
Time-Gated Luminescent In Situ Hybridization (LISH): Highly Sensitive Detection of Pathogenic Staphylococcus aureus

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
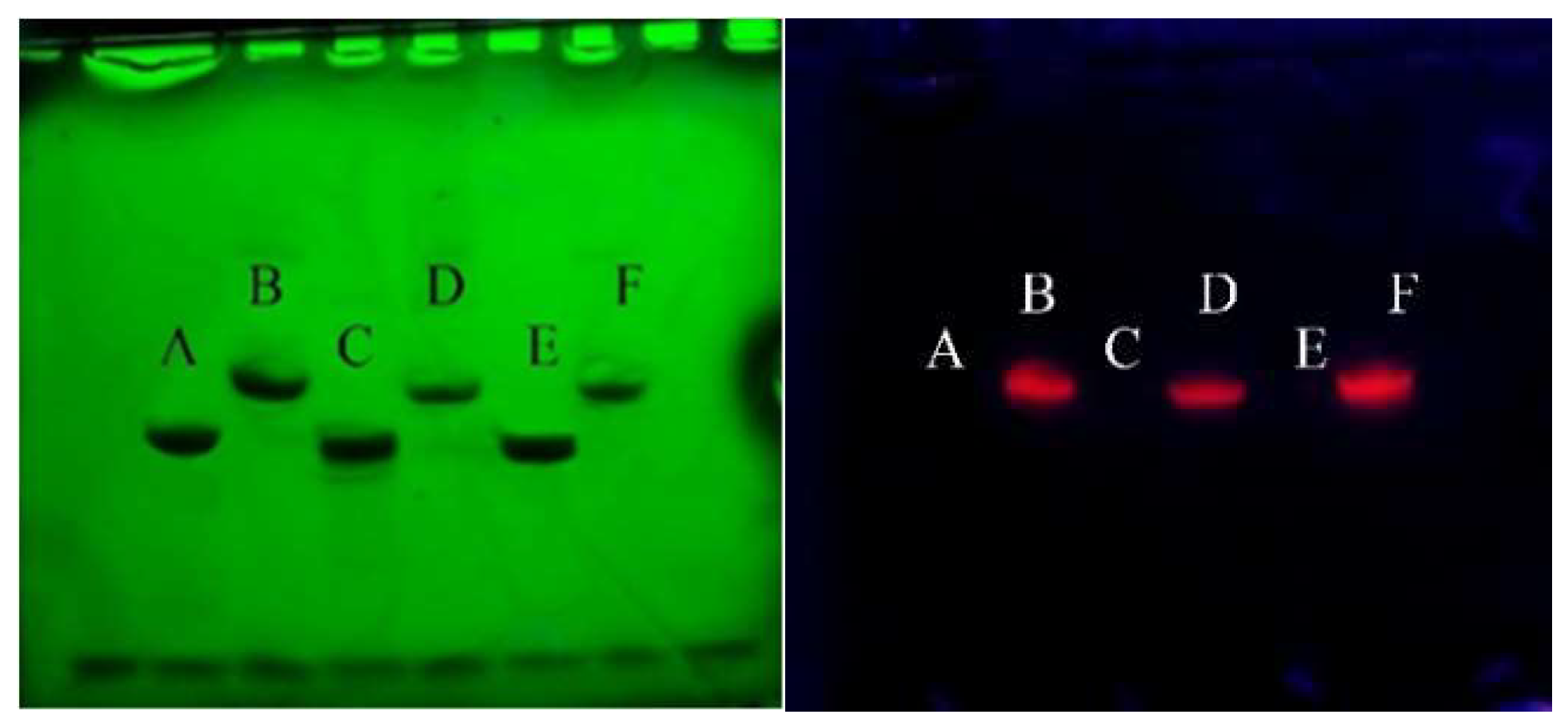
2.1. Preparation of Europium Chelate Probes
2.2. LISH Protocol Optimisation and TGL Imaging
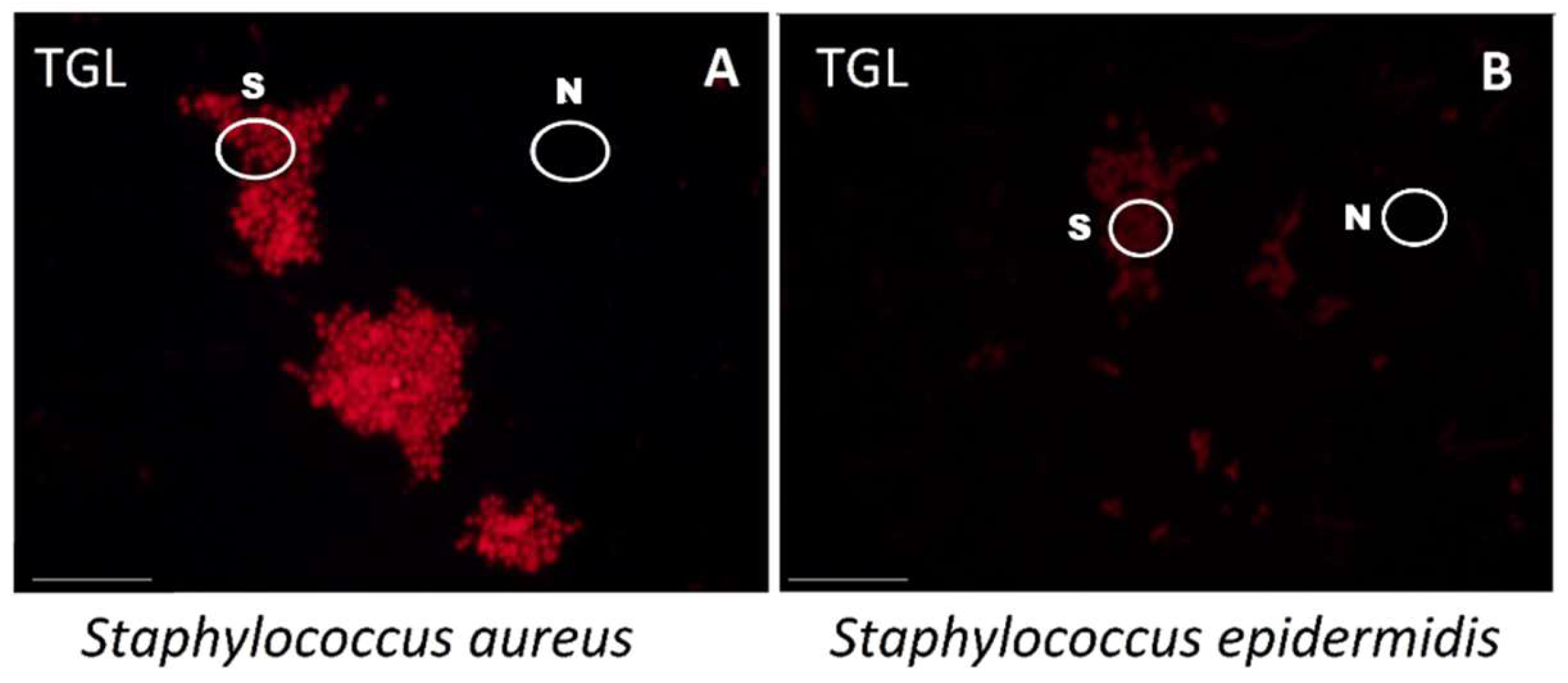
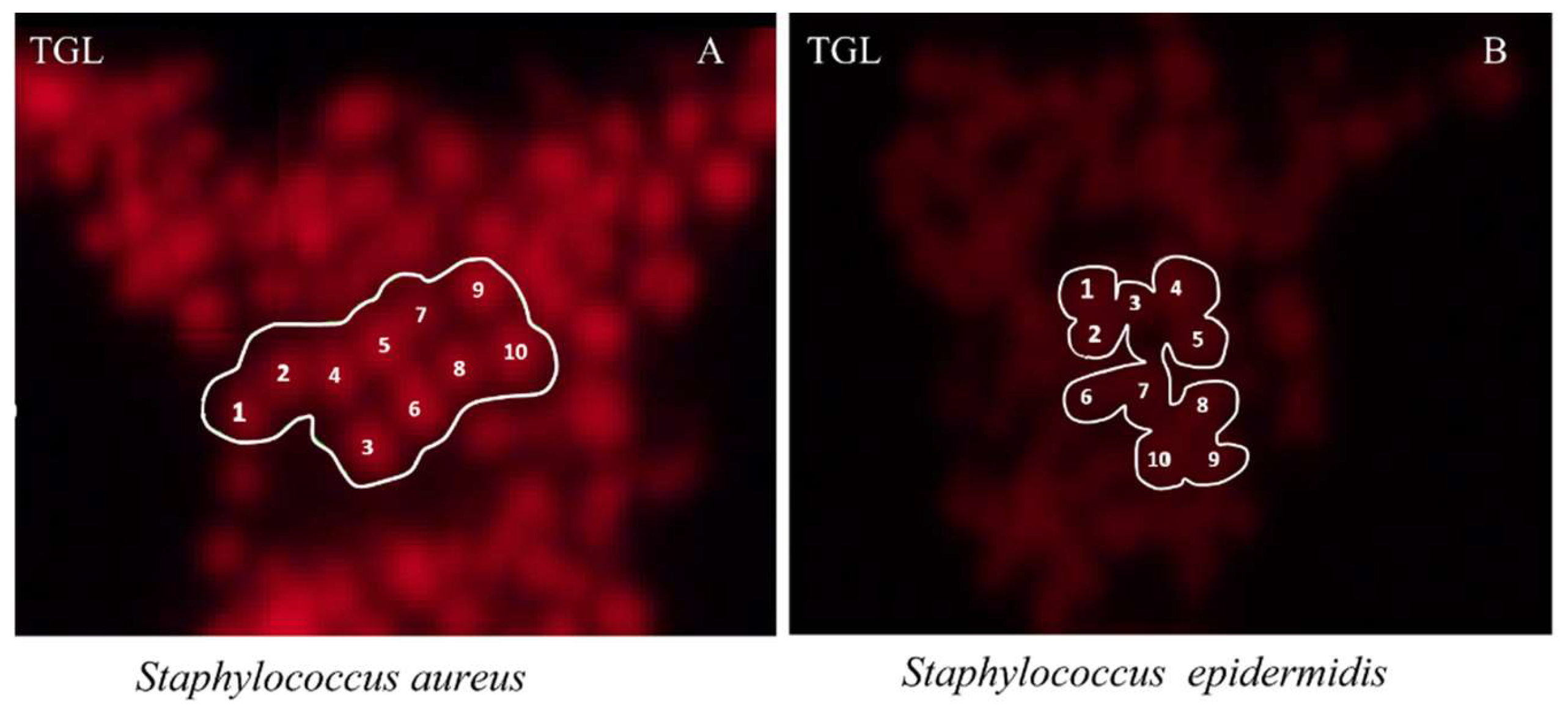
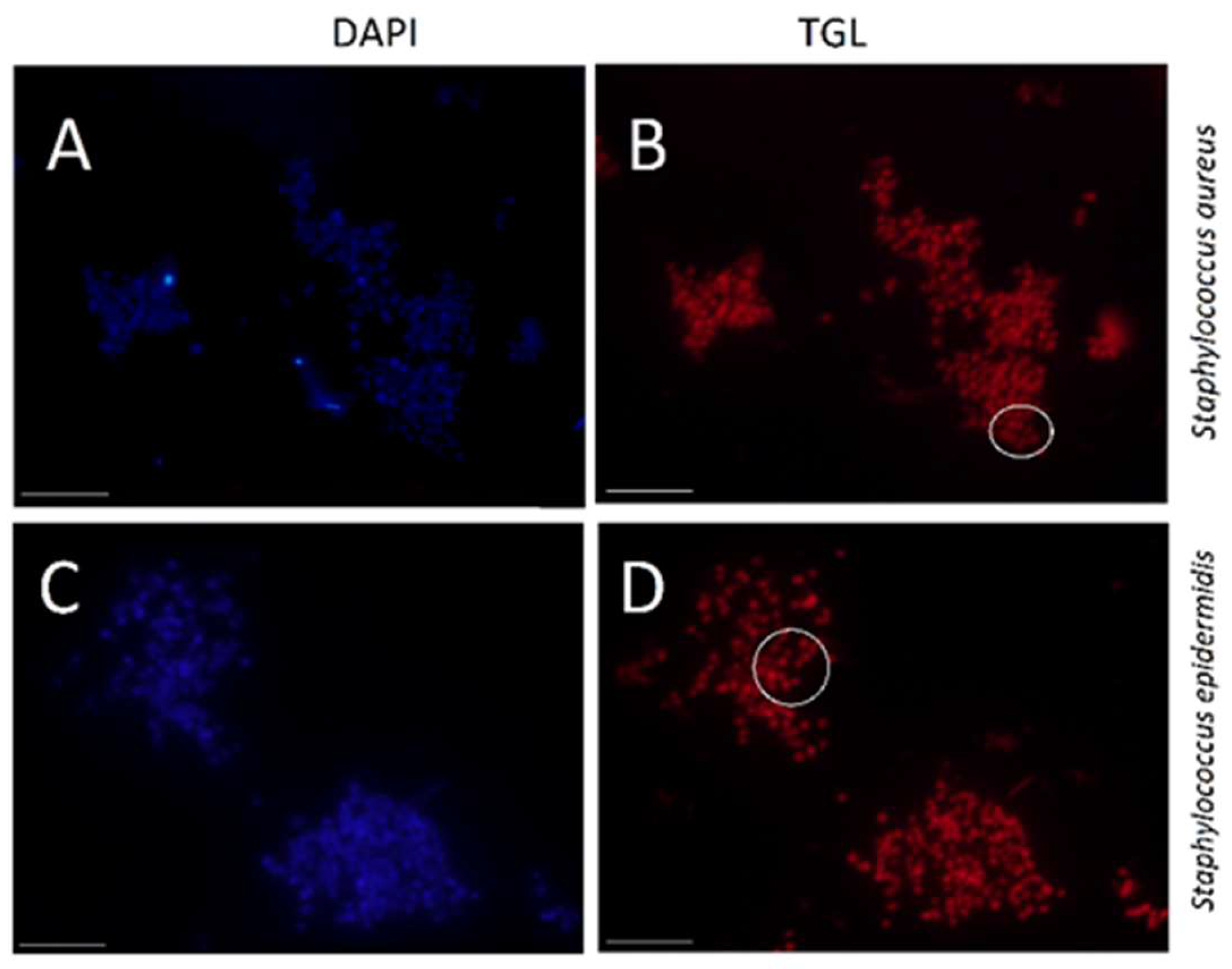
2.3. Differentiation between S. aureus and S. epidermidis
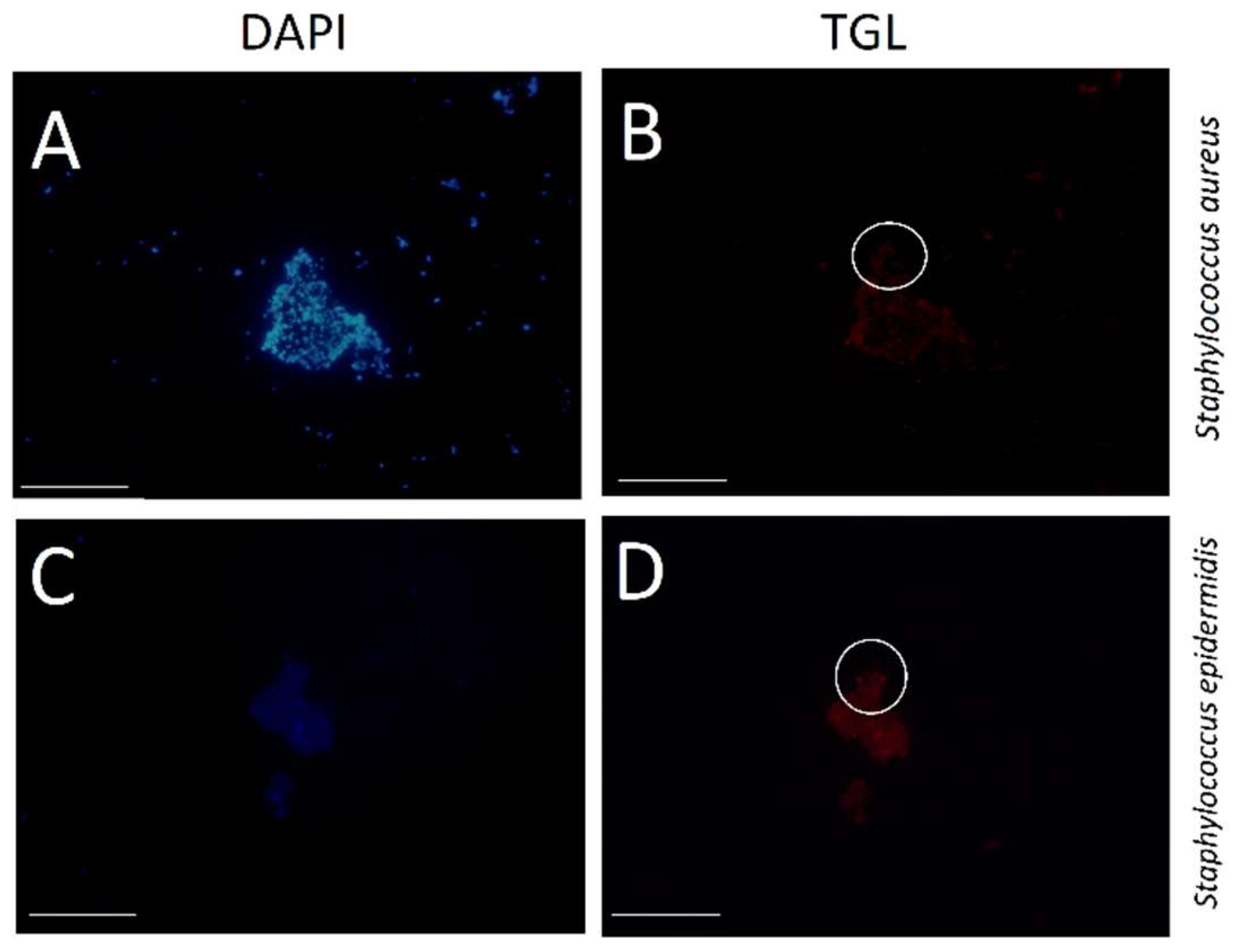
2.4. Positive/Negative Controls
3. Conclusions
4. Experimental Section
4.1. Materials
4.2. The Conjugation of Europium Chelate to DNA
4.3. Electrophoresis Solution and Gel Preparation
4.4. Gel Imaging
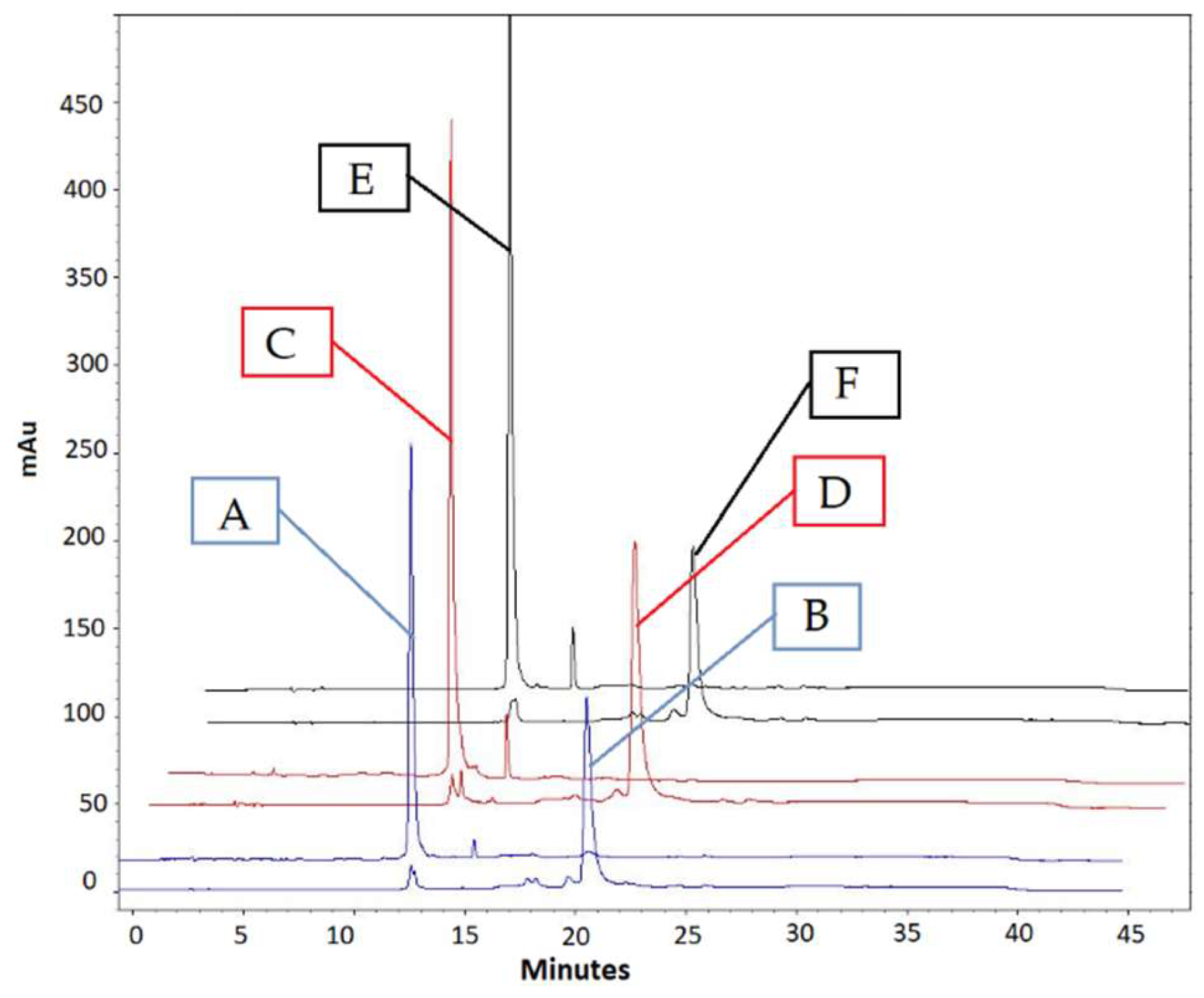
4.5. Analytical HPLC Analysis of DNA Conjugates
4.6. Photophysical Properties of DNA Conjugates
4.7. The Cells (S. aureus and S. epidermidis)
4.8. Buffers
4.9. LISH Procedure
4.10. Imaging
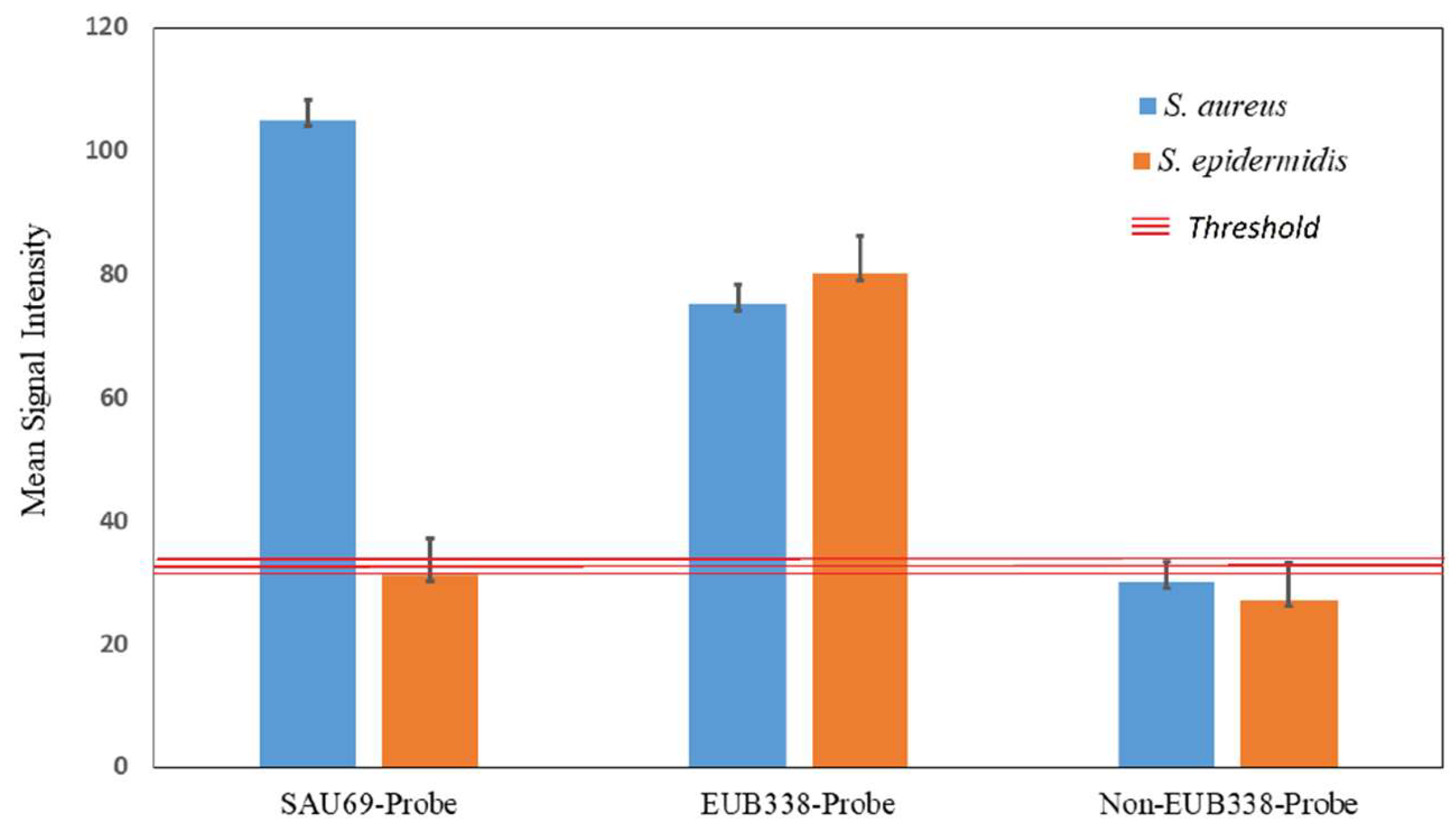
4.11. Quantification of Signal-to-Noise Ratio (SNR)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Gómez-Pavón, J.; Rodríguez, J.S.; de la Puente Fernández, E.; Baztán, J.C.; Ruipérez, I.C.; González, I.; Cortés, R.B. Staphylococcus aureus infection in an acute geriatric unit. Rev. Esp. De Geriatr. Y Gerontol. 2010, 45, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Reighard, A.; Diekema, D.; Wibbenmeyer, L.; Ward, M.; Herwaldt, L. Staphylococcus aureus nasal colonization and colonization or infection at other body sites in patients on a burn trauma unit. Infect. Control Hosp. Epidemiol. 2009, 30, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Bayer, A.; Cosgrove, S.E.; Daum, R.S.; Fridkin, S.K.; Gorwitz, R.J.; Kaplan, S.L.; Karchmer, A.W.; Levine, D.P.; Murray, B.E. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant staphylococcus aureus infections in adults and children. Clin. Infect. Dis. 2011, 52, e18–e55. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y. Pcr in diagnosis of infection: Detection of bacteria in cerebrospinal fluids. Clin. Diagn. Lab. Immunol. 2002, 9, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Ellerbrok, H.; Nattermann, H.; Özel, M.; Beutin, L.; Appel, B.; Pauli, G. Rapid and sensitive identification of pathogenic and apathogenic bacillus anthracis by real-time pcr. FEMS Microbiol. Lett. 2002, 214, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Ratnayake, L.; Olver, W.J. Rapid pcr detection of methicillin-resistant staphylococcus aureus and methicillin-sensitive s. Aureus samples from charcoal-containing blood culture bottles. J. Clin. Microbiol. 2011, 49, 2382. [Google Scholar] [CrossRef]
- Cao, X.; Ye, Y.; Liu, S. Gold nanoparticle-based signal amplification for biosensing. Anal. Biochem. 2011, 417, 1–16. [Google Scholar] [CrossRef]
- Torres-Chavolla, E.; Alocilja, E.C. Nanoparticle based DNA biosensor for tuberculosis detection using thermophilic helicase-dependent isothermal amplification. Biosens. Bioelectron. 2011, 26, 4614–4618. [Google Scholar] [CrossRef]
- Chen, S.-H.; Wu, V.C.; Chuang, Y.-C.; Lin, C.-S. Using oligonucleotide-functionalized au nanoparticles to rapidly detect foodborne pathogens on a piezoelectric biosensor. J. Microbiol. Methods 2008, 73, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; He, X.; Ji, J.; Jia, M.; Wang, Z.; Sun, X. A highly selective and sensitive electrochemical cs–mwcnts/au-nps composite DNA biosensor for staphylococcus aureus gene sequence detection. Talanta 2015, 141, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Vautor, E.; Magnone, V.; Rios, G.; Le Brigand, K.; Bergonier, D.; Lina, G.; Meugnier, H.; Barbry, P.; Thiery, R.; Pepin, M. Genetic differences among staphylococcus aureus isolates from dairy ruminant species: A single-dye DNA microarray approach. Vet. Microbiol. 2009, 133, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Ruan, M.; Niu, C.-G.; Zeng, G.-M.; Qin, P.-Z.; Wang, X.-Y.; Huang, D.-W.; Huang, J.; Fan, C.-Z. Rapid detection of staphylococcus aureus via a sensitive DNA hybridization assay based on a long-lifetime luminescent europium marker. Microchim. ACTA 2011, 175, 105. [Google Scholar] [CrossRef]
- Xia, H.; Wang, F.; Huang, Q.; Huang, J.; Chen, M.; Wang, J.; Yao, C.; Chen, Q.; Cai, G.; Fu, W. Detection of staphylococcus epidermidis by a quartz crystal microbalance nucleic acid biosensor array using au nanoparticle signal amplification. Sensors 2008, 8, 6453–6470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Yan, Y.; Li, Q.; Yu, T.; Cheng, W.; Wang, L.; Ju, H.; Ding, S. Label-free and high-sensitive detection of salmonella using a surface plasmon resonance DNA-based biosensor. J. Biotechnol. 2012, 160, 123–128. [Google Scholar] [CrossRef]
- Qin, P.-Z.; Niu, C.-G.; Ruan, M.; Zeng, G.-M.; Wang, X.-Y. A novel bifunctional europium complex as a potential fluorescent label for DNA detection. Analyst 2010, 135, 2144–2149. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, Y.; Hu, H.; Wang, M.; Wu, Z.; Xu, W. Rapid identification of staphylococcus aureus: Fish versus pcr methods. Lab. Med. 2012, 43, 276–280. [Google Scholar] [CrossRef]
- Tavares, A.; Inácio, J.; Melo-Cristino, J.; Couto, I. Use of fluorescence in situ hybridization for rapid identification of staphylococci in blood culture samples collected in a portuguese hospital. J. Clin. Microbiol. 2008, 46, 3097–3100. [Google Scholar] [CrossRef]
- Hartmann, H.; Stender, H.; Schäfer, A.; Autenrieth, I.B.; Kempf, V.A. Rapid identification of staphylococcus aureus in blood cultures by a combination of fluorescence in situ hybridization using peptide nucleic acid probes and flow cytometry. J. Clin. Microbiol. 2005, 43, 4855–4857. [Google Scholar] [CrossRef]
- Shrestha, N.K.; Scalera, N.M.; Wilson, D.A.; Brehm-Stecher, B.; Procop, G.W. Rapid identification of staphylococcus aureus and methicillin resistance by flow cytometry using a peptide nucleic acid probe. J. Clin. Microbiol. 2011, 49, 3383–3385. [Google Scholar] [CrossRef] [PubMed]
- Frickmann, H.; Zautner, A.E.; Moter, A.; Kikhney, J.; Hagen, R.M.; Stender, H.; Poppert, S. Fluorescence in situ hybridization (fish) in the microbiological diagnostic routine laboratory: A review. Crit. Rev. Microbiol. 2017, 43, 263–293. [Google Scholar] [CrossRef]
- Lawson, T.; Connally, R.; Vemulpad, S.; Piper, J. Dimethyl formamide-free, urea-nacl fluorescence in situ hybridization assay for staphylococcus aureus. Lett. Appl. Microbiol. 2012, 54, 263–266. [Google Scholar] [CrossRef]
- Lawson, T.S.; Connally, R.E.; Iredell, J.R.; Vemulpad, S.; Piper, J.A. Detection of staphylococcus aureus with a fluorescence in situ hybridization that does not require lysostaphin. J. Clin. Lab. Anal. 2011, 25, 142–147. [Google Scholar] [CrossRef]
- Lawson, T.S.; Connally, R.E.; Vemulpad, S.; Piper, J.A. Optimization of a two-step permeabilization fluorescence in situ hybridization (fish) assay for the detection of staphylococcus aureus. J. Clin. Lab. Anal. 2011, 25, 359–365. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 2nd ed.; Plenum Press: New York, NY, USA, 1999; p. 496. [Google Scholar]
- Selvin, P.R. Principles and biophysical applications of lanthanide-based probes. Annu. Rev. Biophys. Biomol. Struct. 2002, 31, 275. [Google Scholar] [CrossRef]
- Yuan, J.; Matsumoto, K.; Kimura, H. A new tetradentate β-diketonate− europium chelate that can be covalently bound to proteins for time-resolved fluoroimmunoassay. Anal. Chem. 1998, 70, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Connally, R.E.; Piper, J.A. Time-gated luminescence microscopy. Ann. N. Y. Acad. Sci. 2008, 1130, 106–116. [Google Scholar] [CrossRef]
- Connally, R.; Veal, D.; Piper, J. Time-resolved fluorescence microscopy using an improved europium chelate bhhst for the in situ detection of cryptosporidium and giardia. Microsc. Res. Tech. 2004, 64, 312–322. [Google Scholar] [CrossRef]
- Sayyadi, N.; Connally, R.E.; Try, A. A novel biocompatible europium ligand for sensitive time-gated immunodetection. Chem. Commun. 2016, 52, 1154–1157. [Google Scholar] [CrossRef]
- Sayyadi, N.; Care, A.; Connally, R.E.; Try, A.C.; Bergquist, P.L.; Sunna, A. A novel universal detection agent for time-gated luminescence bioimaging. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Sayyadi, N.; Justiniano, I.; Connally, R.E.; Zhang, R.; Shi, B.; Kautto, L.; Everest-Dass, A.V.; Yuan, J.; Walsh, B.J.; Jin, D.; et al. Sensitive time-gated immunoluminescence detection of prostate cancer cells using a tegylated europium ligand. Anal. Chem. 2016, 88, 9564–9571. [Google Scholar] [CrossRef]
- Sueda, S.; Yuan, J.; Matsumoto, K. Homogeneous DNA hybridization assay by using europium luminescence energy transfer. Bioconjugate. Chem. 2000, 11, 827–831. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Yuan, J.; Matsumoto, K.; KIMURA, H. Time-resolved fluorometric detection of DNA using a tetradentate β-diketonate europium chelate as a label. Anal. Sci. 1999, 15, 121–124. [Google Scholar] [CrossRef]
- Kempf, V.A.; Trebesius, K.; Autenrieth, I.B. Fluorescent in situ hybridization allows rapid identification of microorganisms in blood cultures. J. Clin. Microbiol. 2000, 38, 830–838. [Google Scholar]
- Connally, R.; Piper, J. Solid-state time-gated luminescence microscope with ultraviolet light-emitting diode excitation and electron-multiplying charge-coupled device detection. J. Biomed. Opt. 2008, 13, 34022–34026. [Google Scholar] [CrossRef]
- Jin, D.; Piper, J.A. Time-gated luminescence microscopy allowing direct visual inspection of lanthanide-stained microorganisms in background-free condition. Anal. Chem. 2011, 83, 2294–2300. [Google Scholar] [CrossRef]
- Connally, R. A device for gated autosynchronous luminescence detection. Anal. Chem. 2011, 83, 4782–4787. [Google Scholar] [CrossRef]
- Amann, R.I.; Binder, B.J.; Olson, R.J.; Chisholm, S.W.; Devereux, R.; Stahl, D.A. Combination of 16s rrna-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 1990, 56, 1919–1925. [Google Scholar]
- Wallner, G.; Amann, R.; Beisker, W. Optimizing fluorescent in situ hybridization with rrna-targeted oligonucleotide probes for flow cytometric identification of microorganisms. Cytom. Part A 1993, 14, 136–143. [Google Scholar] [CrossRef]
- Arnaud, N.; Georges, J. Fluorimetric determination of europium over a large dynamic range using its ternary complex with thenoyltrifluoroacetone and trioctylphosphine oxide in a micellar solution of triton x-100. Analyst 1997, 122, 143–146. [Google Scholar] [CrossRef]
- Lawson, T.S.; Connally, R.E.; Vemulpad, S.; Piper, J.A. In silico evaluation and testing of fluorescence in situ hybridization 16s rrna probes for staphylococcus aureus. Lab. Med. 2015, 42, 729–734. [Google Scholar] [CrossRef]
- Poppert, S.; Riecker, M.; Wellinghausen, N.; Frickmann, H.; Essig, A. Accelerated identification of staphylococcus aureus from blood cultures by a modified fluorescence in situ hybridization procedure. J. Med. Microbiol. 2010, 59, 65–68. [Google Scholar] [CrossRef]
- Li, S.; Spear, R.N.; Andrews, J.H. Quantitative fluorescence in situ hybridization of aureobasidium pullulans on microscope slides and leaf surfaces. Appl. Env. Microbiol. 1997, 63, 3261–3267. [Google Scholar]
- Pernthaler, J.; Pernthaler, A.; Amann, R. Automated enumeration of groups of marine picoplankton after fluorescence in situ hybridization. Appl. Env. Microbiol. 2003, 69, 2631–2637. [Google Scholar] [CrossRef]
- Langendijk, P.S.; Schut, F.; Jansen, G.J.; Raangs, G.C.; Kamphuis, G.R.; Wilkinson, M.; Welling, G.W. Quantitative fluorescence in situ hybridization of bifidobacterium spp. With genus-specific 16s rrna-targeted probes and its application in fecal samples. Appl. Env. Microbiol. 1995, 61, 3069–3075. [Google Scholar]
- Garini, Y.; Gil, A.; Bar-Am, I.; Cabib, D.; Katzir, N. Signal to noise analysis of multiple color fluorescence imaging microscopy. Cytom. J. Int. Soc. Anal. Cytol. 1999, 35, 214–226. [Google Scholar] [CrossRef]
- Zhou, Z.; Pons, M.N.; Raskin, L.; Zilles, J.L. Automated image analysis for quantitative fluorescence in situ hybridization with environmental samples. Appl. Env. Microbiol. 2007, 73, 2956–2962. [Google Scholar] [CrossRef]
- Thomas, L.; Gidding, H.; Ginn, A.; Olma, T.; Iredell, J. Development of a real-time staphylococcus aureus and mrsa (sam-) pcr for routine blood culture. J. Microbiol. Methods 2007, 68, 296–302. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sayyadi, N.; Connally, R.E.; Lawson, T.S.; Yuan, J.; Packer, N.H.; Piper, J.A. Time-Gated Luminescent In Situ Hybridization (LISH): Highly Sensitive Detection of Pathogenic Staphylococcus aureus. Molecules 2019, 24, 2083. https://doi.org/10.3390/molecules24112083
Sayyadi N, Connally RE, Lawson TS, Yuan J, Packer NH, Piper JA. Time-Gated Luminescent In Situ Hybridization (LISH): Highly Sensitive Detection of Pathogenic Staphylococcus aureus. Molecules. 2019; 24(11):2083. https://doi.org/10.3390/molecules24112083
Chicago/Turabian StyleSayyadi, Nima, Russell E. Connally, Thomas S. Lawson, Jingli Yuan, Nicolle H. Packer, and James A. Piper. 2019. "Time-Gated Luminescent In Situ Hybridization (LISH): Highly Sensitive Detection of Pathogenic Staphylococcus aureus" Molecules 24, no. 11: 2083. https://doi.org/10.3390/molecules24112083
APA StyleSayyadi, N., Connally, R. E., Lawson, T. S., Yuan, J., Packer, N. H., & Piper, J. A. (2019). Time-Gated Luminescent In Situ Hybridization (LISH): Highly Sensitive Detection of Pathogenic Staphylococcus aureus. Molecules, 24(11), 2083. https://doi.org/10.3390/molecules24112083