In Silico Identification of Potential Inhibitor Against a Fungal Histone Deacetylase, RPD3 from Magnaporthe Oryzae


Abstract
1. Introduction
2. Results and Discussion
2.1. Target-Template Alignment and Homology Modelling
2.2. Model Validation
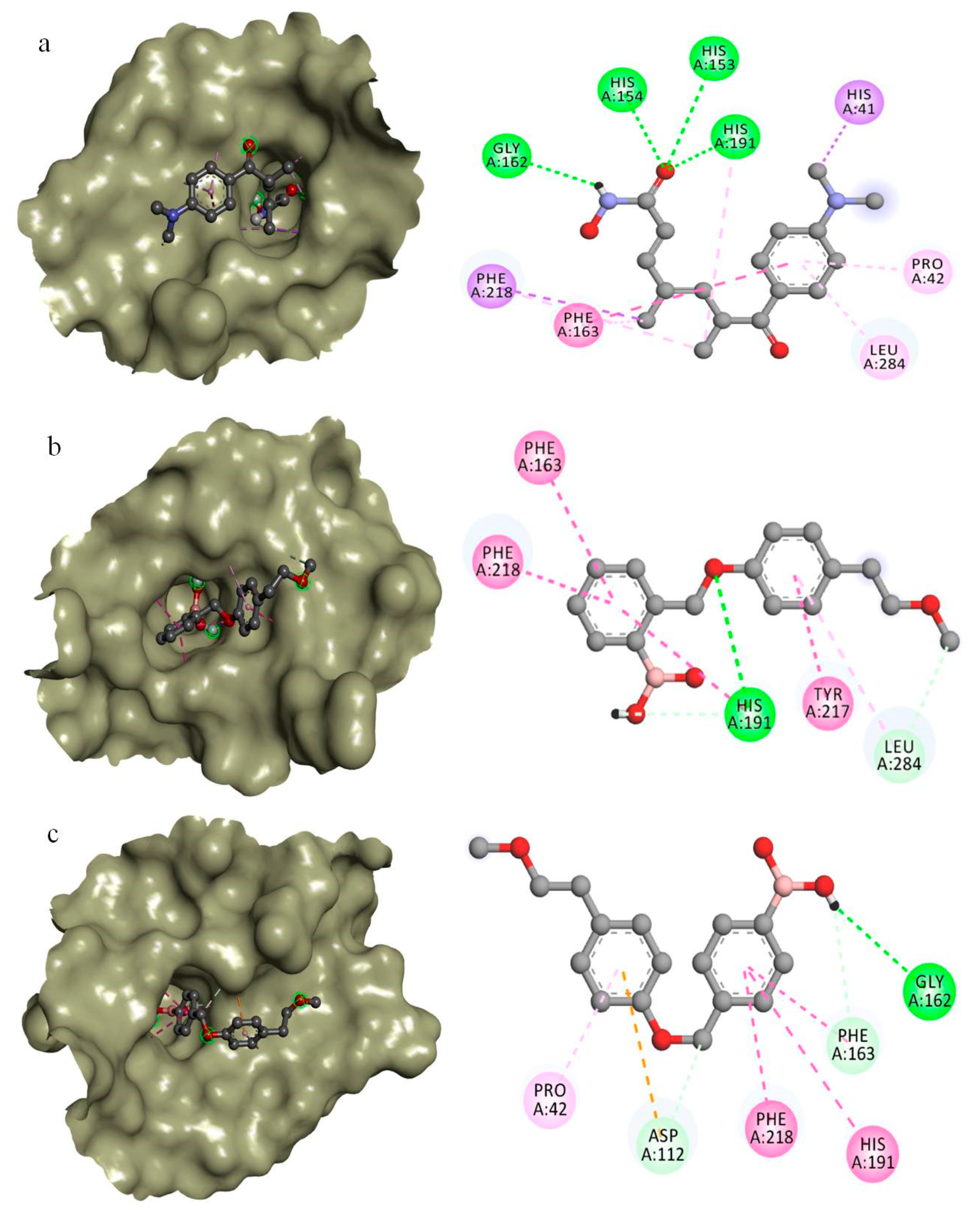
2.3. In Silico Drug Designing
2.4. In Vitro Studies for Appressorium Formation Inhibition
3. Materials and Methods
3.1. Sequence Analysis for Potential Templates
3.2. Homology Modeling
3.3. Model Validation
3.4. Structure-Based Virtual Screening
3.5. Docking Interactions
3.6. In Vitro Studies for Appressorium Formation Inhibition
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, D. Computational studies on the histone deacetylases and the design of selective histone deacetylase inhibitors. Top. Med. Chem. 2009, 9, 241–256. [Google Scholar] [CrossRef]
- Kouzarides, T. Histone acetylases and deacetylases in cell proliferation. Genet. Dev. 1999, 9, 40–48. [Google Scholar] [CrossRef]
- Marks, P.A.; Rifkind, R.A.; Richon, V.M.; Breslow, R.; Miller, T.; Kelly, W.K. Histone deacetylases and cancer: causes and therapies. Nat. Rev. Cancer 2001, 1, 194–202. [Google Scholar] [CrossRef]
- Struhl, K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998, 12, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-F.; Helquist, P.; Wiech, N.L.; Wiest, O. Toward Selective Histone Deacetylase Inhibitor Design: Homology Modeling, Docking Studies, and Molecular Dynamics Simulations of Human Class I Histone Deacetylases. J. Med. Chem. 2005, 48, 6936–6947. [Google Scholar] [CrossRef] [PubMed]
- Vadivelan, S.; Sinha, B.; Rambabu, G.; Boppana, K.; Jagarlapudi, S.A. Pharmacophore modeling and virtual screening studies to design some potential histone deacetylase inhibitors as new leads. J. Mol. Graph. Model. 2008, 26, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Wang, X.S.; Huang, X.-P.; Roth, B.L.; Butler, K.V.; Kozikowski, A.P.; Jung, M.; Tropsha, A. Novel Inhibitors of Human Histone Deacetylase (HDAC) Identified by QSAR Modeling of Known Inhibitors, Virtual Screening, and Experimental Validation. J. Chem. Inf. Model. 2009, 49, 461–476. [Google Scholar] [CrossRef]
- Melagraki, G.; Afantitis, A.; Sarimveis, H.; Koutentis, P.A.; Kollias, G.; Igglessi-Markopoulou, O. Predictive QSAR workflow for the in silico identification and screening of novel HDAC inhibitors. Mol. Divers. 2009, 13, 301–311. [Google Scholar] [CrossRef]
- Nair, S.B.; Teli, M.K.; Pradeep, H.; Rajanikant, G. Computational identification of novel histone deacetylase inhibitors by docking based QSAR. Comput. Boil. Med. 2012, 42, 697–705. [Google Scholar] [CrossRef]
- Park, H.; Kim, S.; Kim, Y.E.; Lim, S.-J. A Structure-Based Virtual Screening Approach toward the Discovery of Histone Deacetylase Inhibitors: Identification of Promising Zinc-Chelating Groups. ChemMedChem 2010, 5, 591–597. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Perspect. Boil. 2014, 6, a018713. [Google Scholar] [CrossRef]
- De Ruijter, A.J.; Van Gennip, A.H.; Caron, H.N.; Kemp, S.; Van Kuilenburg, A.B. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Tsuji, N.; Kobayashi, M. Trichostatin C, a glucopyranosyl hydroxamate. J. Antibiot. 1978, 31, 939–944. [Google Scholar] [CrossRef]
- Yoshida, M.; Kijima, M.; Akita, M.; Beppu, T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Boil. Chem. 1990, 265, 17174–17179. [Google Scholar]
- Yoshida, M.; Nomura, S.; Beppu, T. Effects of trichostatins on differentiation of murine erthroleukemia cells. Cancer Res. 1987, 47, 3688–3691. [Google Scholar]
- Niu, X.; Hao, X.; Hong, Z.; Chen, L.; Yu, X.; Zhu, X. A Putative Histone Deacetylase Modulates the Biosynthesis of Pestalotiollide B and Conidiation in Pestalotiopsis microspora. J. Microbiol. Biotechnol. 2015, 25, 579–588. [Google Scholar] [CrossRef]
- Graessle, S.; Dangl, M.; Haas, H.; Mair, K.; Trojer, P.; Brandtner, E.-M.; Walton, J.D.; Loidl, P.; Brosch, G. Characterization of two putative histone deacetylase genes from Aspergillus nidulans. Biochim. et Biophys. Acta (BBA) - Gene Struct. Expr. 2000, 1492, 120–126. [Google Scholar] [CrossRef]
- Lee, I.; Oh, J.-H.; Shwab, E.K.; Dagenais, T.R.; Andes, D.; Keller, N.P. HdaA, a class 2 histone deacetylase of Aspergillus fumigatus, affects germination and secondary metabolite production. Fungal Genet. Boil. 2009, 46, 782–790. [Google Scholar] [CrossRef]
- Li, Y.; Wang, C.; Liu, W.; Wang, G.; Kang, Z.; Kistler, H.C.; Xu, J.-R. The HDF1 Histone Deacetylase Gene Is Important for Conidiation, Sexual Reproduction, and Pathogenesis in Fusarium graminearum. Mol. Plant-Microbe Interact. 2011, 24, 487–496. [Google Scholar] [CrossRef]
- Liu, W.; Iliuk, A.; Ribot, C.; Vallet, J.; Tao, A.; Wang, Y.; Ding, S.-L.; Lebrun, M.-H.; Xu, J.-R. The tig1 histone deacetylase complex regulates infectious growth in the rice blast fungus Magnaporthe oryzae. Plant Cell 2010, 22, 2495–2508. [Google Scholar]
- Tribus, M.; Bauer, I.; Galehr, J.; Rieser, G.; Trojer, P.; Brosch, G.; Loidl, P.; Haas, H.; Graessle, S. A Novel Motif in Fungal Class 1 Histone Deacetylases Is Essential for Growth and Development ofAspergillus. Mol. Boil. Cell 2010, 21, 345–353. [Google Scholar] [CrossRef]
- Jeon, J.; Park, S.-Y.; Chi, M.-H.; Choi, J.; Park, J.; Rho, H.-S.; Kim, S.; Goh, J.; Yoo, S.; Choi, J.; et al. Genome-wide functional analysis of pathogenicity genes in the rice blast fungus. Nat. Genet. 2007, 39, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Izawa, M.; Takekawa, O.; Arie, T.; Teraoka, T.; Yoshida, M.; Kimura, M.; Kamakura, T. Inhibition of histone deacetylase causes reduction of appressorium formation in the rice blast fungus Magnaporthe oryzae. J. Appl. Microbiol. 2009, 55, 489–498. [Google Scholar] [CrossRef]
- Kuroki, M.; Okauchi, K.; Yoshida, S.; Ohno, Y.; Murata, S.; Nakajima, Y.; Nozaka, A.; Tanaka, N.; Nakajima, M.; Taguchi, H.; et al. Chitin-deacetylase activity induces appressorium differentiation in the rice blast fungus Magnaporthe oryzae. Sci. Rep. 2017, 7, 9697. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Soanes, D.M.; Paszkiewicz, K.H.; Dawe, A.L.; Talbot, N.J. Genome-wide transcriptional profiling of appressorium development by the rice blast fungus Magnaporthe oryzae. PLOS Pathog. 2012, 8, e1002514. [Google Scholar]
- Ryder, L.S.; Talbot, N.J. Regulation of appressorium development in pathogenic fungi. Curr. Opin. Plant Biol. 2015, 26, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xue, C.; Kong, L.; Li, G.; Xu, J.-R. A Pmk1-Interacting Gene Is Involved in Appressorium Differentiation and Plant Infection in Magnaporthe oryzae ▿. Eukaryot. Cell 2011, 10, 1062–1070. [Google Scholar] [CrossRef]
- Li, X.; Gao, C.; Liu, M.; Yin, Z.; Zhang, H.; Zheng, X.; Wang, P. MoEnd3 regulates appressorium formation and virulence through mediating endocytosis in rice blast fungus Magnaporthe oryzae. PLoS Pathog 2017, 13, 1006449. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.S.; Honig, B. An integrated approach to the analysis and modeling of protein sequences and structures. III. A comparative study of sequence conservation in protein structural families using multiple structural alignments. J. Mol. Biol. 2000, 301, 691–711. [Google Scholar] [CrossRef] [PubMed]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. 1999, 12, 85–94. [Google Scholar] [CrossRef] [PubMed]
- SAVES Server. Available online: http://services.mbi.ucla.edu/SAVES/ (accessed on 23 March 2019).
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK a program to check the stereo chemical quality of protein structure. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. [20] VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar] [PubMed]
- Colovos, C.; Yeates, T.O. ERRAT: an empirical atom-based method for validating protein structures. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC – A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; Wang, J.; Yu, B.; Zhang, J.; Bryant, S.H. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Shanmugam, G.; Lee, S.K.; Jeon, J. Identification of Potential Nematicidal Compounds against the Pine Wood Nematode, Bursaphelenchus xylophilus through an In Silico Approach. Molecules 2018, 23, 1828. [Google Scholar] [CrossRef]
- Caracuel-Rios, Z.; Talbot, N.J. Cellular differentiation and host invasion by the rice blast fungus Magnaporthe grisea. Curr. Opin. Microbiol. 2007, 10, 339–345. [Google Scholar] [CrossRef]
- Perfect, S.E.; Hughes, H.; O’Connell, R.J.; Green, J.R. Colletotrichum: A Model Genus for Studies on Pathology and Fungal–Plant Interactions. Fungal Genet. Boil. 1999, 27, 186–198. [Google Scholar] [CrossRef]
- The UniProt Consortium, UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; Fagan, P. The protein data bank. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Boil. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Fiser, A.; Sali, A. Modeller: Generation and Refinement of Homology-Based Protein Structure Models. Methods in Enzymology 2003, 374, 461–491. [Google Scholar]
- Kaplan, W.; Littlejohn, T.G. Swiss-PDB viewer (deep view). Brief. Bioinform. 2001, 2, 195–197. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. Discovery studio 2017 R2 Client; Dassault Systèmes: San Diego, CA, USA, 2017. [Google Scholar]
- Kamakura, T.; Yamaguchi, S.; Saitoh, K.-I.; Teraoka, T.; Yamaguchi, I. A Novel Gene, CBP1, Encoding a Putative Extracellular Chitin-Binding Protein, May Play an Important Role in the Hydrophobic Surface Sensing of Magnaporthe grisea During Appressorium Differentiation. Mol. Plant-Microbe Interact. 2002, 15, 437–444. [Google Scholar] [CrossRef]
Sample Availability: All the compounds are available from Pubchem and ZINC database. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No. | Model | Amino Acids Residues (%) in Ramachandran Plot (PROCHECK) | Verify 3D (%) | ERRAT | |||
|---|---|---|---|---|---|---|---|
| MFA | GAR | AAR | DAR | ||||
| 1. | MoRPD3 Model | 93.0 | 6.7 | 0.3 | 0.0 | 91.21 | 93.25 |
| 2. | 4LXZ Template | 91.7 | 8.2 | 0.1 | 0.0 | 94.11 | 95.83 |
| S.No | Compound ID | IUPAC Name | Binding Affinity (kcal/mol) |
|---|---|---|---|
| 1 | CID444732 | (2E,4E,6R)-7-[4-(dimethylamino)phenyl]-N-hydroxy-4,6-dimethyl-7-oxohepta-2,4-dienamide (Trichostatin A) | −7.0 |
| 2 | CID4962839 | 2-[[4-(2-oxopyrrolidin-1-yl)benzoyl] amino] acetic acid | −7.2 |
| 3 | CID16217875 | [2-[[4-(2-methoxyethyl) phenoxy] methyl] phenyl] boronic acid | −8.7 |
| 4 | CID16218068 | [4-[[4-(2-methoxyethyl) phenoxy]methyl] phenyl] boronic acid | −8.5 |
| 5 | ZINC01753336 | 1-(3-fluorophenyl)-3-(2-methyl-5-nitro-phenyl)urea | −7.9 |
| 6 | ZINC04376856 | 4-(4-methylphenyl)-2-(3-nitrophenyl)-2,3-dihydro-1,5-benzothiazepine | −8.0 |
| 7 | ZINC04692015 | 5,7-dihydroxy-3-((2s,3r,4r,5r,6s)-3,4,5-trihydroxy-6-methyl-tetrahydro-pyran-2-yloxy)-2-(3,4,5-trihydroxy-phenyl)-1-benzopyran-4-one | −7.6 |
| 8 | ZINC05124957 | 4-[4-(carboxymethyl)phenyl]azo-3-hydroxy-naphthalene-2-carboxylic | −7.1 |
| 9 | ZINC01045089 | 2-(4-nitrophenyl)-4-phenyl-2,3-dihydro-1,5-benzothiazepine | −8.1 |
| 10 | ZINC01588812 | 5-(4-Bromo-phenyl)-2-(4-nitro-phenyl)-oxazole | −7.3 |
| 11 | ZINC1726776 | 3-(4-chloro-6-phenoxy-s-triazin-2-yl)-1-phenyl-indole | −7.8 |
| CID444732 | CID16217875 | CID16218068 |
|---|---|---|
| His41 (Pi-Sigma) | - | - |
| Pro42 (Pi-Alkyl) | - | Pro42 (Pi-Alkyl) |
| - | - | # Asp112 * (Pi-Anion) |
| His153 * | - | - |
| His154 * | - | - |
| Gly162 * | - | Gly162 * |
| Phe163 (Pi-Pi) | Phe163 (Pi-Pi-stacked) | # Phe163 * (Pi-Pi-stacked) |
| # His191 * (Pi-Alkyl) | # His191 * (Pi-Pi) | His191 (Pi-Pi-stacked) |
| - | Tyr217 (Pi-Pi-stacked) | - |
| Phe218 (Pi-Alkyl) | Phe218 (Pi-Pi-stacked) | Phe218 (Pi-Pi-stacked) |
| Leu284 (Pi-Alkyl) | # Leu284 * (Pi-Alkyl) | - |
| −7.0 kCal/mol | −8.7 kcal/mol | −8.5 kcal/mol |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shanmugam, G.; Kim, T.; Jeon, J. In Silico Identification of Potential Inhibitor Against a Fungal Histone Deacetylase, RPD3 from Magnaporthe Oryzae. Molecules 2019, 24, 2075. https://doi.org/10.3390/molecules24112075
Shanmugam G, Kim T, Jeon J. In Silico Identification of Potential Inhibitor Against a Fungal Histone Deacetylase, RPD3 from Magnaporthe Oryzae. Molecules. 2019; 24(11):2075. https://doi.org/10.3390/molecules24112075
Chicago/Turabian StyleShanmugam, Gnanendra, Taehyeon Kim, and Junhyun Jeon. 2019. "In Silico Identification of Potential Inhibitor Against a Fungal Histone Deacetylase, RPD3 from Magnaporthe Oryzae" Molecules 24, no. 11: 2075. https://doi.org/10.3390/molecules24112075
APA StyleShanmugam, G., Kim, T., & Jeon, J. (2019). In Silico Identification of Potential Inhibitor Against a Fungal Histone Deacetylase, RPD3 from Magnaporthe Oryzae. Molecules, 24(11), 2075. https://doi.org/10.3390/molecules24112075