
Identification of Novel Potential Inhibitors of Pteridine Reductase 1 in Trypanosoma brucei via Computational Structure-Based Approaches and in Vitro Inhibition Assays

 , , and
, , and 
Abstract
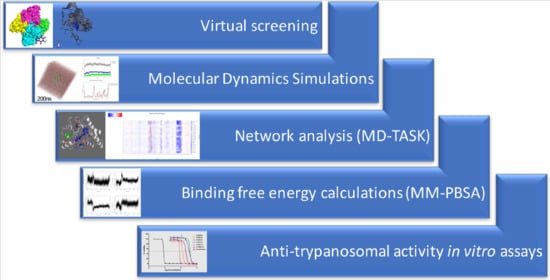
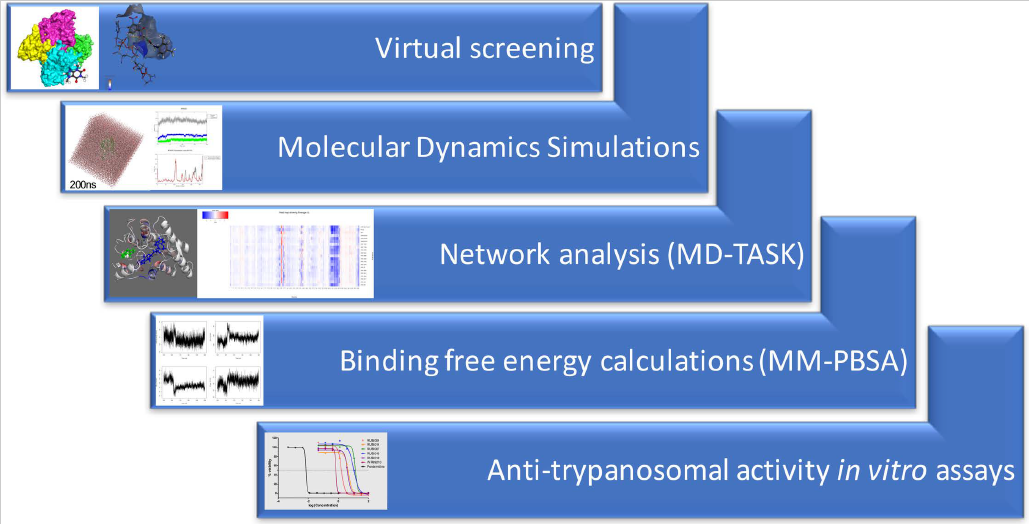
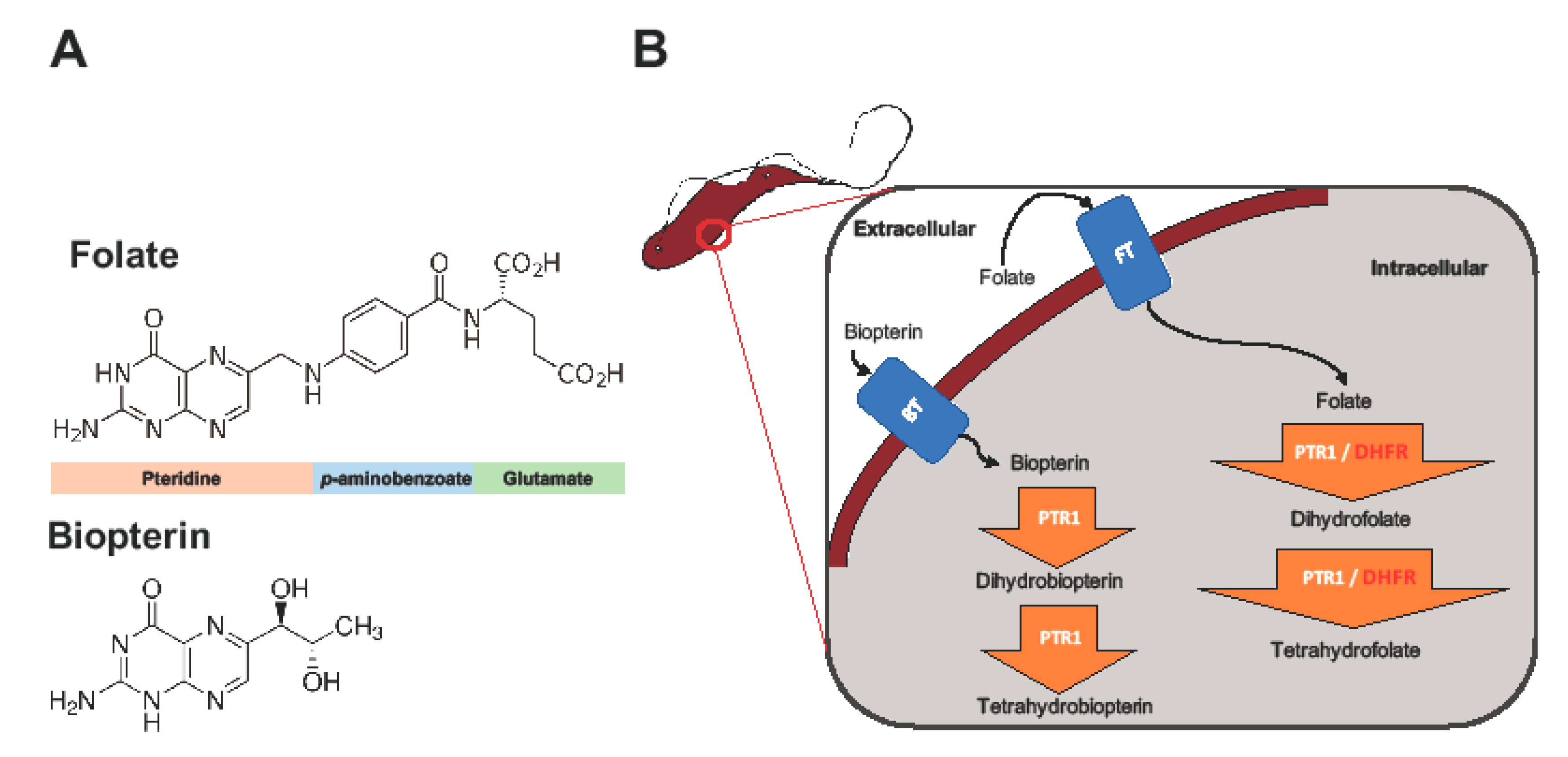
1. Introduction
2. Results
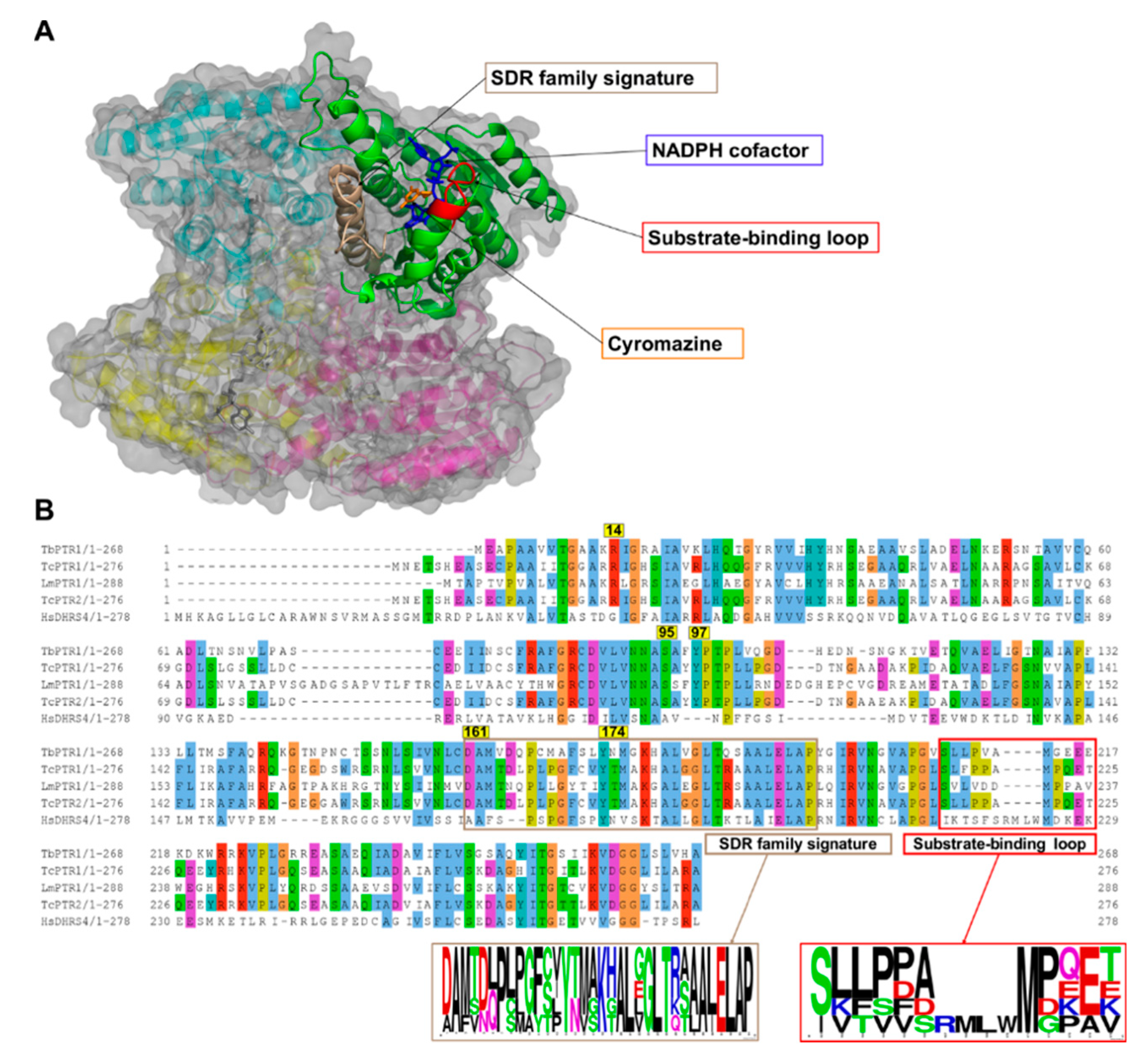
2.1. Overview of PTR1 Structure and Conservation
2.2. Eighteen Potential Hits Out of 5742 Compounds are Identified via Virtual Screening
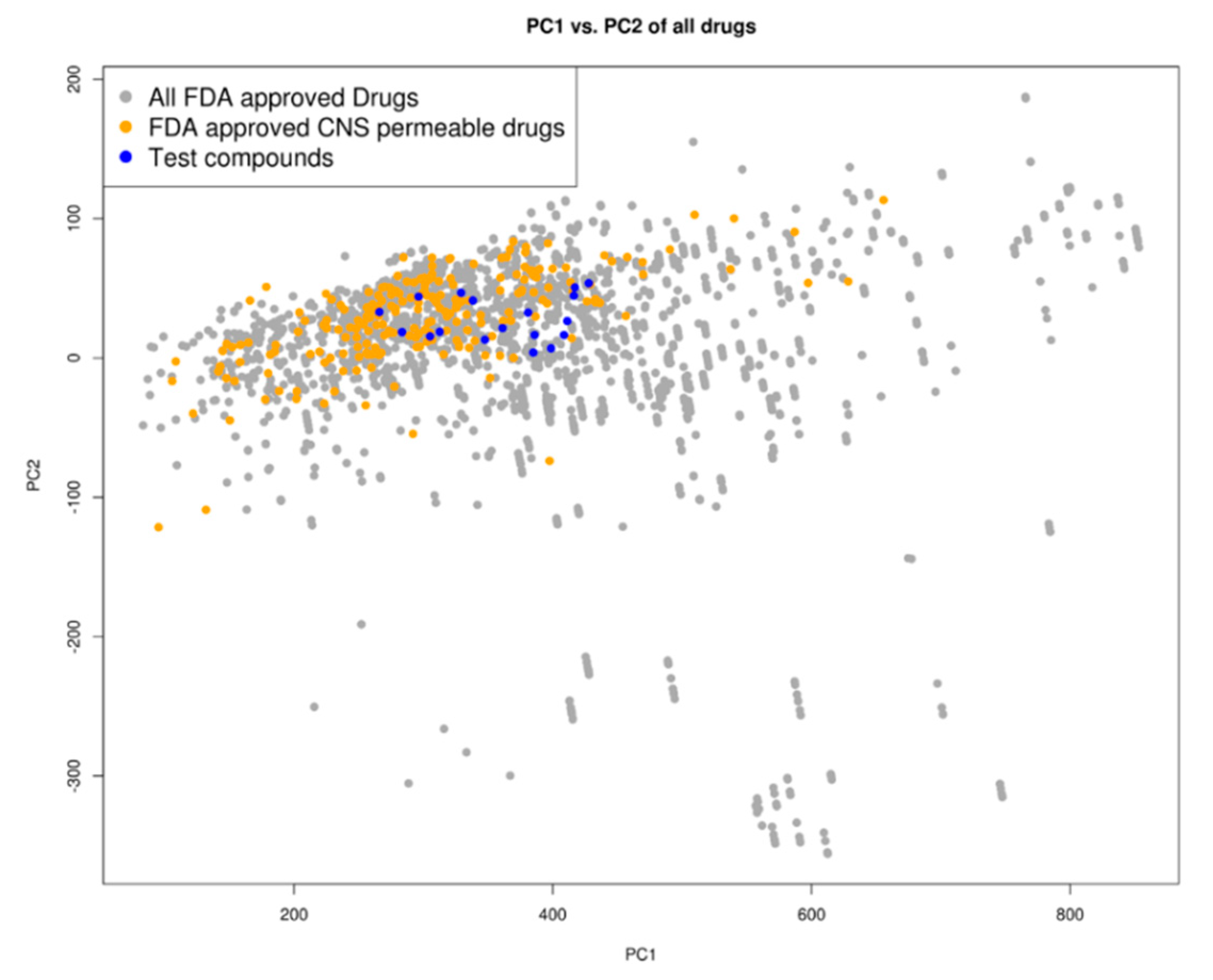
2.3. Drug Likeness
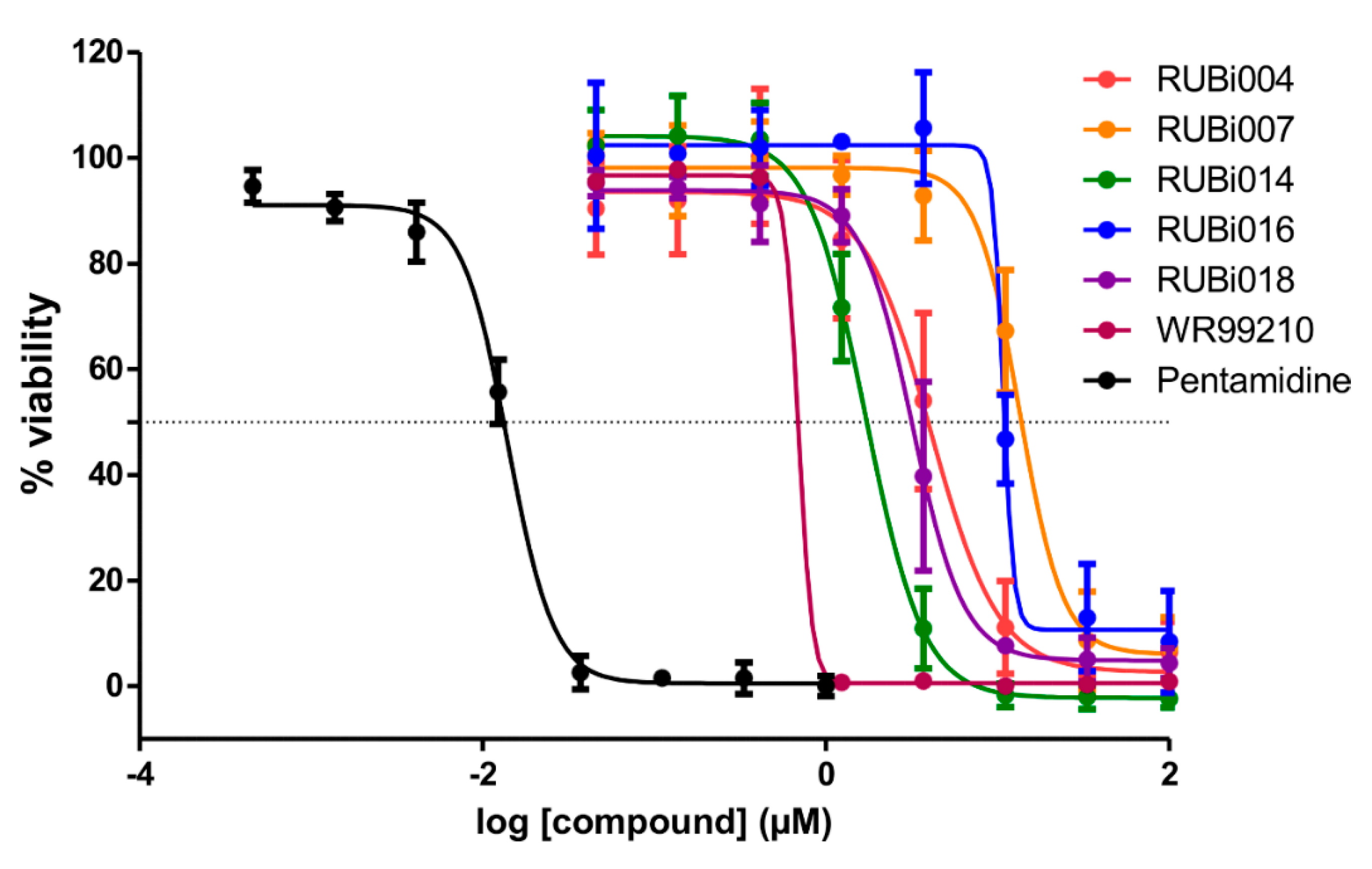
2.4. Five Hit Compounds Show Anti-Trypanosomal Activity In Vitro
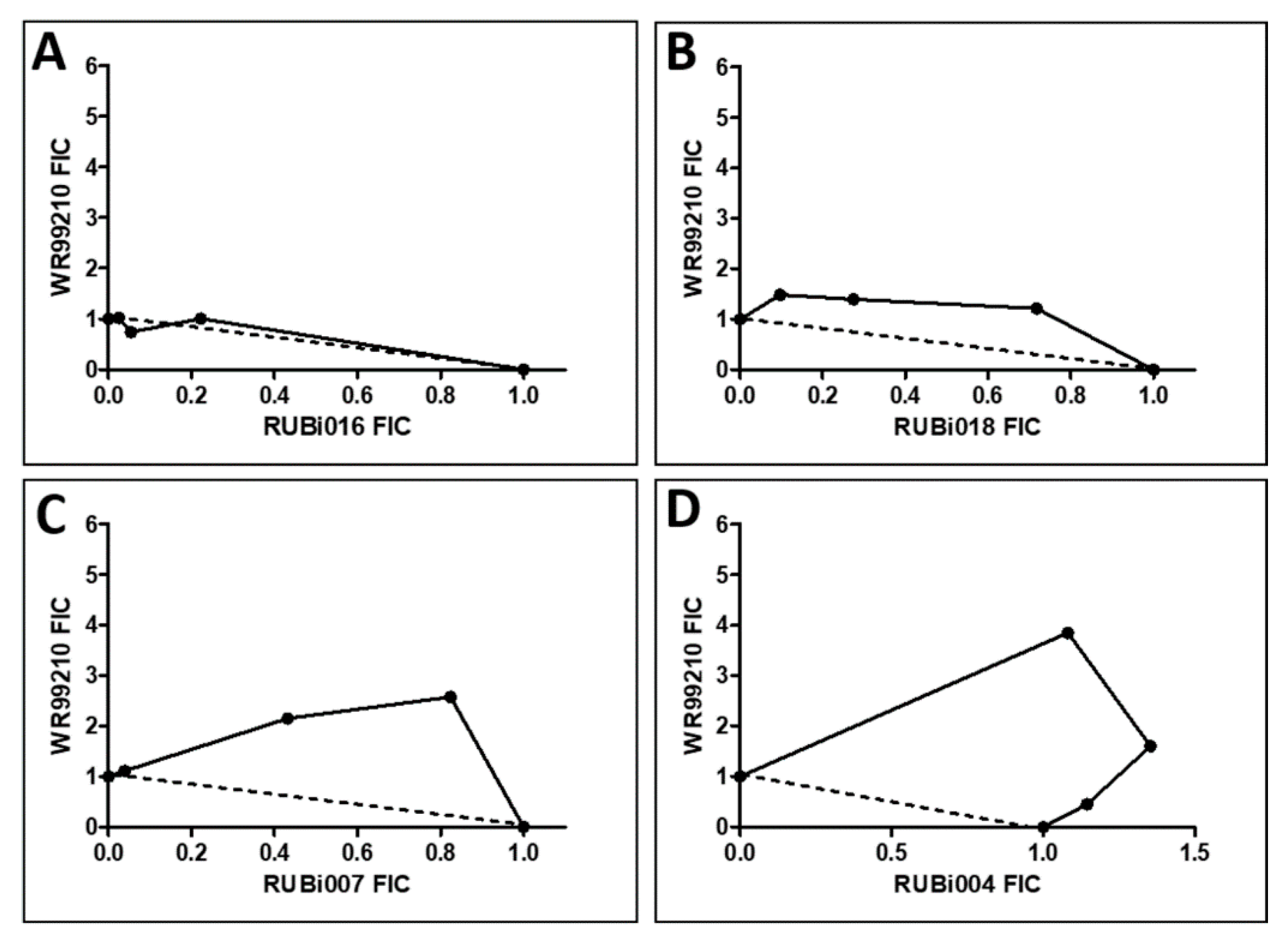
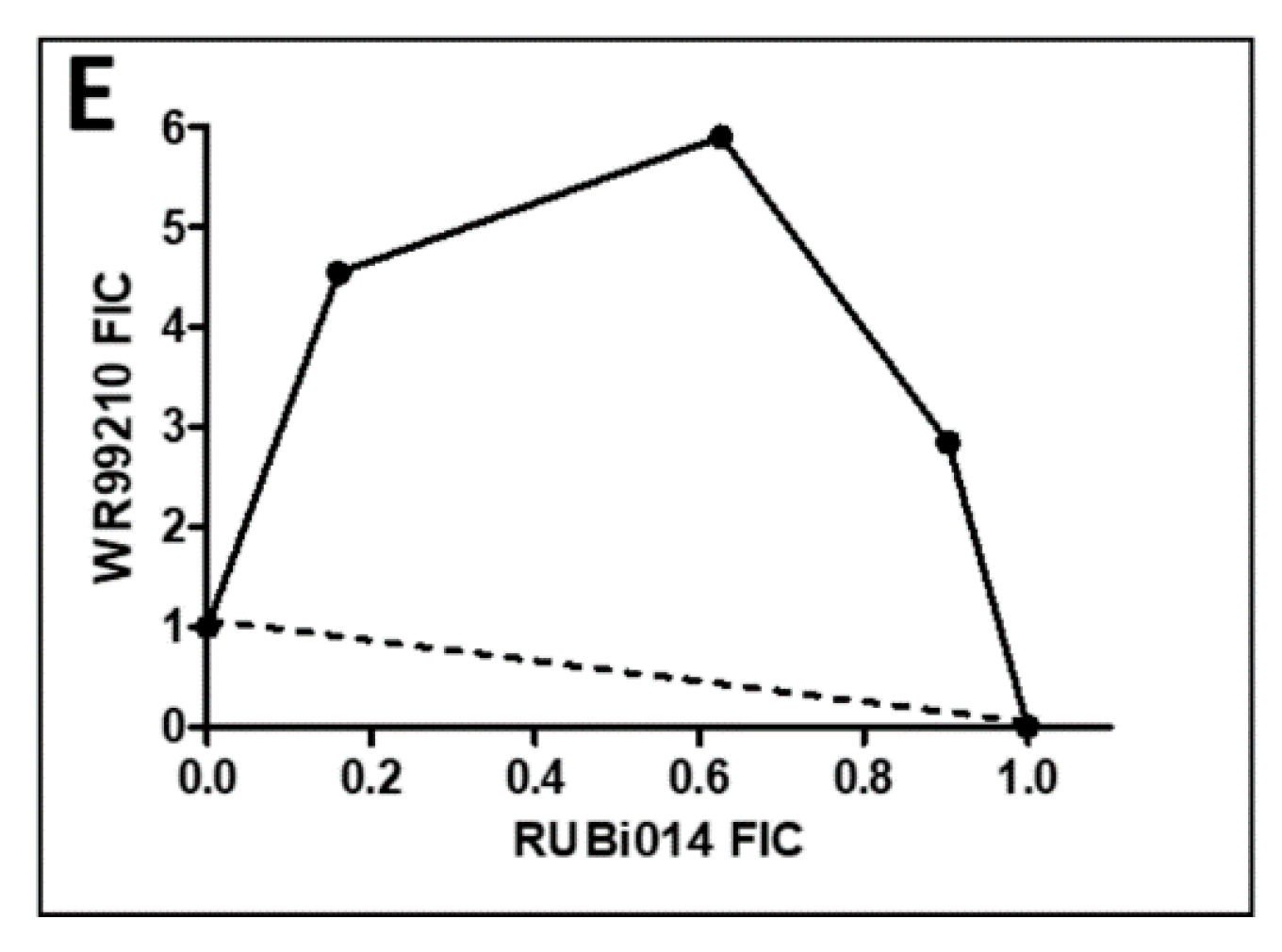
2.4.1. TbPTR1 Hit Compounds Have Either Antagonist or Additive Activity When Used in Combination with a TbDHFR Inhibitor
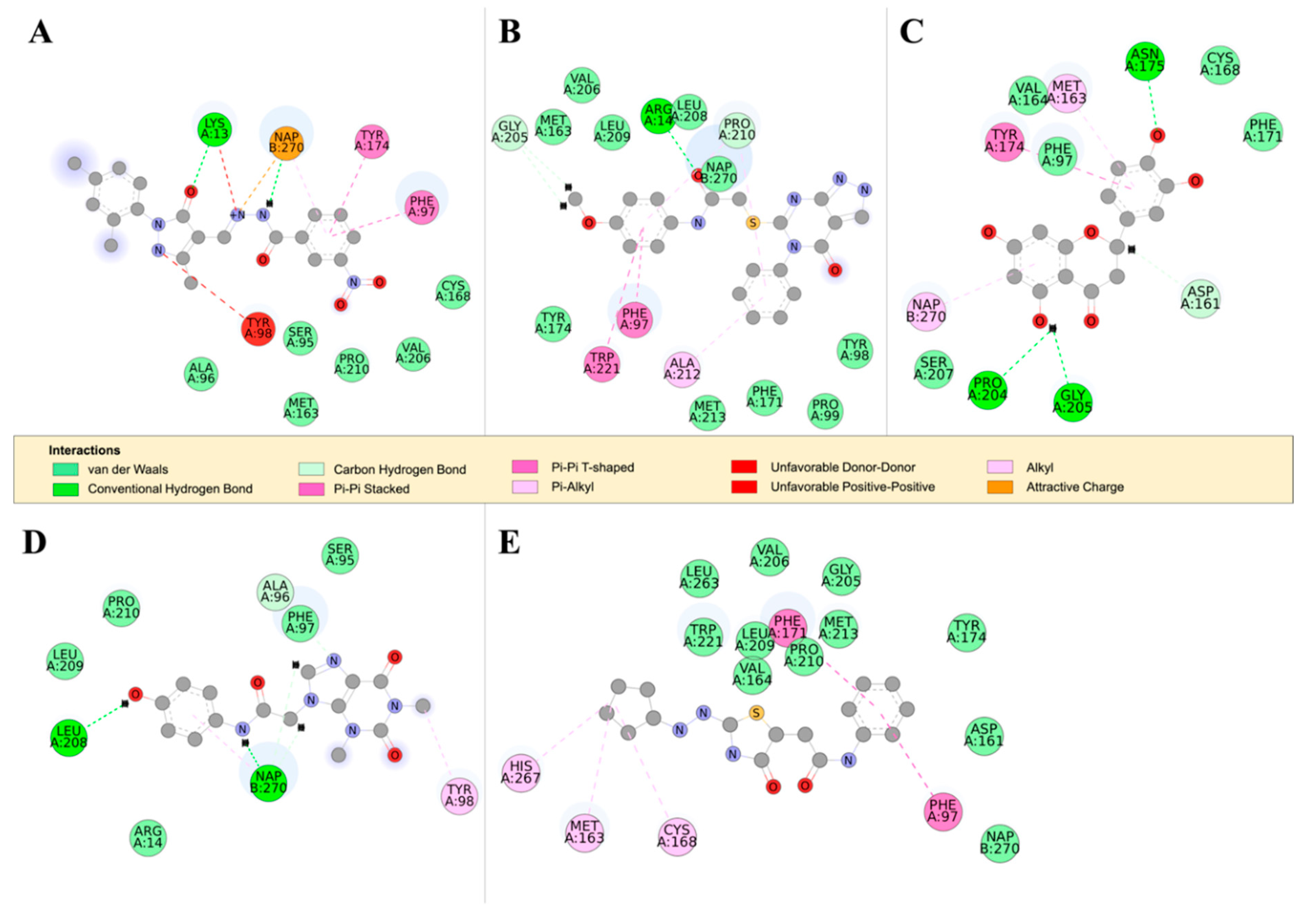
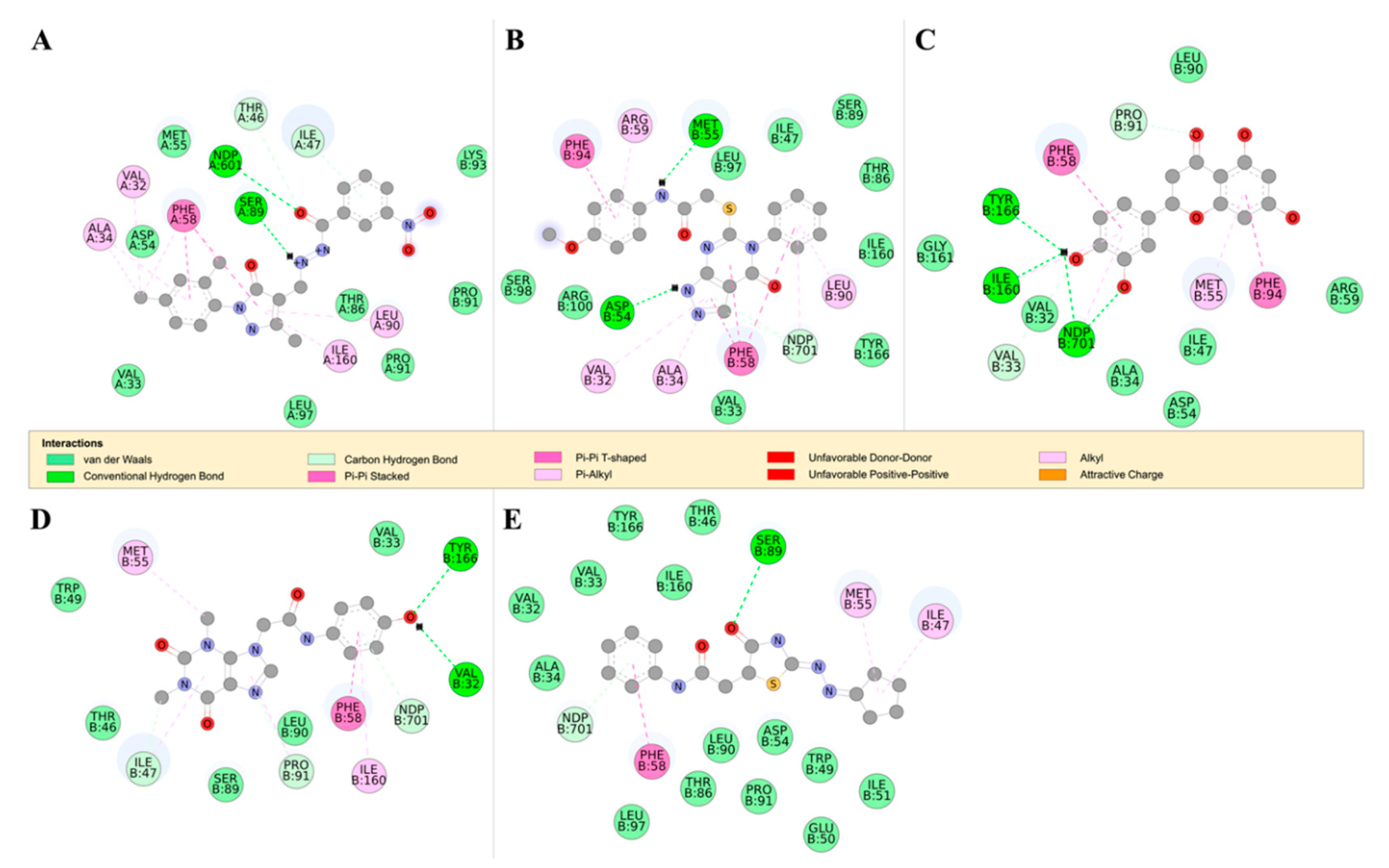
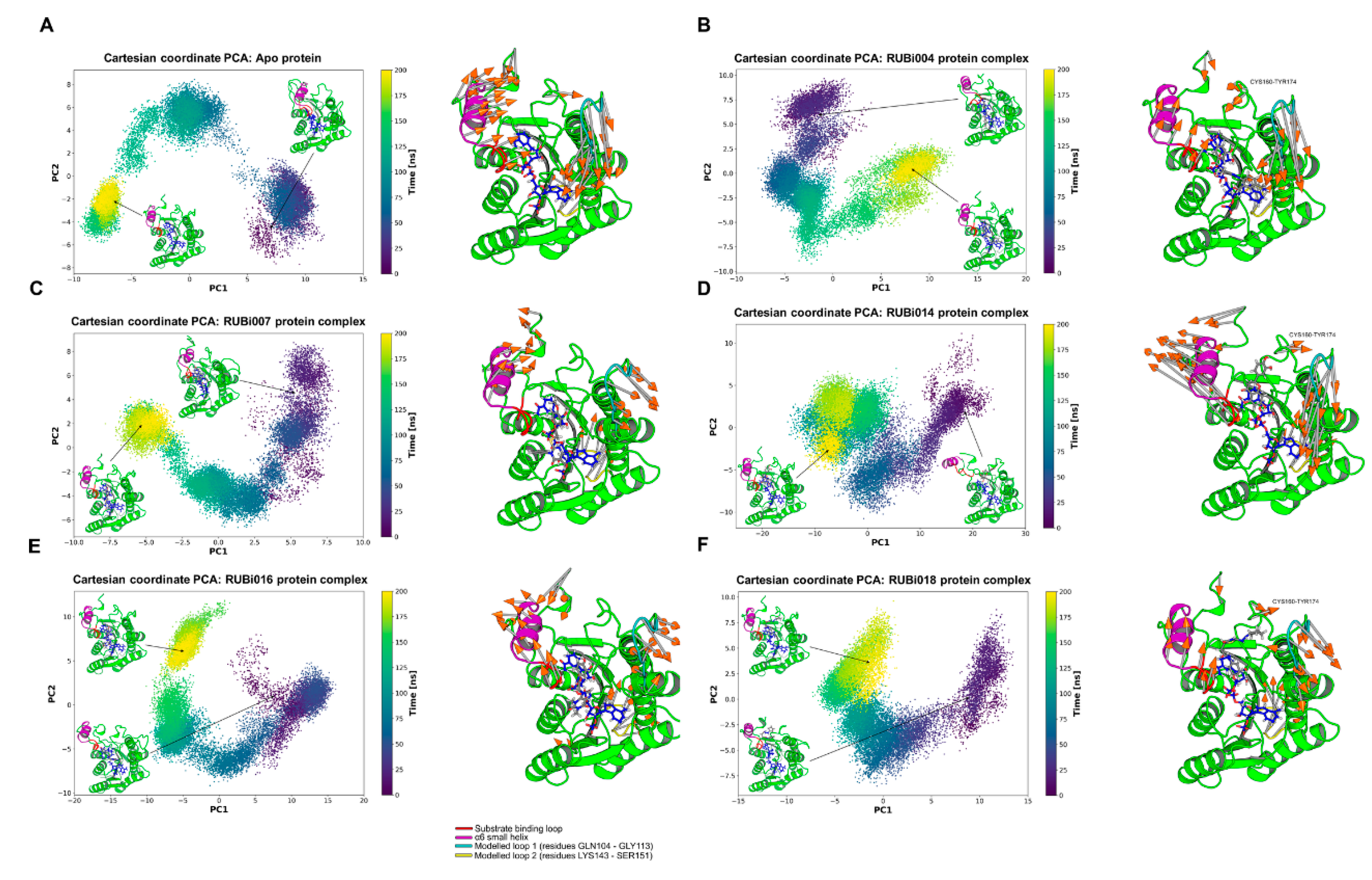
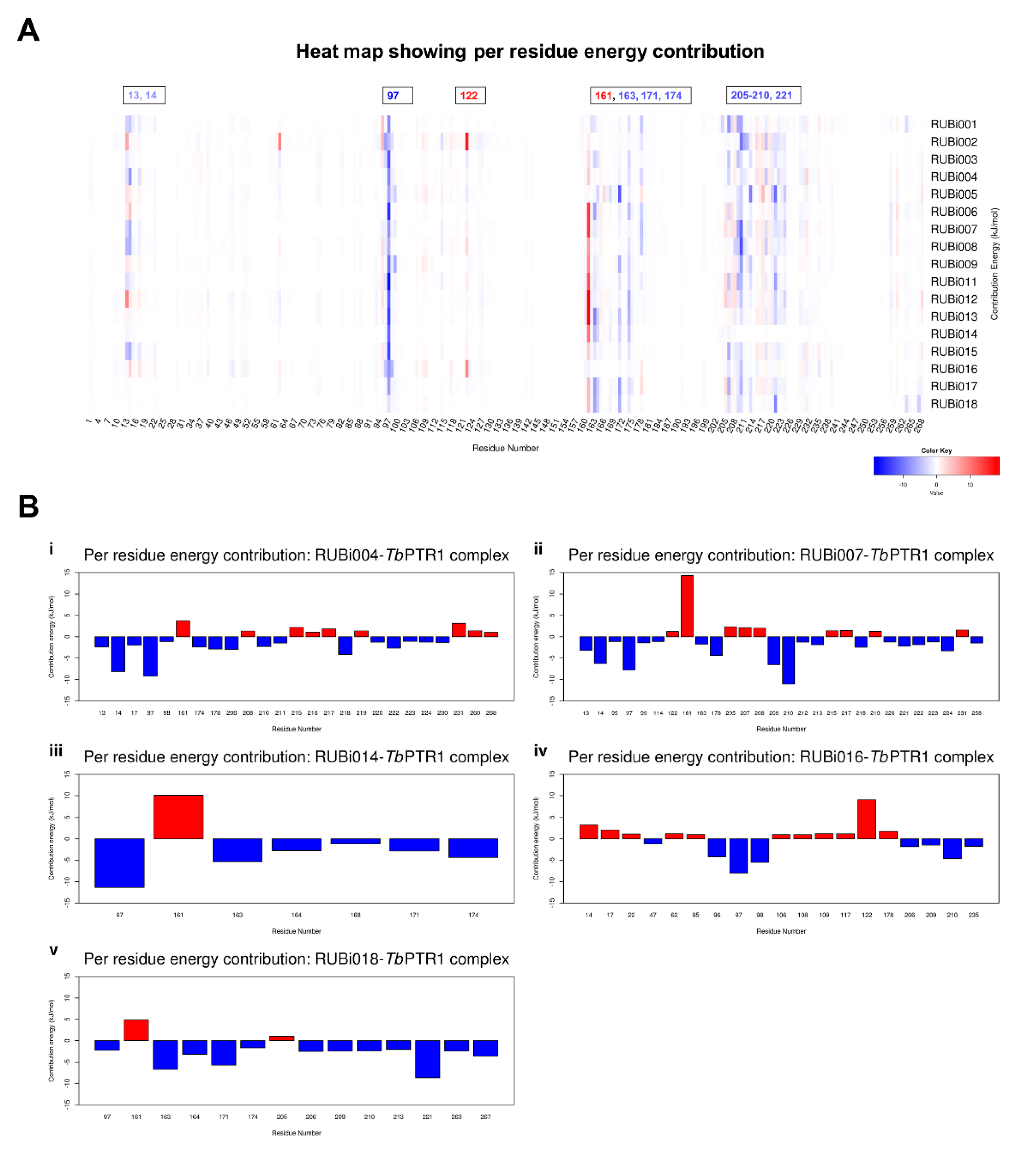
2.4.2. Detailed in Silico Compound Analysis
3. Discussion
4. Materials and Methods
4.1. Ligand Library Preparation
4.2. Preparation of Protein-Ligand Complexes
4.3. Prediction of Blood-Brain Barrier Permeability
4.4. Molecular Dynamics
4.5. MM-PBSA Free Energy Calculations
4.6. Average Shortest Path, and Average Betweenness Centrality
4.7. Trypanosoma In Vitro Inhibition Assay
4.8. In Vitro Human Cytotoxicity Assay
4.9. Pan-Assay Interference Compounds Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Achcar, F.; Kerkhoven, E.J.; Barrett, M.P. Trypanosoma brucei: Meet the system. Curr. Opin. Microbiol. 2013, 3, 78. [Google Scholar] [CrossRef]
- Franco, J.R.; Simarro, P.P.; Diarra, A.; Jannin, J.G. Epidemiology of human African trypanosomiasis. Clin. Epidemiol. 2014, 6, 257–275. [Google Scholar] [PubMed]
- Funk, S.; Nishiura, H.; Heesterbeek, H.; Edmunds, W.J.; Checchi, F. Identifying Transmission Cycles at the Human-Animal Interface: The Role of Animal Reservoirs in Maintaining Gambiense Human African Trypanosomiasis. PLoS Comput. Biol. 2013, 9, 10002855. [Google Scholar] [CrossRef] [PubMed]
- Simarro, P.P.; Diarra, A.; Ruiz Postigo, J.A.; Franco, J.R.; Jannin, J.G. The Human African trypanosomiasis control and surveillance programme of the World Health Organization 2000–2009: The way forward. PLoS Negl. Trop. Dis. 2011, 5, e1007. [Google Scholar] [CrossRef] [PubMed]
- Simarro, P.P.; Cecchi, G.; Paone, M.; Franco, J.R.; Diarra, A.; Ruiz, J.A.; Fèvre, E.M.; Courtin, F.; Mattioli, R.C.; Jannin, J.G. The Atlas of human African trypanosomiasis: A contribution to global mapping of neglected tropical diseases. Int. J. Health Geogr. 2010, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.P.; Gilbert, I.H. Targeting of Toxic Compounds to the Trypanosome’s Interior. Adv. Parasitol. 2006, 63, 125–183. [Google Scholar] [PubMed]
- Fox, J.T.; Stover, P.J. Folate-mediated one-carbon metabolism. Vitam. Horm. 2008, 79, 1–44. [Google Scholar] [CrossRef] [PubMed]
- Bello, A.R.; Nare, B.; Freedman, D.; Hardy, L.; Beverley, S.M. PTR1: A reductase mediating salvage of oxidized pteridines and methotrexate resistance in the protozoan parasite Leishmania major. Proc. Natl. Acad. Sci. USA 1994, 91, 11442–11446. [Google Scholar] [CrossRef] [PubMed]
- Berriman, M.; Ghedin, E.; Hertz-Fowler, C.; Blandin, G.; Renauld, H.; Bartholomeu, D.C.; Lennard, N.J.; Caler, E.; Hamlin, N.E.; Haas, B.; et al. The genome of the African trypanosome Trypanosoma brucei. Science 2005, 309, 416–422. [Google Scholar] [CrossRef]
- Gangjee, A.; Jain, H.D.; Kurup, S. Recent advances in classical and non-classical antifolates as antitumor and antiopportunistic infection agents: Part II. Anticancer Agents Med. Chem. 2008, 8, 205–231. [Google Scholar] [CrossRef]
- Zuccotto, F.; Martin, A.C.R.; Laskowski, R.A.; Thornton, J.M.; Gilbert, I.H. Dihydrofolate reductase: A potential drug target in trypanosomes and leishmania. J. Comput. Aided Mol. Des. 1998, 12, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Sienkiewicz, N.; Jarosławski, S.; Wyllie, S.; Fairlamb, A.H. Chemical and genetic validation of dihydrofolate reductase-thymidylate synthase as a drug target in African trypanosomes. Mol. Microbiol. 2008, 69, 520–533. [Google Scholar] [CrossRef] [PubMed]
- Nare, B.; Hardy, L.W.; Beverley, S.M.; Ptrs, L. The Roles of Pteridine Reductase 1 and Dihydrofolate Reductase-Thymidylate Synthase in Pteridine Metabolism in the Protozoan Parasite Leishmania major. J. Biol. Chem. 1997, 272, 13883–13891. [Google Scholar] [CrossRef] [PubMed]
- Vickers, T.J.; Beverley, S.M. Folate metabolic pathways in Leishmania. Essays Biochem. 2011, 51, 63–80. [Google Scholar] [CrossRef]
- Robello, C.; Navarro, P.; Castanys, S.; Gamarro, F. A pteridine reductase gene ptr1 contiguous to a P-glycoprotein confers resistance to antifolates in Trypanosoma cruzi. Mol. Biochem. Parasitol. 1997, 90, 525–535. [Google Scholar] [CrossRef]
- Gourley, D.G.; Schüttelkopf, A.W.; Leonard, G.A.; Luba, J.; Hardy, L.W.; Beverley, S.M.; Hunter, W.N. Pteridine reductase mechanism correlates pterin metabolism with drug resistance in trypanosomatid parasites. Nat. Struct. Biol. 2001, 8, 521–525. [Google Scholar] [CrossRef]
- Sienkiewicz, N.; Ong, H.B.; Fairlamb, A.H. Trypanosoma brucei pteridine reductase 1 is essential for survival in vitro and for virulence in mice. Mol. Microbiol. 2010, 77, 658–671. [Google Scholar] [CrossRef]
- Cavazzuti, A.; Paglietti, G.; Hunter, W.N.; Gamarro, F.; Piras, S.; Loriga, M.; Allecca, S.; Corona, P.; McLuskey, K.; Tulloch, L.; et al. Discovery of potent pteridine reductase inhibitors to guide antiparasite drug development. Proc. Natl. Acad. Sci. USA 2008, 105, 1448–1453. [Google Scholar] [CrossRef]
- Tulloch, L.B.; Martini, V.P.; Iulek, J.; Huggan, J.K.; Lee, J.H.; Gibson, C.L.; Smith, T.K.; Suckling, C.J.; Hunter, W.N. Structure-based design of pteridine reductase inhibitors targeting African sleeping sickness and the leishmaniases. J. Med. Chem. 2010, 53, 221–229. [Google Scholar] [CrossRef]
- Mpamhanga, C.P.; Spinks, D.; Tulloch, L.B.; Shanks, E.J.; Robinson, D.A.; Collie, I.T.; Fairlamb, A.H.; Wyatt, P.G.; Frearson, J.A.; Hunter, W.N.; et al. One scaffold, three binding modes: Novel and selective pteridine reductase 1 inhibitors derived from fragment hits discovered by virtual screening. J. Med. Chem. 2009, 52, 4454–4465. [Google Scholar] [CrossRef]
- Hardy, L.W.; Matthews, W.; Nare, B.; Beverley, S.M. Biochemical and genetic tests for inhibitors of Leishmania pteridine pathways. Exp. Parasitol. 1997, 87, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Nare, B.; Luba, J.; Hardy, L.W.; Beverley, S. New approaches to Leishmania chemotherapy: Pteridine reductase 1 (PTR1) as a target and modulator of antifolate sensitivity. Parasitology 1997, 114, S101–S110. [Google Scholar] [PubMed]
- Matovu, E.; Seebeck, T.; Enyaru, J.C.K.; Kaminsky, R. Drug resistance in Trypanosoma brucei spp., the causative agents of sleeping sickness in man and nagana in cattle. Microbes Infect. 2001, 3, 763–770. [Google Scholar] [CrossRef]
- Babokhov, P.; Sanyaolu, A.O.; Oyibo, W.; Fagbenro-Beyioku, A.F.; Iriemenam, N.C. A current analysis of chemotherapy strategies for the treatment of human African trypanosomiasis. Pathog. Glob. Health 2013, 107, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Mesu, V.K.B.K.; Kalonji, W.M.; Bardonneau, C.; Mordt, O.V.; Blesson, S.; Simon, F.; Delhomme, S.; Bernhard, S.; Kuziena, W.; Lubaki, J.P.F.; et al. Oral fexinidazole for late-stage African Trypanosoma brucei gambiense trypanosomiasis: A pivotal multicentre, randomised, non-inferiority trial. Lancet 2018, 391, 144–154. [Google Scholar] [CrossRef]
- Dawson, A.; Tulloch, L.B.; Barrack, K.L.; Hunter, W.N. High-resolution structures of Trypanosoma brucei pteridine reductase ligand complexes inform on the placement of new molecular entities in the active site of a potential drug target. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 1334–1340. [Google Scholar] [CrossRef]
- Luba, J.; Nare, B.; Liang, P.H.; Anderson, K.S.; Beverley, S.M.; Hardy, L.W. Leishmania major pteridine reductase 1 belongs to the short chain dehydrogenase family: Stereochemical and kinetic evidence. Biochemistry 1998, 37, 4093–4104. [Google Scholar] [CrossRef]
- Schormann, N.; Pal, B.; Senkovich, O.; Carson, M.; Howard, A.; Smith, C.; DeLucas, L.; Chattopadhyay, D. Crystal structure of Trypanosoma cruzi pteridine reductase 2 in complex with a substrate and an inhibitor. J. Struct. Biol. 2005, 152, 64–75. [Google Scholar] [CrossRef]
- Fiser, A.; Šali, A. MODELLER: Generation and Refinement of Homology-Based Protein Structure Models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar]
- DeLano, W.L. The PyMOL Molecular Graphics System. Schrödinger LLC wwwpymolorg Version 1. Available online: http://www.pymol.org (accessed on 31 December 2018).
- Accelrys Software Inc. Discovery Studio Modeling Environment, Release 3.5; Accelrys Softw Inc.: San Diego, CA, USA, 2012. [Google Scholar]
- Obiol-Pardo, C.; Rubio-Martinez, J. Comparative evaluation of MMPBSA and XSCORE to compute binding free energy in XIAP-peptide complexes. J. Chem. Inf. Model. 2007, 47, 134–142. [Google Scholar] [CrossRef]
- Legros, D.; Ollivier, G.; Gastellu-Etchegorry, M.; Paquet, C.; Burri, C.; Jannin, J.; Büscher, P. Treatment of human African trypanosomiasis—Present situation and needs for research and development. Lancet Infect. Dis. 2002, 2, 437–440. [Google Scholar] [CrossRef]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014. [Google Scholar] [CrossRef]
- Dawson, A.; Gibellini, F.; Sienkiewicz, N.; Tulloch, L.B.; Fyfe, P.K.; McLuskey, K.; Fairlamb, A.H.; Hunter, W.N. Structure and reactivity of Trypanosoma brucei pteridine reductase: Inhibition by the archetypal antifolate methotrexate. Mol. Microbiol. 2006, 61, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R.; Kraut, J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence. Biochemistry 1997, 36, 586–603. [Google Scholar] [CrossRef] [PubMed]
- Schnell, J.R.; Dyson, H.J.; Wright, P.E. Structure, Dynamics, and Catalytic Function of Dihydrofolate Reductase. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 119–140. [Google Scholar] [CrossRef] [PubMed]
- Schüttelkopf, A.W.; Hardy, L.W.; Beverley, S.M.; Hunter, W.N. Structures of Leishmania major pteridine reductase complexes reveal the active site features important for ligand binding and to guide inhibitor design. J. Mol. Biol. 2005, 352, 105–116. [Google Scholar] [CrossRef]
- van Baren, C.; Martino, V.; Di Leo Lira, P.; Debenedetti, S.; Houghton, P.; Croft, S.; Martino, V. Triterpenic Acids and Flavonoids from Satureja parvifolia. Evaluation of their Antiprotozoal Activity. Zeitschrift fur Naturforsch—Sect. C J. Biosci. 2006, 61, 189–192. [Google Scholar] [CrossRef]
- Tasdemir, D.; Kaiser, M.; Brun, R.; Yardley, V.; Schmidt, T.J.; Tosun, F.; Rüedi, P. Antitrypanosomal and antileishmanial activities of flavonoids and their analogues: In vitro, in vivo, structure-activity relationship, and quantitative structure-activity relationship studies. Antimicrob. Agents Chemother. 2006, 50, 1352–1364. [Google Scholar] [CrossRef]
- Hassan, A.A.; Ibrahim, Y.R.; El-Sheref, E.M.; Abdel-Aziz, M.; Braese, S.; Nieger, M. Synthesis and antibacterial activity of 4-Aryl-2-(1-substituted ethylidene)thiazoles. Arch. Pharm. (Weinheim) 2013. [Google Scholar] [CrossRef]
- Vanichtanankul, J.; Taweechai, S.; Yuvaniyama, J.; Vilaivan, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Yuthavong, Y. Trypanosomal dihydrofolate reductase reveals natural antifolate resistance. ACS Chem. Biol. 2011, 6, 905–911. [Google Scholar] [CrossRef]
- Brown, D.K.; Penkler, D.L.; Sheik Amamuddy, O.; Ross, C.; Atilgan, A.R.; Atilgan, C.; Tastan Bishop, Ö. MD-TASK: A software suite for analyzing molecular dynamics trajectories. Bioinformatics 2017. [Google Scholar] [CrossRef] [PubMed]
- Penkler, D.; Atilgan, C.; Tastan Bishop, O. Allosteric Modulation of Human Hsp90α Conformational Dynamics. J. Chem. Inf. Model. 2018. [Google Scholar] [CrossRef] [PubMed]
- Atilgan, A.R.; Akan, P.; Baysal, C. Small-World Communication of Residues and Significance for Protein Dynamics. Biophys. J. 2004, 86, 85–91. [Google Scholar] [CrossRef]
- Olliaro, P.L. Antimalarial compounds: From bench to bedside. J. Exp. Biol. 2003. [Google Scholar] [CrossRef]
- Kenny, P.W. Comment on the Ecstasy and Agony of Assay Interference Compounds. J. Chem. Inf. Model. 2017, 57, 2640–2645. [Google Scholar] [CrossRef]
- Kilchmann, F.; Marcaida, M.J.; Kotak, S.; Schick, T.; Boss, S.D.; Awale, M.; Gonczy, P.; Reymond, J.L. Discovery of a Selective Aurora A Kinase Inhibitor by Virtual Screening. J. Med. Chem. 2016, 59, 7188–7211. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.L.; Walters, M.A. How to Triage PAINS-Full Research. Assay Drug Dev. Technol. 2016, 14, 168–174. [Google Scholar] [CrossRef]
- Hatherley, R.; Brown, D.K.; Musyoka, T.M.; Penkler, D.L.; Faya, N.; Lobb, K.A.; Bishop, Ö.T. SANCDB: A South African natural compound database. J. Cheminform. 2015, 7, 29. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Bacchi, C.J. Chemotherapy of Human African Trypanosomiasis. Interdiscip. Perspect. Infect. Dis. 2009. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization WHO. Trypanosomiasis, Human African (Sleeping Sickness); WHO: Geneva, Switzerland, 2016. [Google Scholar]
- Hitchcock, S.A. Blood-brain barrier permeability considerations for CNS-targeted compound library design. Curr. Opin. Chem. Biol. 2008, 12, 318–323. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Mohammad, A.S.; Adkins, C.E.; Lockman, P.R. Molecular determinants of blood-brain barrier permeation. Ther. Deliv. 2015, 6, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Moving beyond rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Herbertz, T.; Hudkins, R.L.; Dorsey, B.D.; Mallamo, J.P. Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem. Neurosci. 2012. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucl. Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Eswar, N.; Webb, B.; Marti-Renom, M.; Madhusudhan, M.S.; Eramian, D.; Shen, M.Y.; Pieper, U.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 2007. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucl. Acids Res. 2007, 35. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A. AutoDock Vina: Inproving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Clemons, P.A.; Wilson, J.A.; Dancik, V.; Muller, S.; Carrinski, H.A.; Wagner, B.K.; Koehler, A.N.; Schreiber, S.L. Quantifying structure and performance diversity for sets of small molecules comprising small-molecule screening collections. Proc. Natl. Acad. Sci. USA 2011, 108, 6817–6822. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Guha, R.; Giulianotti, M.A.; Pinilla, C.; Houghten, R.A.; Medina-Franco, J.L. Chemoinformatic analysis of combinatorial libraries, drugs, natural products, and molecular libraries Small Molecule Repository. J. Chem. Inf. Model. 2009, 49, 1010–1024. [Google Scholar] [CrossRef] [PubMed]
- Doniger, S.; Hofmann, T.; Yeh, J. Predicting CNS permeability of drug molecules: Comparison of neural network and support vector machine algorithms. J. Comput. Biol. 2002, 9, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Sousa da Silva, A.W.; Vranken, W.F.; Wang, J. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996. [Google Scholar] [CrossRef]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics 2011. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa-A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free energy calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Bowling, T.; Mercer, L.; Don, R.; Jacobs, R.; Nare, B. Application of a resazurin-based high-throughput screening assay for the identification and progression of new treatments for human African trypanosomiasis. Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Doua, F.; Miezan, T.W.; Sanon Singaro, J.R.; Yapo, F.B.; Baltz, T. The efficacy of pentamidine in the treatment of early-late stage Trypanosoma brucei gambiense trypanosomiasis. Am. J. Trop. Med. Hyg. 1996, 55, 586–588. [Google Scholar] [CrossRef] [PubMed]
- Akinboye, E. Biological Activities of Emetine. Open Nat. Prod. J. 2011, 4, 8–15. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds used in this study are available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Information | Docking Binding Energy (kcal/mol) | |||||
|---|---|---|---|---|---|---|
| Code Name | Chemical Structure | Database ID | T. brucei PDB: 2X9N | T. cruzi Homology model | L. major PDB: 1E92 | H. sapiens PDB: 3O4R |
| RUBi001 |  | ZINC00057846 | −10.1 | −9.6 | −9.2 | - |
| RUBi002 |  | ZINC08992677 | −10.2 | −10.1 | −9.8 | - |
| RUBi003 |  | SANC00368 | −9.1 | −8.6 | −8.1 | - |
| RUBi004 |  | ZINC00809143 | −10.3 | −10.1 | −9.1 | - |
| RUBi005 |  | ZINC02690799 | −9.0 | −8.8 | −8.6 | - |
| RUBi006 |  | SANC00470 | −10.2 | −9.5 | −8.6 | −7.7 1 |
| RUBi007 |  | ZINC00630525 | −9.6 | −8.8 | −9.1 | - |
| RUBi008 |  | ZINC06556964 | −8.5 | −8.9 | −8.6 | - |
| RUBi009 |  | ZINC02177983 | −8.9 | −8.3 | −8.2 | - |
| RUBi010 |  | ZINC00359797 | −6.9 | −7.6 | −7.8 | - |
| RUBi011 |  | ZINC00677623 | −9.7 | −9.8 | −9.7 | - |
| RUBi012 |  | ZINC01003765 | −9.1 | −8.1 | −7.9 | - |
| RUBi013 |  | ZINC02184332 | −8.7 | −9.4 | −7.9 | - |
| RUBi014 |  | ZINC0058117/SANC00320 | −9.7 | −9.1 | −9.7 | - |
| RUBi015 |  | ZINC04671320 | −9.1 | −9.1 | −8.3 | - |
| RUBi016 |  | ZINC00612219 | −8.9 | −8.9 | −7.7 | - |
| RUBi017 |  | ZINC04523829 | −8.8 | −9.1 | −8.4 | - |
| RUBi018 |  | ZINC04313814 | −8.4 | −8.4 | −8.8 | - |
| Ligand | Van der Waal Energy (kJ/mol) | Electrostatic Energy (kJ/mol) | Polar Solvation Energy (kJ/mol) | Solvent Accessible Surface Area (SASA) Energy (kJ/mol) | Binding Energy (kJ/mol) |
|---|---|---|---|---|---|
| RUBi001 | −147 ± 10 | −73 ± 8 | 114 ± 7 | −13 ± 1 | −120 ± 9 |
| RUBi002 | −158 ± 11 | −55 ± 17 | 167 ± 25 | −20 ± 1 | −66 ± 18 |
| RUBi003 | −120 ± 10 | −45 ± 16 | 89 ± 15 | −12 ± 1 | −89 ± 10 |
| RUBi004 | −89 ± 9 | −2 ± 20 | 40 ± 29 | −12 ± 1 | −63 ± 14 |
| RUBi005 | −177 ± 13 | −22 ± 11 | 106 ± 13 | −19 ± 1 | −112 ± 15 |
| RUBi006 | −130 ± 9 | −3 ± 7 | 73 ± 7 | −15 ± 1 | −74 ± 9 |
| RUBi007 | −125 ± 8 | −45 ± 7 | 97 ± 10 | −15 ± 1 | −88 ± 10 |
| RUBi008 | −134 ± 8 | −46 ± 11 | 111 ± 15 | −16 ± 1 | −85 ± 11 |
| RUBi009 | −144 ± 19 | 2 ± 20 | 94 ± 13 | −18 ± 2 | −66 ± 22 |
| RUBi010 | −144 ± 11 | −138 ± 32 | 231 ± 48 | −16 ± 1 | −68 ± 23 |
| RUBi011 | −159 ± 10 | −22 ± 13 | 106 ± 16 | −17 ± 1 | −92 ± 12 |
| RUBi012 | −116 ± 12 | 41 ± 9 | 103 ± 21 | −13 ± 1 | 15 ± 13 |
| RUBi013 | −140 ± 11 | −49 ± 28 | 142 ± 31 | −16 ± 1 | −62 ± 13 |
| RUBi014 | −99 ± 17 | −17 ± 20 | 71 ± 28 | −11 ± 2 | −56 ± 12 |
| RUBi015 | −106 ± 11 | −29 ± 11 | 76 ± 17 | −13 ± 1 | −72 ± 12 |
| RUBi016 | −87 ± 9 | 21 ± 8 | 54 ± 11 | −11 ± 1 | −23 ± 10 |
| RUBi017 | −130 ± 12 | −33 ± 14 | 92 ± 19 | −15 ± 1 | −86 ± 12 |
| RUBi018 | −132 ± 12 | −15 ± 9 | 75 ± 11 | −16 ± 1 | −88 ± 12 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimuda, M.P.; Laming, D.; Hoppe, H.C.; Tastan Bishop, Ö. Identification of Novel Potential Inhibitors of Pteridine Reductase 1 in Trypanosoma brucei via Computational Structure-Based Approaches and in Vitro Inhibition Assays. Molecules 2019, 24, 142. https://doi.org/10.3390/molecules24010142
Kimuda MP, Laming D, Hoppe HC, Tastan Bishop Ö. Identification of Novel Potential Inhibitors of Pteridine Reductase 1 in Trypanosoma brucei via Computational Structure-Based Approaches and in Vitro Inhibition Assays. Molecules. 2019; 24(1):142. https://doi.org/10.3390/molecules24010142
Chicago/Turabian StyleKimuda, Magambo Phillip, Dustin Laming, Heinrich C. Hoppe, and Özlem Tastan Bishop. 2019. "Identification of Novel Potential Inhibitors of Pteridine Reductase 1 in Trypanosoma brucei via Computational Structure-Based Approaches and in Vitro Inhibition Assays" Molecules 24, no. 1: 142. https://doi.org/10.3390/molecules24010142
APA StyleKimuda, M. P., Laming, D., Hoppe, H. C., & Tastan Bishop, Ö. (2019). Identification of Novel Potential Inhibitors of Pteridine Reductase 1 in Trypanosoma brucei via Computational Structure-Based Approaches and in Vitro Inhibition Assays. Molecules, 24(1), 142. https://doi.org/10.3390/molecules24010142