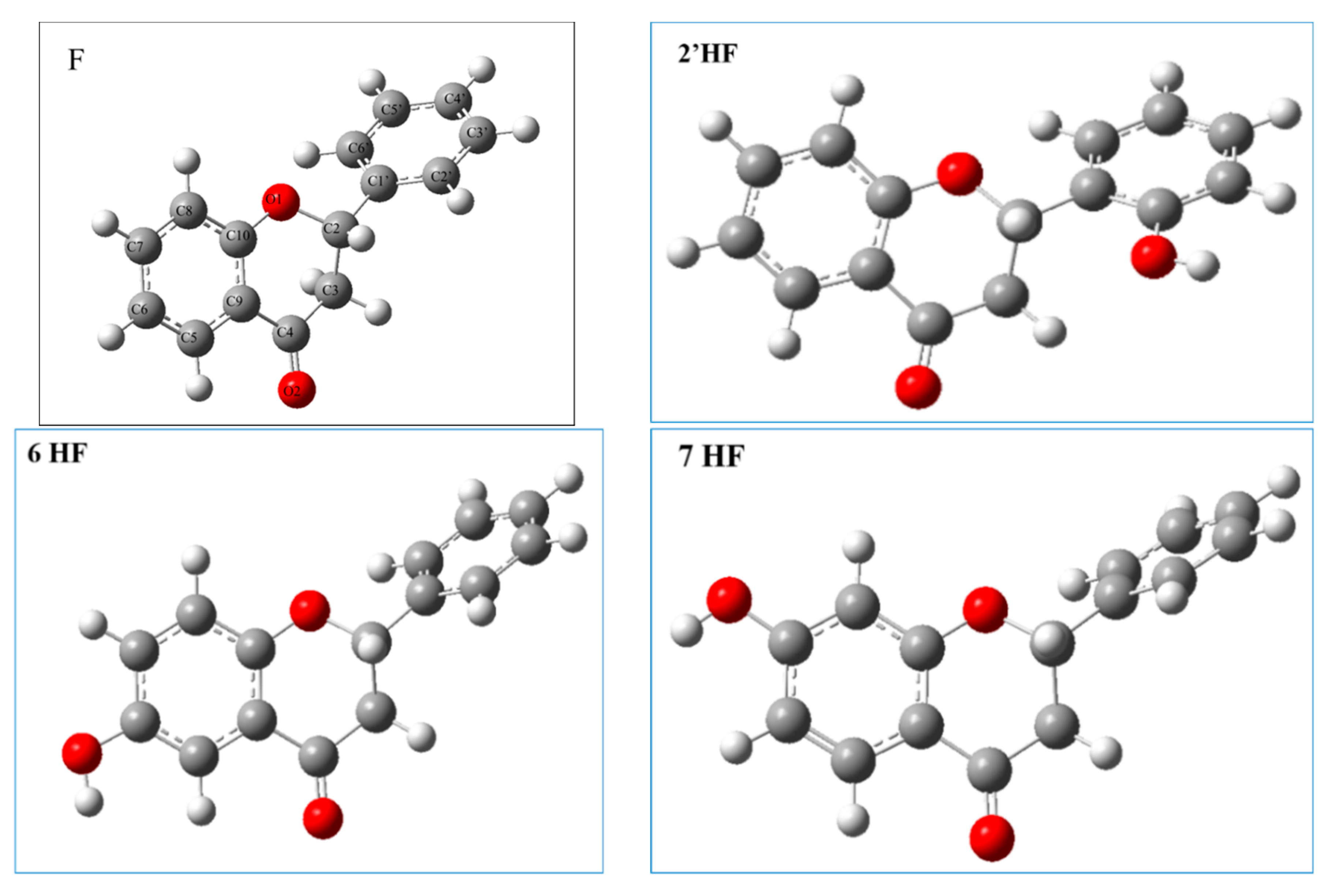
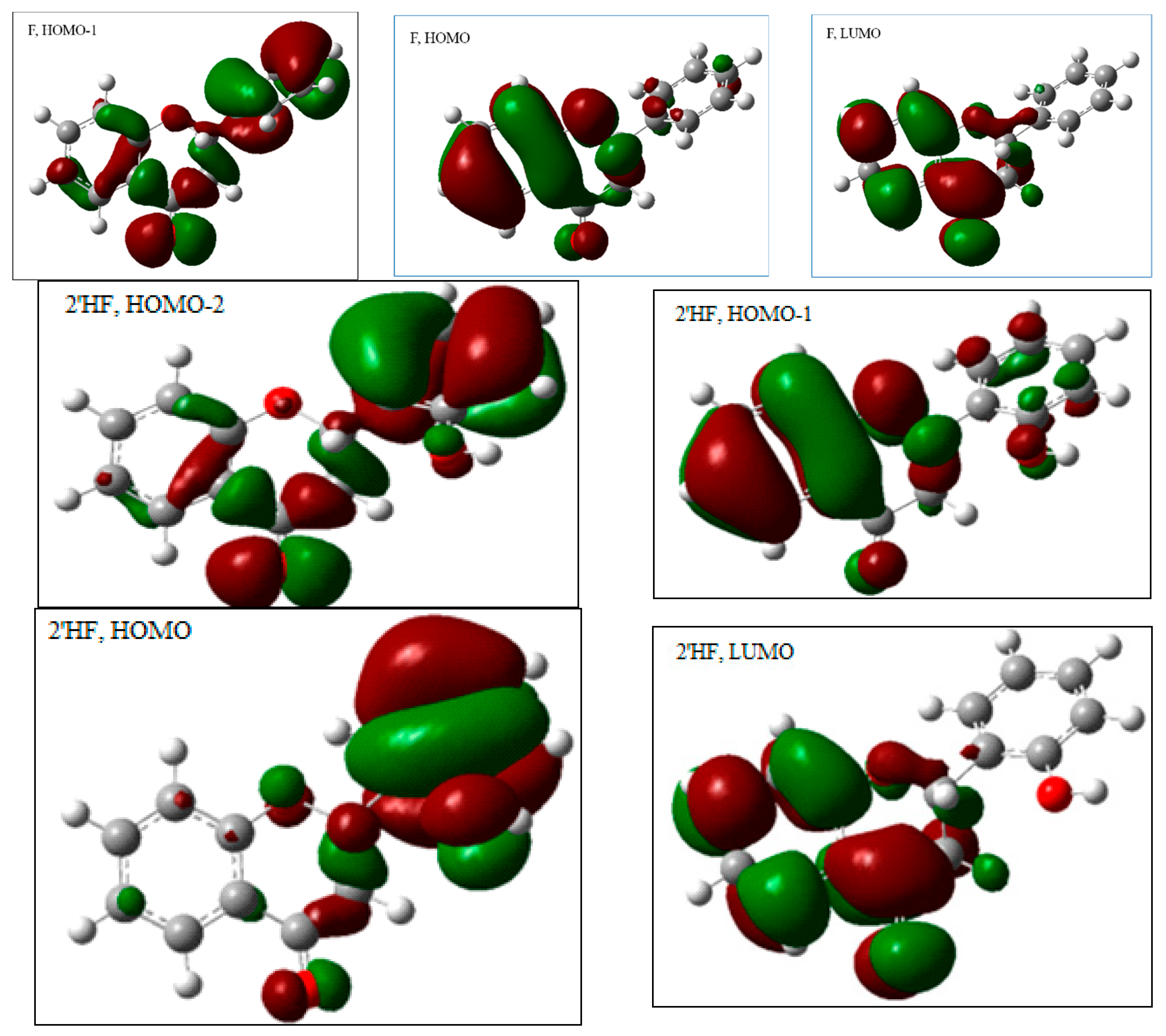
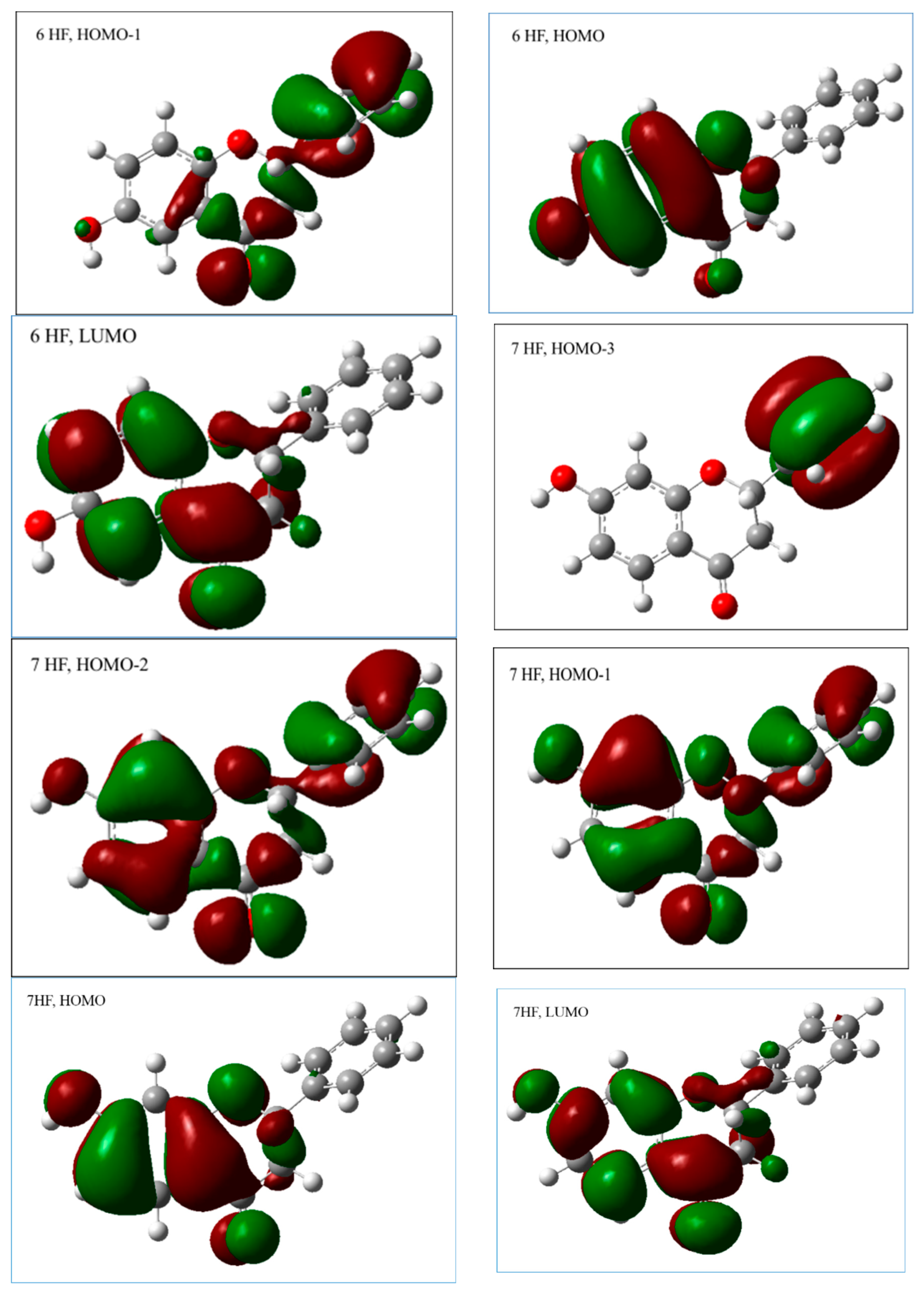
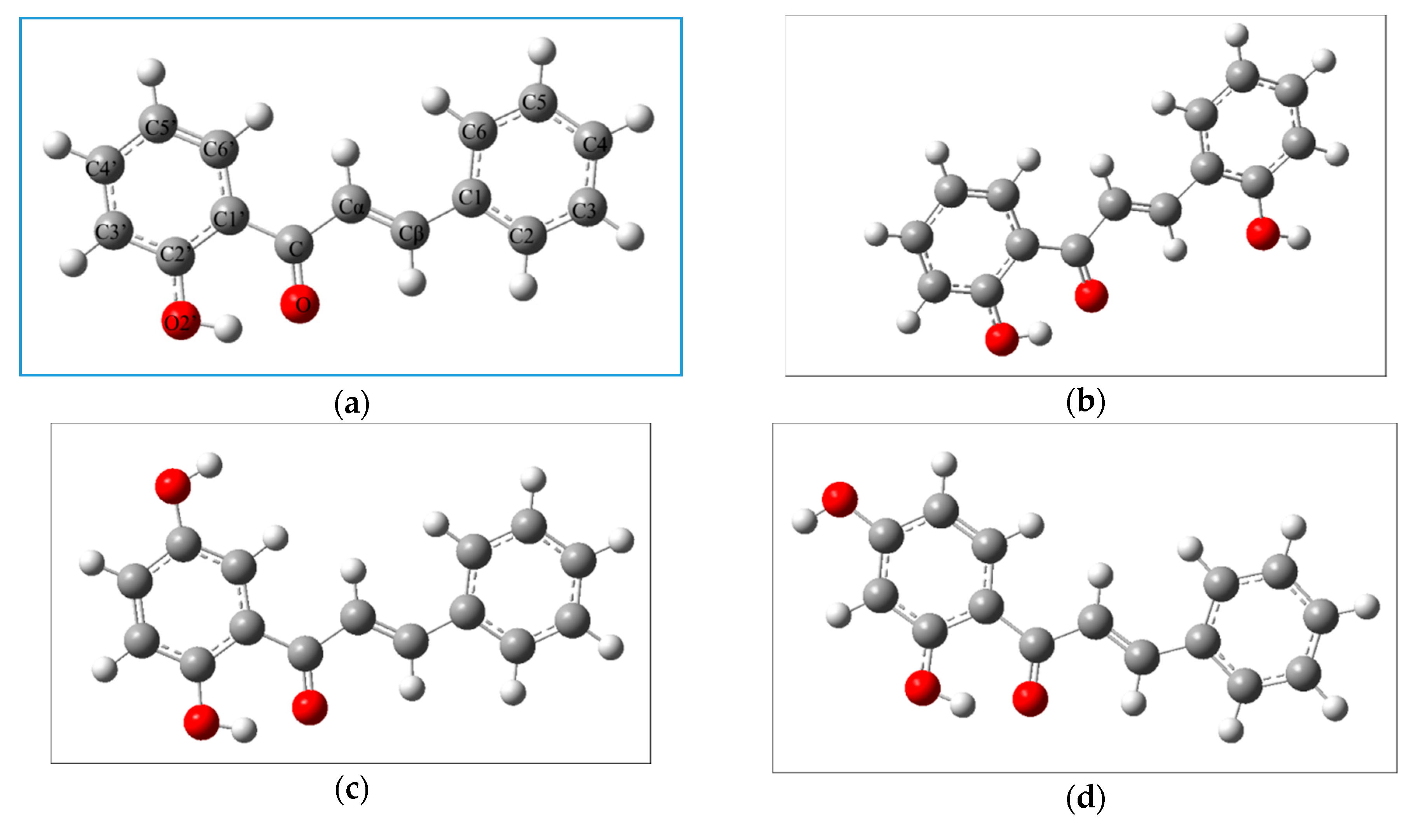
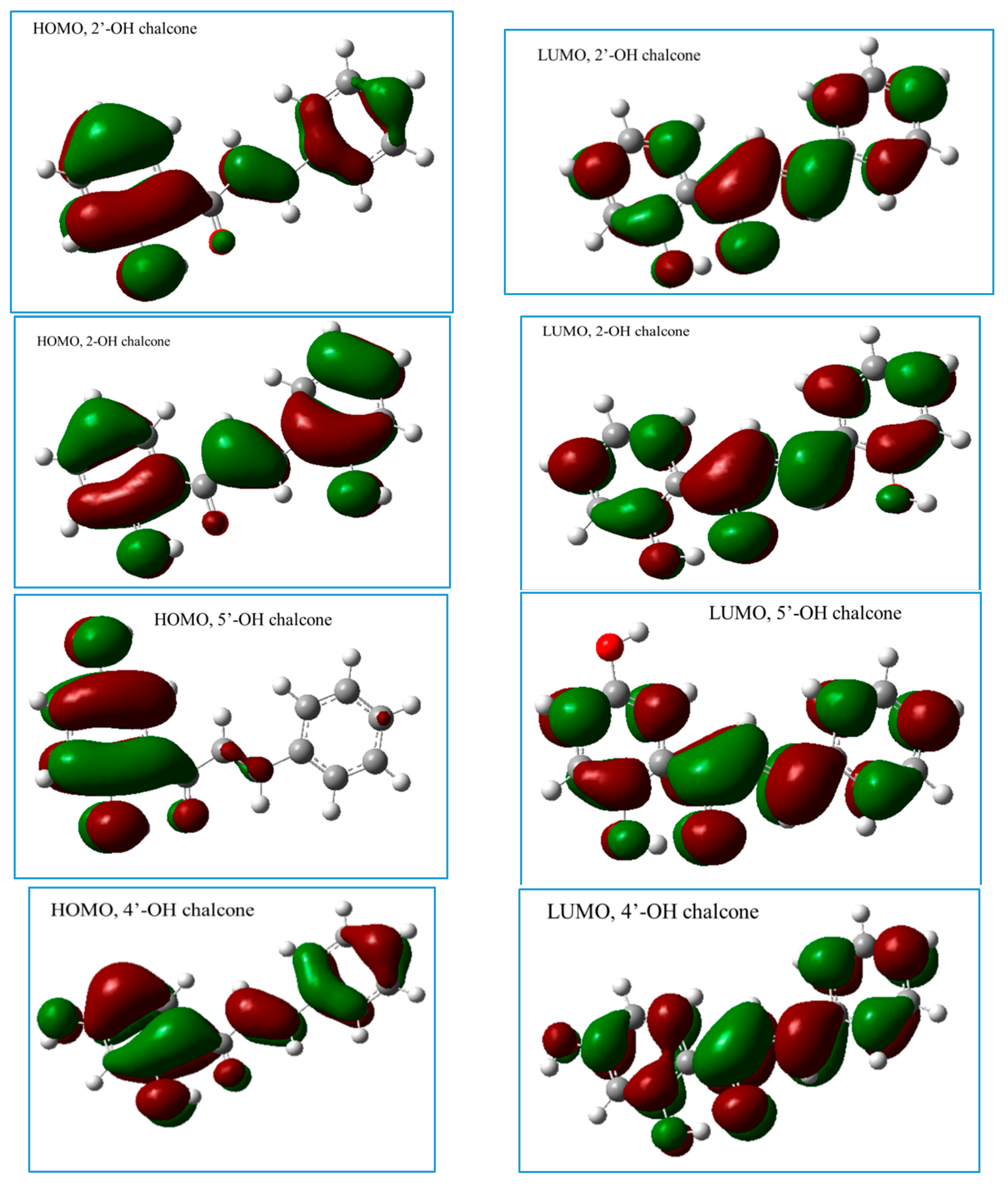
2.2.1. The Electronic Structures of the Compounds Studied Predicted by the DFT(B3LYP)/6-31G(d,p)/PCM Method
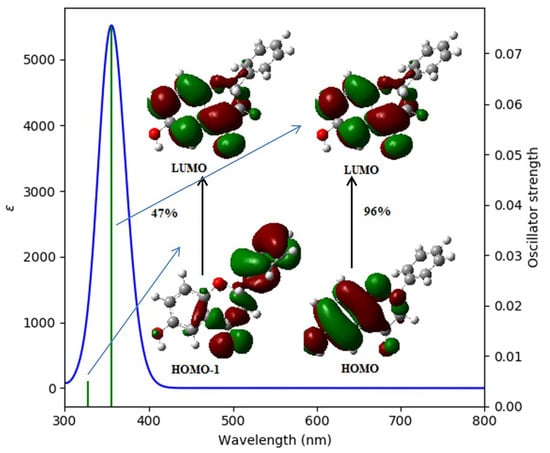
2.2.2. The Theoretically Predicted UV–Vis Characteristic of the Studied Flavanones
2.2.3. The Electronic Structures of the Chalcones Predicted by the DFT(B3LYP)/6-31G(d,p)/PCM Method
2.3. Cyclic Voltammetry
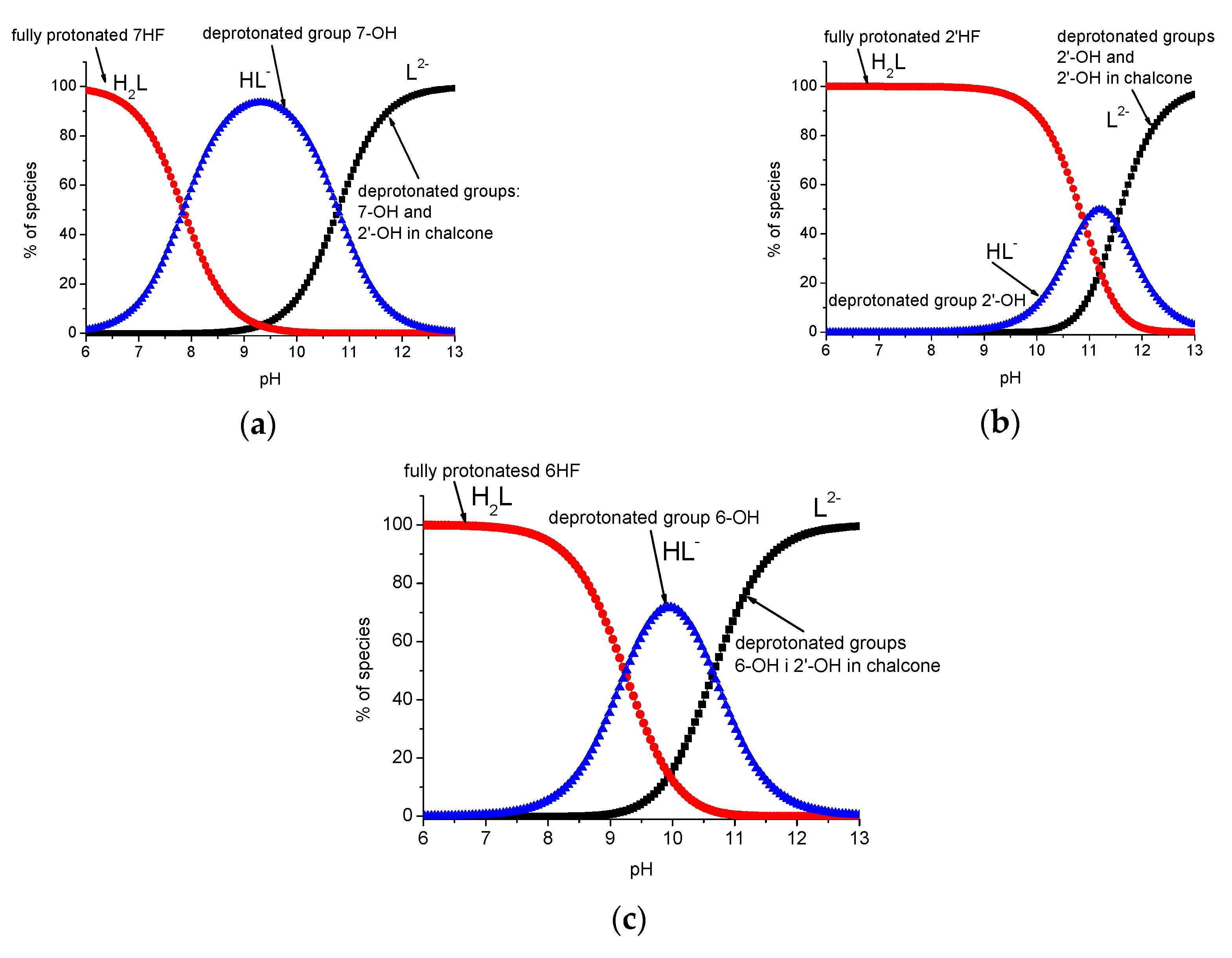
2.4. Acid–Base Profiles of Flavanones Derived from Potentiometric Titration
2.5. Absorption Spectroscopic Measurements of DNA
3. Materials and Methods
3.1. Reagents
3.2. Electrochemical Measurements
3.2.1. Potentiometry
3.2.2. Cyclic Voltammetry
3.3. UV–Vis Spectrophotometric Experiments
3.4. Computational Methods and Softwares
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dixon, R.A.; Dey, P.M.; Lamb, C.J. Phytoalexins: Enzymology and molecular biology. Adv. Enzymol. Relat. Areas Mol. Biol. 1983, 55, 1–136. [Google Scholar] [PubMed]
- Seo, J.; Ryu, J.Y.; Han, J.; Ahn, J.H.; Sadowsky, M.J.; Hur, H.G.; Chong, Y. Amino acid substitutions in naphthalene dioxygenase from Pseudomonas sp. strain NCIB 9816-4 result in regio- and stereo-specific hydroxylation of flavanone and isoflavanone. Appl. Microbiol. Biotechnol. 2013, 97, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.C.; Kuo, W.H.; Chen, P.N.; Chang, H.R.; Lin, T.H.; Yang, W.E.; Hsieh, Y.S.; Chu, S.C. Flavanone and 2′-OH flavanone inhibit metastasis of lung cancer cells via down-regulation of proteinases activities and MAPK pathway. Chem. Biol. Interact. 2007, 167, 193–206. [Google Scholar] [CrossRef]
- HJ Heo, D.O.; Kim, S.C.; Shin, M.J.; Kim, B.G.; Kim, D.H. Shin Effect of antioxidant flavanone, naringenin from Citrus junoson neuroprotection. J. Agric. Food Chem. 2004, 52, 1520–1525. [Google Scholar] [CrossRef]
- Foglia, P.; Giansati, P.; Samperi, R. Flavonoids: Chemical properties and analytical methodologies of identification and quantitation in foods and plants. Nat. Prod. Res. 2011, 5, 469–495. [Google Scholar]
- Tsuchiya, H.; Sato, M.; Miyazaki, T.; Fujiwara, S.; Tanigaki, S.; Ohyama, M.; Tanaka, T.; Iinuma, M. Comparative study on the antibacterial activity of phytochemical flavanones against methicillin-resistant Staphylococcus aureus. J. Ethnopharmacol. 1996, 50, 27–34. [Google Scholar] [CrossRef]
- Ko, C.H.; Shen, S.C.; Lin, H.Y.; Hou, W.C.; Lee, W.R.; Yang, L.L.; Chen, Y.C. Flavanones structure-related inhibition on TPA-induced tumor promotion through suppression of extracellular signal-regulated protein kinases: Involvement of prostaglandin E2 in anti-promotive process. J. Cell Physiol. 2002, 193, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Okuda, T.; Yoshida, T.; Hatano, T. Pharmacologically active tannins isolated from medicinal plants. Basic Life Sci. 1992, 59, 539–569. [Google Scholar]
- Szliszka, E.; Kostrzewa-Susłow, E.; Bronikowska, J.; Jaworska, D.; Janeczko, T.; Czuba, Z.P.; Krol, W. Synthetic Flavanones Augment the Anticancer Effect of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL). Molecules 2012, 17, 11693–11711. [Google Scholar] [CrossRef]
- Wu, K.; Ning, Z.; Zhou, J.; Wang, B.; Fan, J.; Zhu, J.; Gao, Y.; Wang, X.; Hsieh, J.T.; He, D. 2′-hydroxyflavanone inhibits prostate tumor growth through inactivation of AKT/STAT3 signaling and induction of cell apoptosis. Oncol. Rep. 2014, 32, 131–138. [Google Scholar] [CrossRef]
- Nagaprashantha, L.D.; Singhal, J.; Li, H.; Warden, C.; Liu, X.; Horne, D.; Awasthi, S.; Salgia, R.; Singhal, S.S. 2′-Hydroxyflavanone effectively targets RLIP76-mediated drug. Oncotarget 2018, 9, 18053–18068. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pouget, C.; Lauthier, F.; Simon, A.; Fagnere, C.; Basly, J.P.; Delage, C.; Chulia, A.J. Flavonoids: Structural requirements for antiproliferative activity on breast cancer cells. Bioorg. Med. Chem. Lett. 2001, 11, 3095–3097. [Google Scholar] [CrossRef]
- Joray, M.B.; Trucco, L.D.; González, M.L.; Díaz Napal, G.N.; Palacios, S.M.; Bocco, J.L.; Carpinella, M.C. Antibacterial and Cytotoxic Activity of Compounds Isolated from Flourensia oolepis. Evidence-Based. BMC Complement. Altern. Med. 2015, 11. [Google Scholar] [CrossRef]
- Anouar, H.; Gierschner, J.; Duroux, J.L.; Trouillas, P. UV/Visible spectra of natural polyphenols: A time-dependent density functional theory study. Food Chem. 2012, 131, 79–89. [Google Scholar] [CrossRef]
- Wróblewski, T.; Ushakou, D.V. Photophysical properties of 7-hydroxyflavanone: Quantum chemical calculations and experimental studies. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 215, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Serdiuk, I.E.; Wera, M.; Roshal, A.D. Spectral features of 4′-substituted 2′-hydroxychalcones in solutions and crystals: Spectroscopic and theoretical investigations. J. Phys. Chem. A 2018, 122, 2030–2038. [Google Scholar] [CrossRef] [PubMed]
- Amat, A.; Clementi, C.; De Angelis, F.; Sgamellotti, A.; Fantacci, S. Absorption and emission of the apigenin and luteolin flavonoids: A TDDFT investigation. J. Phys. Chem. A 2009, 113, 15118–15126. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, P.; Priyadarsini, K.I.; Naumov, S.; Rao, B.S. Radiation and quantum chemical studies of chalcone derivatives. J. Phys. Chem. A 2010, 114, 7877–7885. [Google Scholar] [CrossRef] [PubMed]
- Casida, M.E. Time dependent density- functional theory of molecules and molecular solids. J. Mol. Struct. THEOCHEM. 2009, 914, 3–18. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Hashmi, M.A.; Khan, A.; Ayub, K.; Farooq, M. Spectroscopic and density functional theory studies of 5,7,30,50-tetrahydroxyflavanone from the leaves of Olea ferruginea. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 128, 225–230. [Google Scholar] [CrossRef]
- Marković, M.; Tošović, J. Application of time-dependent density functional and natural bond orbital theories to the UV−Vis absorption spectra of some phenolic compounds. J. Phys. Chem. A 2015, 119, 9352–9362. [Google Scholar] [CrossRef]
- Arczewska, M.; Kamiński, D.M.; Gieroba, B.; Gagoś, M. Acid−base properties of xanthohumol: A computational and experimental investigation. J. Nat. Prod. 2017, 80, 3194–3202. [Google Scholar] [CrossRef] [PubMed]
- Dreuw, A.; Head-Gordon, M. Single-reference ab initio methods for the calculation of excited states of large molecules. Chem. Rev. 2005, 105, 4009–4037. [Google Scholar] [CrossRef] [PubMed]
- Scalmani, G.; Frisch, M.J. Geometries and properties of excited states in the gas phase and in solution: Theory and application of a time-dependent density functional theory polarizable continuum model. J. Chem. Phys. 2006, 124, 94107–94115. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M.J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110–114125. [Google Scholar] [PubMed]
- Chou, P.T.; Martinez, M.L.; Cooper, W.C. Direct evidence of excited-state intramolecular proton transfer in 2′-hydroxychalcone and photooxygenation forming 3-hydroxyflavone. J. Am. Chem. Soc. 1992, 114, 4943–4944. [Google Scholar] [CrossRef]
- Mabry, T.J.; Markham, K.R.; Thomas, M.B. The Ultraviolet Spectra of Isoflavones, Flavanones and Dihydroflavonols. In The Systematic Identification of Flavonoids, 1st ed.; Springer-Verlag: Basel, Switzerland, 1970; pp. 165–226. [Google Scholar]
- Gattuso, G.; Barreca, D.; Gargiulli, C.; Leuzzi, U.; Caristi, C. Flavonoid Composition of Citrus Juices. Molecules 2007, 12, 1641–1673. [Google Scholar] [CrossRef]
- Mabry, T.J.; Markham, K.R.; Thomas, M.B. The Ultraviolet Spectra of Flavones and Flavonols. In The Systematic Identification of Flavonoids, 1st ed.; Springer: Basel, Switzerland, 1970; pp. 41–57. [Google Scholar]
- Rong, Y.; Wang, Z.W.; Wu, J.H.; Zhao, B. A theoretical study on cellular antioxidant activity of selected flavonoids. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 93, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Chapter VIII; Excited State Dynamics of 6-Hydroxy Flavanone. Available online: http://shodhganga.inflibnet.ac.in/bitstream/10603/108422/8/chapter%208.pdf (accessed on 29 June 2019).
- Błazińska, P.; Sykuła, A. Spectral characteristics of flavanone and its thiocarbohydrazide derivativein. In The Book of Articles—National Scientific Conference, “Understanding Science”; Solarczyk, P., II, Ed.; Promovendi Foundation Publishing: Lodz, Poland, 29 September 2018; pp. 38–45. ISBN 978-83-950109-4-1. [Google Scholar]
- Zhao, G.J.; Han, K.L. Effects of hydrogen bonding on tuning photochemistry: Concerted hydrogen-bond strengthening and weakening. Chemphyschem 2008, 9, 1842–1846. [Google Scholar] [CrossRef] [PubMed]
- Cisak, A.; Mielczarek, C. Practical and theoretical aspects of flavanone–chalcone isomerisations. J. Chem. Soc. Perkin Trans. 1992, 2, 1603–1607. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- GaussView 05 Software Package. GaussView, Version 5; Dennington, R., Keith, T., Millam, J., Eds.; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- The Cambridge Crystallographic Data Centre (CCDC). Available online: www.ccdc.cam.ac.uk/structures (accessed on 29 June 2019).
- Shoja, M.; Samuel, K.; Athanasopoulos, D. Crystal structure of 6-hydroxyflavanone, C15H12O3. Z Krist New Cryst. Struct. 1998, 213, 373. [Google Scholar]
- Ajmala Shireen, P.; Abdul Mujeeb, V.M.; Muraleedharan, K. Theoretical insights on flavanones as antioxidants and UV filters: A TDDFT and NLMO study. J. Photochem. Photobiol. B Biol. 2017, 170, 286–294. [Google Scholar] [CrossRef]
- Mendoza-Wilson, A.M.; Glossman-Mitnik, D. CHIH-DFT study of the electronic properties and chemical reactivity of quercetin. J. Mol. Struct. THEOCHEM 2005, 716, 67–72. [Google Scholar] [CrossRef]
- Ahmed, S.; Shakeel, F. Antioxidant Activity Coefficient, Mechanism, and Kinetics of Different Derivatives of Flavones and Flavanones Towards Superoxide Radical. Czech J. Food Sci. 2012, 30, 153–163. [Google Scholar] [CrossRef]
- Morań Vieyra, F.E.; Boggetti, J.; Zampini, I.C.; Ordoñez, R.M.; Isla, M.A.I.; Alvarez, R.M.S.; De Rosso, V.; Mercadante, A.Z.; Borsarelli, C.D. Singlet oxygen quenching and radical scavenging capacities of structurally-related flavonoids present in Zuccagnia punctata Cav. Free Radic. Res. 2009, 43, 553–564. [Google Scholar] [CrossRef]
- Kocabova, J.; Fiedler, J.; Degano, I.; Sokolova, R. Oxidation mechanism of flavanone taxifolin. Electrochemical and spectroelectrochemical investigation. Electrochim. Acta 2016, 187, 358–363. [Google Scholar] [CrossRef]
- Masek, A.; Chrzescijanska, E.; Latos, M.; Zaborski, M. Influence of hydroxyl substitution on flavanone antioxidants properties. Food Chem. 2017, 215, 501–507. [Google Scholar] [CrossRef]
- Babić, S.; Horvat, A.J.M.; Pavlović, D.M.; Kaštelan-Macan, M. Determination of pKa values of active pharmaceutical ingredients. TrAC Trends Analyt. Chem. 2007, 26, 1043–1061. [Google Scholar] [CrossRef]
- Herrero-Martínez, J.M.; Repollés, C.; Bosch, E.; Rosés, M.; Ràfols, C. Potentiometric determination of aqueous dissociation constants of flavonols sparingly soluble in water. Talanta 2008, 74, 1008–1013. [Google Scholar] [CrossRef] [PubMed]
- Albert, A.; Serjeant, E.P. Determination of ionization constants by potentiometric titration using a glass electrode. In The Determination of Ionization Constants, 3rd ed.; Chapman and Halk: London, UK, 1984; pp. 14–38. [Google Scholar]
- Li, W.H.; Cao, J.J.; Lu, R.; Che, C.X.; Wei, Y.J. Study on Fluorescence Properties of Flavanone and Its Hydroxyl Derivatives. Guang Pu Xue Yu Guang Pu Fen Xi 2016, 36, 1007–1012. [Google Scholar] [PubMed]
- Bianchi, A.; Garcia-España, E. The Use of Calculated Species Distribution Diagrams to Analyze Thermodynamic Selectivity. J. Chem. Educ. 1999, 76, 1727–1732. [Google Scholar] [CrossRef]
- Rimarčík, J.; Lukeš, V.; Klein, E.; Ilčin, M. Study of the solvent effect on the enthalpies of homolytic and heterolytic N–H bond cleavage in p-phenylenediamine and tetracyano-p-phenylenediamine. J. Mol. Struct. (Theochem.) 2010, 952, 25–30. [Google Scholar] [CrossRef]
- Klein, E.; Lukeš, V.; Ilčin, M. DFT/B3LYP study of tocopherols and chromans antioxidant action energetic. Chem. Phys. 2007, 336, 51–57. [Google Scholar] [CrossRef]
- Long, E.C.; Barton, J.K. On demonstrating DNA intercalation. Acc. Chem. Res. 1990, 23, 271–273. [Google Scholar] [CrossRef]
- Sheikh, J.; Parvez, A.; Ingle, V.; Juneja, H.; Dongre, R.; Chohan, Z.H.; Youssoufi, M.H.; Hadda, T.B. Synthesis, biopharmaceutical characterization, antimicrobial and antioxidant activities of 1-(4′-O-β-D-glucopyranosyloxy-2′-hydroxyphenyl)-3-arylpropane-1,3-diones. Eur. J. Med. Chem. 2011, 46, 1390–1399. [Google Scholar] [CrossRef]
- Parvez, A.; Meshram, J.; Tiwari, V.; Sheikh, J.; Dongre, R.; Youssoufi, M.H.; Hadda, T.B. Pharmacophores modeling in terms of prediction of theoretical physico-chemical properties and verification by experimental correlations of novel coumarin derivatives produced via Betti’s protocol. Eur. J. Med. Chem. 2010, 45, 4370–4378. [Google Scholar] [CrossRef]
- Sirajuddin, M.; Ali, S.; Badshah, A. Drug-DNA interactions and their study by UV–Visible, fluorescence spectroscopies and cyclic voltametry. J. Photochem. Photobiol. B Biol. 2013, 124, 1–19. [Google Scholar] [CrossRef]
- Yousuf, S.; Radhika, D.; Enoch, I.V.; Easwaran, M. The influence of β-cyclodextrin encapsulation on the binding of 2′-hydroxyflavanone with calf thymus DNA. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 98, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Meloun, M.; Havel, J.; Högfeldt, E. Computation of Solution Equilibria: A Guide to Methods in Potentiometry, Extraction and Spectrophotometry; Ellis Horwood Limited: Chichester, UK, 1988; (a) pp. 52–62, (b) pp. 84–89, (c) p. 19, (d) pp. 143–144. [Google Scholar]
- Wrobel, R.; Chmurzynski, L. Potentiometric pKa determination of standard substances in binary solvent systems. Anal. Chim. Acta 2000, 405, 303–308. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Irving, H.M.; Miles, M.G.; Pettit, L.D. A study of some problems in determining the stoicheiometric proton dissociation constants of complexes by potentiometric titrations using a glass electrode. Anal. Chim. Acta 1967, 38, 475–488. [Google Scholar] [CrossRef]
- Łodyga-Chruscińska, E.; Pilo, M.; Zucca, A.; Garribba, E.; Klewicka, E.; Rowińska-Żyrek, M.; Symonowicz, M.; Chrusciński, L.; Cheshchevik, V.T. Physicochemical, antioxidant, DNA cleaving properties and antimicrobial activity of fisetin-copper chelates. J. Inorg. Biochem. 2018, 180, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Aslan, N.E.K. Determination of Autoprotolysis Constants of Water-Organic Solvent Mixtures by Potentiometry. Microchim. Acta 2005, 151, 89–92. [Google Scholar]
- Gran, G. Determination of the equivalent point in potentiometric titrations. Acta Chem. Scand. 1950, 4, 559–577. [Google Scholar] [CrossRef]
- Gans, P.; Vacca, A.; Sabatini, A. SUPERQUAD: An improved general program for computation of formation constants from potentiometric data. J. Chem. Soc. Dalton Trans. 1985, 1195–1200. [Google Scholar] [CrossRef]
- Wolfe, A.; Shimer, G.; Meehan, T. Polycyclic aromatic hydrocarbons physically intercalate into duplex regions of denatured DNA. Biochemistry 1987, 26, 6392–6396. [Google Scholar] [CrossRef]
- Li, X.; Frisch, M.J. Energy-represented DIIS within a hybrid geometry optimization method. J. Chem. Theory Comput. 2006, 2, 835–839. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989; pp. 87–99. [Google Scholar]
- Parr, R.G.; Szentpaly, L.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Young, G. Density Functional Calculations. In Recent Progresses of Theory and Application; Young, G., Ed.; IntechOpen: London, UK, 2018; pp. 203–205. [Google Scholar]
- Wright, J.S.; Johnson, E.R.; Dilabio, G.A. Predicting the activity of phenolic antioxidants: Theoretical method, analysis of substituent effects and application to major families of antioxidants. J. Am. Chem. Soc. 2001, 123, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Bryantsev, V.S.; Diallo, M.S.; Goddard, W.A. pKa calculations of aliphatic amines, diamines, and aminoamides via density functional theory with a Poisson−Boltzmann continuum solvent model. J. Phys. Chem. 2007, 111, 4422–4430. [Google Scholar] [CrossRef] [PubMed]
- PerkinElmer Informatics. ChemDraw Version 15.0.0.106; PerkinElmer Informatics: Kraków, Poland, 2016. [Google Scholar]
- OriginLab Corporation. Origin(Pro), Version 2007; OriginLab Corporation: Northampton, MA, USA, 2007. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
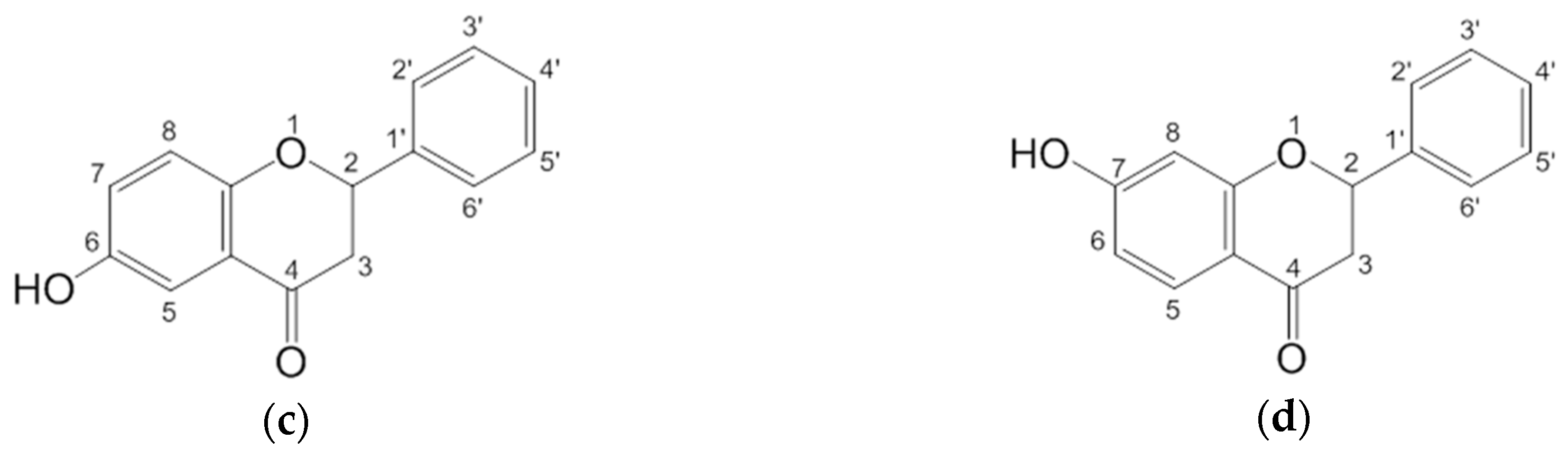{kind=link}
| Compound | C10C9O1C2 [°] | C9C10C4C3 [°] | O1C2C1′C6′ [°] | C3C2C1′C6′ [°] |
|---|---|---|---|---|
| F | −21.055 | −2.945 | −43.348 | 78.770 |
| 2′HF | −14.690 | −7.783 | −23.689 | 98.713 |
| 6HF | −22.449 | −2.431 | −43.553 | 78.464 |
| 6HFexp 1 | −18.892 | −4.058 | −55.323 | 77.974 |
| 7HF | −21.531 | −2.313 | −44.582 | 77.547 |
| Compound | EHOMO 2 [eV] | ELUMO 2 [eV] | HOMO-LUMO gap [eV] | IP [eV] | EA [eV] | η | χ | μ | ω | S |
|---|---|---|---|---|---|---|---|---|---|---|
| F | −6.40 | −1.70 | 4.70 | 6.40 | 1.70 | 2.35 | 4.05 | −4.05 | 4.05 | 0.21 |
| 2′HF | −6.15 | −1.67 | 4.48 | 6.15 | 1.67 | 2.24 | 3.91 | −3.91 | 3.91 | 0.22 |
| 6HF | −5.87 | −1.74 | 4.13 | 5.87 | 1.74 | 2.07 | 3.81 | −3.81 | 3.81 | 0.24 |
| 7 HF | −6.32 | −1.48 | 4.84 | 6.32 | 1.48 | 2.42 | 4.05 | −4.05 | 4.05 | 0.21 |
| 2S Stereoisomer | S0→S1 | S0→S2 | S0→S3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| μ [D] | λ [nm] | f | μ [D] | λ [nm] | f | μ [D] | λ [nm] | f | |
| F | 0.0510 | 327.98 | 0.0047 | 0.6799 | 314.66 | 0.0656 | 0.1213 | 271.61 | 0.0136 |
| 2′HF | 0.0292 | 333.84 | 0.0027 | 0.5159 | 313.93 | 0.0499 | 0.2120 | 304.44 | 0.0208 |
| 6HF | 0.8831 | 356.10 | 0.0753 | 0.0528 | 327.79 | 0.0049 | 0.0794 | 275.33 | 0.0088 |
| 7HF | 0.0463 | 317.88 | 0.0044 | 0.7863 | 300.10 | 0.0793 | 2.7015 | 269.90 | 0.3040 |
| Compound | C2′C1′Cα [°] | CαCβC1C2 [°] | O2′…O [Å] | O2′H…O [°] |
|---|---|---|---|---|
| 2′ OH chalcone (from F) | 180.00 | −179.99 | 2.52 | 150.81 |
| 2′ OH chalcone (from F) | 179.90 | −177.15 | 2.52 | 146.53 |
| exp. Data 3 | 173.48 | −174.67 | 2.53 | 149.76 |
| 2-OH chalcone (from 2′HF) | −179.82 | −179.02 | 2.51 | 151.15 |
| 5′ OH chalcone (from 6HF) | −179.99 | −179.98 | 2.52 | 150.51 |
| 4′ OH chalcone (from 7HF) | 180.0 | −179.97 | 2.51 | 151.74 |
| Compound | Deprotonation Group | This Work | Literature Values | |
|---|---|---|---|---|
| Experimental | Theoretical | |||
| 6HF | 6-OH | 9.24 (±0.03) | 13.43 | 9.90 [51] |
| 5′-OH chalcone | 10.65 (±0.05) | 15.05 | – | |
| 7HF | 7-OH | 7.85 (±0.02) | 8.28 | 7.26 [37]; 7.30 [51] |
| 4′-OH chalcone | 10.78 (±0.04) | 8.85 | ca. 11.2 [37] | |
| 2′HF | 2′-OH | 10.90 (±0.04) | 11.92 | – |
| 2-OH chalcone | 11.50 (±0.05) | 9.86 | – | |
| Spectrum | Wolfe-Shimer Diagram | Kb [M−1] |
|---|---|---|
 |  | 4.58 × 104 |
| (flavanone) | ||
 |  | 4.65 × 104 |
| (2′-hydroxyflavanone) | ||
 |  | 4 × 104 |
| (6-hydroxyflavanone) | ||
 |  | 4.81 × 104 |
| (7-hydroxyflavanone) | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łodyga-Chruścińska, E.; Kowalska-Baron, A.; Błazińska, P.; Pilo, M.; Zucca, A.; Korolevich, V.M.; Cheshchevik, V.T. Position Impact of Hydroxy Groups on Spectral, Acid–Base Profiles and DNA Interactions of Several Monohydroxy Flavanones. Molecules 2019, 24, 3049. https://doi.org/10.3390/molecules24173049
Łodyga-Chruścińska E, Kowalska-Baron A, Błazińska P, Pilo M, Zucca A, Korolevich VM, Cheshchevik VT. Position Impact of Hydroxy Groups on Spectral, Acid–Base Profiles and DNA Interactions of Several Monohydroxy Flavanones. Molecules. 2019; 24(17):3049. https://doi.org/10.3390/molecules24173049
Chicago/Turabian StyleŁodyga-Chruścińska, Elżbieta, Agnieszka Kowalska-Baron, Paulina Błazińska, Maria Pilo, Antonio Zucca, Violetta M. Korolevich, and Vitali T. Cheshchevik. 2019. "Position Impact of Hydroxy Groups on Spectral, Acid–Base Profiles and DNA Interactions of Several Monohydroxy Flavanones" Molecules 24, no. 17: 3049. https://doi.org/10.3390/molecules24173049
APA StyleŁodyga-Chruścińska, E., Kowalska-Baron, A., Błazińska, P., Pilo, M., Zucca, A., Korolevich, V. M., & Cheshchevik, V. T. (2019). Position Impact of Hydroxy Groups on Spectral, Acid–Base Profiles and DNA Interactions of Several Monohydroxy Flavanones. Molecules, 24(17), 3049. https://doi.org/10.3390/molecules24173049