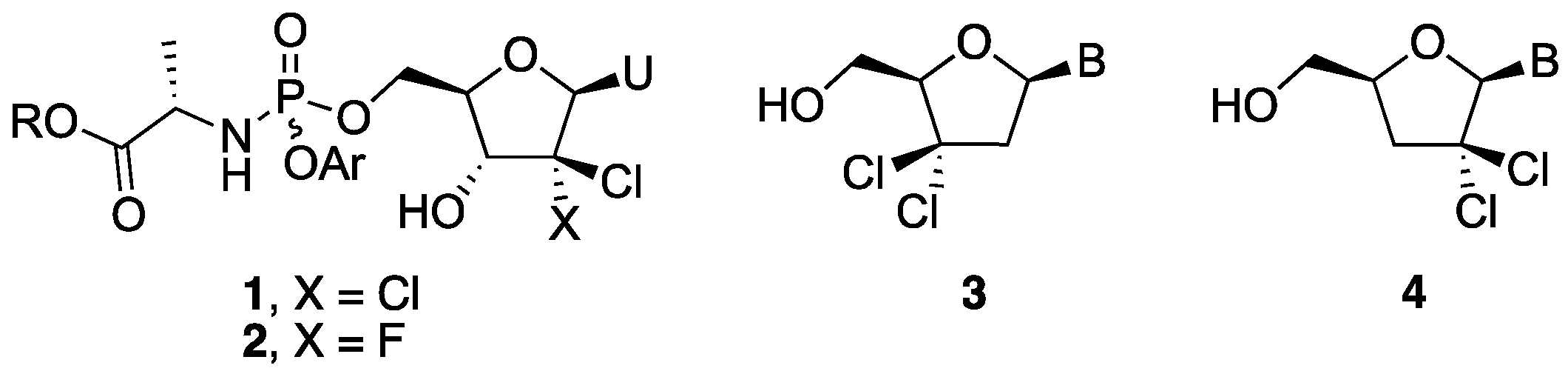
3.5. General Procedure for Sugar-Base Condensation
To a solution of nucleobase (1 equiv.) in dry CH3CN, was added N,O-bis(trimethylsilyl)acetamide (2.5 equiv.) and the resulting mixture was stirred for 20 min at room temperature. A solution of 28 (0.8 equiv.) in dry CH3CN was then added and the reaction mixture was cooled to −20 °C. Next, trimethylsilyl trifluoromethanesulfonate (TMSOTf) (1.05 equiv.) was added dropwise and the mixture was allowed to slowly warm to room temperature over 1 h, heated to 70 °C, and further stirred for 1 h. It was then diluted with EtOAc and washed with saturated aq. NaHCO3, water, and brine. The organic layer was dried over Na2SO4, filtered, and evaporated in vacuo to give a crude residue, which was purified by silica gel column chromatography to afford the tile compound.
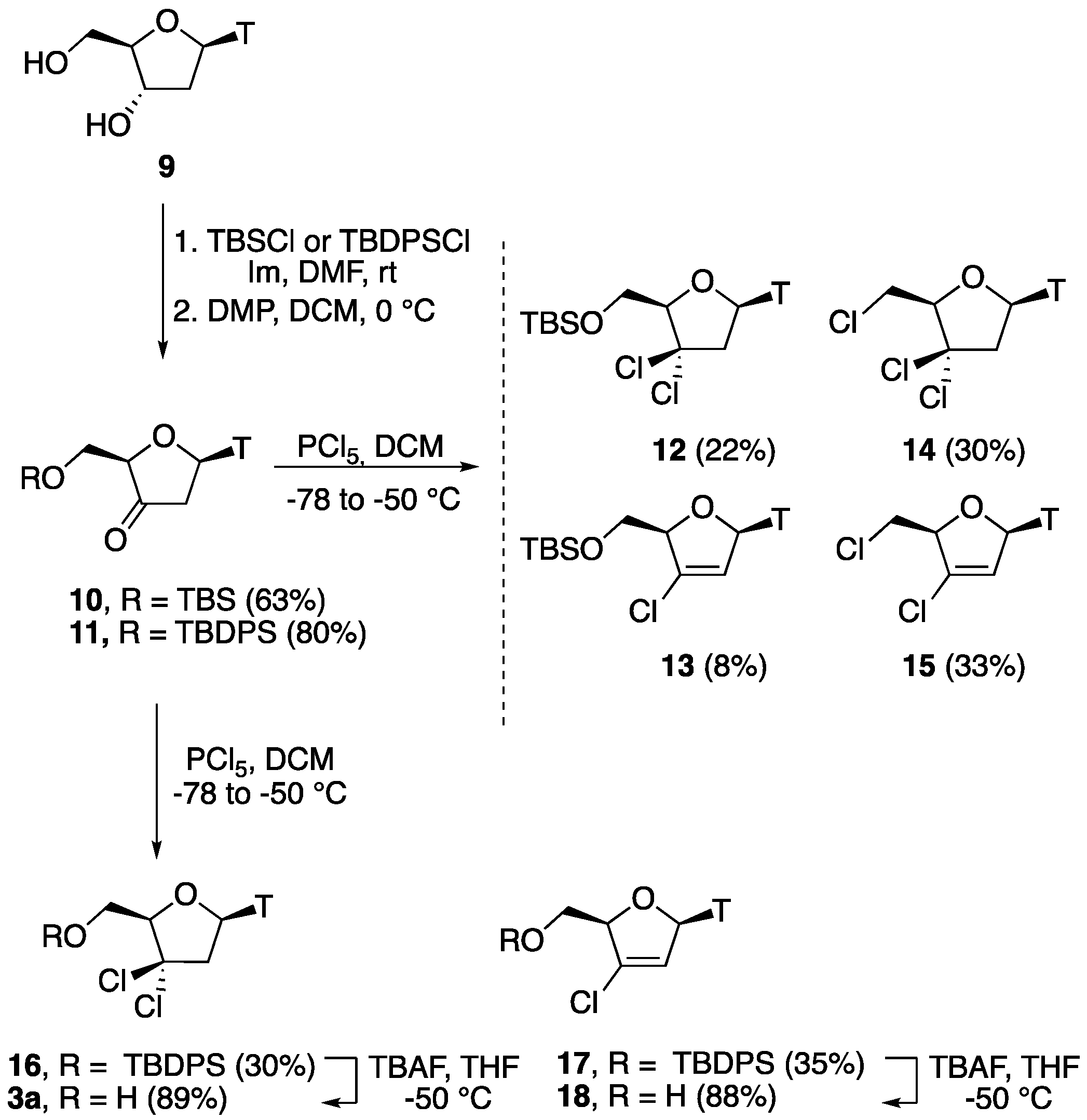
5′-O-(tert-Butyldiphenylsilyl)-3′-keto-2′-deoxythymidine (11). To a solution of 2′-deoxythymidine 9 (1.15 g, 4.78 mmol) and imidazole (0.650 g, 9.56 mmol) in anhydrous DMF (50 mL) was added tert-butyldiphenylsilyl chloride(TBDPSCl) (1.38 g, 5.00 mmol) at −50 °C. The reaction mixture was allowed to warm to room temperature and stirred for 4 h. It was then diluted with EtOAc (200 mL) and washed with water and brine. The organic layer was dried over Na2SO4, filtered, and evaporated in vacuo to give 5′-O-(tert-butyldiphenylsilyl)-2′-deoxythymidine in quantitative yield (2.28 g). 1H-NMR (500 MHz, CDCl3): δ 8.95 (s, 1H, NH), 7.68–7.66 (m, 4H, ArH), 7.54 (d, 1H, J = 1.1 Hz, H-6), 7.41–7.34 (m, 6H, ArH), 6.48 (dd, 1H, J = 8.2, 5.7 Hz, H-1′), 4.61 (br s, 1H, OH), 4.27–4.26 (m, 1H, H-3′), 4.10–4.08 (m, 1H, H-4′), 4.00 (dd, 1H, J = 11.4, 2.2 Hz, H-5′), 3.88 (dd, 1H, J = 11.4, 2.2 Hz, H-5″), 2.49–2.47 (m, 1H, H-2′), 2.21–2.19 (m, 1H, H-2″), 1.58 (d, J = 1.1 Hz, 3H, CH3), 1.09 (s, 9H, 3 × CH3); 13C-NMR (125 MHz, CDCl3): δ 164.2 (C=O), 150.6 (C=O), 135.3 (ArC), 135.2 (ArC), 135.0 (6-C), 132.8 (ArC), 132.1 (ArC), 129.8 (ArC), 129.7 (ArC), 127.7 (ArC), 127.6 (ArC), 110.8 (5-C), 87.1 (1′-CH), 84.5 (4′-CH), 71.3 (3′-CH), 64.0 (5′-CH2), 40.7 (2′-CH2), 31.3 (-C(CH3)3), 26.7 (3 × CH3), 11.8 (5-CH3). Following the general oxidation procedure, 5′-O-(tert-butyldiphenylsilyl)-2′-deoxythymidine (1.54 g, 3.00 mmol) was reacted with DMP (1.70 g, 4.00 mmol) in CH2Cl2 (100 mL). After work-up, the resulting crude residue was then recrystallized from CHCl3 to provide 11 as a white solid (1.14 g, 80%, over two steps), which was used in the next step without any further purification. HRMS: C26H30N2O5 [M + Na+]+ Calc.: 501.1816, found: 501.1886.
5′-O-(tert-Butyldiphenylsilyl)-3′,3′-gem-dichloro-3′-deoxythymidine (16) and 5′-O-(tert-butyldiphenylsilyl)-3′-chloro-2′,3′-didehydro-3′-deoxythymidine (17). Following the general chlorination procedure, a solution of compound 11 (0.500 g, 1.04 mmol) in dry CH2Cl2 (20 mL) was reacted with PCl5 (0.822 g, 3.95 mmol) at −78 °C under an inert atmosphere. After work-up, the resulting crude residue was purified by column chromatography (hexane: EtOAc 4:1) to give 16 (0.165 g, 30%) and 17 (0.180 g, 35%) as pale yellow solids. Data for 16: 1H-NMR (500 MHz, CDCl3): δ 10.3 (s, 1H, NH), 7.68–7.66 (m, 4H, ArH), 7.54 (d, 1H, J = 1.1 Hz, H-6), 7.41–7.35 (m, 6H, ArH), 6.48 (dd, 1H, J = 8.2, 5.7 Hz, H-1′), 4.10 (dd, 1H, J = 4.7, 2.2 Hz, H-4′), 3.99 (dd, 1H, J = 11.5, 2.2 Hz, H-5′), 3.88 (dd, 1H, J = 11.5, 4.7 Hz, H-5″), 3.79 (dd, 1H, J = 11.5, 5.7 Hz, H-2′), 3.35 (dd, 1H, J = 11.5, 8.2 Hz, H-2″), 1.54 (s, 3H, 5-CH3), 0.97 (s, 9H, 3 × CH3); 13C-NMR (150 MHz, CDCl3): δ 163.7 (C=O), 150.2 (C=O), 135.4 (6-C), 135.4 (ArC), 135.3 (ArC), 135.3 (ArC), 135.1 (ArC), 129.7 (ArC), 129.6 (ArC), 127.6 (ArC), 127.5 (ArC), 110.8 (5-C), 87.5 (4′-CH), 85.0 (3′-C(Cl)2), 84.5 (1′-CH), 63.7 (5′-CH2), 41.0 (2′-CH2), 26.6 (3 × CH3), 19.0 (C(CH3)), 11.6 (5-CH3); HRMS: C26H30N2O4SiCl2 [M + H+]+ Calc.: 533.1424, found: 533.1442. Data for 17: 1H-NMR (500 MHz, CDCl3): δ 9.00 (s, 1H, NH), 7.67 (d, 2H, J = 6.7 Hz, ArH), 7.62 (d, 2H, J = 6.7 Hz, ArH), 7.43–7.25 (m, 7H, ArH, H-6), 7.02 (dd, 1H, J = 4.0, 1.3 Hz, H-1′), 5.93 (t, 1H, J = 1.3 Hz, H-2′), 4.78–4.77 (m, 1H, H-4′), 4.09–4.08 (m, 2H, H-5′, H-5″), 1.17 (s, 3H, 5-CH3), 1.09 (s, 9H, 3 × CH3); 13C-NMR (125 MHz, CDCl3): δ 163.7 (C=O), 150.7 (C=O), 136.3 (6-C), (3′-C(Cl)), 135.3 (ArC), 135.2 (ArC), 133.5 (ArC), 132.6 (ArC), 130.1 (ArC), 129.9 (ArC), 127.9 (ArC), 127.8 (ArC), 121.2 (2′-CH), 111.6 (5-C), 87.5 (4′-CH), 86.5 (1′-CH), 62.1 (5′-CH2), 27.1 (3 × CH3), 19.6 (C(CH3)), 11.3 (5-CH3); HRMS: C26H29N2O4SiCl [M + Na+]+ Calc.: 519.14775, found: 519.1468.
3′,3′-gem-Dichloro-3′-deoxythymidine (3a). Following the general desilylation procedure, a solution of compound 16 (0.143 g, 0.27 mmol) in dry THF (2.0 mL) was reacted with TBAF (1 mL, 2 mmol) at −50 °C. After work-up, the resulting crude residue was purified by column chromatography (EtOAc) to give 3a as a yellow solid (76 mg, 89%). 1H-NMR (600 MHz, DMSO-d6): δ 11.4 (s, 1H, NH), 7.70 (d, 1H, J = 1.2 Hz, H-6), 6.27 (dd, 1H, J = 7.0, 6.3 Hz, H-1′), 5.34 (t, J = 5.0 Hz, 1H, OH), 4.38 (dd, 1H, J = 5.1, 3.1 Hz, 4′-H), 3.82 (ddd, 1H, J = 12.1, 5.1, 3.1 Hz, H-5′), 3.78 (ddd, 1H, J = 12.1, 5.0, 3.1 Hz, H-5″), 3.21 (dd, J = 14.7, 6.3 Hz, 1H, H-2′), 3.03 (dd, J = 14.6, 7.0 Hz, 1H, H-2″), 1.78 (d, 3H, J = 1.2 Hz, 5-CH3); 13C-NMR (150 MHz, DMSO): δ 163.7 (C=O), 150.5 (C=O), 135.6 (6-C), 109.8 (5-C), 89.0 (4′-CH), 86.9 (3′-C(Cl)2), 81.8 (1′-CH), 60.7 (5′-CH2), 50.4 (2′-CH2), 12.4 (5-CH3); HRMS: C10H12Cl2N2O4 [M + H+]+ Calc.: 317.0066, found: 317.0074.
3′-Chloro-2′,3′-didehydro-3′-deoxythymidine (18). Following the general desilylation procedure, a solution of compound 17 (0.124 g, 0.25 mmol) in dry THF (2.0 mL) was reacted with TBAF (1 mL, 2 mmol) at −50 °C. After work-up, the resulting crude residue was purified by column chromatography (EtOAc) to give 18 (62.6 mg, 88%) as a yellow solid. 1H-NMR (600 MHz, CDCl3): δ 8.87 (s, 1H, NH), 7.72 (d, 1H, J = 1.4 Hz, H-6), 7.01 (dd, 1H, J = 4.7, 1.7 Hz, H-1′), 5.96 (t, 1H, J = 1.7 Hz, H-2′), 5.30 (s, 1H, OH), 5.00–4.99 (m, 1H, 4′-H), 3.93 (dd, 1H, J = 15.5, 3.0 Hz, H-5′), 3.89 (dd, 1H, J = 15.5, 3.0 Hz, H-5″), 1.91 (d, 3H, J = 1.4 Hz, CH3); 13C-NMR (150 MHz, CDCl3): δ 163.5 (C=O), 150.7 (C=O), 135.9 (3′-C(Cl)), 135.7 (6-C), 122.9 (2′-C), 111.9 (5-C), 87.5 (4′-CH), 84.1 (1′-CH), 44.1 (5′-CH2), 12.4 (5-CH3); HRMS: C10H11Cl1N2O4 [M + H+]+ Calc.: 259.0480, found: 259.0489.
5′-O-(tert-Butyldiphenylsilyl)-3′-keto-2′-deoxyuridine (19). To a solution of uridine (1.09 g, 4.78 mmol) and imidazole (0.650 g, 9.56 mmol) in anhydrous DMF (50 mL) was added TBDPSCl (1.38 g, 5.00 mmol) at −50 °C. The reaction mixture was allowed to warm to room temperature and stirred for 4 h. It was then diluted with EtOAc (200 mL) and washed with water and brine. The organic layer was dried over Na2SO4, filtered, and evaporated in vacuo to give 5′-O-(tert-butyldiphenylsilyl) uridine in quantitative yield (2.21 g). 1H-NMR (500 MHz, CDCl3): δ 10.1 (s, 1H, NH), 7.83 (d, 1H, J = 8.1 Hz, H-6), 7.66–7.63 (m, 4H, ArH), 7.42–7.37 (m, 6H, ArH), 6.37 (t, 1H, J = 6.4 Hz, H-1′), 5.43 (d, 1H, J = 8.1 Hz, H-5), 4.56 (br s, 1H, OH), 4.11–4.10 (m, 1H, H-3′), 4.03–4.02 (m, 1H, H-4′), 3.98 (dd, 1H, J = 11.4, 2.3 Hz, H-5′), 3.85 (dd, 1H, J = 11.4, 2.9 Hz, H-5″), 2.49–2.44 (m, 1H, H-2′), 2.24–2.18 (m, 1H, H-2″), 1.07 (s, 9H, 3 × CH3); 13C-NMR (125 MHz, CDCl3): δ 163.7 (C=O), 150.4 (C=O), 140.0 (6-C), 135.4 (ArC), 135.1 (ArC), 132.6 (ArC), 132.1 (ArC), 129.8 (ArC), 129.8 (ArC), 127.7 (ArC), 127.7 (ArC), 102.0 (5-C), 87.0 (1′-CH), 84.8 (4′-CH), 70.8 (3′-CH), 63.6 (5′-CH2), 41.0 (2′-CH2), 31.3 (-C(CH3)3), 26.7 (3 × CH3); HRMS for C25H30N2O4Si1 [M + Na]+ Calc.: 489.1816, found: 489.1816. Following the general oxidation procedure, 5′-O-(tert-butyldiphenylsilyl) uridine (1.39 g, 3.00 mmol) was reacted with DMP (1.70 g, 4.00 mmol) in CHCl3 (100 mL). After work-up, the resulting crude residue was recrystallized from CHCl3 to provide 19 as a white solid (1.23 g, 87% over two steps), which was used in the next step without any further purification. HRMS: C25H28N2O5 [M + Na+]+ Calc.: 487.1659 found: 487.1686.
5′-O-(tert-Butyldiphenylsilyl)-3′,3′-gem-dichloro-2′,3′-dideoxyuridine (20) and 5′-O-(tert-butyldiphenyl-silyl)-3′-chloro-2′,3′-didehydro-2′,3′-dideoxyuridine (21). Following the general chlorination procedure, a solution of compound 19 (0.482 g, 1.04 mmol) in dry CH2Cl2 (20 mL) was reacted with PCl5 (0.822 g, 3.95 mmol) at −78 °C under an inert atmosphere. After work-up, the resulting crude residue was purified by column chromatography (hexane:EtOAc 4:1) to give 20 (0.227 g, 44%) and 21 (0.175 g, 35%) as yellow pale solids. Data for 20: 1H-NMR (500 MHz, CDCl3): δ 9.57 (s, 1H, NH), 7.70–7.66 (m, 4H, ArH), 7.56 (d, 1H, J = 8.2 Hz, H-6), 7.47–7.38 (m, 6H, ArH), 6.23 (dd, 1H, J = 6.8, 4.9 Hz, H-1′), 5.51 (d, 1H, J = 8.2 Hz, H-5), 4.43 (dd, 1H, J = 5.1, 3.0 Hz, H-4′), 4.15 (dd, 1H, J = 11.8, 3.0 Hz, H-5′), 4.03 (dd, 1H, J = 11.8, 5.2 Hz, H-5″), 3.23 (dd, 1H, J = 14.8, 6.9 Hz, H-2′), 2.96 (dd, 1H, J = 14.8, 4.8 Hz, H-2″), 1.08 (s, 9H, 3 × CH3); 13C-NMR (125 MHz, CDCl3): δ 163.2 (C=O), 150.2 (C=O), 139.4 (6-C), 135.7 (ArC), 135.4 (ArC), 132.7 (ArC), 132.2 (ArC), 130.0 (ArC), 130.0 (ArC), 127.9 (ArC), 127.8 (ArC), 102.1 (5-C), 89.9 (4′-CH), 84.5 (3′-C(Cl)2), 83.3 (1′-CH), 63.1 (5′-CH2), 52.7 (2′-CH2), 29.6 (-C(CH3)3), 26.8 (3 × CH3); HRMS: C25H28Cl2N2O4Si [M + H+]+ Calc.: 519.1268, found: 519.1290. Data for 21: 1H-NMR (600 MHz, CDCl3): δ 8.44 (s, 1H, NH), 7.81 (d, 1H, J = 8.2 Hz, 6-H), 7.66–7.64 (m, 3H, ArH), 7.57–7.55 (m, 2H, ArH), 7.44–7.35 (m, 5H, ArH), 7.03 (dd, 1H, J = 3.8, 1.5 Hz, H-1′), 5.9 (t, 1H, J = 1.5 Hz, H-2′), 4.86 (d, 1H, J = 8.2 Hz, 5-H), 4.80 (dddd, 1H, J = 3.8, 2.7, 1.5, 1.3 Hz, H-4′), 4.07 (dd, 1H, J = 12.3, 1.3 Hz, H-5′), 4.07 (dd, 1H, J = 12.3, 2.7 Hz, H-5″), 1.10 (s, 9H, 3 × CH3); 13C-NMR (150 MHz, CDCl3): δ 158.9 (C=O), 149.7 (C=O), 136.9 (6-C), 136.5 (3′-C(Cl)), 136.5 (ArC), 135.5 (ArC), 135.2 (ArC), 132.9 (ArC), 132.3 (ArC), 130.0 (ArC), 129.9 (ArC), 127.8 (ArC), 127.7 (ArC), 121.7 (2′-CH), 109.9 (5-C), 88.3 (4′-CH), 86.9 (1′-CH), 62.9 (5′-CH2), 29.6 (C(CH3), 27.1 (3 × -CH3); HRMS: C25H27ClN2O4Si [M + Na+]+ Calc.: 505.1321, found: 505.1321.
5′-O-(tert-Butyldiphenylsilyl)-3′,3′-dichloro-2′,3′-dideoxy-4-(1H-1,2,4-triazol-1-yl)uridine (22). To a stirred solution of 20 (1.04 g, 2.00 mmol) in dry CH3CN (10 mL) was added POCl3 (0.28 mL, 3 mmol) at −50 °C. Triethylamine (0.7 mL, 5.00 mmol) was then added dropwise over 10 min, followed by a solution of 1,2,4-triazole (0.414 g, 6.00 mmol) and triethylamine (0.8 mL, 6.00 mmol) in dry CH3CN (15 mL) over 30 min. The reaction mixture was stirred at room temperature for two days. It was then diluted with EtOAc (200.0 mL) and quenched with a NaH2PO4/Na2HPO4 buffer (pH = 7, 150.0 mL). The organic layer was washed with water and brine, dried over Na2SO4, and evaporated in vacuo to give a crude residue, which was purified by column chromatography (EtOAc:hexane 7:3) to afford compound 22 as a white solid (0. 967 mg, 85%). 1H-NMR (300 MHz, CDCl3): δ 9.19 (s, 1H, N-CH=N), 8.06 (s, 1H, N-CH=N), 7.98 (d, 1H, J = 7.3 Hz, H-6), 7.68–7.62 (m, 4H, ArH), 7.43–7.42 (m, 6H, ArH), 6.81 (d, 1H, J = 7.3 Hz, H-5), 6.10 (dd, 1H, J = 7.3, 3.0 Hz, H-1′), 4.49 (dd, 1H, J = 6.0, 3.0 Hz, H-4′), 4.19 (dd, 1H, J = 11.8, 3.0 Hz, H-5′), 4.01 (dd, 1H, J = 11.8, 6.0 Hz, H-5″), 3.38 (dd, 1H, J = 15.3, 7.3 Hz, H-2′), 3.05 (dd, 1H, J = 15.3, 3.0 Hz, H-2″), 1.03 (s, 9H, 3 × CH3); 13C-NMR (75 MHz, CDCl3): δ 159.8 (C=O), 154.4 (4-CN), 146.6 (N-CH=N), 143.6 (N-CH=N), 136.1 (6-C), 136.0 (ArC), 135.8 (ArC), 132.9 (ArC), 132.7 (ArC), 130.4 (ArC), 130.3 (ArC), 128.2 (ArC), 128.1 (ArC), 94.4 (5-C), 90.9 (4′-CH), 86.4 (1′-CH), 84.3 (3′-C(Cl)2), 63.2 (5′-CH2), 53.3 (2′-CH2), 30.0 (C(CH3)3), 27.1 (3 × CH3); HRMS: C27H29Cl2N5O3Si [M + H+]+ Calc.: 570.1489, found: 570.1489.
3′,3′-gem-Dichloro-2′,3′-dideoxycytidine (3b). Following the general desilylation procedure, 22 (0.142 g, 0.25 mmol) was reacted with a solution of TBAF in THF (0.4 mL, 0.375 mmol) in dry THF (2.0 mL). After work-up, the resulting crude residue was purified by column chromatography (EtOAc) to give 3′,3′-gem-dichloro-2′,3′-dideoxy-4-(1H-1,2,4-triazol-1-yl)uridine as a yellow solid (66 mg, 80%). 1H-NMR (500 MHz, DMSO-d6): δ 9.44 (s, 1H, N-CH=N), 8.61 (d, 1H, J = 7.3 Hz, H-6), 8.41 (s, 1H, N-CH=N), 7.03 (d, 1H, J = 7.3 Hz, H-5), 6.15 (dd, 1H, J = 7.4, 3.6 Hz, H-1′), 5.35 (s, 1H, OH), 4.56 (dd, 1H, J = 5.9, 3.1 Hz, H-4′), 3.91 (dd, 1H, J = 12.3, 3.1 Hz, H-5′), 3.88 (dd, 1H, J = 12.3, 5.8 Hz, H-5″), 3.50 (dd, J = 14.9, 7.4 Hz, 1H, H-2′), 3.06 (dd, J = 14.9, 3.6 Hz, 1H, H-2″); 13C-NMR (125 MHz, DMSO-d6): δ 159.9 (2-C=O), 154.2 (4-CN), 153.5 (N-CH=N), 148.1 (N-CH=N), 143.8 (6-C), 140.1 (5-C), 93.8 (4′-CH), 90.0 (1′-CH), 85.9 (3′-C(Cl)2), 60.3 (5′-CH2), 51.7 (2′-CH2); HRMS: C11H11Cl2N5O3 [M + Na+]+ Calc.: 354.0131, found: 354.0132. To a stirred solution of the above compound (0.100 g, 0.30 mmol,) in EtOH (3 mL) at −20 °C was added a solution of NH3 in EtOH, and then the reaction was allowed to warm to room temperature. After stirring for 4 h, the volatiles were removed in vacuo, and the crude residue was purified by column chromatography (EtOAc:EtOH 75:25) to give 3b as a pale green solid (0.753 g, 90%). 1H-NMR (600 MHz, DMSO-d6): δ 7.78 (d, 1H, J = 7.5 Hz, H-6), 7.34 (s, 1H, NH2), 7.19 (s, 1H, NH2), 6.19 (dd, 1H, J = 7.1, 5.2 Hz, H-1′), 5.78 (d, 1H, J = 7.5 Hz, H-5), 5.32 (t, 1H, J = 5.3 Hz, OH), 4.37 (dd, 1H, J = 5.0, 3.0 Hz, H-4′), 3.83 (ddd, 1H, J = 12.1, 5.3, 3.0 Hz, H-5′), 3.77 (ddd, 1H, J = 12.1, 5.3, 5.0 Hz, H-5″), 3.25 (dd, 1H, J = 14.7, 7.2 Hz, H-2′), 2.87 (dd, 1H, J = 14.6, 5.2 Hz, H-2″); 13C-NMR (150 MHz, DMSO-d6): δ 165.7 (4-C-NH2), 154.9 (2-C=O), 140.65 (6-C), 94.2 (5-C), 89.1 (4′-C), 86.80 (3′-C(Cl)2), 83.1 (1′-CH), 60.6 (5′-CH2), 51.6 (2′-CH2); HRMS: C9H10Cl1N3O3 [M + H+]+ Calc.: 280.0250, found: 280.0250.
3′-Chloro-2′,3′-didehydro-2′,3′-dideoxycytidine (23). To a stirred solution of 21 (0.95 g, 2.00 mmol) in dry CH3CN (10 mL) was added POCl3 (0.28 mL, 3 mmol) at −50 °C. Triethylamine (0.7 mL, 5.00 mmol) was then added dropwise over 10 min, followed by a solution of 1,2,4-triazole (0.414 g, 6.00 mmol) and triethylamine (0.8 mL, 6.00 mmol) in dry CH3CN over 30 min. The reaction mixture was stirred at room temperature for two days. It was then diluted with ethyl acetate (200 mL) and quenched with a NaH2PO4/Na2HPO4 buffer (pH = 7, 150.0 mL). The organic layer was washed with water and brine, dried over Na2SO4, and evaporated in vacuo to give a crude residue, which was purified by column chromatography (EtOAc:hexane 7:3) to give 5′-O-(tert-butyldiphenylsilyl)-3′-chloro-2′,3′-didehydro-2′,3′-dideoxy-4-(1H-1,2,4-triazol-1-yl)uridine as a white solid (0.884 mg, 80%). 1H-NMR (300 MHz, CDCl3): δ 9.24 (s, 1H, N-CH=N), 8.55 (d, 1H, J = 7.25 Hz, H-6), 8.07 (s, 1H, N-CH=N), 8.07–7.08 (m, 10H, ArH), 7.09 (dd, 1H, J = 3.3, 1.5 Hz, H-1′), 6.28 (d, 1H, J = 7.25 Hz, H-5), 6.16 (t, 1H, J = 1.5 Hz, H-2′), 4.89–4.87 (m, 1H, H-4′), 4.13 (dd, 1H, J = 12.2, 1.6 Hz, H-5′), 4.11 (dd, 1H, J = 12.2, 2.0 Hz, H-5″), 1.11 (s, 9H, 3 × CH3); 13C-NMR (75 MHz, CDCl3): δ 154.0 (C=O), 147.8 (4-CN), 147.6 (N-CH=N), (6-C) 143.2, 141.8 (N-CH=N), 136.3 (3′-C(Cl)), 135.5 (ArC), 135.0 (ArC), 130.5 (ArC), 130.4 (ArC), 128.3 (ArC), 128.2 (ArC), 123.0 (2′-C), 94.9 (5-C), 90.3 (4′-CH), 87.5 (1′-CH), 62.5 (5′-CH2), 29.9 (C(CH3)3), 27.3 (3 × CH3); HRMS: C27H28ClN5O3Si [M + H+]+ Calc.: 534.1722, found: 534.1718. Following the general desilylation procedure, a solution of 5′-O-(tert-butyldiphenylsilyl)-3′-chloro-2′,3′-didehydro-2′,3′-dideoxy-4-(1H-1,2,4-triazol-1-yl)uridine (0.53 g, 1.00 mmol) in dry THF (2.00 mL) was reacted with TBAF (1.5 mL, 1.5 mmol) at −50 °C. After work-up, the resulting crude residue was purified by column chromatography (EtOAc) to give 3′-chloro-2′,3′-didehydro-2′,3′-dideoxy-4-(1H-1,2,4-triazol-1-yl)uridine as a white solid (0.221 mg, 75%). 1H-NMR (500 MHz, MeOD): δ 9.38 (s, 1H, N-CH=N), 8.87 (d, 1H, J = 7.2 Hz, H-6), 8.24 (s, 1H, N-CH=N), 7.12 (d, 1H, J = 7.2 Hz, H-5), 7.02 (dd, 1H, J = 2.5, 1.5 Hz, H-1′), 6.19 (t, 1H, J = 1.5 Hz, H-2′), 4.89 (br s, 1H, H-4′), 3.89 (dq, 2H, J = 12.8, 1.5 Hz, H-5′, H-5″); 13C-NMR (125 MHz, DMSO): δ 161.1 (4-C=N), 157.1 (2-C=O), 154.8 (N-CH=N), 150.5 (N-CH=N), 144.7 (6-C), 137.7 (3′-C(Cl)), 123.3 (2′-CH) 96.0 (5-C), 92.0 (4′-CH), 89.3 (1′-CH), 62.9 (5′-CH2); HRMS: C11H10ClN5O3 [M + H+]+ Calc.: 296.0544, found: 296.0542. To a stirred solution of 3′-chloro-2′,3′-didehydro-2′,3′-dideoxy-4-(1H-1,2,4-triazol-1-yl)uridine (0.295 g 0.01 mmol,) in EtOH (3.00 mL) at −20 °C was added a solution of NH3 in EtOH. The reaction mixture was allowed to warm to room temperature and stirred for 4 h. After removal of all the volatiles in vacuo, the crude residue was purified by column chromatography (EtOAc:EtOH 75:25) to give 23 as a pale green solid (0.194 g, 80%). 1H-NMR (600 MHz, MeOD): δ 8.12 (d, 1H, J = 7.5 Hz, H-6), 7.01 (dd, 1H, J = 3.3, 1.5 Hz, H-1′), 6.06 (t, 1H, J = 1.5, H-2′), 5.93 (d, 1H, J = 7.5 Hz, H-5), 4.87–4.77 (m, 1H, H-4′), 3.86 (dq, 2H, J = 12.8, 2.0 Hz, H-5′, H-5″); 13C-NMR (150 MHz, MeOD): δ 166.7 (4-C), 157.1 (C=O), 144.2 (6-C), 137.1 (3′-CCl), 123.7 (2′-C), 96.0 (5-C), 90.2 (1′-C), 88.4 (4′-CH), 61.2 (5′-CH2); HRMS: C9H10Cl1N3O3 [M + Na+]+ Calc.: 266.0303, found: 266.0308.
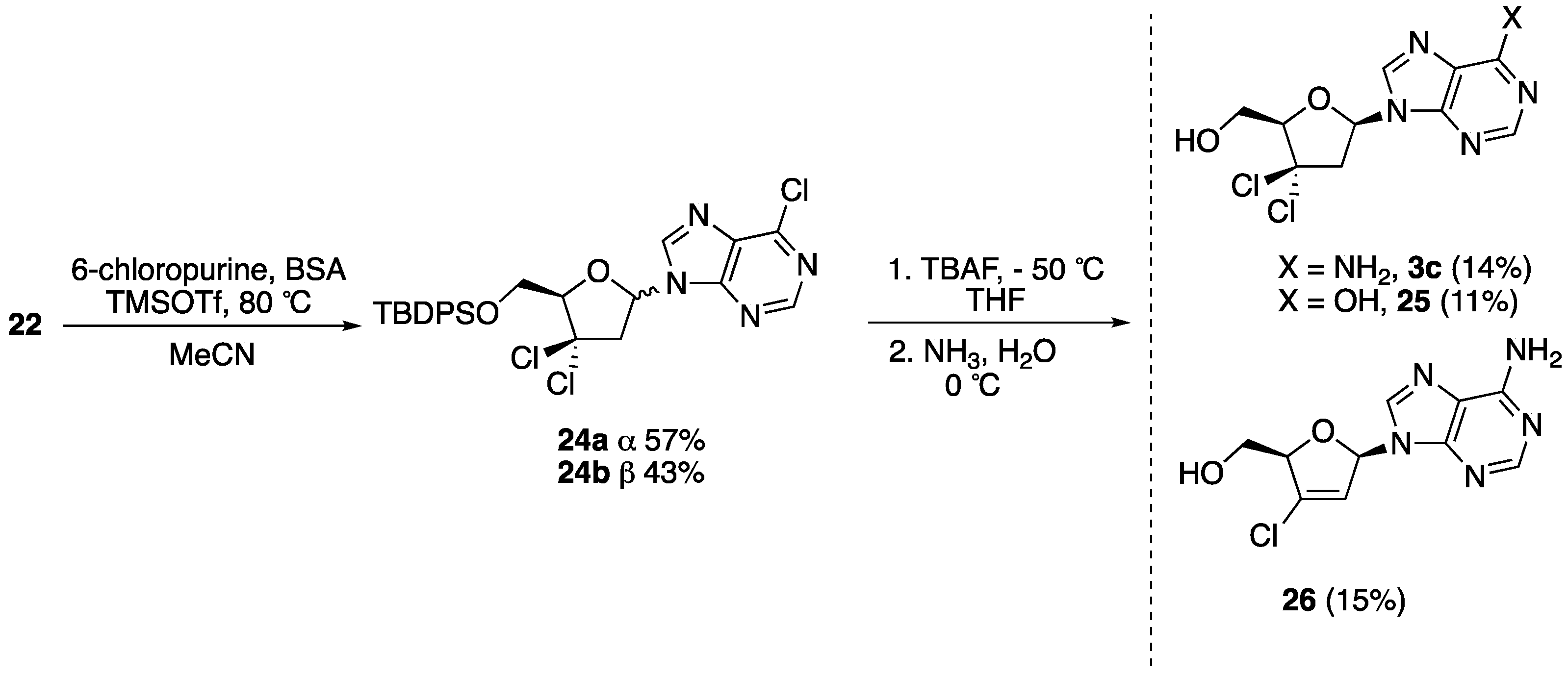
5′-O-(tert-Butyldiphenylsilyl)-3′,3′-gem-dichloro-2′,3′-dideoxy-6-chloropurine (24). To a stirred solution of 6-chloropurine (0.616 g, 4.00 mmol) in dry CH3CN (10 mL), N,O-bistrimethylsilylacetamide (1.09 mL, 4.50 mmol) was added, and then the reaction mixture was heated at 78 °C for 1 h. After cooling to room temperature, a solution of compound 22 (0.569 g, 1.00 mmol) in dry CH3CN (15 mL) was added. The reaction mixture was then cooled to −20 °C and TMSOTf (0.92 mL, 5.00 mmol) was added. The mixture was allowed to slowly warm to 20 °C and it was then heated at 78 °C and stirred for 5 h. It was then diluted with ethyl acetate and washed with saturated aq. NaHCO3 (50 mL), water (50 mL), and brine (50 mL). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by column chromatography (hexane:EtOAc 8.5:1.5 to 7:3) to give 24 as an anomeric mixture (0.448 g, 80%). This mixture was further purified by column chromatography to give 57% of 24a (α-anomer) and 43% of 24b (β-anomer). The unreacted excess of 6-chloropurine was recovered. Data for 24b: 1H-NMR (300 MHz, CDCl3): δ 8.71 (s, 1H, H-2), 8.28 (s, 1H, H-8), 7.69–7.26 (m, 10H, ArH), 6.54 (dd, 1H, J = 6.3, 5.3 Hz, H-1′), 4.55 (dd, 1H, J = 5.5, 3.4 Hz, H-4′), 4.20 (dd, 1H, J = 11.7, 3.3 Hz, H-5′), 4.09 (dd, 1H, J = 11.7, 5.5 Hz, H-5″), 3.44–3.42 (m, 2H, H-2′, H-2″), 1.06 (s, 9H, 3 × CH3); 13C-NMR (75 MHz, CDCl3): δ 152.4 (6-C), 151.8 (2-C), 151.3 (4-C), 144.0 (5-C), 135.9 (ArC), 135.8 (ArC), 133.0 (ArC), 132.9 (ArC), 130.2 (8-C), 128.0 (ArC), 128.0 (ArC), 90.1 (4′-CH), 85.5 (3′-C(Cl2)), 84.4 (1′-CH), 63.5 (5′-CH2), 52.3 (2′-CH2), 29.9 (C(CH3)3), 27.0 (3 × CH3); HRMS: C26H27Cl3N4O2Si [M + H+]+ Calc.: 561.1041, found: 561.1046.
3′,3′-gem-Dichloro-2′,3′-dideoxyadenosine (3c). Following the general desilylation procedure, 24b (0.028 g, 0.05 mmol) was reacted with a solution of TBAF in THF (0.5 mL, 0.5 mmol) in dry THF (5 mL). After work-up, the resulting crude material was dissolved in dry MeOH (5 mL) and cooled to −20 °C. A saturated solution of NH3 in H2O (10 mL, 25%) was then added; the reaction mixture was allowed to warm to room temperature and stirred overnight. After removal of all the volatiles in vacuo, the crude residue was purified by column chromatography (MeOH:EtOAc 0:10 to 2.5:7.5) to give 3c as a pale yellow solid (4 mg, 14%). Compounds 25 and 26 were also isolated as an oil (11%) and white solid (15%), respectively. Data for 3c: 1H-NMR (600 MHz, DMSO-d6): δ 8.33 (s, 1H, H-2), 8.16 (s, 1H, H-8), 7.38 (s, 2H, NH2), 6.47 (t, 1H, J = 6.8 Hz, H-1′), 5.48 (t, 1H, J = 5.3 Hz, OH), 4.46 (dd, 1H, J = 5.0, 3.3 Hz, H-4′), 3.83 (ddd, 1H, J = 10.4, 5.0, 3.3 Hz, H-5′), 3.79 (ddd, 1H, J = 10.4, 5.3, 5.0 Hz, H-5″), 3.65 (dd, 1H, J = 14.6, 6.8 Hz, H-2′), 3.42 (dd, 1H, J = 14.6, 6.8 Hz, H-2″); 13C-NMR (150 MHz, DMSO-d6): δ 156.2 (6-C), 152.8 (2-C), 149.2 (4-C), 139.1 (8-C), 118.9 (5-CH), 89.7 (4′-CH), 87.0 (3′-C(Cl2)), 81.2 (1′-CH), 61.2 (5′-CH2), 49.9 (2′-CH2); HRMS: C10H11Cl2N5O2 [M + H+]+ Calc.: 304.0362, found: 304.0357. Data for 25: 1H-NMR (600 MHz, DMSO-d6): δ 8.56 (s, 1H, H-2), 8.55 (s, 1H, H-8), 6.57 (dd, 1H, J = 7.1, 6.2 Hz, H-1′), 5.32 (t, 1H, J = 4.9 Hz, OH), 4.48 (dd, 1H, J = 5.3, 3.4 Hz, 1H, H-4′), 3.84 (ddd, 1H, J = 12.2, 5.0, 3.4 Hz, H-5′), 3.80 (ddd, 1H, J = 12.2, 5.3, 5.0 Hz, H-5″), 3.66 (dd, 1H, J = 14.6, 6.2 Hz, H-2′), 3.48 (dd, 1H, J = 14.6, 7.1 Hz, H-2″); 13C-NMR (150 MHz, DMSO-d6): δ 160.1 (6-C), 152.0 (2-C), 151.8 (4-C), 141.7 (8-C), 120.9 (5-CH), 89.7 (1′-CH), 86.7 (3′-C(Cl2)), 81.4 (4′-CH), 60.9 (5′-CH2), 50.0 (2′-CH2). Data for 26: 1H-NMR (300 MHz, DMSO-d6): δ 8.23 (s, 1H, H-2), 8.15 (s, 1H, H-8), 7.29 (s, 2H, NH2), 6.96 (dd, 1H, J = 5.1, 1.6 Hz, H-1′), 6.37 (t, 1H, J = 1.6 Hz, H-2′), 5.06–5.03 (m, 1H, H-4′), 3.71 (dd, 1H, J = 12.7, 1.9 Hz, H-5″), 3.59 (dd, 1H, J = 12.7, 3.0 Hz, H-5′); 13C-NMR (75 MHz, DMSO-d6): δ 156.1 (6-C), 152.9 (2-C), 149.2 (4-C), 139.1 (8-C), 134.8 (3′-C), 122.0 (2′-CH), 119.0 (5-CH), 87.1 (1′-CH), 87.0 (4′-CH), 60.4 (5′-CH2); HRMS: C10H10ClN5O2 [M + H+]+ Calc.: 268.0595, found: 268.0587.
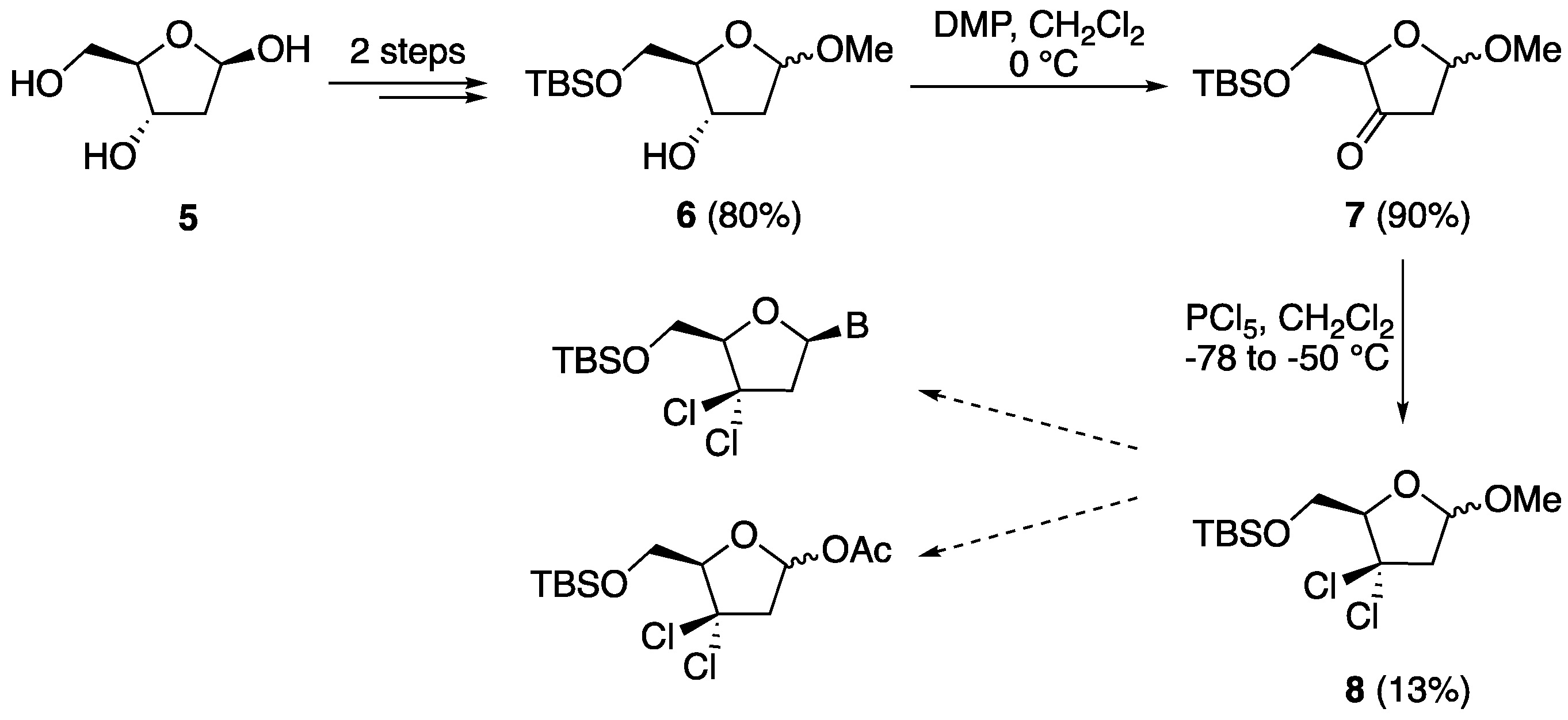
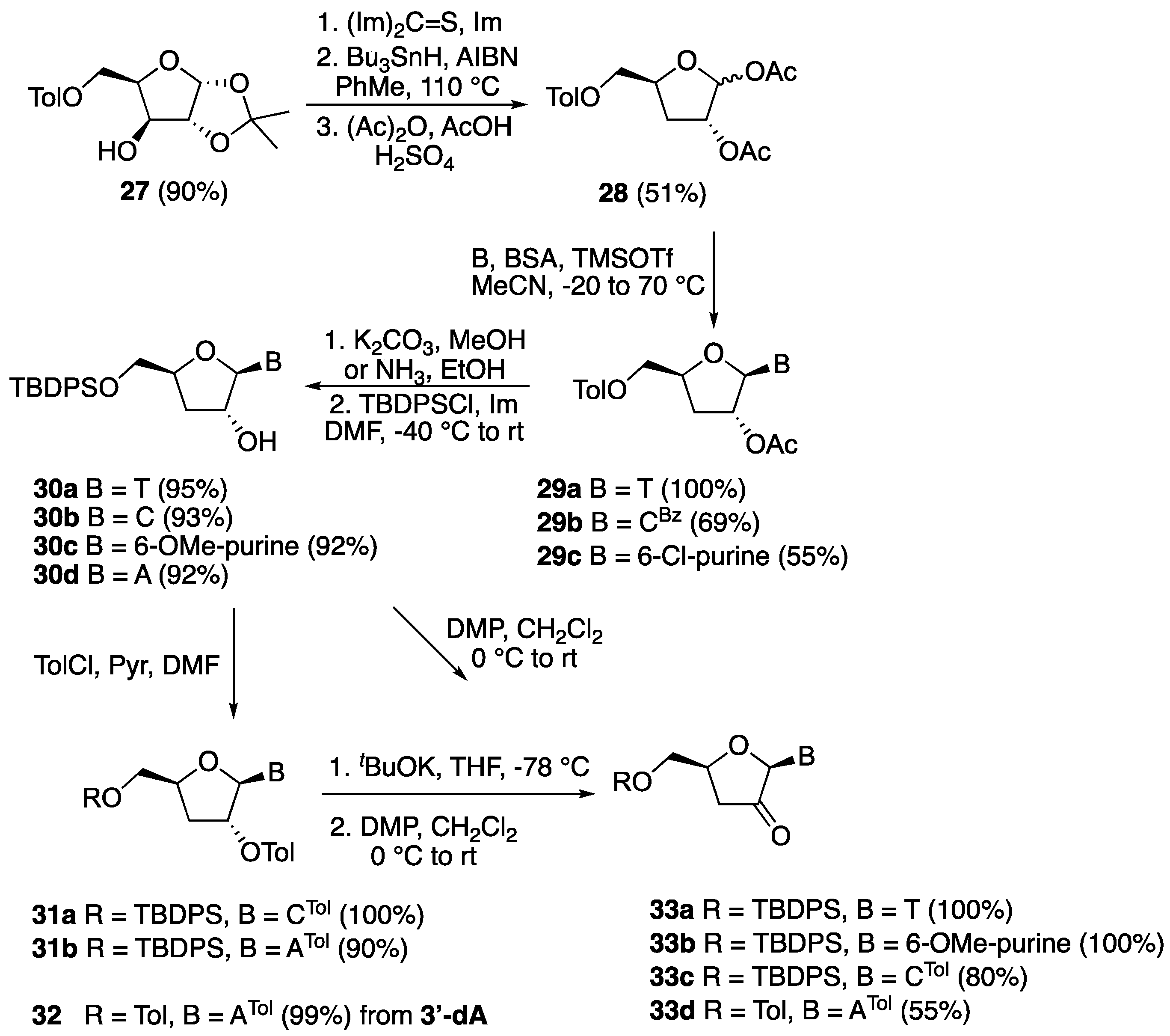
5-O-(o-Toluoyl)-1,2-isopropylidene-α-d-xylofuranose (27). d-xylose (10.0 g, 0.067 mol) was dissolved in acetone (260 mL) containing H2SO4 (0.66 M, 10.0 mL) and the solution was stirred for 30 min. A solution of Na2CO3 (13.0 g, 0.123 mol) in water (112 mL) was carefully added to the above cooled mixture, which was then stirred for further 2.5 h at 20 °C. Then, solid Na2CO3 (7.00 g, 0.066 mol) was added, Na2SO4 (22.3 g) was filtered off and washed with acetone, and the filtrate was evaporated in vacuo to afford a crude residue (14 g). This residue was resolubilized in a 9:1 mixture of EtOAc (270 mL) and methanol (30 mL), filtered, and evaporated in vacuo to give a yellow oil (12 g, 96%). This residue was dissolved in dry DMF (150 mL) under an inert atmosphere, cooled in an ice bath, and then o-toluoyl chloride (9.85 g, 8.31 mL, 0.064 mol) was added, followed by imidazole (4.35 g, 0.064 mol). The reaction mixture was allowed to warm to room temperature and stirred for 5 h. It was then diluted with EtOAc (300 mL) and washed with water (300 mL) and brine (100 mL). The organic layer was dried over Na2SO4, filtered, and evaporated in vacuo to give a crude residue, which was purified by column chromatography (hexane:EtOAc 7:3) to afford compound 27 (16.5 g, 84%) as a colorless oil. 1H-NMR (300 MHz, CDCl3): δ 7.92 (dd, 1H, J = 8.2, 1.6 Hz, ArH), 7.41–7.20 (m, 3H, ArH), 5.97 (d, J = 3.6 Hz, 1H, H-1′), 4.69 (dd, 1H, J = 13.4, 8.4 Hz, H-5′), 4.57 (d, 1H, J = 3.6 Hz, H-2′), 4.43 (dd, 1H, J = 13.4, 6.0 Hz, H-5″), 4.42 (ddd, 1H, J = 8.4, 6.0, 2.1 Hz, H-4′), 4.24 (d, 1H, J = 2.1, H-3′), 3.73 (s, 1H, OH), 2.57 (s, 3H, CH3), 1.49 (s, 3H, CH3), 1.30 (s, 3H, CH3); 13C-NMR (75 MHz, CDCl3): δ 168.2 (CO), 140.7 (ArC), 132.7 (ArC), 132.0 (ArC), 131.1 (ArC), 129.0 (ArC), 126.0 (ArC), 112.0 (OCO) 105.1 (1′-C), 85.4 (4′-C), 78.8 (3′-CH), 74.8 (2′-CH), 62.0 (5′-CH2), 27.0 (CH3), 26.4 (CH3), 22.0 (CH3); HRMS for C16H20O6 [M + Na+]+ Calc.: 331.1152, found: 331.1156.
5-O-(o-Toluoyl)-1,2-diacetyl-3-deoxyxylofuranose (28). Compound 27 (1.00 g, 3.10 mmol) and N,N-thiocarbonyldiimidazole (0.55 g, 3.10 mmol) were dissolved in dry DMF (20 mL) and then imidazole (0.034 g, 0.50 mmol) was added under a nitrogen atmosphere. The reaction mixture was stirred overnight, and it was then diluted with EtOAc (100 mL) and washed with water (100 mL) and brine (500 mL). The organic layer was dried over Na2SO4, filtered, and evaporated in vacuo to give a crude residue which was purified by column chromatography (hexane:EtOAc 7:3) to afford 5-O-(o-toluoyl)-3-O-imidazolylthiocarbonyl-1,2-isopropylidene-d-xylofuranose (1.10 g, 89%) as a pale yellow oil. 1H-NMR (300 MHz, CDCl3): δ 8.32 (s, 1H, NCH) 7.88–7.01 (m, 6H, ArH, NCH), 6.06 (d, 1H, J = 3.8 Hz, H-1′), 5.99 (d, 1H, J = 2.9, H-3′), 4.81–4.76 (m, 1H, H-4′), 4.78 (d, 1H, J = 3.9 Hz, H-2′), 4.59–4.56 (m, 2H, H-5′, H-5″), 2.53 (s, 3H, ArCH3), 1.57 (s, 3H, CH3), 1.35 (s, 3H, CH3); 13C-NMR (75 MHz, CDCl3): δ 182.2 (CS), 168.8 (CO), 140.7 (C=N), 137.2 (C=N), 132.6 (ArC), 131.9 (ArC), 131.5 (ArC), 130.9 (ArC), 126.0 (ArC), 117.9 (ArC), 112.9 (OCO), 105.1 (1′-C), 84.5 (4′-C), 83.0 (3′-CH), 76.9 (2′-CH), 61.1 (5′-CH2), 26.8 (CH3), 26.4 (CH3), 21.9 (CH3). 5-O-(o-Toluoyl)-3-O-imidazolylthiocarbonyl-1,2-isopropylidene-α-d-xylofuranose (1.05 g, 2.50 mmol) and AIBN (0.254 g, 1.50 mmol) were solubilized in toluene (50 mL). The resulting solution was stirred at 105 °C for 25 min, and then tri-n-butyltin hydride (0.87 mL, 3.00 mmol) was added dropwise over 1 h under a nitrogen atmosphere. The reaction mixture was stirred for an additional 2.5 h at 105 °C, and then it was cooled to room temperature and evaporated in vacuo. The crude residue was dissolved in EtOAc (200 mL) and washed with water (200 mL) and brine (100 mL). The organic layer was dried over Na2SO4, filtered, and evaporated in vacuo to give a crude product that was purified by column chromatography (hexane:EtOAc 4:1) to afford 5-O-(o-toluoyl)-1,2-isopropylidene-3-deoxyxylofuranose (0.50 g, 60%) as a colorless oil. 1H-NMR (300 MHz, CDCl3): δ 7.95–7.92 (m, 2H, ArH), 7.42–7.37 (m, 2H, ArH), 7.26–7.21 (m, 4H, ArH), 5.96 (d, 1H, J = 3.8 Hz, H-1′), 5.87 (d, 1H, J = 3.7 Hz, H-1′), 4.79–4.76 (m, 1H, H-2′), 4.59 (dd, 1H, J = 10.5, 4.1 Hz, H-5′), 4.79–4.45 (m, 5H, H-4′, H-4′, H-5′, H-5″, H-5″), 2.21–2.15 (m, 1H, H-3′), 1.77 (ddd, 1H, J = 13.1, 10.7, 4.9 Hz, H-3″), 2.60 (s, 3H, ArCH3), 2.59 (s, 3H, ArCH3), 1.54 (s, 3H, CH3), 1.50 (s, 3H, CH3), 1.33 (s, 6H, 2 × CH3); 13C-NMR (75 MHz, CDCl3): δ 167.5 (CO), 140.5 (ArC), 132.4 (ArC), 132.3 (ArC), 131.9 (ArC), 131.0 (ArC), 125.9 (ArC), 112.0 (OCO), 105.5 (1′-C), 84.6 (4′-C), 76.0 (2′-CH), 62.5 (5′-CH2), 35.7 (3′-CH), 27.0 (CH3), 26.5 (CH3), 22.0 (CH3); HRMS for C16H20O5 [M + Na+]+ Calc.: 315.1203, found: 315.1195. To a round-bottom flask containing 5-O-(o-toluoyl)-1,2-isopropylidene-3-deoxyxylofuranose (0.292 g, 1.00 mmol) were added acetic acid (5.0 mL) and acetic anhydride (3.0 mL). The reaction mixture was then cooled to 0 °C and then H2SO4 (0.5 mL) was added the stirring was continued overnight. The reaction mixture was then diluted with EtOAc (50 mL) and washed with a saturated solution of NaHCO3 (200 mL), water (50 mL), and brine (50 mL). The organic layer was dried over Na2SO4, filtered, and evaporated in vacuo to give a crude residue that was purified by column chromatography (hexane:EtOAc 7:3) to afford compound 28 (0.32 g, 95%) as a colorless oil and anomeric mixture. 1H-NMR (300 MHz, CDCl3): δ 7.95 (d, 1H, J = 8.0 Hz, ArH), 7.43–7.38 (m, 1H, ArH), 7.27–7.21 (m, 2H, ArH), 6.19 (s, 1H, H-1′), 5.23 (dd, 1H, J = 4.2, 1.5 Hz, H-2′), 4.73–4.67 (m, 1H, H-4′), 4.47 (dd, 1H, J = 11.8, 3.9 Hz, H-5′), 4.33 (dd, 1H, J = 11.8, 5.6 Hz, H-5″), 2.61 (s, 3H, ArCH3), 2.25–2.20 (m, 2H, H-3′, H-3″), 2.09 (s, 3H, CH3), 1.96 (s, 3H, CH3); 13C-NMR (75 MHz, CDCl3): δ 170.2 (ArCO), 169.5 (1′-CO), 167.3 (2′-CO), 140.7 (ArC), 132.5 (ArC), 132.0 (ArC), 130.9 (ArC), 125.9 (ArC), 121.6 (ArC), 99.6 (1′-CH), 78.9 (4′-CH), 76.9 (2′-CH), 66.1.0 (5′-CH2), 31.9 (3′-CH2), 22.0 (ArCH3), 21.3 (COCH3), 21.1 (COCH3); HRMS for C17H20O7 [M − H]− Calc.: 335.1136, found: 335.1121.
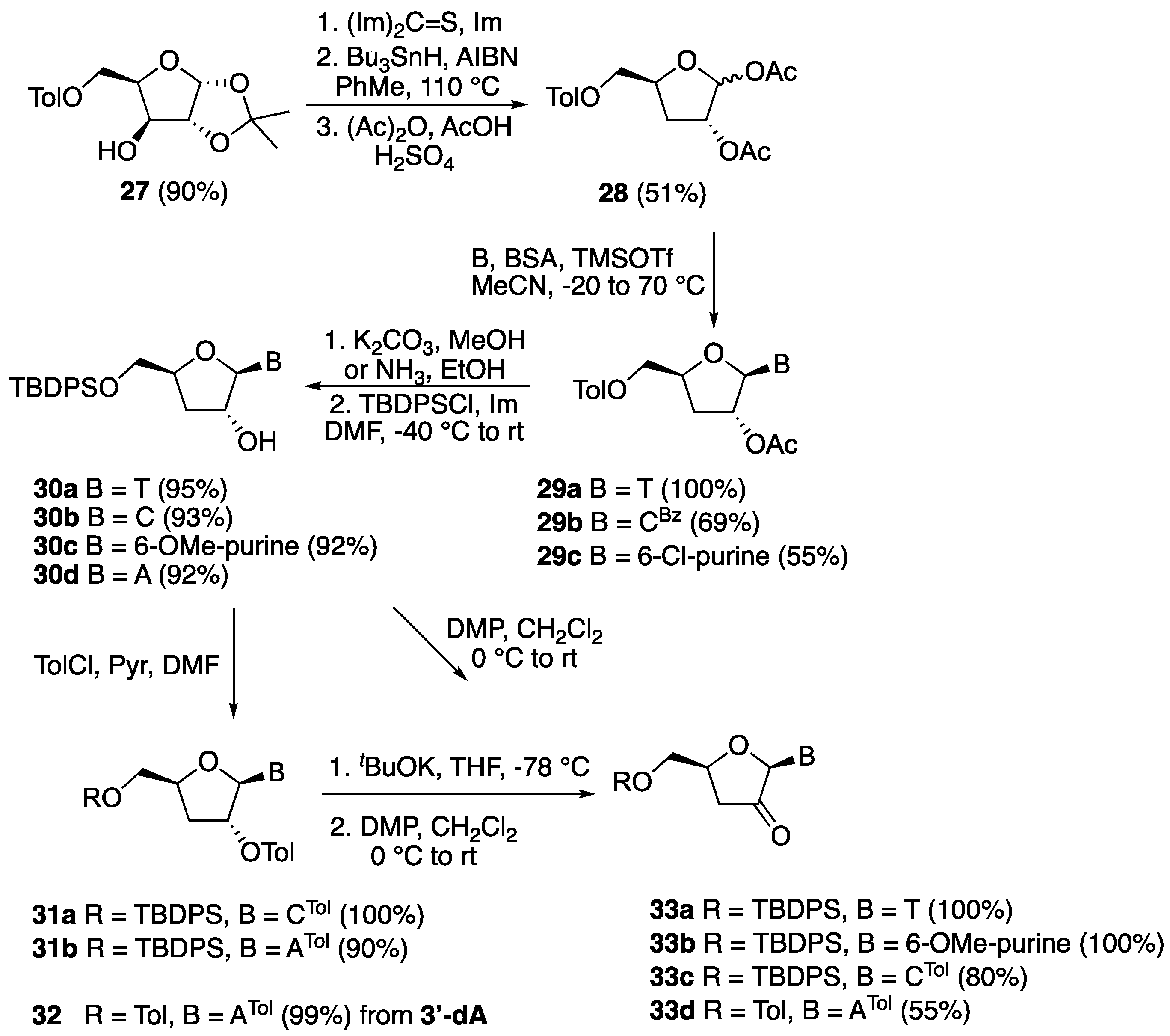
5′-O-(o-Toluoyl)-2′-O-acetyl-3′-deoxythymidine (29a). Following the general procedure for sugar-base condensation, a solution of thymine (0.302 g, 2.40 mmol) in dry CH3CN (10 mL) was reacted with N,O-bis(trimethylsilyl)acetamide (1.46 mL, 6.00 mmol), a solution of 28 (0.672 g, 2.00 mmol) in dry CH3CN (10 mL), and TMSOTf (0.46 mL, 2.50 mmol). After work-up, the crude residue was purified by silica gel column chromatography (hexane:EtOAc 3:2) to afford compound 29a (0.803 g) as a white solid in quantitative yield. 1H-NMR (300 MHz, CDCl3): δ 8.76 (s, 1H, NH), 7.92–7.14 (m, 1H, ArH), 7.45–7.40 (m, 1H, ArH), 7.28–7.22 (m, 2H, ArH), 7.14 (d, 1H, J = 1.2 Hz, H-6), 5.85 (d, 1H, J = 2.3 Hz, H-1′), 5.35 (dt, 1H, J = 9.5, 2.3 Hz, H-2′), 4.65 (dd, J = 12.6, 2.7 Hz, 1H, H-5′), 4.60 (dddd, J = 6.6, 5.6, 5.0, 2.7 Hz, 1H, H-4′), 4.47 (dd, 1H, J = 12.8, 5.0 Hz, H-5″), 2.60 (s, 3H, CH3), 2.40 (ddd, 1H, J = 14.0, 9.5, 6.6 Hz, H-3′), 2.19 (ddd, 1H, J = 14.0, 5.6, 2.2 Hz, H-3″), 2.12 (s, 3H, CH3), 1.65 (d, J = 1.1 Hz, 3H, 6-CH3); 13C-NMR (75 MHz, CDCl3): δ 170.3 (CO), 167.2 (CO), 163.7 (4-C), 150.2 (2-C), 140.8 (6-C), 135.8 (ArC), 132.7 (ArC), 132.5 (ArC), 130.5 (ArC), 126.2 (ArC), 111.5 (5-CH), 91.3 (1′-CH), 77.9 (4′-CH), 77.6 (2′-CH), 64.6 (5′-CH2), 33.1 (3′-CH), 21.9 (CH3), 21.1 (CH3), 12.1 (6-CH3); HRMS for C20H22N2O7 [M + H+]+ Calc.: 403.1499, found: 403.1495.
5′-O-(o-Toluoyl)-2′-O-acetyl-3′-deoxy-4-N-benzoylcytidine (29b). Following the general procedure for sugar-base condensation, a solution of N4-benzoylcytosine (0.538 g, 2.40 mmol) in dry CH3CN (10 mL) was reacted with N,O-bis(trimethylsilyl)acetamide (0.73 mL, 3.00 mmol), a solution of 28 (0.672 g, 2.00 mmol) in dry CH3CN (10 mL), and TMSOTf (0.46 mL, 2.50 mmol). After work-up, the crude residue was purified by silica gel column chromatography (hexane:EtOAc 1:1) to afford product 29b (0.677 g) as a white solid in 69% yield. The corresponding α-anomer was also isolated as a white solid (0.098 g, 10%). 1H-NMR (300 MHz, CDCl3): δ 8.00–7.99 (m, 1H, ArH), 7.90–7.89 (m, 2H, ArH), 7.79 (d, J = 7.8 Hz, 1H, H-6), 7.64–7.62 (m, 1H, ArH), 7.54–7.52 (m, 2H, ArH), 7.36–7.20 (m, 3H, ArH), 6.38 (d, 1H, J = 7.8 Hz, H-5), 5.58–5.55 (m, 1H, H-4′), 5.51 (d, 1H, J = 4.7 Hz, H-1′), 4.42–4.40 (m, 1H, H-2′), 4.56 (dd, 1H, J = 12.0, 4.2 Hz, H-5′), 4.40 (dd, 1H, J = 12.0, 6.2 Hz, H-5″), 2.23–2.17 (m, 1H, H-3′), 2.10–2.06 (m, 1H, H-3″), 2.57 (s, 3H, CH3), 2.28 (s, 3H, CH3); 13C-NMR (75 MHz, CDCl3): δ 186.4 (NHCO), 167.7 (ArCO), 166.6 (CH3CO), 162.2 (4-CN), 145.5 (2-CO), 140.6 (6-C), 133.3 (ArC), 132.3 (ArC), 131.7 (ArC), 130.5 (ArC), 129.1 (ArC), 128.5 (ArC), 127.5 (ArC), 125.8 (ArC), 97.3 (5-C), 92.0 (1′-CH), 73.8 (4′-C), 70.3 (2′-C), 62.2 (5′-CH2), 36.5 (3′-CH2), 21.9 (CH3), 21.7 (CH3); HRMS for C26H25N3O7 [M + H+]+ Calc.: 492.1765, found: 492.1763.
5′-O-(o-Toluoyl)-2′-O-acetyl-3′-deoxy-6-chloropurine (29c). Following the general procedure for sugar-base condensation, a solution of 6-chloropurine (0.371 g, 2.40 mmol) in dry CH3CN (10 mL) was reacted with N,O-bis(trimethylsilyl)acetamide (1.46 mL, 6.00 mmol), a solution of 28 (0.672 g, 2.00 mmol) in dry CH3CN (10 mL), and TMSOTf (0.46 mL, 2.50 mmol) After work-up, the crude residue was purified by silica gel column chromatography (hexane:EtOAc 3:2) to afford product 29c (0.473 g) as a white solid in 55% yield. 1H-NMR (300 MHz, CDCl3): δ 8.65 (s, 1H, H-2), 8.24 (s, 1H, H-8), 7.78–7.16 (m, 4H, ArH), 6.10 (d, 1H, J = 1.3 Hz, H-1′), 5.84 (dt, 1H, J = 6.1, 1.3 Hz, H-2′), 4.78 (dddd, 1H, J = 10.4, 5.6, 5.3, 3.1 Hz, H-4′), 4.66 (dd, 1H, J = 12.2, 3.1 Hz, H-5′), 4.50 (dd, 1H, J = 12.2, 5.3 Hz, H-5″), 2.85 (ddd, 1H, J = 14.0, 10.4, 6.2 Hz, H-3′), 2.54 (s, 3H, CH3), 2.34 (ddd, 1H, J = 14.0, 5.6, 1.3 Hz, H-3″), 2.16 (s, 3H, CH3); 13C-NMR (75 MHz, CDCl3): δ 170.4 (ArCO), 167.1 (CO), 152.3 (6-C), 151.6 (2-C), 151.0 (4-C), 144.4 (8-C), 140.7 (ArC), 132.7 (ArC), 132.6 (ArC), 132.1 (ArC), 130.6 (ArC), 128.8 (ArC), 126.0 (5-CH), 90.9 (1′-CH), 79.4 (4′-CH), 78.0 (2′-CH), 64.6 (5′-CH2), 33.3 (3′-CH2), 21.9 (CH3), 21.1 (CH3); HRMS for C20H19Cl1N4O5 [M + H+]+ Calc.: 431.1116, found: 431.1115.
5′-O-(tert-Butyldiphenylsilyl)-3′-deoxythymidine (30a). To a solution of 29a (0.402 g, 1.00 mmol) in dry MeOH (20 mL) was added K2CO3 (0.276 g, 2.00 mmol) and the reaction mixture was stirred at room temperature for 4 h. The solvent was then removed in vacuo and the resulting crude residue was purified by silica gel column chromatography (CH2Cl2:MeOH 9:1) to give 3′-deoxythymidine (0.241 g) as a white solid in quantitative yield. 1H-NMR (500 MHz, MeOD): δ 7.97 (d, 1H, J = 1.1 Hz, H-6), 5.68 (d, 1H, J = 1.8 Hz, H-1′), 4.41 (dddd, 1H, J = 9.9, 3.2, 2.6, 2.5 Hz, H-4′), 4.33–4.32 (dt, 1H, J = 5.6, 1.8 Hz, H-2′), 3.93 (dd, 1H, J = 12.4, 2.6 Hz, H-5′), 3.67 (dd, 1H, J = 12.4, 3.2 Hz, H-5″), 2.13 (ddd, 1H, J = 13.4, 9.9, 5.6 Hz, H-3′), 1.87 (ddd, 1H, J = 13.4, 5.6, 2.5 Hz, H-3″), 1.86 (d, 3H, J = 1.1 Hz, CH3); 13C-NMR (125 MHz, MeOD): δ 166.5 (4-C), 152.4 (2-C), 138.2 (6-C), 110.6 (5-CH), 93.6 (1′-CH), 82.5 (4′-CH), 77.0 (2′-CH), 63.1 (5′-CH2), 34.0 (3′-CH), 12.3 (6-CH3); HRMS for C10H14N2O5 [M + Na+]+ Calc.: 265.0795, found: 265.0796. To a stirred solution of 3′-deoxythymidine (0.242 g, 1.00 mmol) and imidazole (0.070 g, 1.00 mmol) in dry DMF (5.0 mL) was added TBDPSCl (0.275 g, 1.00 mmol) at −50 °C. The reaction mixture was allowed to warm to room temperature and stirred for 4 h. It was then diluted with EtOAc (100 mL) and washed with saturated aq. NaHCO3 (100 mL), water, and brine. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by silica gel column chromatography (hexane:EtOAc 3:2) to give product 30a (0.456 g) as a white solid in 95% yield. 1H-NMR (600 MHz, CDCl3): 1H-NMR (600 MHz, CDCl3): δ 9.08 (s, 1H, NH), 7.66–7.64 (m, 4H, ArH), 7.60 (d, 1H, J = 1.2 Hz, H-6), 7.45–7.37 (m, 6H, ArH), 5.68 (d, 1H, J = 2.1 Hz, H-1′), 4.54–4.51 (m, 1H, H-4′), 4.46–4.44 (m, 1H, H-2′), 4.10 (dd, 1H, J = 11.8, 2.5 Hz, H-5′), 3.75 (dd, 1H, J = 11.8, 3.4 Hz, H-5″), 2.20 (ddd, 1H, J = 13.2, 8.6, 6.4 Hz, H-3′), 1.99 (ddd, 1H, J = 13.2, 6.1, 3.3 Hz, H-3′), 1.62 (d, J = 1.2 Hz, 3H, CH3), 1.08 (s, 9H, 3 × CH3); 13C-NMR (150 MHz, CDCl3): δ 163.7 (4-C), 150.9 (2-C), 135.5 (ArC), 135.4 (ArC), 135.1 (6-C), 133.0 (ArC), 132.6 (ArC), 130.0 (ArC), 130.0 (ArC), 127.9 (ArC), 127.9 (ArC), 110.3 (5-CH), 93.6 (1′-CH), 82.5 (4′-CH), 77.0 (2′-CH), 63.1 (5′-CH2), 34.0 (3′-CH), 29.6 (C(CH3)3), 26.9 (3 × CH3), 12.2 (6-CH3); HRMS for C26H32N2O5Si1 [M + H+]+ Calc.: 481.2153, found: 481.2154.
5′-O-(tert-Butyldiphenylsilyl)-3′-deoxycytidine (30b). To a solution of 29b (0.491 g, 1.00 mmol) in dry MeOH (20 mL) was added K2CO3 (0.415 g, 3.00 mmol) and the reaction mixture was stirred at room temperature for 4 h. The solvent was then removed in vacuo and the resulting crude residue was purified by silica gel column chromatography (CH2Cl2:MeOH 9:1) to give 3′-deoxycytidine (0.225 g) as a white solid in quantitative yield. 1H-NMR (500 MHz, MeOD): δ 8.16 (d, 1H, J = 7.5 Hz, H-6), 5.86 (d, 1H, J = 7.5 Hz, H-5), 5.75 (s, 1H, H-1′), 4.45 (dddd, 1H, J = 10.7, 5.4, 3.4, 2.7 Hz, H-4′), 4.28 (dd, 1H, J = 5.2, 1.6 Hz, H-2′), 3.97 (dd, 1H, J = 12.4, 2.7 Hz, H-5′), 3.71 (dd, 1H, J = 12.4, 3.4 Hz, H-5′), 2.02 (ddd, 1H, J = 13.3, 10.7, 5.2 Hz, H-3′), 1.85 (ddd, 1H, J = 13.3, 5.4, 1.6 Hz, H-3″); 13C-NMR (150 MHz, MeOD): δ 167.8 (4-C), 158.4 (2-C), 142.6 (6-C), 118.6 (5-CH), 94.9 (1′-CH), 83.1 (4′-CH), 77.6 (2′-CH), 63.0 (5′-CH2), 33.5 (3′-CH2); HRMS for C9H13N3O4 [M + H+]+ Calc.: 228.0978, found: 228.0988. To a stirred solution of 3′-deoxycytidine (0.227 g, 1.00 mmol) and imidazole (0.070 g, 1.00 mmol) in dry DMF (5.0 mL) was added TBDPSCl (0.275 g, 1.00 mmol) at −50 °C. The reaction mixture was allowed to warm to room temperature and stirred for 4 h. It was then diluted with EtOAc (100 mL) and washed with saturated aq. NaHCO3 (100 mL), water, and brine. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by silica gel column chromatography (CH2Cl2:MeOH 9:1) to give compound 30b (0.432 g) as a white solid in 93% yield. 1H-NMR (600 MHz, DMSO-d6): δ 7.80 (d, 1H, J = 7.4 Hz, H-6), 7.73–7.72 (m, 1H, ArH), 7.65–7.62 (m, 3H, ArH), 7.49–7.44 (m, 3H, ArH), 7.35–7.33 (m, 1H, ArH), 7.23–7.21 (m, 2H, ArH), 7.01 (s, 2H, NH2), 5.72 (d, 1H, J = 1.1 Hz, H-1′), 5.51 (d, 1H, J = 7.4 Hz, H-5), 4.36 (dddd, 1H, J = 10.5, 3.5, 2.6, 1.5 Hz, H-4′), 4.12 (dt, 1H, J = 5.1, 1.1 Hz, H-2′), 4.02 (dd, 1H, J = 11.7, 2.6 Hz, H-5′), 3.72 (dd, 1H, J = 11.8, 3.5 Hz, H-5″), 3.50 (s, 1H, OH), 2.01 (ddd, 1H, J = 12.9, 5.2, 1.5 Hz, H-3′), 1.75 (ddd, 1H, J = 12.9, 10.5, 5.2 Hz, H-3″), 1.02 (s, 9H, 3 × CH3); 13C-NMR (150 MHz, DMSO-d6): δ 165.6 (4-C), 155.2 (2-C), 140.3 (6-C), 138.0 (ArC), 135.2 (ArC), 135.0 (ArC), 132.7 (ArC), 132.4 (ArC), 131.1 (ArC), 130.4 (ArC), 130.1 (ArC), 130.0 (ArC), 129.7 (ArC), 128.0 (ArC), 125.5 (5-CH), 92.2 (1′-CH), 80.2 (4′-CH), 75.4 (2′-CH), 64.4 (5′-CH2), 33.1 (3′-CH), 29.6 (C(CH3)3), 26.7 (CH3); HRMS for C25H31N3O4Si1 [M + Na+]+ Calc.: 488.1976, found: 488.1976.
5′-O-(tert-Butyldiphenylsilyl)-3′-deoxy-6-methoxy-adenosine (30c). To a solution of 29c (0.430 g, 1.00 mmol) in dry MeOH (20 mL) was added K2CO3 (0.415 g, 3.00 mmol) and the reaction mixture was stirred at room temperature for 4 h. The solvent was then removed in vacuo and the resulting crude residue was purified by silica gel column chromatography (CH2Cl2/MeOH 9:1) to give product 3′-deoxy-6-methoxy-adenosine (0.260 g) as a white solid in quantitative yield. 1H-NMR (500 MHz, MeOD): δ 8.62 (s, 1H, H-2), 8.52 (s, 1H, H-8), 6.08 (d, 1H, J = 2.2 Hz, H-1′), 4.74 (ddt, 1H, J = 5.8, 3.2, 2.2 Hz, H-2′), 4.56 (dddd, 1H, J = 8.7, 6.4, 3.5, 2.7 Hz, H-4′), 4.19 (s, 3H, OCH3), 3.95 (dd, J = 12.4, 2.7 Hz, 1H, H-5′), 3.70 (dd, 1H, J = 12.4, 3.5 Hz, H-5″), 2.40 (ddd, 1H, J = 13.4, 8.7, 5.8 Hz, H-3′), 2.07 (ddd, 1H, J = 13.4, 6.4, 3.2 Hz, H-3″); 13C-NMR (125 MHz, MeOD): δ 152.8 (6-C), 143.7 (2-C), 142.8 (4-C), 133.7 (8-C), 113.1 (5-CH), 84.2 (1′-CH), 73.4 (4′-CH), 67.4 (2′-CH), 54.5 (OCH3), 45.4 (5′-CH2), 45.4 (3′-CH2); HRMS for C11H14N4O4 [M + H+]+ Calc.: 267.1087, found: 267.1092. To a stirred mixture of 3′-deoxy-6-methoxy-adenosine (0.266 g, 1.00 mmol) and imidazole (0.070 g, 1.00 mmol) in dry DMF (5.0 mL) was added TBDPSCl (0.275 g, 1.00 mmol) at −50 °C. The reaction mixture was allowed to warm to room temperature and stirred for 4 h. It was then diluted with EtOAc (100 mL), and then washed with water and brine. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by silica gel column chromatography (hexane:EtOAc 7:3) to give compound 30c (0.463 g) as a white solid in 92% yield. 1H-NMR (300 MHz, CDCl3): δ 8.50 (s, 1H, H-2), 8.31 (s, 1H, H-8), 7.66–7.60 (m, 4H, ArH), 7.45–7.32 (m, 6H, ArH), 5.98 (d, 1H, J = 2.7 Hz, H-1′), 5.13 (s, 1H, OH), 4.72 (m, 1H, H-2′), 4.64–4.59 (m, 1H, H-4′), 4.19 (s, 3H, OCH3), 4.00 (dd, 1H, J = 11.5, 3.1 Hz, H-5′), 3.73 (dd, 1H, J = 11.5, 3.5 Hz, H-5″), 2.39 (ddd, 1H, J = 13.1, 7.2, 5.7 Hz, H-3′), 2.13 (ddd, 1H, J = 13.1, 6.6, 4.5 Hz, H-3″), 1.02 (s, 9H, 3 × CH3); 13C-NMR (150 MHz, CDCl3): δ 161.4 (6-C), 152.0 (2-C), 151.0 (4-C), 140.5 (8-C), 135.8 (ArC), 135.8 (ArC), 133.0 (ArC), 133.0 (ArC), 130.1 (ArC), 128.1 (ArC), 128.0 (ArC), 115.5 (5-CH), 93.2 (1′-CH), 81.7 (4′-CH), 76.7 (2′-CH), 65.2 (5′-CH2), 54.6 (OCH3), 33.4 (3′-CH2), 29.9 (C(CH3)3), 27.1 (CH3); HRMS: C27H32N4O4Si [M + H+]+ Calc.: 505.2265, found: 505.2269.
5′-O-(tert-Butyldiphenylsilyl)-3′-deoxy-adenosine (30d). A 50 mL round-bottomed flask was charged with 29c (0.430 g, 1.00 mmol) and dry EtOH (10 mL) and the mixture was cooled to −20 °C. Then, a solution of NH3 in MeOH (10 mL, 7 N) was added and the mixture was stirred for 48 h. After removal of all the volatiles in vacuo, the resulting crude residue was purified by silica gel chromatography CH2Cl2/MeOH (7:3) to afford product 3′-deoxyadenosine (0.250 g) as a white solid in quantitative yield. 1H-NMR (600 MHz, D2O): δ 8.26 (s, 1H, H-2), 8.14 (s, 1H, H-8), 6.01 (d, 1H, J = 2.2 Hz, H-1′), 4.76 (m, 2H, H-2′, H-4′), 3.91 (dd, 2H, J = 12.6, 2.7 Hz, H-5′), 3.71 (dd, 1H, J = 12.6, 4.5 Hz, H-5″), 2.28 (ddd, 1H, J = 13.6, 8.7, 5.7 Hz, H-3′), 2.20 (ddd, 1H, J = 13.6, 6.6, 3.2 Hz, H-3″); 13C-NMR (150MHz, D2O): δ 155.3 (6-C), 152.3 (2-C), 148.0 (4-C), 139.6 (8-C), 118.6 (5-CH), 90.8 (1′-CH), 81.1 (4′-CH), 74.8 (2′-CH), 62.6 (5′-CH2), 32.9 (3′-CH2); HRMS for C10H13N5O3 [M + H+]+ Calc.: 252.1091, found: 252.1085. To a stirred mixture of 3′-deoxyadenosine (0.05 g, 0.25 mmol) and imidazole (0.070 g, 1.0 mmol) in anhydrous DMF (5 mL) was added TBDPSCl (0.275 g, 1.0 mmol) at −50 °C. The reaction mixture was allowed to warm to room temperature and stirred for 4 h. It was then diluted with EtOAc (100 mL) and washed with water and brine. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue was then purified by silica gel column chromatography (EtOAc) to afford compound 30d (0.112 g, 92%) as a white solid. 1H-NMR (600 MHz, MeOD): δ 8.32 (s, 1H, H-2), 8.18 (s, 1H, H-8), 7.67–7.32 (m, 10H, ArH), 6.02 (d, 1H, J = 1.5 Hz, H-1′), 4.71 (dt, 1H, J = 5.5, 1.6 Hz, H-2′), 4.57 (dddd, 1H, J = 8.9, 3.9, 2.8, 2.1 Hz, H-4′), 4.05 (dd, 1H, J = 11.6, 2.8 Hz, H-5′), 3.78 (dd, 1H, J = 11.7, 3.9 Hz, H-5″), 2.46 (ddd, 1H, J = 14.4, 8.9, 5.5 Hz, H-3′), 2.01 (ddd, 1H, J = 14.4, 5.6, 2.1 Hz, H-3″), 1.28 (s, 9H, 3 × CH3); 13C-NMR (150 MHz, MeOD): δ 157.3 (6-C), 153.8 (2-C), 150.0 (4-C), 140.3 (8-C), 136.7 (ArC), 136.6 (ArC), 134.2 (ArC), 134.0 (ArC), 131.0 (ArC), 131.0 (ArC), 128.8 (ArC), 120.4 (5-CH), 93.2 (1′-CH), 82.7 (4′-CH), 77.0 (2′-CH), 66.0 (5′-CH2), 34.4 (3′-CH2), 31.6 (CCH3)3), 27.4 (CH3); HRMS for C26H31N5O3Si1 [M + H+]+ Calc.: 490.2268, found: 490.2273.
5′-O-(tert-Butyldiphenylsilyl)-2′-O-o-toluoyl-4-N-o-toluoyl-3′-deoxycytidine (31a). To a stirred solution of 30b (0.93 g, 2.00 mmol) in dry DMF (30 mL), ortho-toluoyl chloride (0.386 g, 0.325 mL, 2.50 mmol) was added at 0 °C under an inert atmosphere. Next, imidazole (0.17 g, 2.50 mmol) was added and the mixture was stirred at room temperature for 3 h. It was then diluted with EtOAc (100 mL) and washed with water (100 mL) and brine (100 mL). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by silica gel column chromatography (hexane:EtOAc 7:3) to afford compound 31a as a colorless oil (1.17 g, 84%). 1H-NMR (300 MHz, CDCl3): δ 8.52 (d, 1H, J = 7.5 Hz, H-6), 8.01–7.14 (m, 18H, ArH), 6.26 (s, 1H, H-1′), 5.58 (d, 1H, J = 7.5 Hz, H-5), 5.27 (s, 1H, H-2′), 4.56–4.51 (m, 1H, H-4′), 4.27 (dd, 1H, J = 12.0, 2.0 Hz, H-5′), 3.79 (dd, 1H, J = 12.0, 2.5 Hz, H-5″), 2.62 (s, 3H, CH3) 2.51 (s, 3H, CH3) 2.14 (dd, 1H, J = 14.0, 4.9 Hz, H-3′), 2.13 (dd, J = 14.0, 4.9 Hz, 1H, H-3″), 1.15 (s, 9H, 3 × CH3); 13C-NMR (75 MHz, CDCl3): δ 171.1 (CO), 166.7 (CO), 163.2 (4-C), 154.9 (2-C), 145.0 (6-C), 138.3 (ArC), 137.7 (ArC), 135.8 (ArC), 135.7 (ArC), 132.1 (ArC), 131.7 (ArC), 131.2 (ArC), 128.5 (ArC), 128.3 (ArC), 126.1 (ArC), 125.8 (ArC), 117.6 (5-CH), 91.3 (1′-CH), 82.0 (4′-CH), 79.2 (2′-CH), 63.6 (5′-CH2), 31.1 (3′-CH2), 29.9 (C(CH3)3), 27.2 (3 × CH3), 22.1 (ArCH3), 22.1 (ArCH3); HRMS: C41H43N3O6Si [M + Na+]+ Calc.: 724.2813, found: 724.2947.
5′-O-(tert-Butyldiphenylsilyl)-2′-O-o-toluoyl-6-N-O-o-toluoyl-3′-deoxyadenosine (31b). To a stirred solution of 30d (0.489 g, 1.00 mmol) in dry pyridine (30 mL), ortho-toluoyl chloride (0.386 g, 0.325 mL, 2.5 mmol) was added at 0 °C under an inert atmosphere. The mixture was stirred at room temperature overnight. It was diluted with EtOAc (100 mL) and washed with water (100 mL) and brine (100 mL). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by silica gel column chromatography (hexane:EtOAc 7:3) to give compound 31b as a colorless oil (0.580 g, 80%). 1H-NMR (600 MHz, CDCl3): δ 8.74 (s, 1H, H-2), 8.41 (s, 1H, H-8), 7.73–7.09 (m, 18H, ArH), 5.96 (d, 1H, J = 3.1 Hz, H-1′), 4.78 (ddd, 1H, J = 5.8, 3.1, 2.4 Hz, H-2′), 4.62 (ddt, 1H, J = 6.9, 3.9, 3.5 Hz, H-4′), 3.95 (dd, 1H, J = 11.5, 3.5 Hz, H-5′), 3.73 (dd, 1H, J = 11.5, 3.9 Hz, H-5″), 2.49 (s, 6H, ArCH3), 2.38 (ddd, 1H, J = 13.1, 6.9, 5.8 Hz, H-3′), 2.17 (ddd, 1H, J = 13.1, 6.9, 2.4 Hz, H-3″), 1.02 (s, 9H, 3 × CH3); 13C-NMR (150 MHz, CDCl3): δ 172.3 (CO), 152.2 (6-C), 151.8 (2-C), 151.6 (4-C), 143.1 (8-C), 139.2 (ArC), 135.5 (ArC), 134.9 (ArC), 132.7 (ArC), 132.7 (ArC), 131.3 (ArC), 131.3 (ArC), 129.9 (ArC), 129.3 (ArC), 128.2 (ArC), 127.7 (ArC), 127.7 (ArC), 125.4 (5-CH), 93.0 (1′-CH), 81.3 (4′-CH), 76.1 (2′-CH), 65.1 (5′-CH2), 33.5 (3′-CH2), 29.6 (C(CH3)3), 26.8 (3 × CH3), 19.9 (CH3), 19.1 (CH3); HRMS: C42H43N5O5Si [M + H+]+ Calc.: 726.3106, found: 726.3123.
3′,5′-Bis-O-o-toluoyl-6-N-O-o-toluoyl-3′-deoxyadenosine (32). To a stirred solution of 3′-deoxyadenosine (0.251 g, 1.00 mmol) in dry pyridine (30 mL), o-toluoyl chloride (0.539 g, 0.453 mL, 3.50 mmol) was added at 0 °C under an inert atmosphere and stirred overnight at room temperature. The mixture was diluted with EtOAc (100 mL) and washed with water (100 mL) and brine (100 mL). The organic layer was dried over Na2SO4, filtered, and evaporated in vacuo. The crude residue was purified by column chromatography (EtOAc:hexane 3:7) to afford 32 as a colorless oil (0.600 g, 99%). 1H-NMR (600 MHz, CDCl3): δ 8.49 (s, 1H, H-2), 8.10 (s, 1H, H-8), 7.99–7.19 (m, 12H, ArH), 6.26 (d, 1H, J = 1.3 Hz, H-1′), 6.07 (dt, 1H, J = 6.0, 1.3 Hz, H-2′), 4.85 (dddd, 1H, J = 9.2, 5.6, 5.4, 3.1 Hz, H-4′), 4.68 (dd, 1H, J = 12.1, 3.1 Hz, H-5′), 4.53 (dd, 1H, J = 12.1, 5.4 Hz, H-5″), 3.18 (s, 3H, ArCH3), 2.98 (ddd, 1H, J = 12.9, 9.2, 6.0 Hz, H-3′), 2.60 (s, 3H, ArCH3), 2.56 (s, 3H, ArCH3), 2.46 (ddd, 1H, J = 12.9, 5.6, 1.3 Hz, H-3″); 13C-NMR (150 MHz, CDCl3): δ 171.1 (NHCO), 166.9 (CO), 166.3 (CO), 161.0 (6-C), 152.2 (2-C), 151.0 (4-C), 141.1 (8-C), 140.8 (ArC), 140.4 (ArC), 132.7 (ArC), 131.9 (ArC), 131.7 (ArC), 130.8 (ArC), 130.4 (ArC), 128.6 (ArC), 128.0 (ArC), 125.8 (ArC), 125.7 (ArC), 122.1 (5-CH), 90.4 (1′-CH), 78.9 (4′-CH), 78.1 (2′-CH), 64.6 (5′-CH2), 33.4 (3′-CH2), 21.9 (CH3), 21.8 (CH3), 21.6 (CH3).
5′-O-(tert-Butyldiphenylsilyl)-2′-keto-3′-deoxythymidine (33a). Following the general oxidation procedure, a solution of 30a (0.480 g, 1.00 mmol) in CH2Cl2 (15 mL) was reacted with DMP (0.425 g, 1.00 mmol). After work-up, the crude residue was purified by silica gel column chromatography (hexane:EtOAc 7:3) to afford compound 33a (0.476 g, 100%) as a white solid in quantitative yield. 1H-NMR (300 MHz, CDCl3): δ 10.2 (s, 1H, NH), 8.45–7.96 (m, 11H, ArH, H-6), 6.02 (s, 1H, H-1′), 5.10 (dddd, 1H, J = 8.4, 7.3, 4.8, 3.6 Hz, H-4′), 4.67 (dd, 1H, J = 11.3, 3.6 Hz, H-5′), 4.59 (dd, 1H, J = 11.2, 4.8 Hz, H-5″), 3.60 (dd, 1H, J = 18.8, 8.4 Hz, H-3′), 3.22 (dd, 1H, J = 18.7, 7.3 Hz, H-3″), 2.44 (d, 3H, J = 1.0 Hz, CH3), 1.77 (s, 9H, 3 × CH3); 13C-NMR (75 MHz, CDCl3): δ 207.0 (2′-CO), 163.9 (4-C), 150.3 (2-C), 138.4 (6-C), 135.8 (ArC), 135.7 (ArC), 133.2 (ArC), 133.2 (ArC), 130.1 (ArC), 128.0 (ArC), 112.0 (5-CH), 85.9 (1′-CH), 76.2 (4′-CH), 65.4 (5′-CH2), 36.6 (3′-CH), 29.9 (C(CH3)), 27.1 (6-CH3) 12.3 (6-CH3); HRMS for C26H30N2O5Si1 [M + Na+]+ Calc.: 501.1816, found: 501.1817.
5′-O-(tert-Butyldiphenylsilyl)-2′-keto-3′-deoxy-6-methoxy-adenosine (33b). Following the general oxidation procedure, a solution of 30c (0.504 g, 1.00 mmol) in CH2Cl2 (15 mL) was reacted with DMP (0.425 g, 1.00 mmol). After work-up, the crude residue was purified by silica gel column chromatography (hexane:EtOAc 7:3) to afford compound 33b (0.500 g, 99%) as a white solid in quantitative yield. 1H-NMR (300 MHz, CDCl3): δ 8.40 (s, 1H, H-2), 7.96 (s, 1H, H-8), 7.62–7.29 (m, 10H, ArH), 5.92 (s, 1H, H-1′), 4.61 (dddd, 1H, J = 8.7, 6.7, 4.4, 3.9 Hz, H-4′), 4.18 (s, 3H, OCH3), 4.03 (dd, 1H, J = 11.3, 3.9 Hz, H-5′), 3.90 (dd, 1H, J = 11.3, 4.4 Hz, H-5″), 3.24 (dd, 1H, J = 18.7, 8.7 Hz, H-3′), 2.79 (dd, 1H, J = 18.7, 6.7 Hz, H-3″), 1.03 (s, 9H, 3 × CH3); 13C-NMR (75 MHz, CDCl3): δ 206.9 (2′-CO), 161.4 (6-C), 152.7 (2-C), 151.7 (4-C), 141.5 (8-C), 135.8 (ArC), 135.7 (ArC), 133.0 (ArC), 130.1 (ArC), 128.0 (ArC), 127.9 (ArC), 122.0.0 (5-CH), 82.3 (1′-CH), 76.5 (4′-CH), 65.1 (5′-CH2), 54.6 (OCH3), 37.5 (3′-CH), 29.9 (C(CH3)), 27.0 (3 × CH3); HRMS for C27H30N4O4Si1 [M + H+]+ Calc.: 503.2108, found: 503.2102.
5′-O-(tert-Butyldiphenylsilyl)-2′-keto-4-N-o-toluoyl-3′-deoxycytidine (33c). To a solution of 31a (0.701 g, 1.00 mmol) in dry THF (30 mL) at −78 °C under an inert atmosphere was added potassium tert-butoxide (0.112 g, 1.00 mmol), and the mixture was stirred for 1 h. It was then diluted with EtOAc (100 mL) and washed with water (100 mL) and brine (100 mL). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by silica gel column chromatography (EtOAc:hexane 1:1) to afford 5′-O-(tert-butyldiphenylsilyl)-4-N-o-toluoyl-3′-deoxycytidine as a colorless oil (0.466 g, 80%). 1H-NMR (300 MHz, CDCl3): δ 8.42 (d, 1H, J = 7.3 Hz, H-6), 7.66–7.26 (m, 14H, ArH), 5.77 (s, 1H, H-1′), 4.59 (d, 1H, J = 7.3 Hz, H-5), 4.59 (m, 1H, H-2′), 4.45 (br s, 1H, OH), 4.15–4.08 (m, 2H, H-4′, H-5′), 3.73 (d, 1H, J = 9.9 Hz, H-5″), 2.31–2.17 (m, 2H, H-3′,H-3″), 2.50 (s, 3H, CH3), 1.25 (s, 9H, 3 × CH3); 13C-NMR (75 MHz, CDCl3): δ 168.9 (CO), 162.7 (4-C), 156.2 (2-C), 144.8 (6-C), 137.6 (ArC), 135.8 (ArC), 135.7 (ArC), 135.7 (ArC), 134.4 (ArC), 133.0 (ArC), 132.8 (ArC), 131.9 (ArC), 131.7 (ArC), 130.3 (ArC), 128.2 (ArC), 127.4 (ArC), 126.3 (ArC), 96.5 (5-CH), 95.5 (1′-CH), 82.5 (4′-CH), 77.5 (2′-CH), 64.4 (5′-CH2), 32.7 (3′-CH2), 29.9 (C(CH3)3), 27.2 (3 × CH3), 20.3 (CH3); HRMS: C33H37N3O5Si [M + H+]+ Calc.: 584.2575, found: 584.2586. Following the general oxidation procedure, a solution of 5′-O-(tert-butyldiphenylsilyl)-4-N-o-toluoyl-3′-deoxycytidine (0.466 g, 0.80 mmol) in CH2Cl2 (15 mL) was reacted with DMP (0.425 g, 1.00 mmol). After work-up, the crude residue was purified by silica gel column chromatography (hexane:EtOAc 7:3) to afford compound 33c (0.46 g, 100%) as a white solid in quantitative yield. 1H-NMR (300 MHz, CDCl3): 7.66–7.26 (m, 16H, ArH, H-5, H-6), 5.40 (s, 1H, H-1′), 4.57–4.51 (m, 1H, H-4′), 3.99–3.97 (m, 2H, H-5′, H-5″), 3.03 (dd, 1H, J = 18.6, 8.1 Hz, H-3′), 2.63 (dd, 1H, J = 18.6, 7.6 Hz, H-3″), 2.51 (s, 3H, CH3), 1.25 (s, 9H, 3 × CH3); 13C-NMR (75 MHz, CDCl3): δ 206.0 (2′-CO), 163.6 (4-C), 154.6 (2-C), 148.2 (6-C), 137.9 (ArC), 135.8 (ArC), 135.8 (ArC), 133.3 (ArC), 132.0 (ArC), 131.9 (ArC), 130.1 (ArC), 128.0 (ArC), 128.0 (ArC), 127.4 (ArC), 126.4 (ArC), 117.7 (5-CH), 97.4 (1′-CH), 87.7 (4′-CH), 66.0 (5′-CH2), 37.0 (3′-CH2), 29.9 (C(CH3)3), 27.1 (3 × CH3), 20.4 (CH3); HRMS: C33H35N3O5Si [M + H+]+ Calc.: 582.2418, found: 582.2429.
5′-O-o-Toluoyl-2′-keto-6-N-o-toluoyl-3′-deoxyadenosine (33d). To a stirred suspension of 32 (0.302 g, 0.50 mmol) in dry THF (10 mL) at −78 °C under an inert atmosphere was added potassium tert-butoxide (0.112 g, 1.00 mmol) and the mixture was stirred for 1 h. It was then diluted with EtOAc (100 mL) and washed with water (100 mL) and brine (100 mL). After work-up, the crude residue was purified by silica gel column chromatography (hexane:EtOAc 1:1) to afford 5′-O-o-toluoyl-6-N-o-toluoyl-3′-deoxyadenosine as a colorless oil (0.133 g, 55%). 1H-NMR (600 MHz, CDCl3): δ 9.34 (s, 1H, H-2), 8.94 (s, 1H, H-8), 8.61–7.73 (m, 8H, ArH), 6.42 (d, 1H, J = 5.8 Hz, H-1′), 5.01 (m), 1H, H-4′), 4.75 (m, 1H, H-2′), 4.64 (m, 2H, H-5′, H-5″), 3.65–3.55 (m, 1H, H-3′), 3.43–3.37 (m, 1H, H-3″), 3.27 (s, 3H, ArCH3), 3.11 (s, 3H, ArCH3); 13C-NMR (75 MHz, CDCl3): δ 172.5 (NHCO), 166.8 (CO), 153.1 (6-C), 152.6 (2-C), 151.8 (4-C), 143.2 (8-C), 140.8 (ArC), 139.5 (ArC), 135.9 (ArC), 135.8 (ArC), 133.0 (ArC), 131.6 (ArC), 130.2 (ArC), 128.5 (ArC), 128.2 (ArC), 125.7 (5-CH), 85.8 (1′-CH), 84.8 (4′-CH), 75.1 (2′-CH), 64.2 (5′-CH2), 38.6 (3′-CH2), 27.2 (CH3), 20.2 (CH3). Following the general oxidation procedure, a solution of 5′-O-o-toluoyl-6-N-o-toluoyl-3′-deoxyadenosine (0.097 g, 0.20 mmol) in CH2Cl2 (10 mL) was reacted with DMP (0.106 g, 0.25 mmol). After work-up, the crude residue was purified by silica gel column chromatography (hexane:EtOAc 3:2) to afford compound 33d (0.077 g, 80%) as a white solid. 1H-NMR (300 MHz, CDCl3): δ 8.40 (s, 1H, H-2), 7.94 (s, 1H, H-8), 8.05–7.18 (m, 8H, ArH), 5.88 (s, 1H, H-1′), 4.89 (dddd, 1H, J = 9.2, 6.7, 5.4, 3.3 Hz, H-4′), 4.72 (dd, 1H, J = 12.0, 3.3 Hz, H-5′), 4.60 (dd, 1H, J = 12.1, 5.4 Hz, H-5″), 3.31 (dd, 1H, J = 18.5, 9.2 Hz, H-3′), 2.94 (dd, 1H, J = 18.6, 6.7 Hz, H-3″), 2.55 (s, 6H, CH3); 13C-NMR (75 MHz, CDCl3): δ 206.8 (2′-CO), 167.1 (CO), 161.5 (CO), 152.7 (6-C), 151.5 (2-C), 142.0 (4-C), 141.8 (8-C), 140.9 (ArC), 132.7 (ArC), 132.1 (ArC), 130.9 (ArC), 126.0 (ArC), 122.0 (5-CH), 82.4 (1′-CH), 74.2 (4′-CH), 65.2 (5′-CH2), 37.9 (3′-CH2), 21.9 (CH3), 21.8 (CH3).
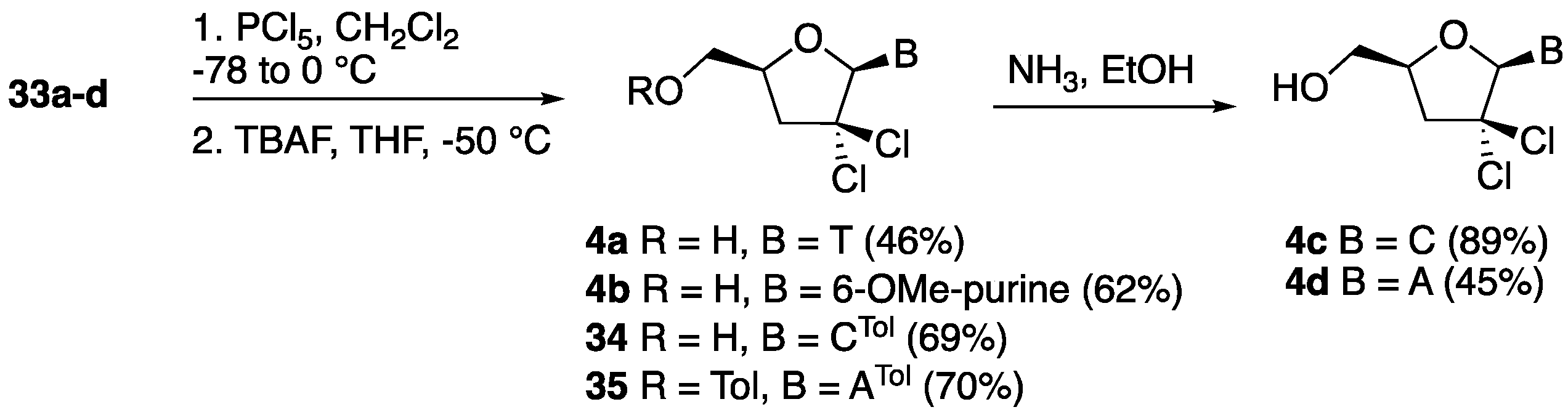
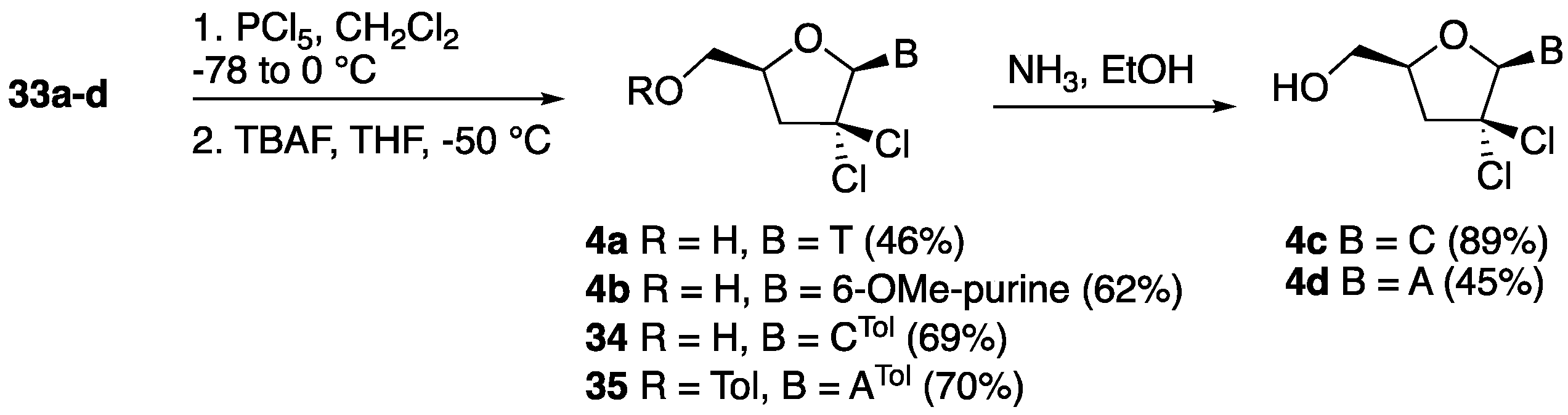
2′,2′-gem-Dichloro-3′-deoxythymidine (4a). Following the general chlorination procedure, a solution of compound 33a (0.500 g, 1.04 mmol) in dry CH2Cl2 (20 mL) was reacted with PCl5 (0.822 g, 3.95 mmol) at −78 °C under an inert atmosphere. After work-up, the resulting crude residue was purified by column chromatography (hexane:EtOAc 4:1) to give 5′-O-(tert-butyldiphenylsilyl)-2′,2′-gem-dichloro-3′-deoxythymidine (0.283 g, 51%) as a major compound and 5′-O-(tert-butyldiphenylsilyl)-2′-chloro-2′,3′-didehydro-3′-deoxythymidine as a minor side product (0.049 g, 10%). 1H-NMR (500 MHz, CDCl3): δ 8.91 (s, 1H, NH), 7.66–7.37 (m, 11H, ArH, H-6), 6.55 (s, 1H, H-1′), 4.37 (dddd, 1H, J = 12.4, 6.0, 3.2, 2.8 Hz, H-4′), 4.15 (dd, 1H, J = 14.3, 3.2 Hz, H-5′), 3.84 (dd, 1H, J = 14.3, 2.8 Hz, H-5″), 2.90 (dd, 1H, J = 16.7, 12.4 Hz, H-3′), 2.82 (dd, 1H, J = 16.7, 6.0 Hz, H-3″), 1.61 (s, 3H, CH3), 1.11 (s, 9H, 3 × CH3); 13C-NMR (125 MHz, CDCl3): δ 163.4 (4-C), 150.3 (2-C), 135.4 (6-C), 135.2 (ArC), 134.2 (ArC), 132.7 (ArC), 132.3 (ArC), 130.1 (ArC), 131.1 (ArC), 128.0 (ArC), 111.0 (5-CH), 92.8 (1′-CH), 89.2 (2′-C(Cl)2), 78.5 (4′-CH), 62.8 (5′-CH2), 47.2 (3′-CH), 29.6 (C(CH3)), 26.9 (3 × CH3) 12.0 (6-CH3); HRMS: C26H30N2O4SiCl2 [M + H+]+ Calc.: 533.1424, found: 533.1428. Data for 5′-O-(tert-butyldiphenylsilyl)-2′-chloro-2′,3′-didehydro-3′-deoxythymidine: 1H-NMR (300 MHz, CDCl3): δ 8.23 (s, 1H, NH), 7.66–7.60 (m, 4H, ArH), 7.46–7.344 (m, 6H, ArH), 7.06 (d, 1H, J = 1.3 Hz, H-6), 6.89 (dd, 1H, J = 3.8, 1.7 Hz, H-1′), 6.30 (t, 1H, J = 1.7 Hz, H-3′), 4.95–4.93 (m, 1H, H-4′), 3.90–3.89 (m, 2H, H-5′, H-5″),1.46 (d, 3H, J = 1.3 Hz, CH3), 1.09 (s, 9H, 3 × CH3);HRMS: C26H29N2O4SiCl [M + Na+]+ Calc.: 519.1477, found: 519.1477. Following the general desilylation procedure, a solution of 5′-O-(tert-butyldiphenylsilyl)-2′,2′-gem-dichloro-3′-deoxythymidine (0.266 g, 0.50 mmol) in dry THF (10 mL) was reacted with TBAF (0.75 mL, 0.75 mmol). After work-up, the resulting crude residue was purified by column chromatography (EtOAc) to give 4a as a white solid (0.132 g, 90%). 1H-NMR (600 MHz, MeOD): δ 8.18 (d, 1H, J = 2.4 Hz, H-6), 6.51 (s, 1H, H-1′), 4.44 (ddt, 1H, J = 10.8, 5.5, 2.4, Hz, H-4′), 4.06 (dd, 1H, J = 12.8, 2.4 Hz, H-5′), 3.76 (dd, 1H, J = 12.8, 2.4 Hz, H-5″), 2.86–2.85 (m, 2H, H-3′, H-3″), 1.88 (d, 3H, J = 2.3 Hz, CH3); 13C-NMR (150 MHz, MeOD): δ 166.1 (4-C), 152.5 (2-C), 136.8 (6-C), 111.4 (5-CH), 93.9 (1′-CH), 91.2 (2′-C(Cl)2), 81.6 (4′-CH), 61.1 (5′-CH2), 46.4 (3′-CH), 12.4 (6-CH3); HRMS: C10H12Cl2N2O4 [M + H+]+ Calc.: 295.0246, found: 295.0244.
2′,2′-gem-Dichloro-2′,3′-dideoxy-6-methoxy-adenosine (4b). Following the general chlorination procedure, a solution of compound 33b (0.500 g, 1.00 mmol) in dry CH2Cl2 (20 mL) was reacted with PCl5 (0.790 g, 3.80 mmol) at −78 °C under an inert atmosphere. After work-up, the resulting crude residue was purified by column chromatography (hexane:EtOAc 4:1) to give 5′-O-(tert-butyldiphenylsilyl)-2′,2′-gem-dichloro-2′,3′-dideoxy-6-methoxy-adenosine (0.359 g, 65%) as a pale yellow solid. 1H-NMR (300 MHz, CDCl3): δ 8.55 (s, 1H, H-2), 8.47 (s, 1H, H-8), 7.70–7.38 (m, 10H, ArH), 6.67 (s, 1H, H-1′), 4.55 (dddd, 1H, J = 10.0, 5.3, 4.8, 3.2 Hz, H-4′), 4.20 (s, 3H, OCH3), 4.13 (dd, 1H, J = 11.9, 3.2 Hz, H-5′), 3.87 (dd, 1H, J = 8.5, 4.8 Hz, H-5″), 3.24 (dd, 1H, J = 13.9, 10.0 Hz, H-3′), 2.86 (dd, 1H, J = 13.9, 5.3 Hz, H-3″), 1.13 (s, 9H, 3 × CH3); 13C-NMR (75 MHz, CDCl3): δ 161.4 (6-C), 152.7 (2-C), 149.2 (4-C), 140.2 (8-C), 135.8 (ArC), 135.7 (ArC), 132.6 (ArC), 130.3 (ArC), 128.2 (ArC), 128.0 (ArC), 121.9 (5-CH), 93.0 (1′-CH), 89.0 (2′-C(Cl)2), 80.0 (4′-CH), 63.7 (5′-CH2), 54.5 (OCH3), 45.9 (3′-CH), 29.9 (C(CH3)), 27.0 (3 × CH3); HRMS: C27H30N4O3SiCl2 [M + H+]+ Calc.: 557.1536, found: 557.1219. Following the general desilylation procedure, a solution of 5′-O-(tert-butyldiphenylsilyl)-2′,2′-gem-dichloro-2′,3′-dideoxy-6-methoxy-adenosine (0.278 g, 0.50 mmol) in dry THF (10 mL) was reacted with TBAF (0.75 mL, 0.75 mmol). After work-up, the resulting crude residue was purified by column chromatography (EtOAc) to give compound 4b as a white solid (151 mg, 95%). 1H-NMR (600 MHz, MeOD): δ 8.95 (s, 1H, H-2), 8.58 (s, 1H, H-8), 6.77 (s, 1H, H-1′), 4.62 (dddd, 1H, J = 10.5, 5.1, 2.9, 2.6 Hz, H-4′), 4.21 (s, 3H, OCH3), 4.09 (dd, 1H, J = 12.6, 2.6 Hz, H-5′), 3.87 (dd, 1H, J = 12.6, 2.9 Hz, H-5″), 3.20 (dd, 1H, J = 14.1, 10.5 Hz, H-3′), 2.98 (dd, 1H, J = 14.1, 5.1 Hz, H-3″); 13C-NMR (150 MHz, MeOD): δ 162.4 (6-C), 153.7 (2-C), 153.1 (4-C), 142.4 (8-C), 121.8 (5-CH), 94.2 (1′-CH), 90.2 (2′-C(Cl)2), 82.4 (4′-CH), 61.7 (5′-CH2), 54.9 (OCH3), 45.7 (3′-CH); HRMS: C11H12Cl2N4O3 [M + H+]+ Calc.: 319.0359, found: 319.0361.
2′,2′-gem-Dichloro-4-N-o-toluoyl-2′,3′-dideoxycytidine (34). Following the general chlorination procedure, a solution of compound 33c (0.46 g, 0.80 mmol) in dry CH2Cl2 (20 mL) was reacted with PCl5 (0.632 g, 3.04 mmol) at −78 °C under an inert atmosphere. After work-up, the resulting crude residue was purified by column chromatography (hexane:EtOAc 4:1) to give 5′-O-(tert-butyldiphenylsilyl)-2′,2′-gem-dichloro-4-N-o-toluoyl-2′,3′-dideoxycytidine (0.350 g, 69%) as a pale yellow solid. 1H-NMR (300 MHz, CDCl3): 8.40 (d, 1H, J = 7.5 Hz, H-6), 7.68–7.26 (m, 15H, ArH, H-5), 6.67 (s, 1H, H-1′), 4.45 (ddt, 1H, J = 10.8, 4.7, 2.3 Hz, H-4′), 4.24 (dd, 1H, J = 12.2, 2.3 Hz, H-5′), 3.80 (dd, 1H, J = 12.2, 2.3 Hz, H-5″), 2.94 (dd, 1H, J = 13.4, 10.8 Hz, H-3′), 2.76 (dd, 1H, J = 13.4, 4.7 Hz, H-3″), 2.52 (s, 3H, CH3), 1.15 (s, 9H, 3 × CH3); 13C-NMR (75 MHz, CDCl3): δ 162.7 (4-C), 155.3 (2-C), 144.1 (6-C), 137.7 (ArC), 135.8 (ArC), 135.6 (ArC), 134.3 (ArC), 132.6 (ArC), 132.3 (ArC), 132.0 (ArC), 131.8 (ArC), 130.6 (ArC), 130.5 (ArC), 128.4 (ArC), 128.3 (ArC), 127.2 (ArC), 126.4 (ArC), 96.8 (5-CH), 93.4 (1′-CH), 89.0 (2′-C(Cl)2), 79.7 (4′-CH), 62.7 (5′-CH2), 46.0 (3′-CH2), 29.5 (C(CH3)3), 27.2 (3 × CH3), 20.4 (CH3); HRMS: C33H35Cl2N3O4Si [M + H+]+ Calc.: 636.1846, found: 636.1848. Following the general desilylation procedure, a solution of 5′-O-(tert-butyldiphenylsilyl)-2′,2′-gem-dichloro-4-N-o-toluoyl-2′,3′-dideoxycytidine (0.350 g, 0.55 mmol) in dry THF (10 mL) was reacted with TBAF (0.82 mL, 0.82 mmol). After work-up, the resulting crude residue was purified by column chromatography (EtOAc) to give compound 34 as a white solid (0.217 g, 100%). 1H-NMR (600 MHz, MeOD): 8.76 (d, 1H, J = 7.5 Hz, H-6), 7.59 (d, 1H, J = 7.5 Hz, H-5), 7.53–7.29 (m, 4H, ArH), 6.67 (s, 1H, H-1′), 4.53 (ddt, 1H, J = 9.7, 6.2, 2.5, Hz, H-4′), 4.06 (dd, 1H, J = 12.8, 2.5 Hz, H-5′), 3.79 (dd, 1H, J = 12.8, 2.5 Hz, H-5″), 2.88–2.86 (m, 2H, H-3′, H-3′), 2.46 (s, 3H, CH3); 13C-NMR (150 MHz, MeOD): δ 171.7 (CO), 165.0 (4-C), 158.0 (2-C), 145.7 (6-C), 138.0 (ArC), 136.2 (ArC), 132.2 (ArC), 132.2 (ArC), 128.6 (ArC), 126.9 (ArC), 98.3 (5-CH), 94.8 (1′-CH), 90.6 (2′-C(Cl)2), 82.0 (4′-CH), 61.1 (5′-CH2), 46.4 (3′-CH2), 19.9 (CH3); HRMS: C17H17Cl2N3O4 [M + H+]+ Calc.: 398.0668, found: 398.0665.
5′-O-o-Toluoyl-2′,2′-gem-dichloro-6-N-o-toluoyl-2′,3′-dideoxyadenosine (35). Following the general chlorination procedure, a solution of compound 33d (0.048 g, 0.10 mmol) in dry CH2Cl2 (5 mL) was reacted with PCl5 (0.079 g, 0.38 mmol) at −78 °C under an inert atmosphere. After work-up, the resulting crude residue was purified by column chromatography (hexane:EtOAc 7.5:2.5) to give compound 35 (0.037 g, 70%) as a pale yellow solid. 1H-NMR (600 MHz, MeOD): δ 9.08 (s, 1H, H-2), 8.76 (s, 1H, H-8), 7.67–7.08 (m, 8H, ArH), 6.76 (s, 1H, H-1′), 4.60 (dddd, 1H, J = 10.5, 5.2, 3.0, 2.7 Hz, H-4′), 4.06 (dd, 1H, J = 12.6, 2.7 Hz, H-5′), 3.84 (dd, 1H, J = 12.6, 3.0 Hz, H-5″), 3.14 (dd, 1H, J = 14.2, 10.5 Hz, H-3′), 2.95 (dd, 1H, J = 14.2, 5.2 Hz, H-3″), 2.45 (s, 6H, 2 × CH3); 13C-NMR (150 MHz, MeOD): δ 173.5 (NHCO), 170.2 (CO), 154.6 (6-C), 153.5 (2-C), 152.5 (4-C), 145.5 (8-C), 140.1 (ArC), 136.1 (ArC), 132.6 (ArC), 132.3 (ArC), 129.9 (ArC), 129.1 (ArC), 126.5 (ArC), 126.5 (5-CH), 94.2 (1′-CH), 90.6 (2′- C(Cl)2), 82.5 (4′-CH), 61.5 (5′-CH2), 45.7 (3′-CH2), 20.1 (CH3), 14.4 (CH3); HRMS: C26H23Cl2N5O4 [M + H+]+ Calc.: 540.1199, found: 540.1211.
2′,2′-gem-Dichloro-2′,3′-dideoxycytidine (4c). To a stirred solution of 34 (0.050 g, 0.125 mmol) in EtOH (3.0 mL) at −20 °C was added a saturated solution of NH3 in EtOH. The reaction mixture was then stirred for 2 days at room temperature. After removal of all the volatiles in vacuo, the crude residue was purified by silica gel column chromatography (EtOAc:MeOH 7.5:2.5) to give 4c as a white solid (0.031 g, 89%). HRMS: 1H-NMR (600 MHz, MeOD): 8.21 (d, 1H, J = 7.6 Hz, H-6), 6.63 (s, 1H, H-1′), 5.91 (d, 1H, J = 7.6 Hz, H-5), 4.43 (ddt, 1H, J = 10.4, 5.1, 2.6 Hz, H-4′), 4.02 (dd, 1H, J = 12.8, 2.6 Hz, H-5′), 3.76 (dd, 1H, J = 12.7, 2.7 Hz, H-5″), 2.84 (dd, 1H, J = 13.7, 10.4 Hz, H-3′), 2.79 (dd, 1H, J = 13.8, 5.1 Hz, H-3′); 13C-NMR (150 MHz, MeOD): δ 167.6 (4-C), 158.2 (2-C), 141.7 (6-C), 96.1 (5-CH), 94.5 (1′-CH), 91.2 (2′-(CCl)2), 81.2 (4′-CH), 61.3 (5′-CH2), 46.7 (3′-CH2); C9H11Cl2N3O3 [M + H+]+ Calc.: 280.0250, found: 280.0250.
2′,2′-gem-Dichloro-2′,3′-dideoxyadenosine (4d). To a stirred solution of 35 (0.037 g, 0.068 mmol) in dry MeOH (5.0 mL) was added K2CO3 (0.138 g, 0.10 mmol), and then the reaction mixture was stirred for 48 h. After removal of all the volatiles, the crude residue was purified by silica gel column chromatography (MeOH:CHCl3 1.5:8.5 to 2.5:7.5) to give 4d as a white solid (0.0092 mg, 45%). 1H-NMR (300 MHz, DMSO-d6): δ 8.88 (s, 1H, H-2), 8.62 (s, 1H, H-8), 6.70 (s, 1H, H-1′), 5.52 (s, 1H, OH), 4.51 (dddd, 1H, J = 8.7, 7.7, 3.9, 3.2, H-4′), 3.92 (dd, 1H, J = 12.5, 8.7 Hz, H-5′), 3.76 (dd, 1H, J = 12.5, 3.2 Hz, H-5′), 3.17 (dd, 1H, J = 11.1, 7.7 Hz, H-3′), 3.06 (dd, 1H, J = 11.1, 3.9 Hz, H-3″); 13C-NMR (75 MHz, DMSO-d6): δ 160.6 (6-C), 152.3 (2-C), 151.9 (4-C), 141.0 (8-C), 120.7 (5-CH), 92.1 (1′-CH), 89.8 (2′-C(CCl)2), 81.0 (4′-CH), 60.5 (5′-CH2), 44.4 (3′-CH2); HRMS: C10H11Cl2N5O2 [M + NH4+]+ Calc.: 321.0395, found: 321.0328.



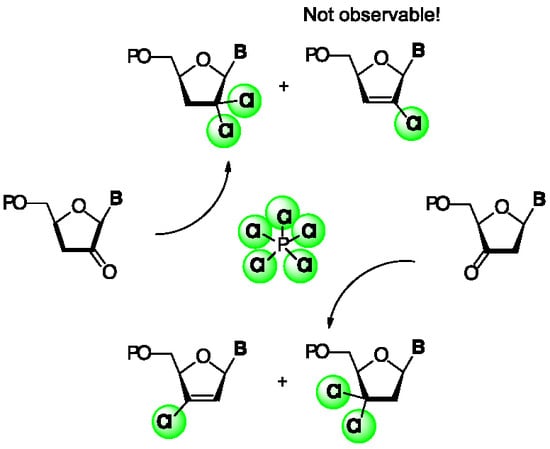
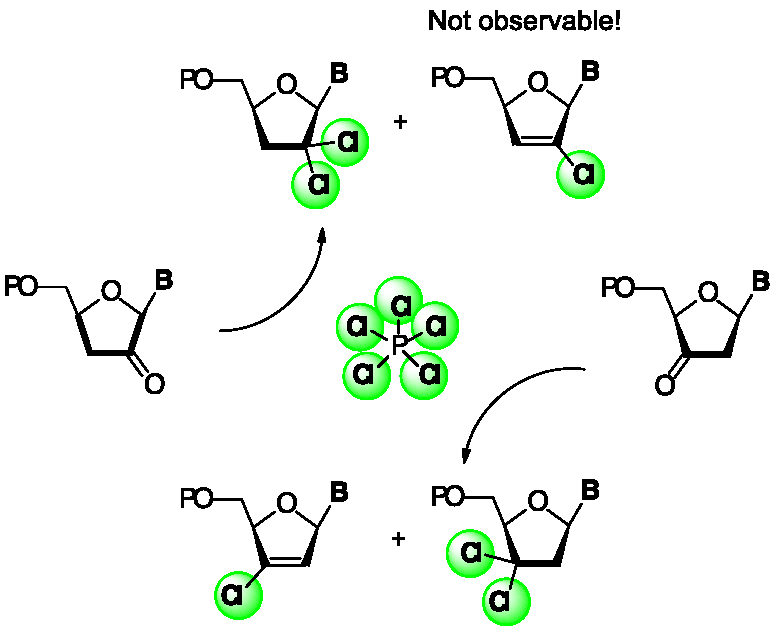{kind=link}
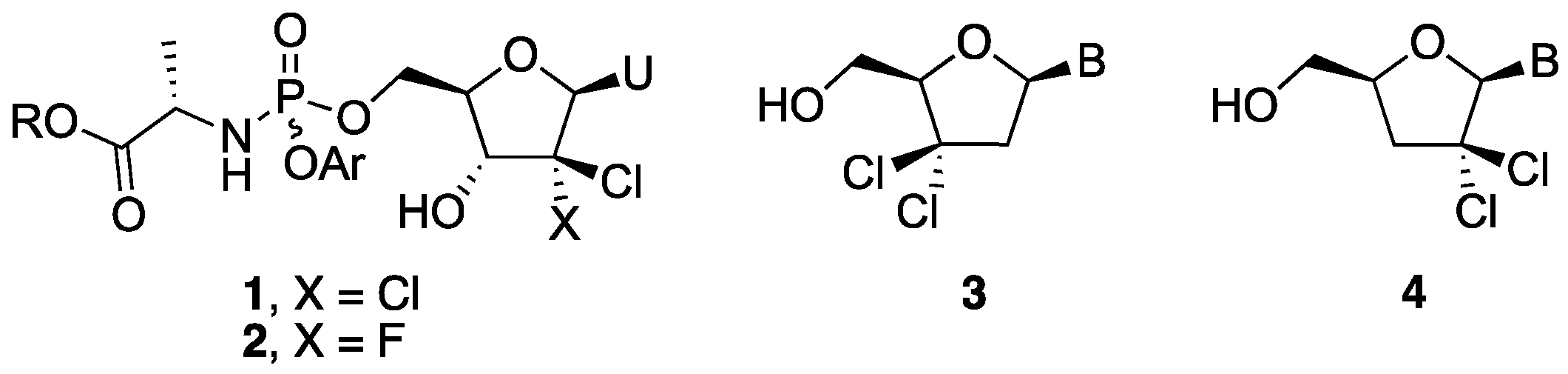{kind=link}
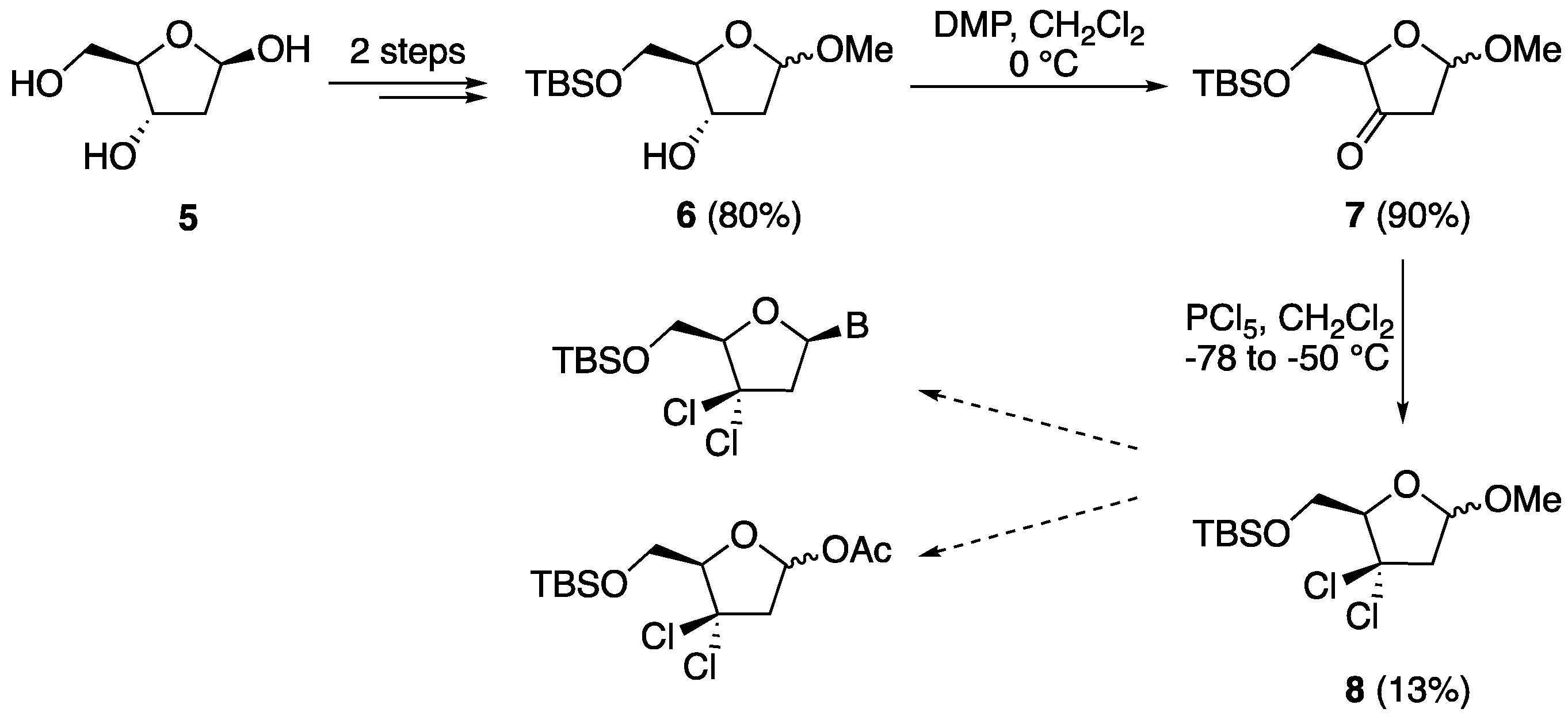{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}