
Amorphous Carbon Generation as a Photocatalytic Reaction on DNA-Assembled Gold and Silver Nanostructures



Abstract
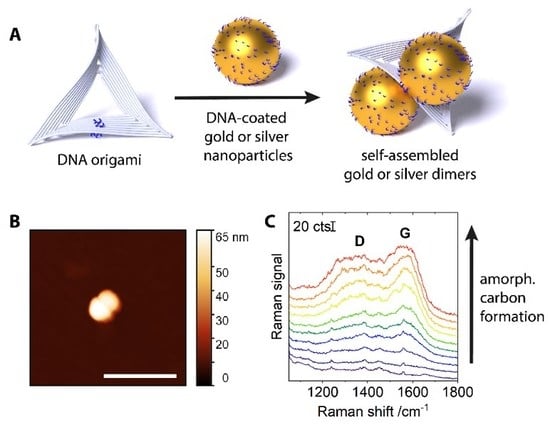
{kind=link}
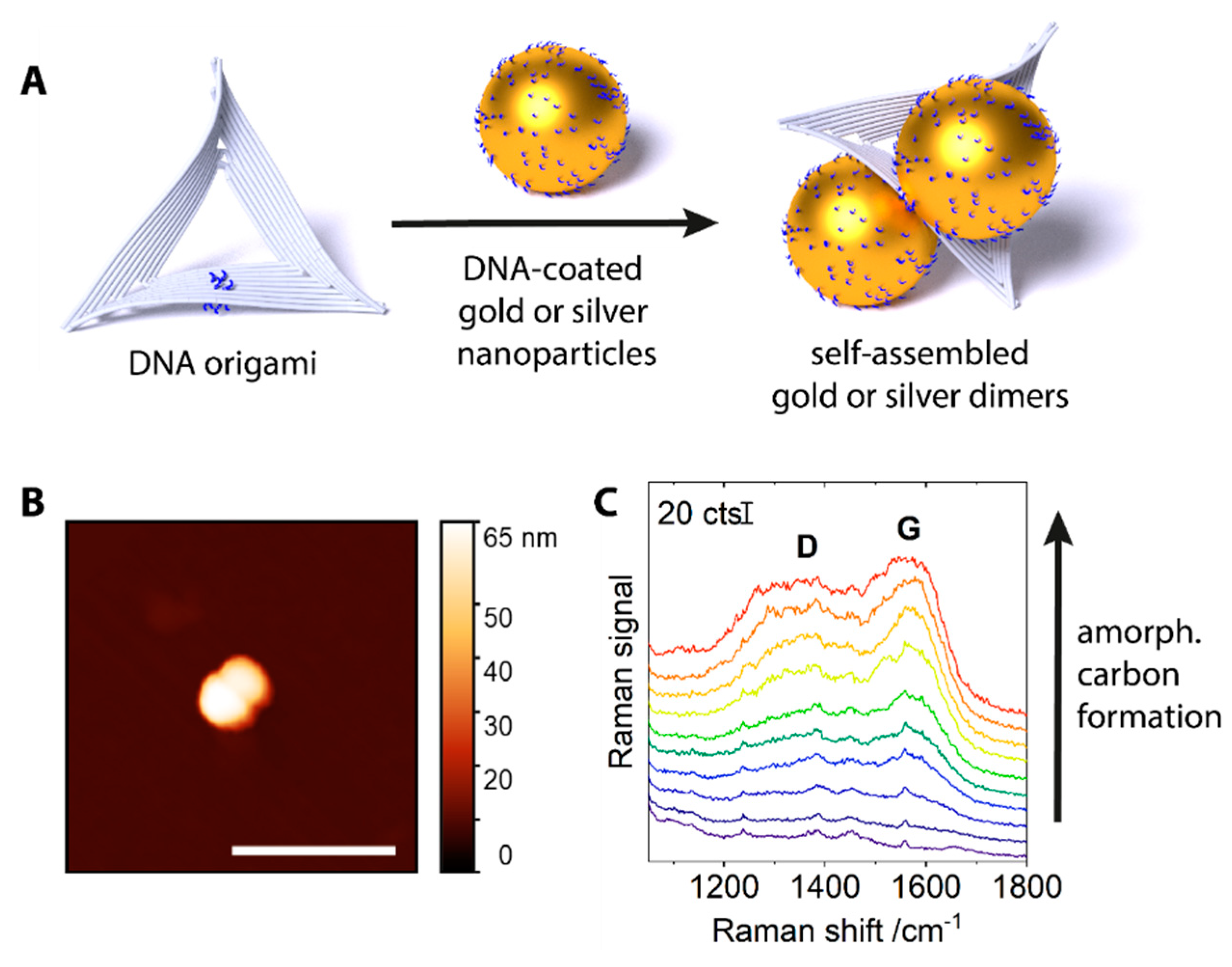{kind=link}
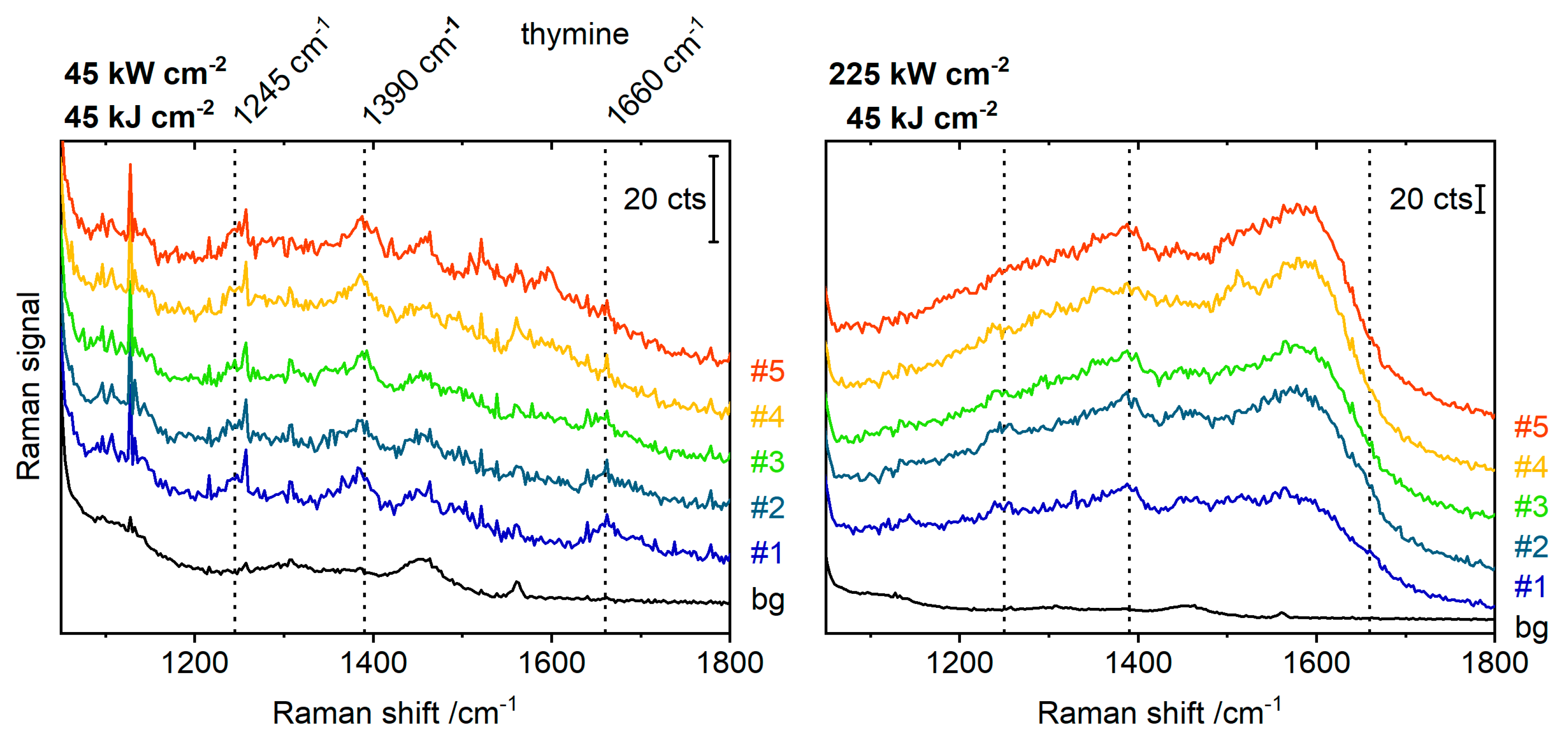{kind=link}
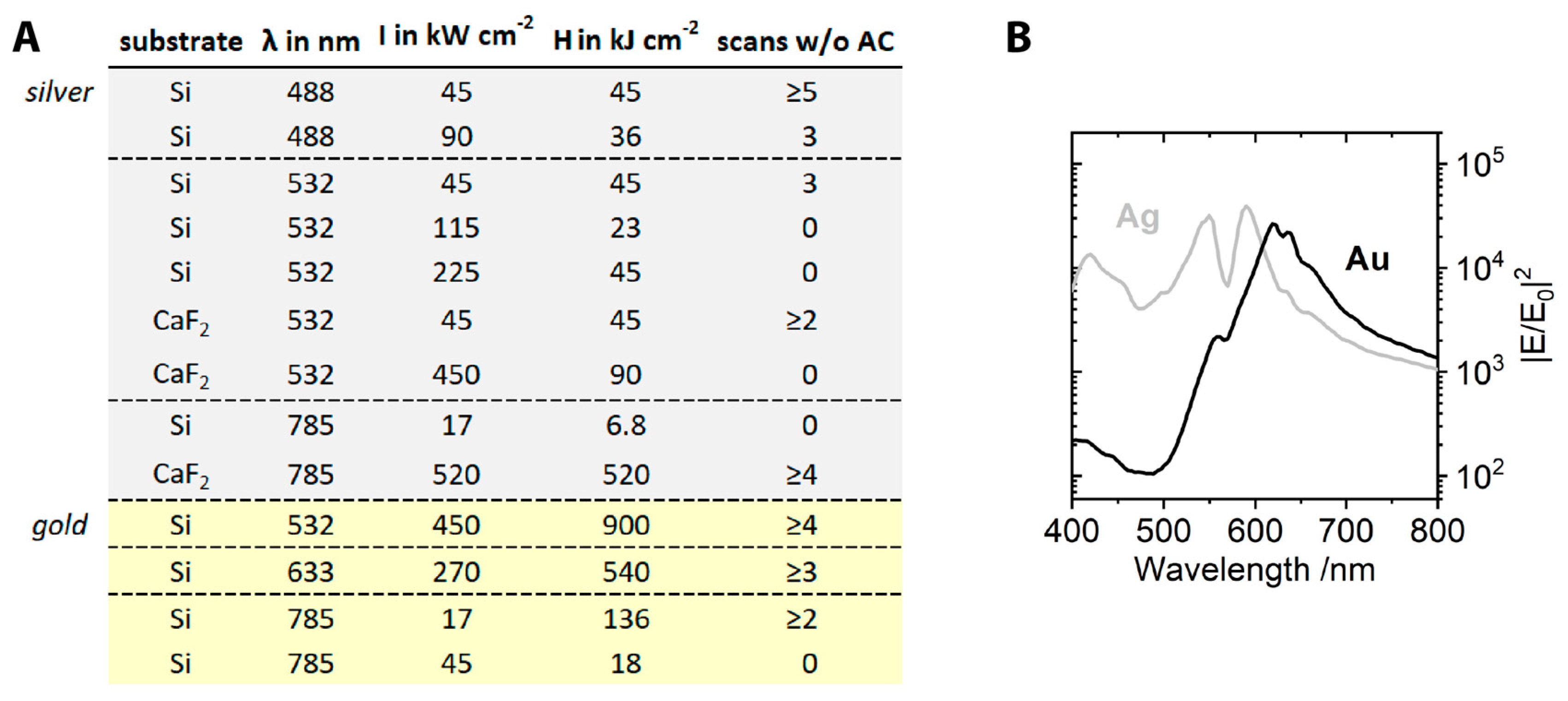{kind=link}
{kind=link}
1. Introduction
2. Results
3. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tsang, J.C.; Demuth, J.E.; Sanda, P.N.; Kirtley, J.R. Enhanced Raman scattering from carbon layers on silver. Chem. Phys. Lett. 1980, 76, 54–57. [Google Scholar] [CrossRef]
- Goncher, G.M.; Parsons, C.A.; Harris, C.B. Photochemistry on rough metal surfaces. J. Phys. Chem. 1984, 88, 4200–4209. [Google Scholar] [CrossRef]
- Pieczonka, N.P.W.; Aroca, R.F. Inherent complexities of trace detection by surface-enhanced Raman scattering. ChemPhysChem 2005, 6, 2473–2484. [Google Scholar] [CrossRef] [PubMed]
- Domke, K.F.; Zhang, D.; Pettinger, B. Enhanced Raman spectroscopy: Single molecules or carbon? J. Phys. Chem. C 2007, 111, 8611–8616. [Google Scholar] [CrossRef]
- Taylor, C.E.; Garvey, S.D.; Pemberton, J.E. Carbon Contamination at Silver Surfaces: Surface Preparation Procedures Evaluated by Raman Spectroscopy and X-ray Photoelectron Spectroscopy. Anal. Chem. 1996, 68, 2401–2408. [Google Scholar] [CrossRef]
- Suh, J.S.; Moskovits, M.; Shakhesemampour, J. Photochemical decomposition at colloid surfaces. J. Phys. Chem. 1993, 97, 1678–1683. [Google Scholar] [CrossRef]
- Tuinstra, F.; Koenig, J.L. Raman Spectrum of Graphite. J. Chem. Phys. 1970, 53, 1126–1130. [Google Scholar] [CrossRef]
- Kudelski, A.; Pettinger, B. SERS on carbon chain segments: Monitoring locally surface chemistry. Chem. Phys. Lett. 2000, 321, 356–362. [Google Scholar] [CrossRef]
- Zrimsek, A.B.; Chiang, N.; Mattei, M.; Zaleski, S.; McAnally, M.O.; Chapman, C.T.; Henry, A.I.; Schatz, G.C.; Van Duyne, R.P. Single-Molecule Chemistry with Surface- and Tip-Enhanced Raman Spectroscopy. Chem. Rev. 2017, 117, 7583–7613. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Resonant Raman spectroscopy of disordered, amorphous, and diamondlike carbon. Phys. Rev. B 2001, 64, 075414. [Google Scholar] [CrossRef]
- Heck, C.; Prinz, J.; Dathe, A.; Merk, V.; Stranik, O.; Fritzsche, W.; Kneipp, J.; Bald, I. Gold Nanolenses Self-Assembled by DNA Origami. ACS Photonics 2017, 4, 1123–1130. [Google Scholar] [CrossRef]
- Liu, N.; Liedl, T. DNA-Assembled Advanced Plasmonic Architectures. Chem. Rev. 2018, 118, 3032–3053. [Google Scholar] [CrossRef] [PubMed]
- Simoncelli, S.; Roller, E.M.; Urban, P.; Schreiber, R.; Turberfield, A.J.; Liedl, T.; Lohmüller, T. Quantitative Single-Molecule Surface-Enhanced Raman Scattering by Optothermal Tuning of DNA Origami-Assembled Plasmonic Nanoantennas. ACS Nano 2016, 10, 9809–9815. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Wen, T.; Wang, Z.G.; He, Y.; Shi, J.; Wang, T.; Liu, X.; Lu, G.; Ding, B. DNA Origami Directed Assembly of Gold Bowtie Nanoantennas for Single-Molecule Surface-Enhanced Raman Scattering. Angew. Chem. Int. Ed. 2018, 57, 2846–2850. [Google Scholar] [CrossRef] [PubMed]
- Tanwar, S.; Haldar, K.K.; Sen, T. DNA Origami Directed au Nanostar Dimers for Single-Molecule Surface-Enhanced Raman Scattering. J. Am. Chem. Soc. 2017, 139, 17639–17648. [Google Scholar] [CrossRef] [PubMed]
- Heck, C.; Kanehira, Y.; Kneipp, J.; Bald, I. Placement of Single Proteins within the SERS Hot Spots of Self-Assembled Silver Nanolenses. Angew. Chem. Int. Ed. 2018, 57, 7444–7447. [Google Scholar] [CrossRef] [PubMed]
- Prinz, J.; Heck, C.; Ellerik, L.; Merk, V.; Bald, I. DNA origami based Au-Ag-core-shell nanoparticle dimers with single-molecule SERS sensitivity. Nanoscale 2016, 8, 5612–5620. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.; Robertson, J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 2000, 61, 14095–14107. [Google Scholar] [CrossRef]
- Movileanu, L.; Benevides, J.M.; Thomas, G.J., Jr. Temperature dependence of the raman spectrum of DNA. Part I—Raman signatures of premelting and melting transitions of poly (dA–dT)·poly (dA–dT). J. Raman Spectrosc. 1999, 30, 637–649. [Google Scholar] [CrossRef]
- Torres-Nuñez, A.; Faulds, K.; Graham, D.; Alvarez-Puebla, R.A.; Guerrini, L. Silver colloids as plasmonic substrates for direct label-free surface-enhanced Raman scattering analysis of DNA. Analyst 2016, 141, 5170–5180. [Google Scholar] [CrossRef]
- Christopher, P.; Xin, H.; Marimuthu, A.; Linic, S. Singular characteristics and unique chemical bond activation mechanisms of photocatalytic reactions on plasmonic nanostructures. Nat. Mater. 2012, 11, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Swearer, D.F.; Zhang, C.; Robatjazi, H.; Zhao, H.; Henderson, L.; Dong, L.; Christopher, P.; Carter, E.A.; Nordlander, P.; et al. Quantifying hot carrier and thermal contributions in plasmonic photocatalysis. Science 2018, 362, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Schürmann, R.; Bald, I. Real-time monitoring of plasmon induced dissociative electron transfer to the potential DNA radiosensitizer 8-bromoadenine. Nanoscale 2017, 9, 1951–1955. [Google Scholar] [CrossRef] [PubMed]
- Christopher, P.; Moskovits, M. Hot Charge Carrier Transmission from Plasmonic Nanostructures. Annu. Rev. Phys. Chem. 2017, 68, 379–398. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Seong, N.H.; Dlott, D.D. Measurement of the distribution of site enhancements in surface-enhanced raman scattering. Science 2008, 321, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Pilo-Pais, M.; Watson, A.; Demers, S.; Labean, T.H.; Finkelstein, G. Surface-enhanced Raman scattering plasmonic enhancement using DNA origami-based complex metallic nanostructures. Nano Lett. 2014, 14, 2099–2104. [Google Scholar] [CrossRef] [PubMed]
- Kudelski, A. Role of O2 in inducing intensive fluctuations of surface-enhanced raman scattering spectra. J. Phys. Chem. B 2006, 110, 12610–12615. [Google Scholar] [CrossRef]
- Kudelski, A. Some aspects of SERS temporal fluctuations: Analysis of the most intense spectra of hydrogenated amorphous carbon deposited on silver. J. Raman Spectrosc. 2007, 38, 1494–1499. [Google Scholar] [CrossRef]
- Szczerbiński, J.; Gyr, L.; Kaeslin, J.; Zenobi, R. Plasmon-driven photocatalysis leads to products known from e-beam and x-ray-induced surface chemistry. Nano Lett. 2018, 18, 6740–6749. [Google Scholar] [CrossRef]
- Sellers, H.; Ulman, A.; Shnidman, Y.; Eilers, J.E. Structure and Binding of Alkanethiolates on Gold and Silver Surfaces: Implications for Self-Assembled Monolayers. J. Am. Chem. Soc. 1993, 115, 9389–9401. [Google Scholar] [CrossRef]
- Li, K.; Stockman, M.I.; Bergman, D.J. Self-similar chain of metal nanospheres as an efficient nanolens. Phys. Rev. Lett. 2003, 91, 227402. [Google Scholar] [CrossRef] [PubMed]
- Emory, S.R.; Jensen, R.A.; Wenda, T.; Han, M.; Nie, S. Re-examining the origins of spectral blinking in single-molecule and single-nanoparticle SERS. Faraday Discuss. 2006, 132, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Kneipp, K.; Wang, Y.; Kneipp, H.; Perelman, L.T.; Itzkan, I.; Dasari, R.R.; Feld, M.S. Single molecule detection using surface-enhanced raman scattering (SERS). Phys. Rev. Lett. 1997, 78, 1667–1670. [Google Scholar] [CrossRef]
- Xu, H.; Bjerneld, E.J.; Käll, M.; Börjesson, L. Spectroscopy of single hemoglobin molecules by surface enhanced raman scattering. Phys. Rev. Lett. 1999, 83, 4357–4360. [Google Scholar] [CrossRef]
- Bjerneld, E.J.; Johansson, P.; Käll, M. Single Molecule Vibrational Fine-structure of Tyrosine Adsorbed on Ag Nano-Crystals. Single Mol. 2000, 1, 239–248. [Google Scholar] [CrossRef]
- Otto, A. What is observed in single molecule SERS, and why? J. Raman Spectrosc. 2002, 33, 593–598. [Google Scholar] [CrossRef]
- Neacsu, C.C.; Dreyer, J.; Behr, N.; Raschke, M.B. Scanning-probe Raman spectroscopy with single-molecule sensitivity. Phys. Rev. B 2006, 73, 193406. [Google Scholar] [CrossRef]
- Domke, K.F.; Pettinger, B. Comment on “Scanning-probe Raman spectroscopy with single-molecule sensitivity”. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 75, 236401. [Google Scholar] [CrossRef]
- Kneipp, K.; Kneipp, H.; Dresselhaus, M.S.; Lefrant, S. Surface-enhanced Raman scattering on single-wall carbon nanotubes. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2004, 362, 2361–2373. [Google Scholar] [CrossRef]
- Bjerneld, E.J.; Svedberg, F.; Johansson, P.; Käll, M. Direct observation of heterogeneous photochemistry on aggregated Ag nanocrystals using Raman spectroscopy: The case of photoinduced degradation of aromatic amino acids. J. Phys. Chem. A 2004, 108, 4187–4193. [Google Scholar] [CrossRef]
- Etchegoin, P.; Liem, H.; Maher, R.C.; Cohen, L.F.; Brown, R.J.C.; Hartigan, H.; Milton, M.J.T.; Gallop, J.C. A novel amplification mechanism for surface enhanced Raman scattering. Chem. Phys. Lett. 2002, 366, 115–121. [Google Scholar] [CrossRef]
- Prinz, J.; Matković, A.; Pešić, J.; Gajić, R.; Bald, I. Hybrid Structures for Surface-Enhanced Raman Scattering: DNA Origami/Gold Nanoparticle Dimer/Graphene. Small 2016, 12, 5458–5467. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.; Chen, X.-J.; Yi, J.; Li, J.-F.; Wu, D.-Y.; Tian, Z.-Q. From plasmon-enhanced molecular spectroscopy to plasmon-mediated chemical reactions. Nat. Rev. Chem. 2018, 2, 216–230. [Google Scholar] [CrossRef]
- Zhang, Y.; He, S.; Guo, W.; Hu, Y.; Huang, J.; Mulcahy, J.R.; Wei, W.D. Surface-Plasmon-Driven Hot Electron Photochemistry. Chem. Rev. 2018, 118, 2927–2954. [Google Scholar] [CrossRef] [PubMed]
- Zwick, A.; Carles, R. Multiple-order Raman scattering in crystalline and amorphous silicon. Phys. Rev. B 1993, 48, 6024–6032. [Google Scholar] [CrossRef]
- Lehmann, A.; Schumann, L.; Hübner, K. Optical Phonons in Amorphous Silicon Oxides. I. Calculation of the Density of States and Interpretation of Lo—To Splittings of Amorphous Sio2. Phys. Status Solidi 1983, 117, 689–698. [Google Scholar] [CrossRef]
- Weber, A.; McGinnis, E.A. The Raman spectrum of gaseous oxygen. J. Mol. Spectrosc. 1960, 4, 195–200. [Google Scholar] [CrossRef]
- Johnson, P.B.; Christy, R.W. Optical constants of the noble metals. Phys. Rev. B 1972, 6, 4370–4379. [Google Scholar] [CrossRef]
- Palik, E.D. Handbook of Optical Constants of Solids; Academic Press: Cambridge, MA, USA, 2012; Volume 1. [Google Scholar]
- Thacker, V.V.; Herrmann, L.O.; Sigle, D.O.; Zhang, T.; Liedl, T.; Baumberg, J.J.; Keyser, U.F. DNA origami based assembly of gold nanoparticle dimers for surface-enhanced Raman scattering. Nat. Commun. 2014, 5, 3448. [Google Scholar] [CrossRef]
Sample Availability: Samples of the assembled gold and silver nanostructures are available from the authors. |




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heck, C.; Kanehira, Y.; Kneipp, J.; Bald, I. Amorphous Carbon Generation as a Photocatalytic Reaction on DNA-Assembled Gold and Silver Nanostructures. Molecules 2019, 24, 2324. https://doi.org/10.3390/molecules24122324
Heck C, Kanehira Y, Kneipp J, Bald I. Amorphous Carbon Generation as a Photocatalytic Reaction on DNA-Assembled Gold and Silver Nanostructures. Molecules. 2019; 24(12):2324. https://doi.org/10.3390/molecules24122324
Chicago/Turabian StyleHeck, Christian, Yuya Kanehira, Janina Kneipp, and Ilko Bald. 2019. "Amorphous Carbon Generation as a Photocatalytic Reaction on DNA-Assembled Gold and Silver Nanostructures" Molecules 24, no. 12: 2324. https://doi.org/10.3390/molecules24122324
APA StyleHeck, C., Kanehira, Y., Kneipp, J., & Bald, I. (2019). Amorphous Carbon Generation as a Photocatalytic Reaction on DNA-Assembled Gold and Silver Nanostructures. Molecules, 24(12), 2324. https://doi.org/10.3390/molecules24122324