Theoretical Study of the Photolysis Mechanisms of Methylpentaphenyldimetallanes (Ph3MM′Ph2Me; M, M′ = Si and Ge)


Abstract
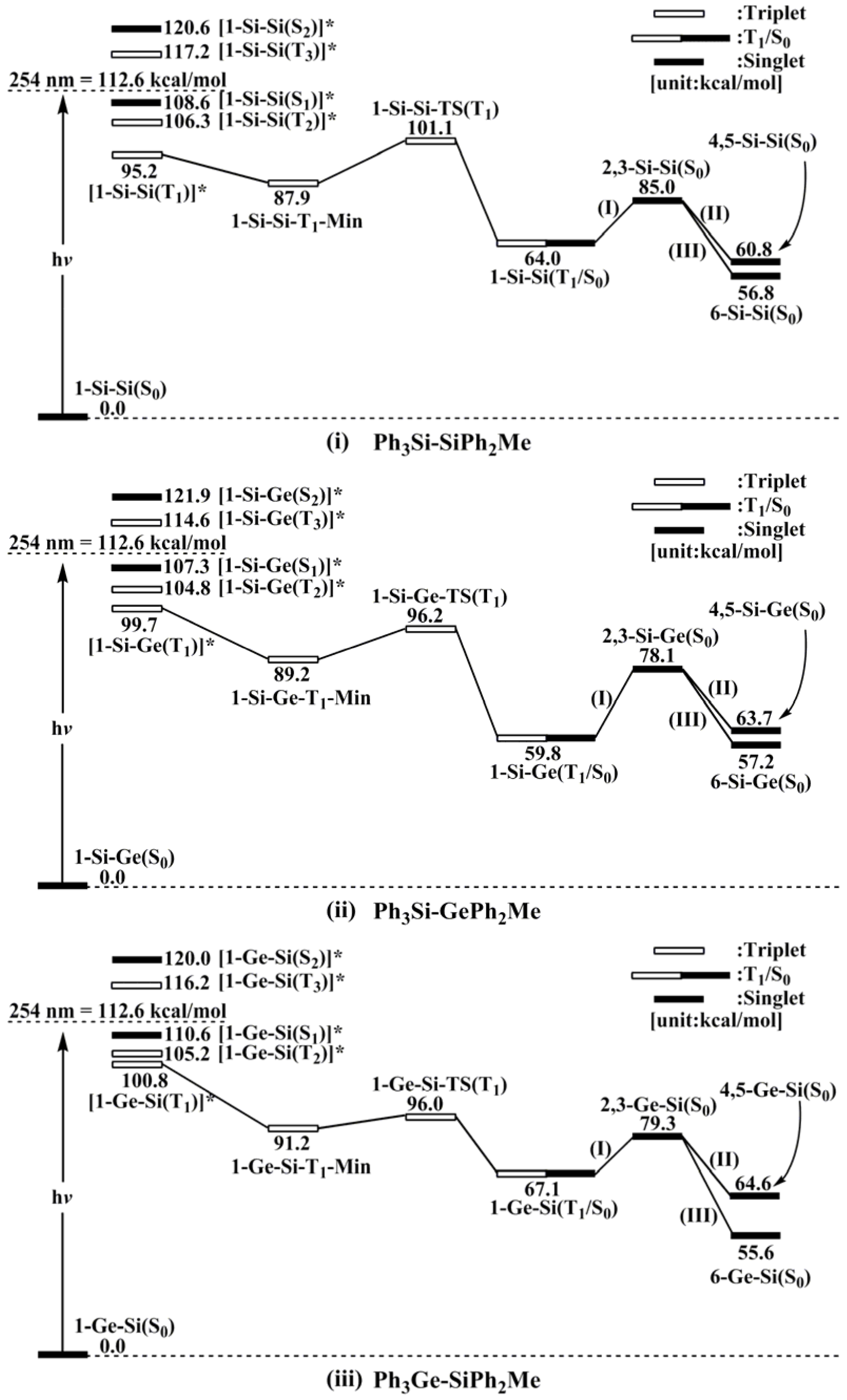
- (a)
- For 1-Si-Si: [1-Si-Si(T1)]* (95.2) < [1-Si-Si(T2)]* (106.3) < [1-Si-Si(S1)]* (108.6) < [1-Si-Si(T3)]* (117.2) < [1-Si-Si(S2)]* (120.6).
- (b)
- For 1-Si-Ge: [1-Si-Ge(T1)]* (99.7) < [1-Si-Ge(T2)]* (104.8) < [1-Si-Ge(S1)]* (107.3) < [1-Si-Ge(T3)]* (114.6) < [1-Si-Ge(S2)]* (121.9).
- (c)
- For 1-Ge-Si: [1-Ge-Si(T1)]* (100.8) < [1-Ge-Si(T2)]* (105.2) < [1-Ge-Si(S1)]* (110.6) < [1-Ge-Si(T3)]* (116.2) < [1-Ge-Si(S2)]* (120.0).
- (d)
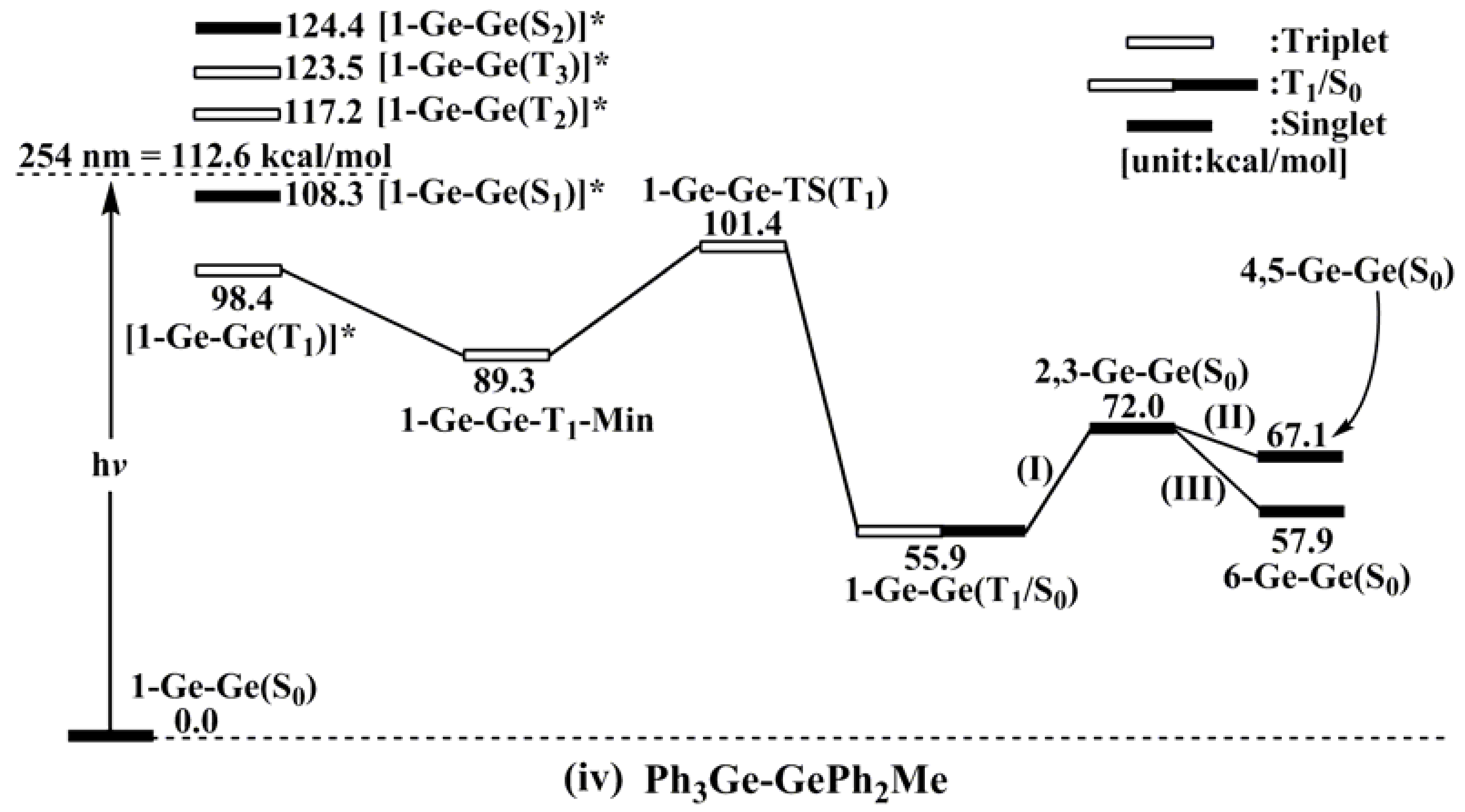
- For 1-Ge-Ge: [1-Ge-Ge(T1)]* (98.4) < [1-Ge-Ge(S1)]* (108.3) < [1-Ge-Ge(T2)]* (117.2) < [1-Ge-Ge(T3)]* (123.5) < [1-Ge-Ge(S2)]* (124.4).
- (1)
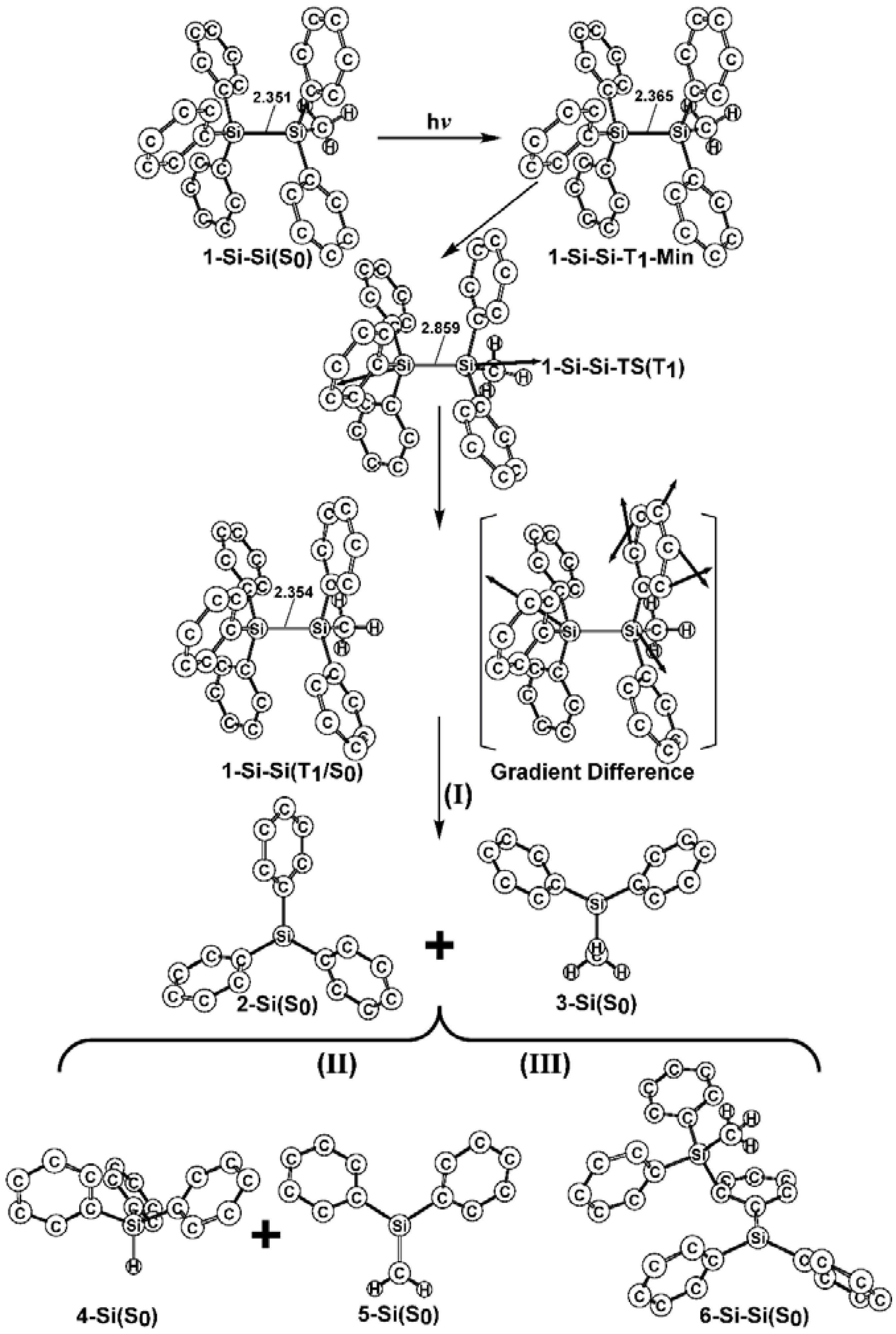
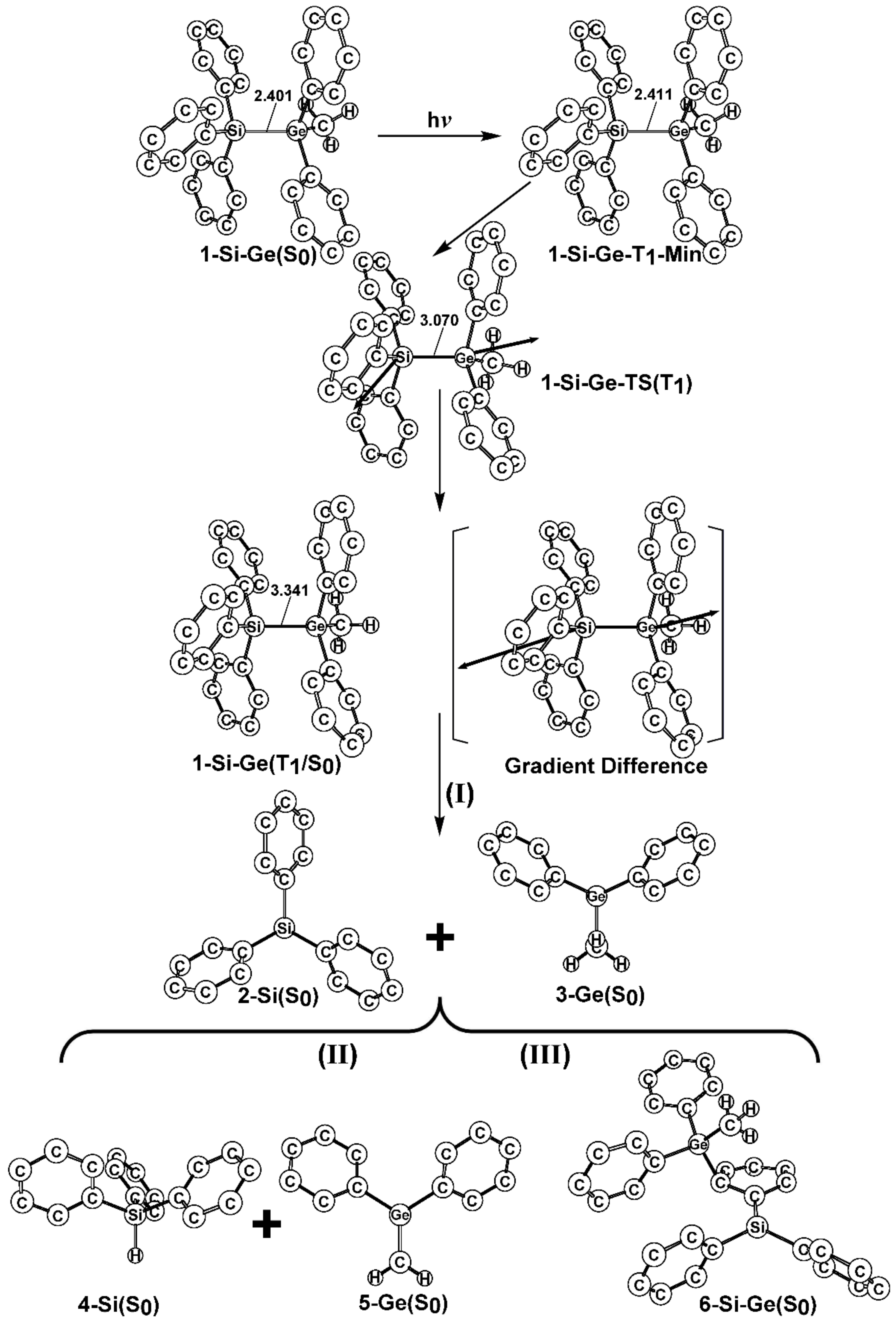
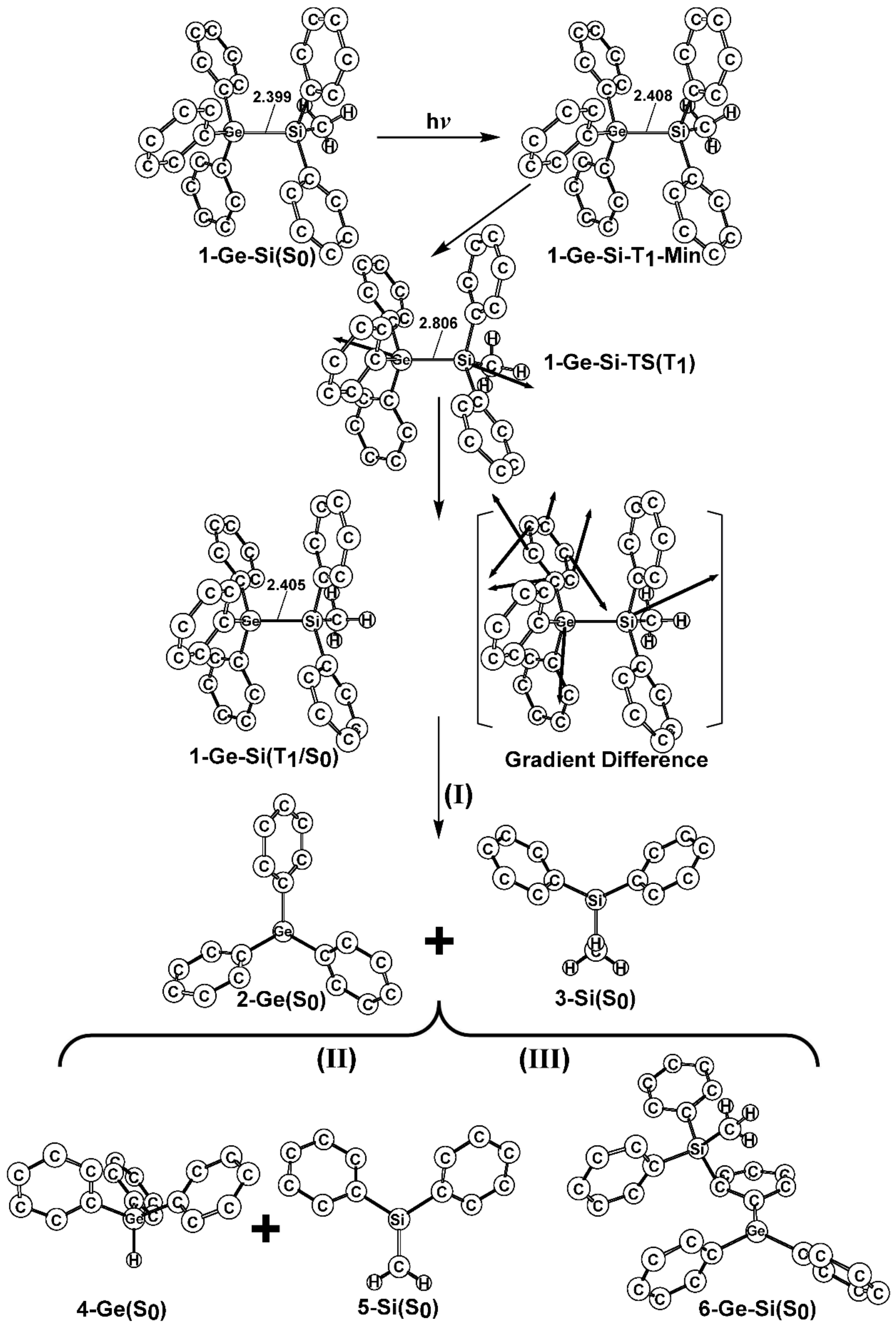
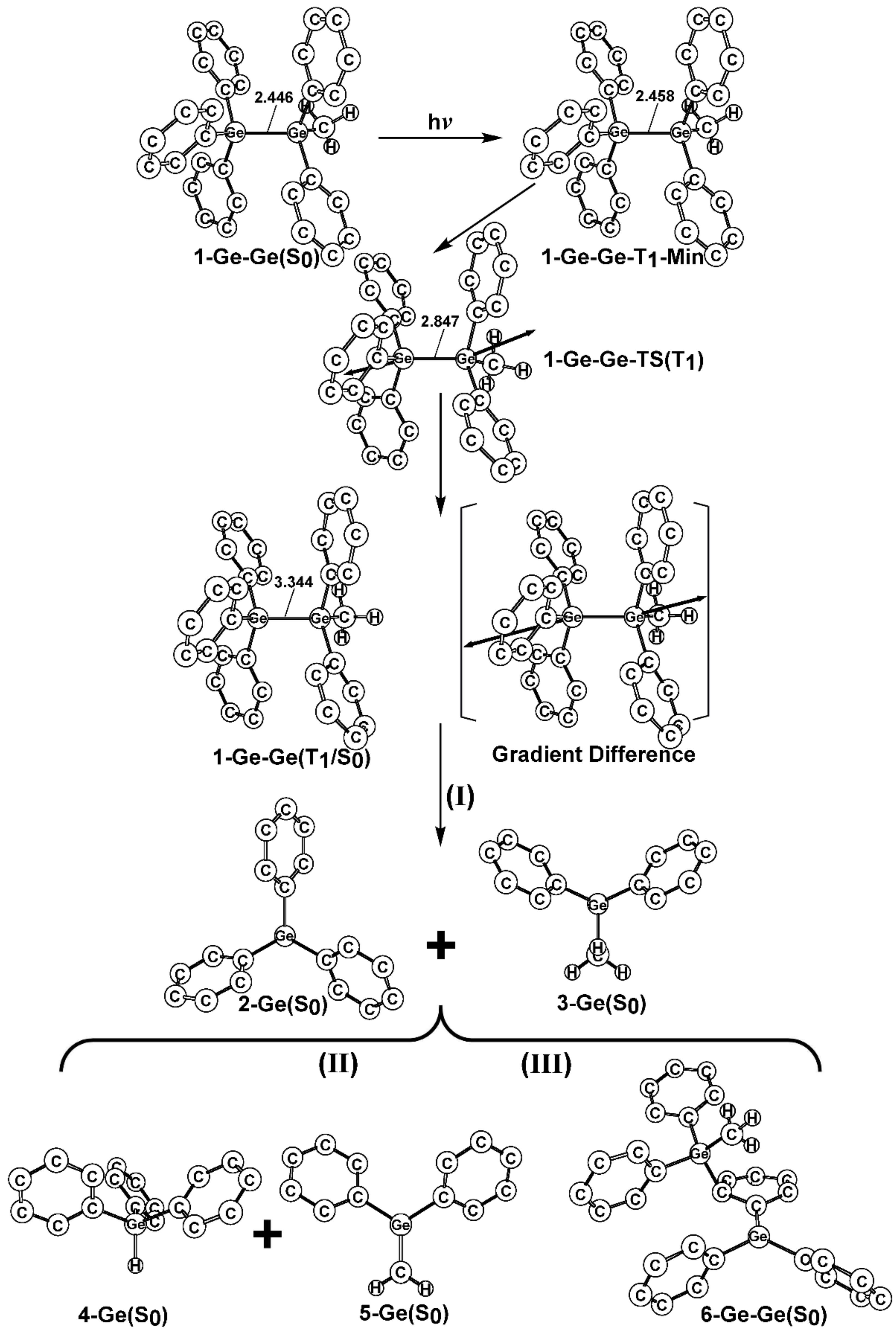
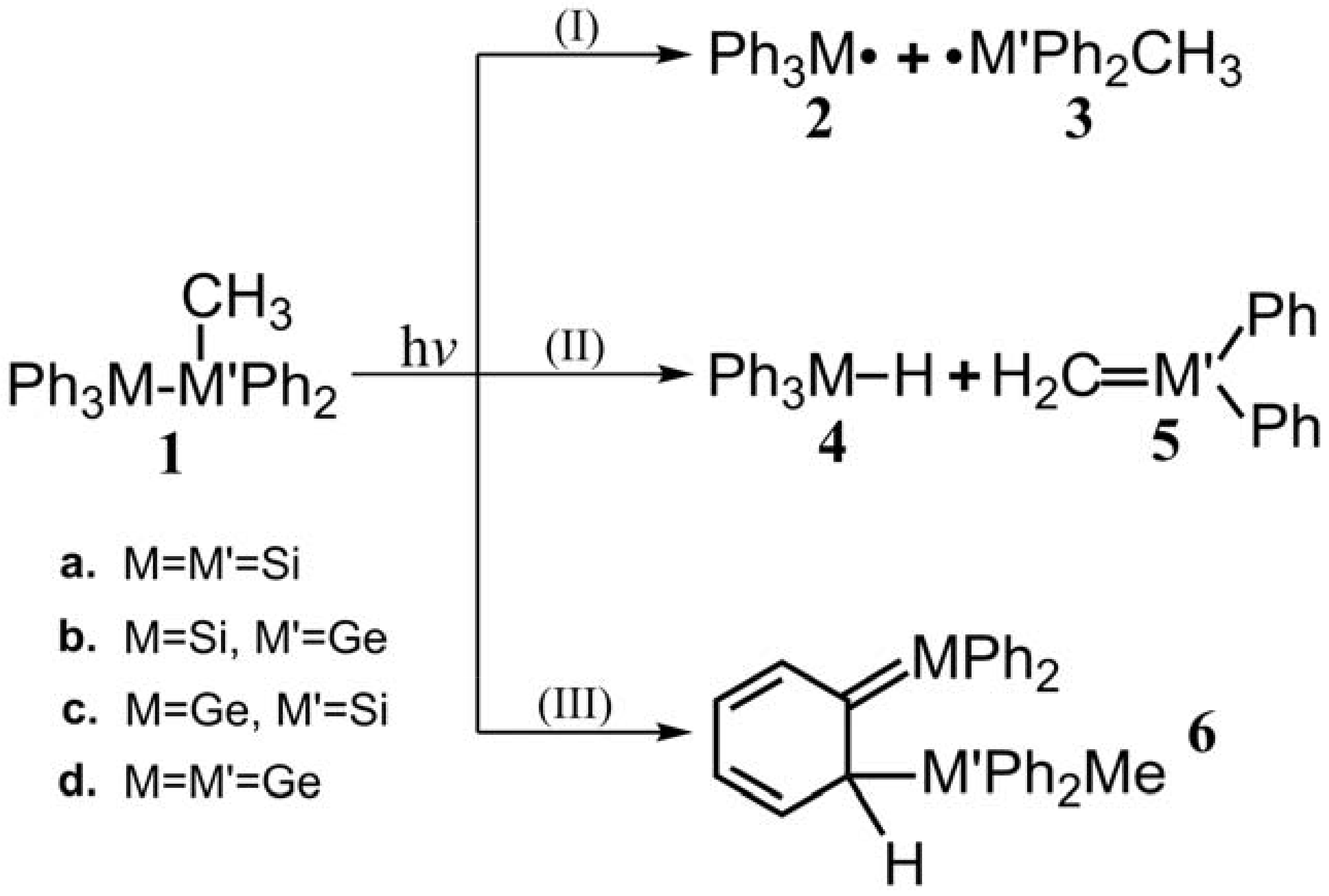
- The theoretical studies using the M06-2X/6-311G(d) level of theory show that after the direct irradiation of aryldisilanes, arylgermasilanes, and aryldigermanes of the general structure, Ph3M–M′Ph2Me (1), they occupy the lowest excited triplet state and then proceed to M–M′ bond homolysis via the intersystem crossing, to acquire two doublet radicals, 2 and 3 (overall singlet). After the subsequent dispropotionation and recombination procedures, they finally generate a mixture of singlet photoproducts (4–6). It should be emphasized that this is the first theoretical verification that radicals are formed as a result of reactions of the triplet states of Ph3M–M′Ph2Me (1).
- (2)
- Figure 1 clearly shows that the reactants 1 have an excess of energy, because of the vertical excitation to the Frank-Condon point at the triplet state (1-T1)*, which is larger than the subsequent activation barriers. That is to say, when Ph3M–M′Ph2Me (1) absorbs light, this molecule has sufficient internal energy to overcome the energy barriers to produce various types of photoproducts (2–6) without any difficulty. This theoretical finding for the 1-Si-Si molecule is in good agreement with the experimental evidence [24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41]. Since there are no pertinent experimental or theoretical reports on the photolysis reactions of the compounds, 1-Si-Ge, 1-Ge-Si, and 1-Ge-Ge, this result is a prediction.
- (3)
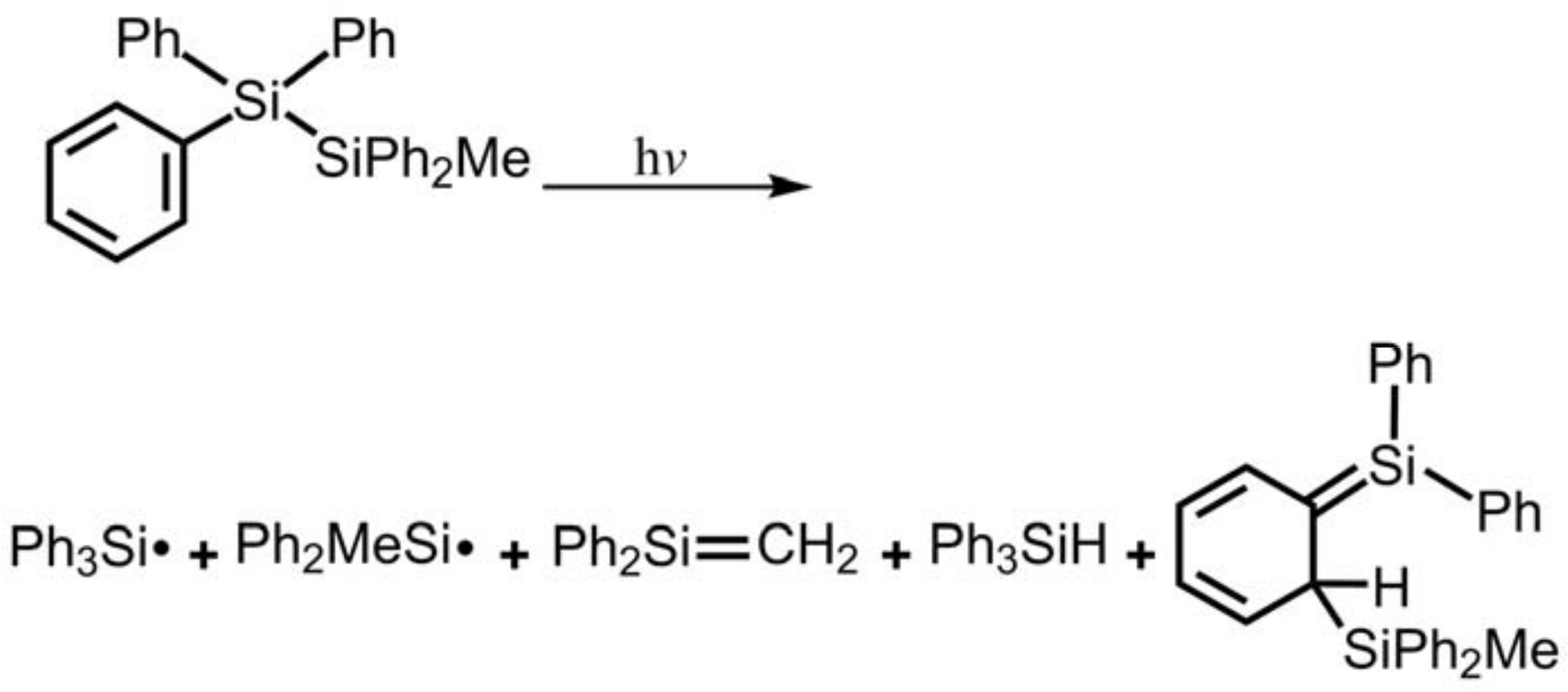
- The theoretical observations that are shown in Figure 1 indicate that the relative energies of two monoradical species (Ph3M·and·M′Ph2Me) are higher than those of the other singlet photoproducts (4–6). The quantum yields of the former are estimated to be lower than those of the latter. This prediction is again consistent with the available experimental observations for the 1-Si-Si molecule, in which it is reported that the formation of the corresponding silyl radicals (2 and 3), 1,1-diphenylsilene (5), and the silatriene (6) give respective yields of ca. 20%, 35%, and 42% [30]. Therefore, in the photolysis reactions for compounds 1-Si-Ge, 1-Ge-Si and 1-Ge-Ge, the quantum yields for their singlet photoproducts 4–6 should be larger than those of the other two doublet radicals, 2 and 3 [50].

- (4)
- The theoretical conclusions shown in Figure 1 also apply to other group 14 dimetallane analogues, such as 1,1,1-trimethyl-2,2,2-triphenyldimetallanes (7; Ph3M–M′Me3; M and M′ = Si and Ge) and 1,1,1,2,2-pentamethyl-2-phenyldimetallanes (8; PhMe2M–M′Me3; M and M′ = Si and Ge). That is to say, the direct irradiation of these compounds produces the triplet species, which then mainly results in M–M′ bond homolysis to generate the formation of a radical and other singlet photoproducts (similar to 4–6), via radical disproportionation and recombination. Indeed, some earlier experimental studies of 7d by Mochida and Hayashi [51] and a related those for series of molecules (8b–8d), as reported by Mochida and Gaspar [52,53] and their co-workers, show similar results that are in reasonable agreement with these theoretical predictions.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Author Contributions
Acknowledgments
Conflicts of Interest
References and Notes
- Steinmetz, M.G. Organosilane Photochemistry. Chem. Rev. 1995, 95, 1527–1588. [Google Scholar] [CrossRef]
- Brook, A.G.; Brook, M.A. The Chemistry of Silenes. Adv. Organomet. Chem. 1996, 39, 71–158. [Google Scholar]
- Brook, A.G. The Chemistry of Organic Silicon Compounds; Rappoport, Z., Apeloig, Y., Eds.; John Wiley and Sons: New York, NY, USA, 1998; pp. 1233–1310. [Google Scholar]
- Kira, M.; Miyazawa, T. The Chemistry of Organic Silicon Compounds; Rappoport, Z., Apeloig, Y., Eds.; John Wiley & Sons: New York, NY, USA, 1998; pp. 1311–1337. [Google Scholar]
- Ohshita, J.; Niwa, H.; Ishikawa, M. Silicon−Carbon Unsaturated Compounds. 59. Stereochemistry in Addition of Carbonyl Compounds to Silenes Generated Photochemically from meso- and rac-1,2-Diethyl-1,2-dimethyldiphenyldisilane. Organometallics 1996, 15, 4632–4638. [Google Scholar] [CrossRef]
- Ohshita, J.; Niwa, H.; Ishikawa, M.; Yamabe, T.; Yoshii, T.; Nakamura, K. Silicon−Carbon Unsaturated Compounds. 57. Photolysis of meso- and racemic-1,2-Diethyl-1,2-dimethyldiphenyldisilane, Direct Evidence for a Concerted 1,3-Silyl Shift to ortho-Carbon in the Phenyl Ring. J. Am. Chem. Soc. 1996, 118, 6853–6859. [Google Scholar] [CrossRef]
- Ishikawa, M.; Fuchikami, T.; Sugaya, T.; Kumada, M. Photolysis of Organopolysilanes. Novel Addition Reaction of Aryl Substituted Disilanes to Olefins. J. Am. Chem. Soc. 1975, 97, 5923–5924. [Google Scholar] [CrossRef]
- Ishikawa, M.; Fuchikami, T.; Kumada, M. Photochemically Generated Silicon-carbon Double-bonded Intermidiates: II. Photolysis of Aryldisilanes in the Presence of Dienes. J. Organomet. Chem. 1976, 118, 139–153. [Google Scholar] [CrossRef]
- Ishikawa, M.; Fuchikami, T.; Kumada, M. Photochemically Generated Silicon—Carbon Double-bonded Intermediates: V. Photolysis of Arylpentamethyldisilanes in the Presence of Carbonyl Compounds. J. Organomet. Chem. 1977, 133, 19–28. [Google Scholar] [CrossRef]
- Sluggett, G.W.; Leigh, W.J. Photochemistry of 1,2-Di-tert-butyl-1,1,2,2-tetraphenyldisilane, a Clean, Direct Source of Arylalkylsilyl Radicals. Organometallics 1992, 11, 3731–3736. [Google Scholar] [CrossRef]
- Miller, R.D.; Michl, J. Polysilane High Polymers. Chem. Rev. 1989, 89, 1359–1410. [Google Scholar] [CrossRef]
- West, R. The Chemistry of Organic Silicon Compounds; Patai, S., Rappoport, Z., Eds.; John Wiley & Sons: New York, NY, USA, 1989; pp. 1207–1240. [Google Scholar]
- Michl, J. Solution Photophysics and Electronic Structure of Polysilanes. Synthet. Met. 1992, 50, 367–386. [Google Scholar] [CrossRef]
- Mazieres, S.; Raymond, M.K.; Raabe, G.; Prodi, A.; Michl, J. [2]Staffane Rod as a Molecular Rack for Unraveling Conformer Properties: Proposed Singlet Excitation Localization Isomerism in anti,anti,anti-Hexasilanes. J. Am. Chem. Soc. 1997, 119, 6682–6683. [Google Scholar] [CrossRef]
- Kira, M.; Sakamoto, K.; Sakurai, H. Photochemistry of Dibenzo-1,1,2,2-tetramethyl-1,2-disilacyclohexa-3,5-diene and the Germanium Analog. Exclusive Extrusion of the Divalent Species. J. Am. Chem. Soc. 1983, 105, 7469–7470. [Google Scholar] [CrossRef]
- Leigh, W.J.; Sluggett, G.W. Aryldisilane Photochemistry. A Kinetic and Product Study of the Mechanism of Alcohol Additions to Transient Silenes. J. Am. Chem. Soc. 1994, 116, 10468–10476. [Google Scholar] [CrossRef]
- Toltl, N.P.; Leigh, W.J. Synthesis and isolation of stable 1,2-siloxetanes from reaction of transient silenes with acetone. Organometallics 1996, 15, 2554–2561. [Google Scholar] [CrossRef]
- Toltl, N.P.; Leigh, W.J. Direct Detection of 1,1-Diphenylgermene in Solution and Absolute Rate Constants for Germene Trapping Reactions. J. Am. Chem. Soc. 1998, 120, 1172–1178. [Google Scholar] [CrossRef]
- Leigh, W.J. Kinetics and Mechanisms of the Reactions of Si=C and Ge=C Double Bonds. Pure Appl. Chem. 1999, 71, 453–462. [Google Scholar] [CrossRef]
- Ishikawa, M.; Fuchikami, T.; Kumada, M. Photochemically Generated Silicon—Carbon Double-bonded Intermediates: III. The Reaction of p-Tolylpentamethyldisilane with Methanol and Methanol-d1. J. Organomet. Chem. 1976, 118, 155–160. [Google Scholar] [CrossRef]
- Shizuka, H.; Okazaki, K.; Tanaka, M.; Ishikawa, M.; Sumitani, M.; Yoshihara, K. Intramolecular 2pπ* → 3dπ Charge Transfer in the Excited State of Phenyldisilane Studied by Picosecond and Nanosecond Laser Spectroscopy. Chem. Phys. Lett. 1985, 113, 89–92. [Google Scholar] [CrossRef]
- Sakurai, H.; Nakadaira, Y.; Kira, M.; Sugiyama, H.; Yoshida, K.; Takiguchi, T. Chemistry of Organosilicon Compounds: CXXIX. Evidence for Formation of Free Silyl Radicals in the Photolysis of Aryldisilanes. J. Organomet. Chem. 1980, 184, C36–C40. [Google Scholar] [CrossRef]
- Boudjouk, P.; Roberts, J.R.; Golino, C.M.; Sommer, L.H. Photochemical Dehydrosilylation of Pentaphenylmethyldisilane. Generation and Trapping of an Unstable Intermediate Containing a Silicon-carbon Double Bond or Its Equivalent. J. Am. Chem. Soc. 1972, 94, 7926–7927. [Google Scholar] [CrossRef]
- Ishikawa, M. Photolysis of Organopolysilanes. Generation and Reactions of Silicon-carbon Double-Bonded Intermediates. Pure Appl. Chem. 1978, 50, 11–18. [Google Scholar] [CrossRef]
- Sakurai, H. Spectra and Some Reactions of Organopolysilanes. J. Organomet. Chem. 1980, 200, 261–286. [Google Scholar] [CrossRef]
- Ishikawa, M.; Kumada, M. Photochemistry of Organopolysilanes. Adv. Organomet. Chem. 1981, 19, 51–95. [Google Scholar]
- Shizuka, H.; Hiratsuka, H. Photochemistry and Photophysics of Organosilicon Compounds. Res. Chem. Intermed. 1992, 18, 131–182. [Google Scholar] [CrossRef]
- Kira, M.; Miyazawa, T.; Sugiyama, H.; Yamaguchi, M.; Sakurai, H. sp* Orthogonal Intramolecular Charge-transfer (OICT) Excited States and Photoreaction Mechanism of Trifluoromethyl-substituted Phenyldisilanes. J. Am. Chem. Soc. 1993, 115, 3116–3124. [Google Scholar] [CrossRef]
- Sluggett, G.W.; Leigh, W.J. Reactive Intermediates from the Photolysis of Methylpentaphenyl- and pentamethylphenyldisilane. J. Am. Chem. Soc. 1992, 114, 1195–1201. [Google Scholar] [CrossRef]
- Sluggett, G.W.; Leigh, W.J. Aryldisilane Photochemistry. The Role of Silyl Free Radicals in Silene Formation from Photolysis of Methylpentaphenyldisilane. Organometallics 1994, 13, 1005–1013. [Google Scholar] [CrossRef]
- Leigh, W.J.; Sluggett, G.W. Triplet-state Photoreactivity of Phenyldisilanes. J. Am. Chem. Soc. 1993, 115, 7531–7532. [Google Scholar] [CrossRef]
- Leigh, W.J.; Sluggett, G.W. Aryldisilane Photochemistry. Substituent and Solvent Effects on the Photochemistry of Aryldisilanes and the Reactivity of 1,3,5-(1-sila)Hexatrienes. Organometallics 1994, 13, 269–281. [Google Scholar] [CrossRef]
- Sakurai, H. Silicon Chemistry; Corey, J.Y., Corey, E.Y., Gaspar, P.P., Eds.; Ellis Horwood: Chichester, UK, 1988; pp. 163–172. [Google Scholar]
- Brook, A.G. The Chemistry of Organic Silicon Compounds; Patai, S., Rappoport, Z., Eds.; Wiley: New York, NY, USA, 1989; pp. 965–1005. [Google Scholar]
- Braddock-Wilking, J.; Chiang, M.Y.; Gaspar, P.P. Photolysis of 1,3-Dimesitylhexamethyltrisilane and 1,2-Dimesityltetramethyldisilane. Organometallics 1993, 12, 197–209. [Google Scholar] [CrossRef]
- Ishikawa, M.; Kikuchi, M.; Kunai, A.; Takeuchi, T.; Tsukihara, T.; Kido, M. Silicon-carbon Unsaturated Compounds. 47. Dimerization of Silenes Generated from 1,4-bis(pentamethyldisilanyl)Benzene and 1-(pentamethyldisilanyl)-4-(trimethylsilyl)Benzene and the Molecular Structure of a Dimer. Organometallics 1993, 12, 3474–3479. [Google Scholar] [CrossRef]
- Ishikawa, M.; Oda, M.; Nishimura, K.; Kumada, M. Silicon–Carbon Unsaturated Compounds. XVI. Photorearrangement of an Adduct Derived from Reaction of a Silicon–Carbon Unsaturated Compound with t-Butyl Alcohol. Bull. Chem. Soc. Jpn. 1983, 56, 2795–2798. [Google Scholar] [CrossRef]
- Ishikawa, M.; Sakamoto, H. Silicon-carbon Unsaturated Compounds: XXXII. Photolysis of Tolyl- and xylylpentamethyldisilanes. J. Organomet. Chem. 1991, 414, 1–10. [Google Scholar] [CrossRef]
- Ishikawa, M.; Fuchikami, T.; Kumada, M. Photochemically Generated Silicon—Carbon Double-bonded Intermediates IV. Photolysis of 1-Alkenyldisilanes. J. Organomet. Chem. 1976, 117, C58–C62. [Google Scholar] [CrossRef]
- Ishikawa, M.; Fuchikami, T.; Kumada, M. Photochemically Generated Silicon-carbon Double-bonded Intermediates: VII. A New Route to Silaethene Derivatives from 1-Alkenyldisilanes. J. Organomet. Chem. 1978, 149, 37–48. [Google Scholar] [CrossRef]
- Leigh, W.J.; Toltl, N.P.; Apodaca, P.; Castruita, M.; Pannell, K.H. Photochemistry of Group 14 1,1,1-Trimethyl-2,2,2-triphenyldimetallanes (Ph3MM‘Me3; M, M′ = Si, Ge). Direct Detection and Characterization of Silene and Germene Reactive Intermediates. Organometallics 2000, 19, 3232–3241. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A. 02; Gaussian, Inc.: Wallingford CT, USA, 2013. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Casida, M.E.; Jamorski, C.; Casida, K.C.; Salahub, D.R. Molecular Excitation Energies to High-lying Bound States from Time-dependent Density-functional Response Theory: Characterization and Correction of the Time-dependent Local Density Approximation Ionization Threshold. J. Chem. Phys. 1998, 108, 4439–4449. [Google Scholar] [CrossRef]
- Stramann, R.E.; Scuseria, G.E.; Frisch, M.J. An Efficient Implementation of Time-dependent Density-functional Theory for the Calculation of Excitation Energies of Large Molecules. J. Chem. Phys. 1998, 109, 8218–8224. [Google Scholar] [CrossRef]
- Harvey, J.N.; Aschi, M.; Schwarz, H.; Koch, W. The Singlet and Triplet States of Phenyl Cation. A Hybrid Approach for Locating Minimum Energy Crossing Points between Non-interacting Potential Energy Surfaces. Theor. Chem. Acc. 1998, 99, 95–99. [Google Scholar] [CrossRef]
- Harvey, J.N.; Aschi, M. Spin-forbidden Dehydrogenation of Methoxy Cation: A Statistical View. Phys. Chem. Chem. Phys. 1999, 1, 5555–5563. [Google Scholar] [CrossRef]
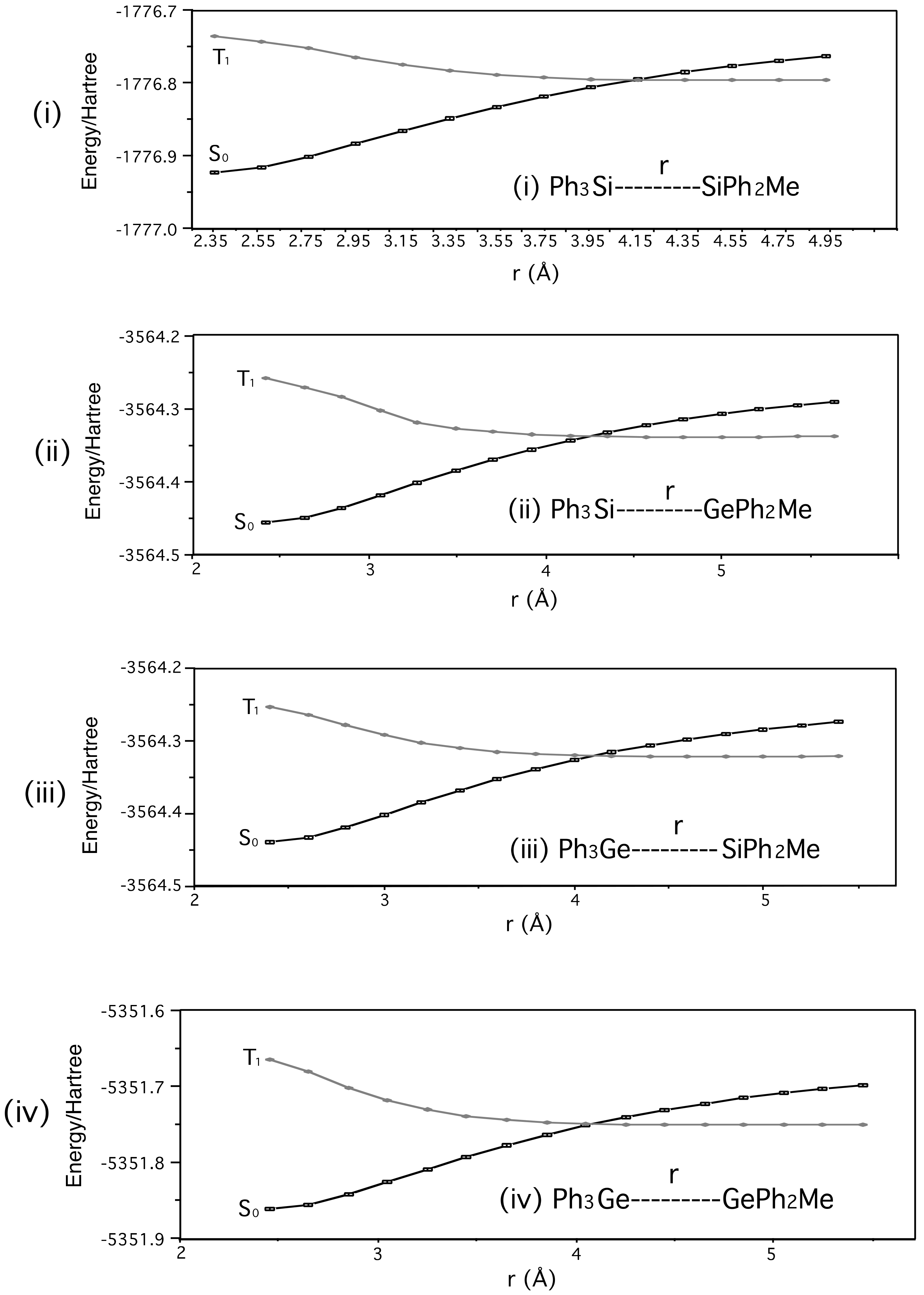
- The points in the graphs of Figure 2 were obtained using fixed potential energy surface scans.
- However, the absorption, fluorescence, and phosphorescence spectra in Reference 27 indicate S1 and T1 energies of ca. 105 and 78 kcal mol−1 for PhMe2Si SiMe3. UV absorption and fluorescence spectra are available for 1-Si-Si in Reference 29, and place its S1 (0-0) energy at ca. 95 kcal mol−1.
- However, it is believed that photolysis of 1-Si-Ge, 1-Ge-Si, and 1-Ge-Ge in more polar solvents can lead to high yields of the corresponding silyl and/or germyl radicals, rather than other singlet photoproducts.
- Mochida, K.; Wakasa, M.; Sakaguchi, Y.; Hayashi, H. Photochemical Reactions of Aryl-Substituted Digermanes through a Pair of Organogermyl Radicals. Bull. Chem. Soc. Jpn. 1991, 64, 1889–1895. [Google Scholar] [CrossRef]
- Mochida, K.; Kikkawa, H.; Nakadaira, Y. Photochemical Reactions of Aryl-substituted Catenates of Group 4B Elements, PhMe2E-E’Me3 (E, E’ = Si and Ge). Formation of a Radical Pair. J. Organomet. Chem. 1991, 412, 9–19. [Google Scholar] [CrossRef]
- Bobbitt, K.L.; Maloney, V.M.; Gaspar, P.P. New Photochemical Routes to Germylenes and Germenes and Kinetic Evidence Concerning the Germylene-diene Addition Mechanism. Organometallics 1991, 10, 2772–2777. [Google Scholar] [CrossRef]
- Zumdahl, S.S.; DeCoste, D.J. Chemical Principles; Mary Finch: New York, NY, USA, 2013; Chapter 18; pp. 893–903. [Google Scholar]
- It should be mentioned here that triplet/ground state singlet conversions are common in photoreactions that produce two non-coupled radical centers. See, for example: Mohamed, R.K.; Mondal, S.; Jorner, K.; Delgado, T.F.; Lobodin, V.V.; Ottosson, H.; Alabugin, I.V. The Missing C1–C5 Cycloaromatization Reaction: Triplet State Antiaromaticity Relief and Self-Terminating Photorelease of Formaldehyde for Synthesis of Fulvenes from Enynes. J. Am. Chem. Soc. 2015, 137, 15441–15450.
Sample Availability: Samples of the compounds are not available from the authors. |









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, S.-H.; Su, M.-D. Theoretical Study of the Photolysis Mechanisms of Methylpentaphenyldimetallanes (Ph3MM′Ph2Me; M, M′ = Si and Ge). Molecules 2018, 23, 1351. https://doi.org/10.3390/molecules23061351
Su S-H, Su M-D. Theoretical Study of the Photolysis Mechanisms of Methylpentaphenyldimetallanes (Ph3MM′Ph2Me; M, M′ = Si and Ge). Molecules. 2018; 23(6):1351. https://doi.org/10.3390/molecules23061351
Chicago/Turabian StyleSu, Shih-Hao, and Ming-Der Su. 2018. "Theoretical Study of the Photolysis Mechanisms of Methylpentaphenyldimetallanes (Ph3MM′Ph2Me; M, M′ = Si and Ge)" Molecules 23, no. 6: 1351. https://doi.org/10.3390/molecules23061351
APA StyleSu, S.-H., & Su, M.-D. (2018). Theoretical Study of the Photolysis Mechanisms of Methylpentaphenyldimetallanes (Ph3MM′Ph2Me; M, M′ = Si and Ge). Molecules, 23(6), 1351. https://doi.org/10.3390/molecules23061351