Comparative Transcriptome Analysis Identifies Genes Involved in Diosgenin Biosynthesis in Trigonella foenum-graecum L.

Abstract
1. Introduction
2. Results and Discussion
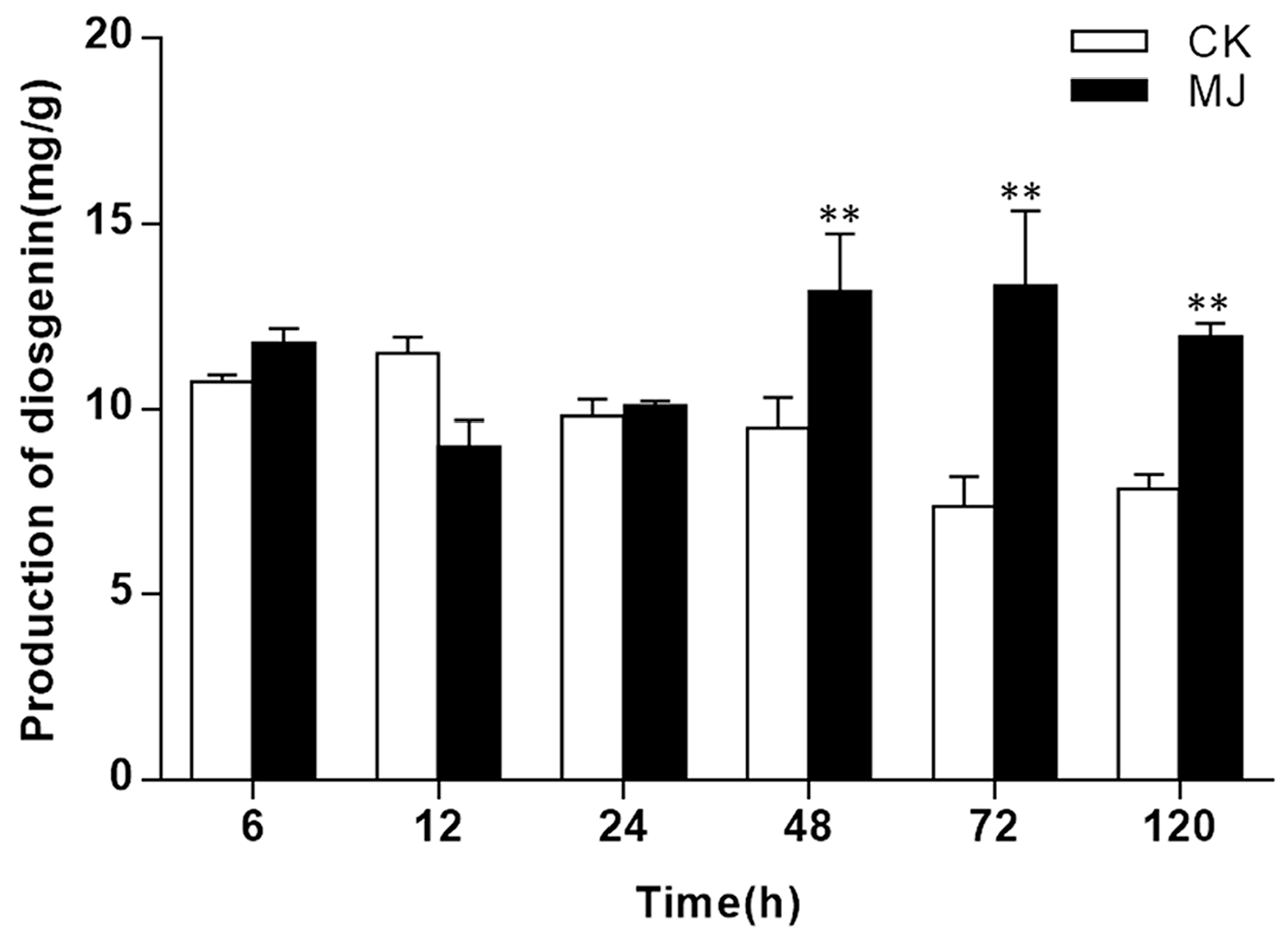
2.1. Diosgenin Biosynthesis in Fenugreek was Improved by MeJA Stimuli
2.2. De Novo Assembly and Functional Annotation
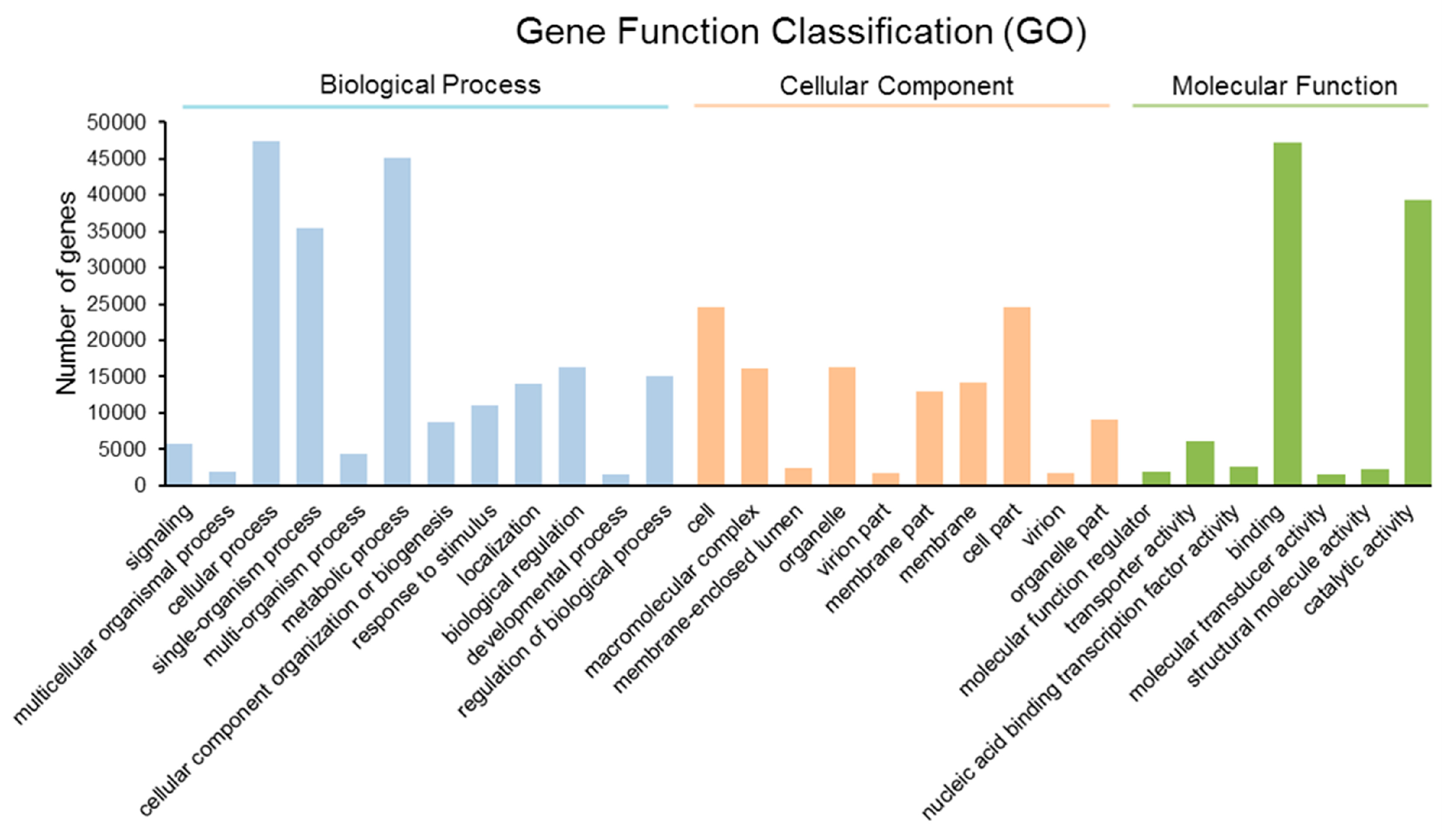
2.3. Gene Ontology (GO) Annotation
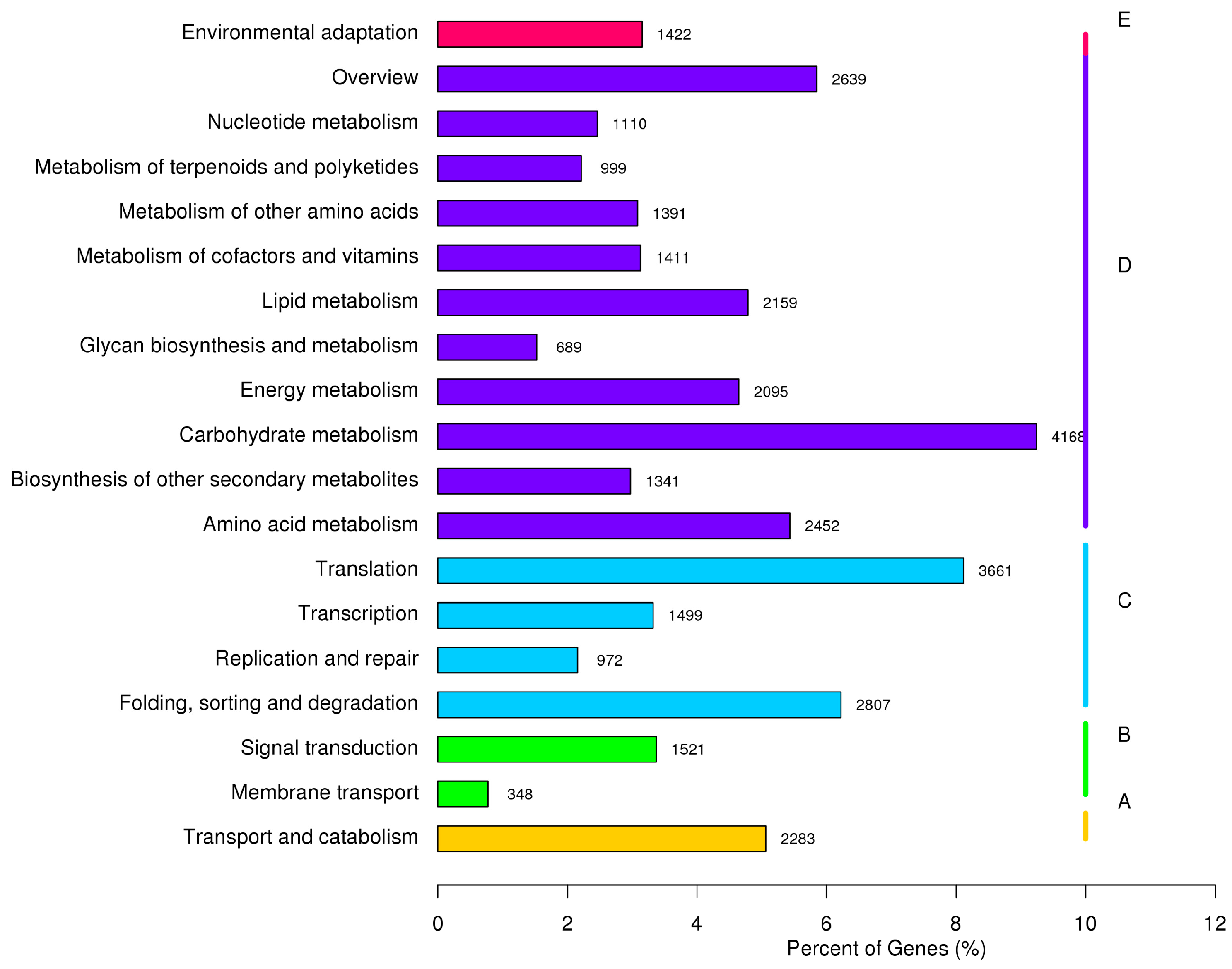
2.4. Metabolic Pathway Analysis by KEGG
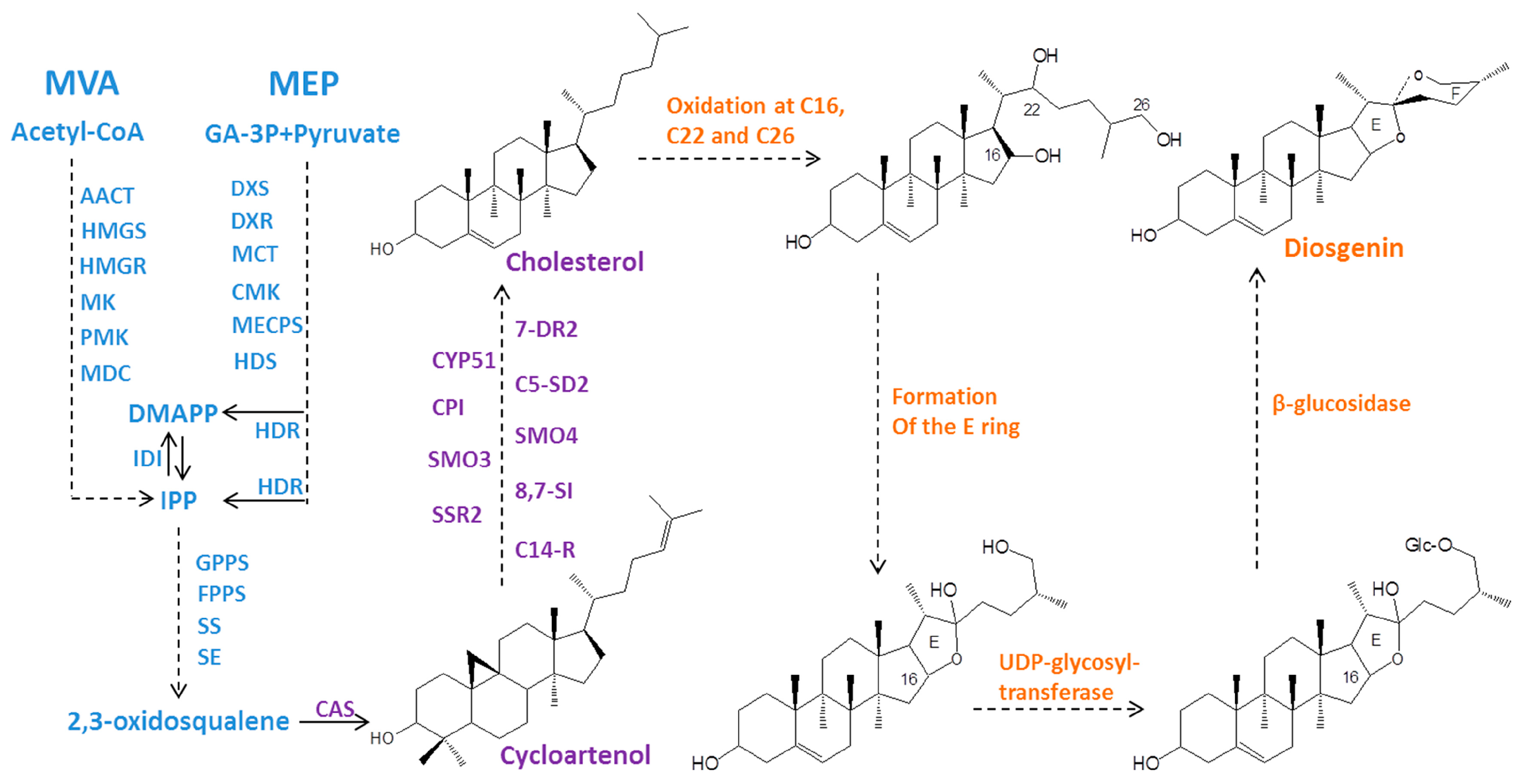
2.5. Identification of Genes Related to Steroidal Backbone Biosynthesis
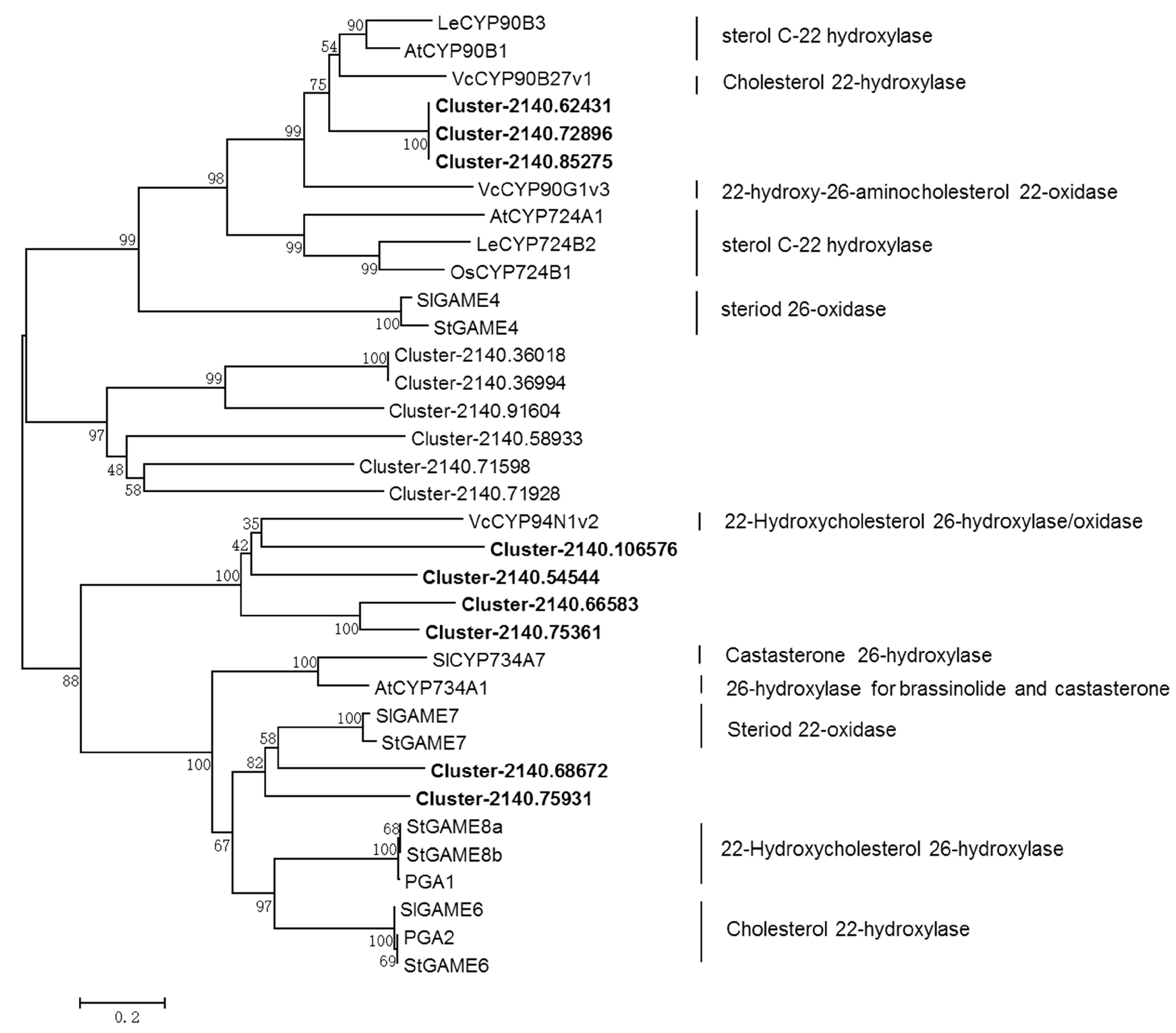
2.6. Identification of the CYP- and UGT-encoding Genes in the Downstream Diosgenin Pathway
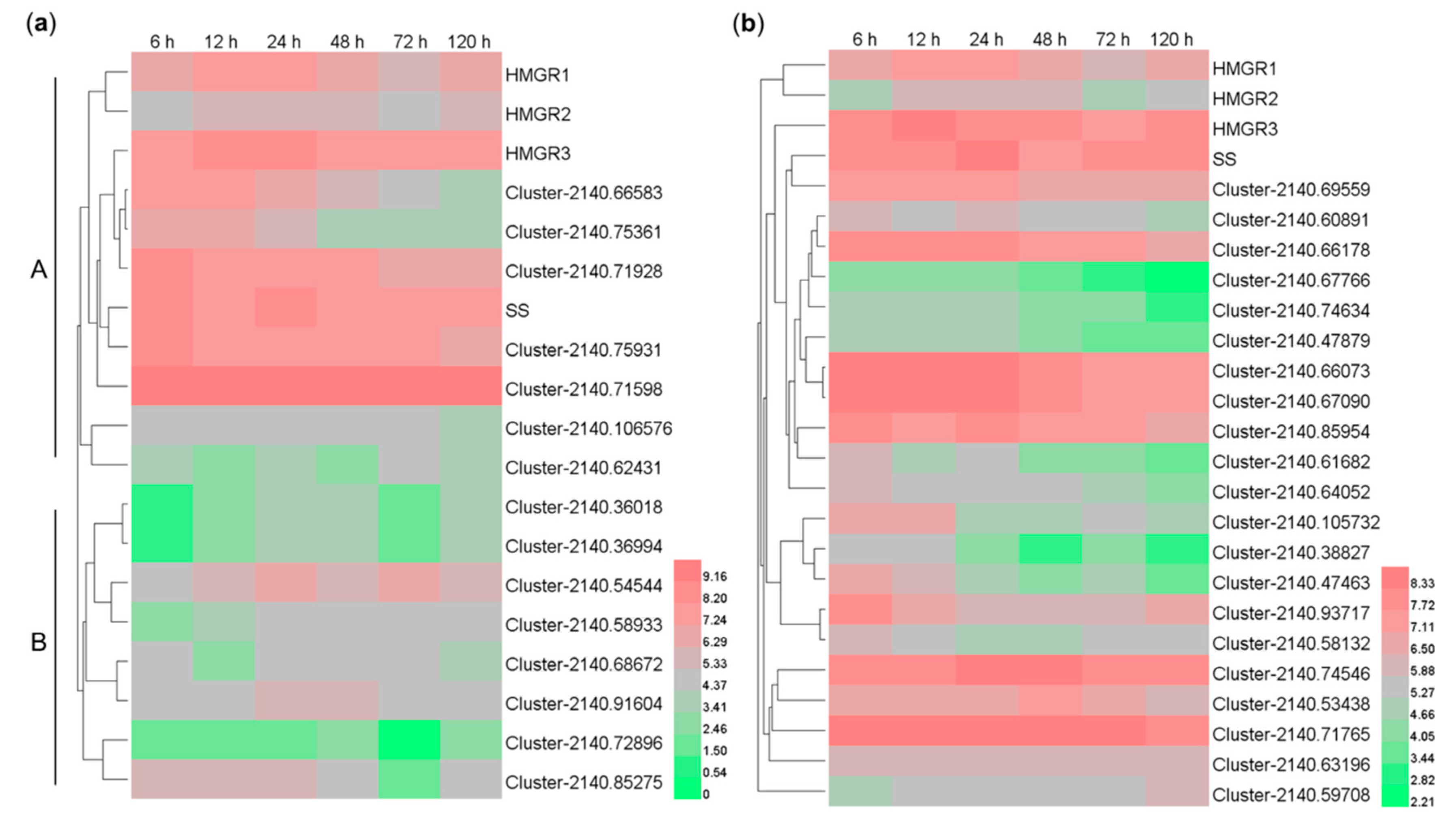
2.7. Co-expression Analysis of the MeJA-induced CYP- and UGT-encoding Genes with the HMGR and SS
2.8. Identification of the MeJA-induced Transcription Factors
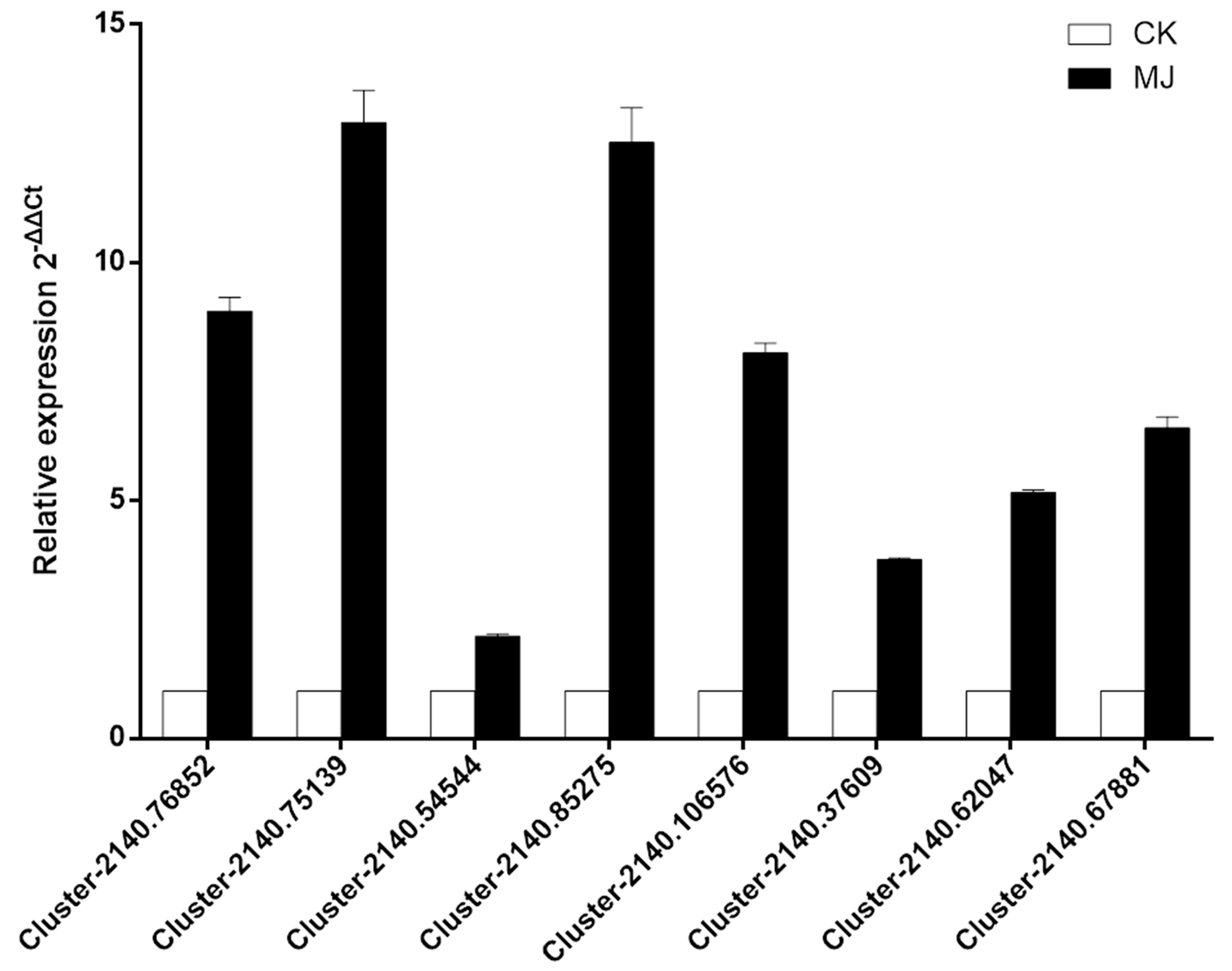
2.9. Validation of Selected Transcripts by qRT-PCR
3. Materials and Methods
3.1. Plant Material
3.2. MeJA Treatment
3.3. Diosgenin Extraction and Analysis
3.4. RNA Extraction and Sequencing
3.5. Digital Differential Gene Expression Analysis
3.6. Validation of transcripts by Quantitative Real-Time PCR (qRT-PCR)
3.7. Gene Cluster Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Martin, E.; Akan, H.; Ekici, M.; Aytac, Z. New chromosome numbers in the genus Trigonella L. (Fabaceae) from Turkey. Afr. J. Biotechnol. 2011, 10, 116–125. [Google Scholar]
- Acharya, S.N.; Thomas, J.E.; Basu, S.K. Fenugreek, an Alternative Crop for Semiarid Regions of North America. Crop Sci. 2008, 48, 841–853. [Google Scholar] [CrossRef]
- Sagar, P. De Novo RNA Seq Assembly and Annotation of Trigonella foenum-graecum L. (SRR066197). Legume Genom. Genet. 2014, 5, 1–7. [Google Scholar] [CrossRef]
- Basch, E.; Ulbricht, C.; Kuo, G.; Szapary, P.; Smith, M. Therapeutic applications of Fenugreek. (Fenugreek). Altern. Med. Rev. 2003, 8, 20–28. [Google Scholar] [PubMed]
- Chaudhary, S.; Chaudhary, P.S.; Chikara, S.K.; Sharma, M.C.; Iriti, M. Review on Fenugreek (Trigonella foenum-graecum L.) and its Important Secondary Metabolite Diosgenin. Not. Bot. Horti. Agrobo. 2018, 46, 22–31. [Google Scholar] [CrossRef]
- Mehrafarin, A.; Rezazadeh, S.; Naghdi Badi, H.; Noormohammadi, G.; Zand, E.; Qaderi, A. A Review on Biology, Cultivation and Biotechnology of Fenugreek (Trigonella foenum-graecum L.) as a Valuable Medicinal Plant and Multipurpose. J. Med. Plants 2011, 1, 6–24. [Google Scholar]
- Aasim, M.; Hussain, N.; Umer, E.M.; Zubair, M.; Hussain, S.B.; Saeed, S.; Rafique, T.S.; Sancak, C. In vitro shoot regeneration of fenugreek (Trigonella foenum-graecum L.) using different cytokinins. Afr. J. Biotechnol. 2010, 9, 7174–7179. [Google Scholar]
- Devi, B.A.; Kamalakkannan, N.; Prince, P.S. Supplementation of fenugreek leaves to diabetic rats. Effect on carbohydrate metabolic enzymes in diabetic liver and kidney. Phytother. Res. 2003, 17, 1231–1233. [Google Scholar] [CrossRef]
- Aumsuwan, P.; Khan, S.I.; Khan, I.A.; Ali, Z.; Avula, B.; Walker, L.A.; Shariat-Madar, Z.; Helferich, W.G.; Katzenellenbogen, B.S.; Dasmahapatra, A.K. The anticancer potential of steroidal saponin, dioscin, isolated from wild yam (Dioscorea villosa) root extract in invasive human breast cancer cell line MDA-MB-231 in vitro. Arch. Biochem. Biophys. 2016, 591, 98–110. [Google Scholar] [CrossRef]
- Wu, S.; Xu, H.; Peng, J.; Wang, C.; Jin, Y.; Liu, K.; Sun, H.; Qin, J. Potent anti-inflammatory effect of dioscin mediated by suppression of TNF-alpha-induced VCAM-1, ICAM-1 and EL expression via the NF-kappa B pathway. Biochimie 2015, 110, 62–72. [Google Scholar] [CrossRef]
- Tong, Q.; Qing, Y.; Wu, Y.; Hu, X.; Jiang, L.; Wu, X. Dioscin inhibits colon tumor growth and tumor angiogenesis through regulating VEGFR2 and AICT/MAPK signaling pathways. Toxicol. Appl. Pharmacol. 2014, 281, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Gadewar, M.; Tahilyani, V.; Patel, D.K. A review on pharmacological and analytical aspects of diosgenin: a concise report. Nat. Prod. Bioprospect 2012, 2, 46–52. [Google Scholar] [CrossRef]
- Ciura, J.; Szeliga, M.; Grzesik, M.; Tyrka, M. Next-generation sequencing of representational difference analysis products for identification of genes involved in diosgenin biosynthesis in fenugreek (Trigonella foenum-graecum). Planta 2017, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mehrafarin, A.; Qaderi, A.; Rezazadeh, S.; Naghdi Badi, H.; Noormohammadi, G.; Zand, E. Bioengineering of Important Secondary Metabolites and Metabolic Pathways in Fenugreek (Trigonella foenum-graecum L.). J. Med. Plants 2010, 9, 1–18. [Google Scholar]
- Vaidya, K.; Ghosh, A.; Kumar, V.; Chaudhary, S.; Srivastava, N.; Katudia, K.; Tiwari, T.; Chikara, S.K. De Novo Transcriptome Sequencing in L. to Identify Genes Involved in the Biosynthesis of Diosgenin. Plant Genome 2013, 6, 1–11. [Google Scholar] [CrossRef]
- Vranova, E.; Coman, D.; Gruissem, W. Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu. Rev. Plant Biol. 2013, 64, 665–700. [Google Scholar] [CrossRef]
- Volkman, J.K. Sterols and other triterpenoids: Source specificity and evolution of biosynthetic pathways. Org. Geochem. 2005, 36, 139–159. [Google Scholar] [CrossRef]
- Sonawane, P.D.; Pollier, J.; Panda, S.; Szymanski, J.; Massalha, H.; Yona, M.; Unger, T.; Malitsky, S.; Arendt, P.; Pauwels, L.; et al. Plant cholesterol biosynthetic pathway overlaps with phytosterol metabolism. Nat. Plants 2016, 3. [Google Scholar] [CrossRef]
- Inoue, K.; Ebizuka, Y. Purification and characterization of furostanol glycoside 26-O-β-glucosidase from Costus speciosus rhizomes. FEBS Lett. 1996, 378, 157–160. [Google Scholar] [CrossRef]
- Nakayasu, M.; Kawasaki, T.; Lee, H.J.; Sugimoto, Y.; Onjo, M.; Muranaka, T.; Mizutani, M. Identification of furostanol glycoside 26-O-β-glucosidase involved in steroidal saponin biosynthesis from Dioscorea esculenta. Plant Biotechnol. 2015, 32, 299–308. [Google Scholar] [CrossRef]
- Kumar, S.; Kalra, S.; Singh, B.; Kumar, A.; Kaur, J.; Singh, K. RNA-Seq mediated root transcriptome analysis of Chlorophytum borivilianum for identification of genes involved in saponin biosynthesis. Funct. Integr. Genom. 2016, 16, 37–55. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Wang, Y.; Hua, W.; Zhang, Y.; Wang, Z. De Novo Sequencing of Hypericum perforatum Transcriptome to Identify Potential Genes Involved in the Biosynthesis of Active Metabolites. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.; Zhang, Y.; Song, J.; Zhao, L.; Wang, Z. De novo transcriptome sequencing in Salvia miltiorrhiza to identify genes involved in the biosynthesis of active ingredients. Genomics 2011, 98, 272–279. [Google Scholar]
- Chaudhary, S.; Chikara, S.K.; Sharma, M.C.; Chaudhary, A.; Alam Syed, B.; Chaudhary, P.S.; Mehta, A.; Patel, M.; Ghosh, A.; Iriti, M. Elicitation of Diosgenin Production in Trigonella foenum-graecum (Fenugreek) Seedlings by Methyl Jasmonate. Int. J. Mol. Sci. 2015, 16, 29889–29899. [Google Scholar] [CrossRef] [PubMed]
- Friesen, J.A.; Rodwell, V.W. The 3-hydroxy-3-methylglutaryl coenzyme-A (HMG-CoA) reductases. Genome Biol. 2004, 5. [Google Scholar] [CrossRef] [PubMed]
- Tal, B.; Tamir, I.; Rokem, J.S.; Goldberg, I. Isolation and characterization of an intermediate steroid metabolite in diosgenin biosynththesis in suspension-cultures of Dioscorea-deltoidea cells. Biochem. J. 1984, 219, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.; Dilkes, B.P.; Fujioka, S.; Takatsuto, S.; Sakurai, A.; Feldmann, K.A. The DWF4 Gene of Arabidopsis Encodes a Cytochrome P450 That Mediates Multiple 22a-Hydroxylation Steps in Brassinosteroid Biosynthesis. Plant Cell 1998, 10, 231–243. [Google Scholar] [CrossRef]
- Ohnishi, T.; Watanabe, B.; Sakata, K.; Mizutani, M. CYP724B2 and CYP90B3 function in the early C-22 hydroxylation steps of brassinosteroid biosynthetic pathway in tomato. Biosci. Biotechnol. Biochem. 2006, 70, 2071–2080. [Google Scholar] [CrossRef]
- Augustin, M.M.; Ruzicka, D.R.; Shukla, A.K.; Augustin, J.M.; Starks, C.M.; O’Neil-Johnson, M.; McKain, M.R.; Evans, B.S.; Barrett, M.D.; Smithson, A.; et al. Elucidating steroid alkaloid biosynthesis in Veratrum californicum: production of verazine in Sf9 cells. Plant J. 2015, 82, 991–1003. [Google Scholar] [CrossRef]
- Itkin, M.; Heinig, U.; Tzfadia, O.; Bhide, A.J.; Shinde, B.; Cardenas, P.D.; Bocobza, S.E.; Unger, T.; Malitsky, S.; Finkers, R.; et al. Biosynthesis of antinutritional alkaloids in solanaceous crops is mediated by clustered genes. Science 2013, 341, 175–179. [Google Scholar] [CrossRef]
- Nakayasu, M.; Umemoto, N.; Ohyama, K.; Fujimoto, Y.; Lee, H.J.; Watanabe, B.; Muranaka, T.; Saito, K.; Sugimoto, Y.; Mizutani, M. A Dioxygenase Catalyzes Steroid 16alpha-Hydroxylation in Steroidal Glycoalkaloid Biosynthesis. Plant Physiol. 2017, 175, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Kalinowska, M.; Zimowski, J.; Pączkowski, C.; Wojciechowski, Z.A. The Formation of Sugar Chains in Triterpenoid Saponins and Glycoalkaloids. Phytochem. Rev. 2005, 4, 237–257. [Google Scholar] [CrossRef]
- Sugiyama, A.; Yamazaki, Y.; Yamashita, K.; Takahashi, S.; Nakayama, T.; Yazaki, K. Developmental and nutritional regulation of isoflavone secretion from soybean roots. Biosci. Biotechnol. Biochem. 2016, 80, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.-S.; Lee, H.; Choi, Y.E. Transcriptomic analysis of Siberian ginseng (Eleutherococcus senticosus) to discover genes involved in saponin biosynthesis. BMC Genom. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Li, X.; Zhan, Y.; Li, Y.; Qu, Z.; Sun, L.; Wang, S.; Yang, J.; Xiao, J. Cloning and expression of BpMYC4 and BpbHLH9 genes and the role of BpbHLH9 in triterpenoid synthesis in birch. BMC Plant Biol. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Singh, S.K.; Patra, B.; Sui, X.; Pattanaik, S.; Yuan, L. A differentially regulated AP2/ERF transcription factor gene cluster acts downstream of a MAP kinase cascade to modulate terpenoid indole alkaloid biosynthesis in Catharanthus roseus. New Phytol. 2017, 213, 1107–1123. [Google Scholar] [CrossRef] [PubMed]
- Schluttenhofer, C.; Yuan, L. Regulation of Specialized Metabolism by WRKY Transcription Factors. Plant Physiol. 2015, 167, 295–306. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef]
- Deng, W.; Wang, Y.; Liu, Z.; Cheng, H.; Xue, Y. HemI: A Toolkit for Illustrating Heatmaps. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raw Reads | Clean Reads | Q20 (%) | Q30 (%) | GC (%) | |
|---|---|---|---|---|---|
| MJ6 | 68803276 | 67560716 | 97.54 | 93.60 | 41.63 |
| MJ12 | 48826160 | 47953056 | 97.62 | 93.77 | 41.98 |
| MJ24 | 50687102 | 49648026 | 97.72 | 94.02 | 41.71 |
| MJ48 | 56366906 | 55413478 | 97.55 | 93.65 | 41.85 |
| MJ72 | 56903406 | 55842268 | 97.60 | 93.78 | 41.88 |
| MJ120 | 62961696 | 61951984 | 97.50 | 93.59 | 41.93 |
| CK24 | 52183650 | 51110028 | 97.43 | 93.47 | 42.06 |
| Annotation | Number of Genes | Percentage (%) |
|---|---|---|
| Annotated in NR | 118,631 | 68.64 |
| Annotated in NT | 137,541 | 79.58 |
| Annotated in KO | 45,112 | 26.10 |
| Annotated in Swiss-Prot | 89,801 | 51.96 |
| Annotated in Pfam | 82,876 | 47.95 |
| Annotated in GO | 83,000 | 48.02 |
| Annotated in KOG | 30,687 | 17.75 |
| Total Unigenes | 172,825 | 100 |
| Gene Name | EC Number | Gene ID | Total Unigenes |
|---|---|---|---|
| AACT | 2.3.1.9 | Cluster-2140.52353, 2140.68129, 2140.31592,2140.74635, 2140.75440, 2140.31592, 2140.63638 | 7 |
| HMGS | 2.3.3.10 | Cluster-2140.76852, 2140.76851, 2140.124037 | 3 |
| HMGR | 1.1.1.34 | Cluster-2140.36411, 2140.78950, 2140.104061 | 3 |
| MK | 2.7.1.36 | Cluster-2140.53808, 2140.53807 | 2 |
| PMK | 2.7.4.2 | Cluster-2140.37776 | 1 |
| MDC | 4.1.1.33 | Cluster-2140.84671 | 1 |
| DXS | 2.2.1.7 | Cluster-2140.115496, 2140.69916, 2140.52884, 2140.108949, 2140.69095 | 5 |
| DXR | 1.1.1.267 | Cluster-2140.80427 | 1 |
| MCT | 2.7.7.60 | Cluster-2140.48880, 2140.39394, 2140.69765 | 3 |
| CMK | 2.7.1.148 | Cluster-2140.108677 | 1 |
| MECPS | 4.6.1.12 | Cluster-2140.49224, 2140.56745 | 2 |
| HDS | 1.17.7.1 | Cluster-2140.79758, 2140.66344 | 2 |
| HDR | 1.17.1.4 | Cluster-2140.76895 | 1 |
| IDI | 5.3.3.2 | Cluster-2140.68527, 2140.68885 | 2 |
| GPPS | 2.5.1.29 | Cluster-2140.83026 | 1 |
| FPPS | 2.5.1.10 | Cluster-2140.77163 | 1 |
| SS | 2.5.1.21 | Cluster-2140.75139 | 1 |
| SE | 1.14.14.17 | Cluster-2140.68704, 2140.63913, 2140.63916, 2140.55508, 2140.65244, 2140.63914 | 6 |
| CAS | 5.4.99.8 | Cluster-2140.68038, 2140.75982, 2140.67914, 2140.68038 | 3 |
| SSR | 1.3.1.72 | Cluster-2140.73628, 2140.75526, 2140.71445, 2140.65744, 2140.65030 | 5 |
| SMO | 1.14.13.72 | Cluster-2140.88745, 2140.50156, 2140.82687, 2140.51993, 2140.62857, 2140.62855, 2140.43204, 2140.62858 | 8 |
| CPI | 5.5.1.9 | Cluster-2140.77401, 2140.52812 | 2 |
| CYP51 | 1.14.13.70 | Cluster-2140.73183, 2140.70550, 2140.65854, 2140.71941, 2140.84199, 2140.65644 | 6 |
| C14-R | 1.3.1.70 | Cluster-2140.69242, 2140.69243, 2140.69244, 2140.56203 | 4 |
| 8,7 SI | 5.3.3.5 | Cluster-2140.71662 | 1 |
| C5-SD | 1.14.19.20 | Cluster-2140.68714, 2140.72000 | 2 |
| 7-DR | 1.3.1.21 | Cluster-2140.70707 | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, C.; Li, X.; Zhou, Z.; Li, C.; Zhang, Y. Comparative Transcriptome Analysis Identifies Genes Involved in Diosgenin Biosynthesis in Trigonella foenum-graecum L. Molecules 2019, 24, 140. https://doi.org/10.3390/molecules24010140
Zhou C, Li X, Zhou Z, Li C, Zhang Y. Comparative Transcriptome Analysis Identifies Genes Involved in Diosgenin Biosynthesis in Trigonella foenum-graecum L. Molecules. 2019; 24(1):140. https://doi.org/10.3390/molecules24010140
Chicago/Turabian StyleZhou, Chen, Xiaohua Li, Zilin Zhou, Changfu Li, and Yansheng Zhang. 2019. "Comparative Transcriptome Analysis Identifies Genes Involved in Diosgenin Biosynthesis in Trigonella foenum-graecum L." Molecules 24, no. 1: 140. https://doi.org/10.3390/molecules24010140
APA StyleZhou, C., Li, X., Zhou, Z., Li, C., & Zhang, Y. (2019). Comparative Transcriptome Analysis Identifies Genes Involved in Diosgenin Biosynthesis in Trigonella foenum-graecum L. Molecules, 24(1), 140. https://doi.org/10.3390/molecules24010140