Irinotecan and Δ9-Tetrahydrocannabinol Interactions in Rat Liver: A Preliminary Evaluation Using Biochemical and Genotoxicity Markers



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. Serum Parameters of Liver Function
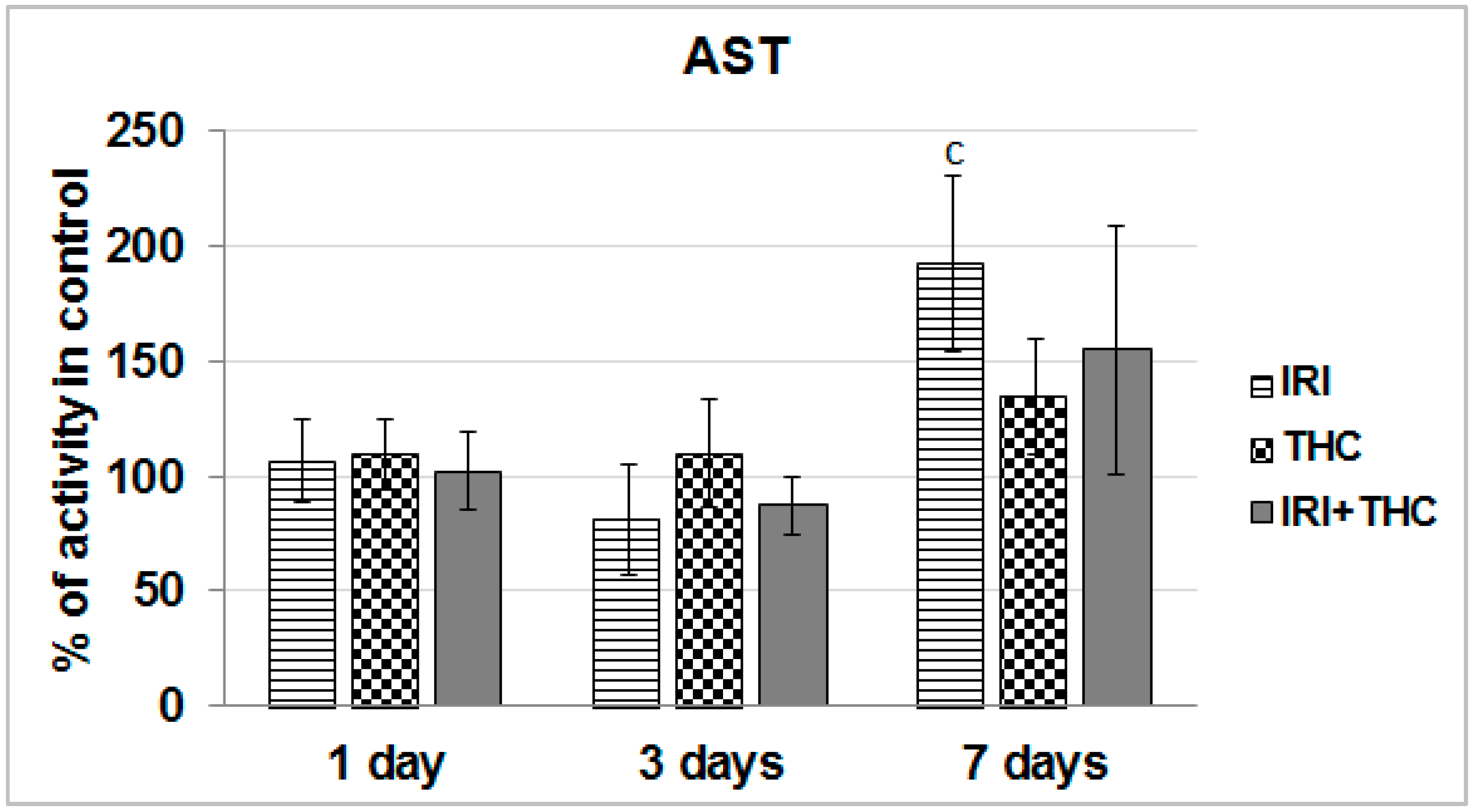
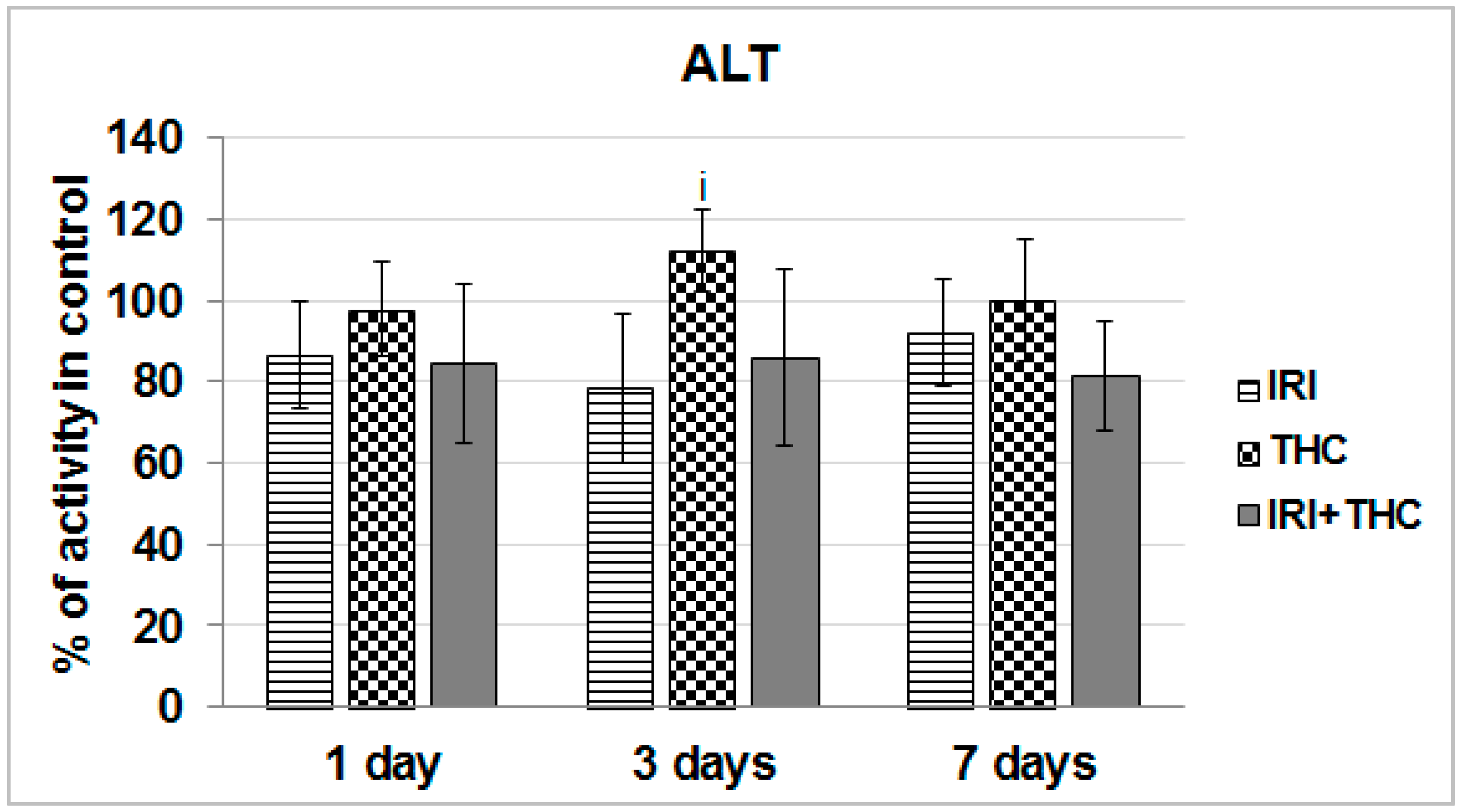
2.1.1. AST, ALT, and ALP Activity
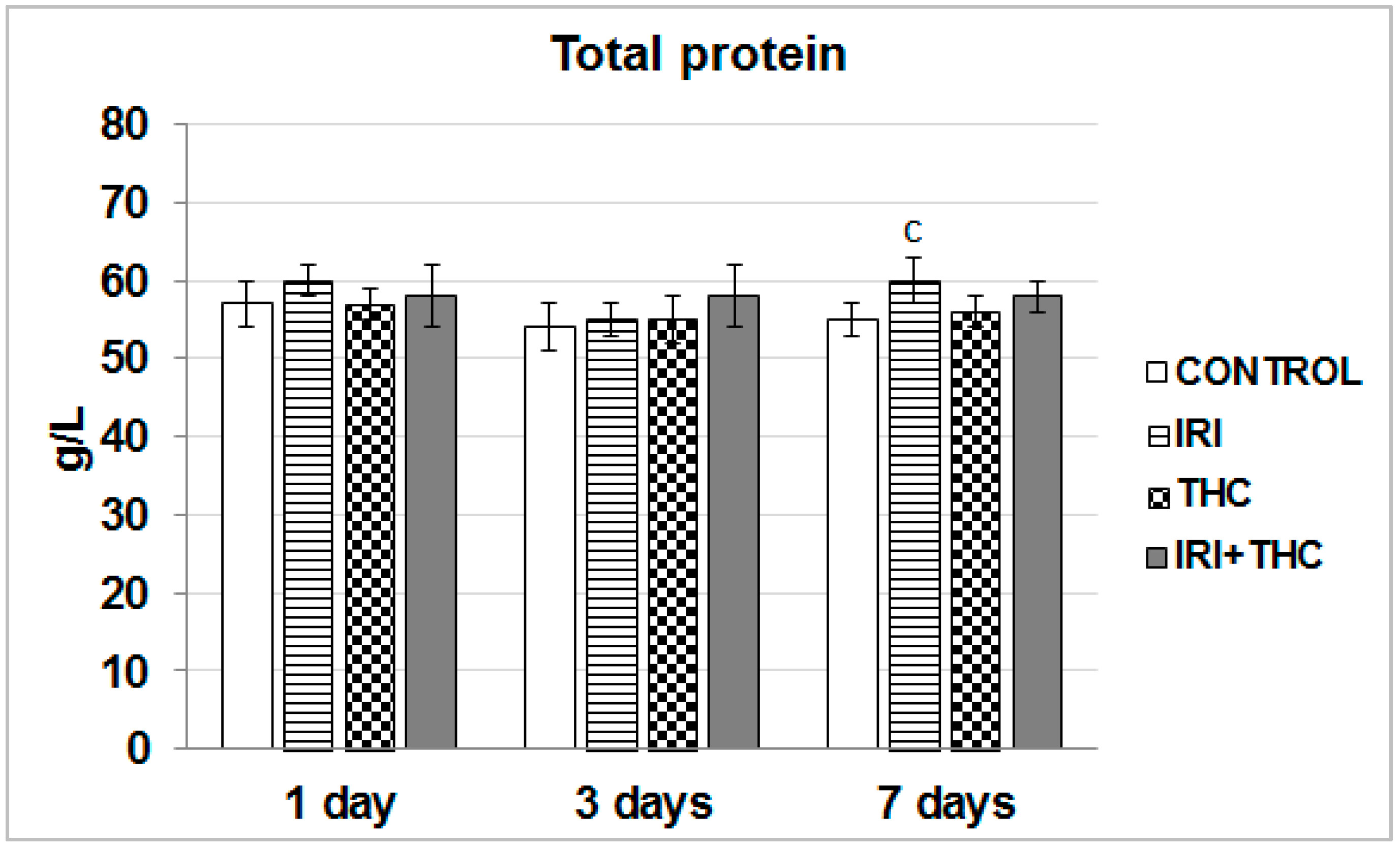
2.1.2. Total Protein in Serum
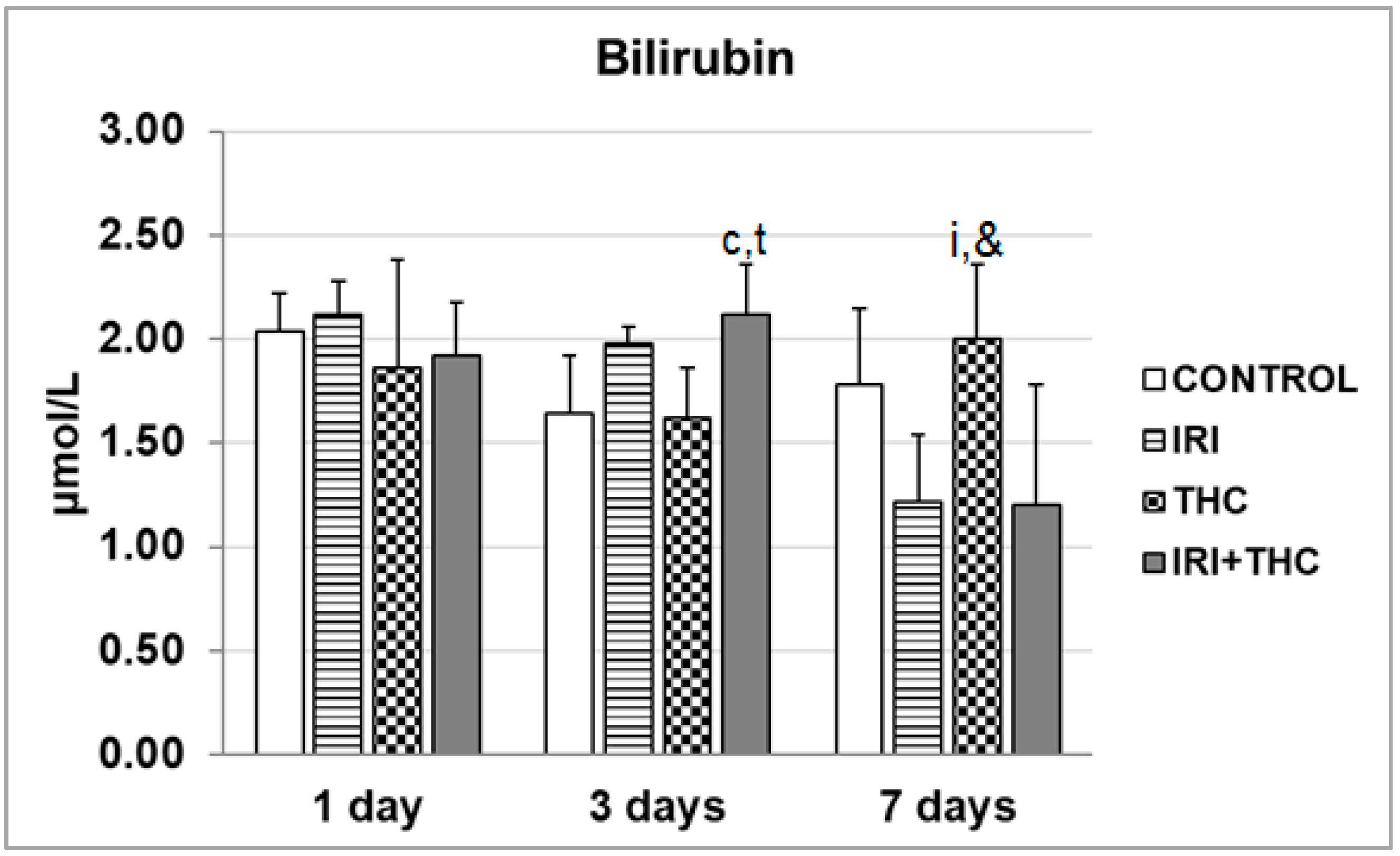
2.1.3. Bilirubin
2.2. Biochemical Markers of Oxidative Stress
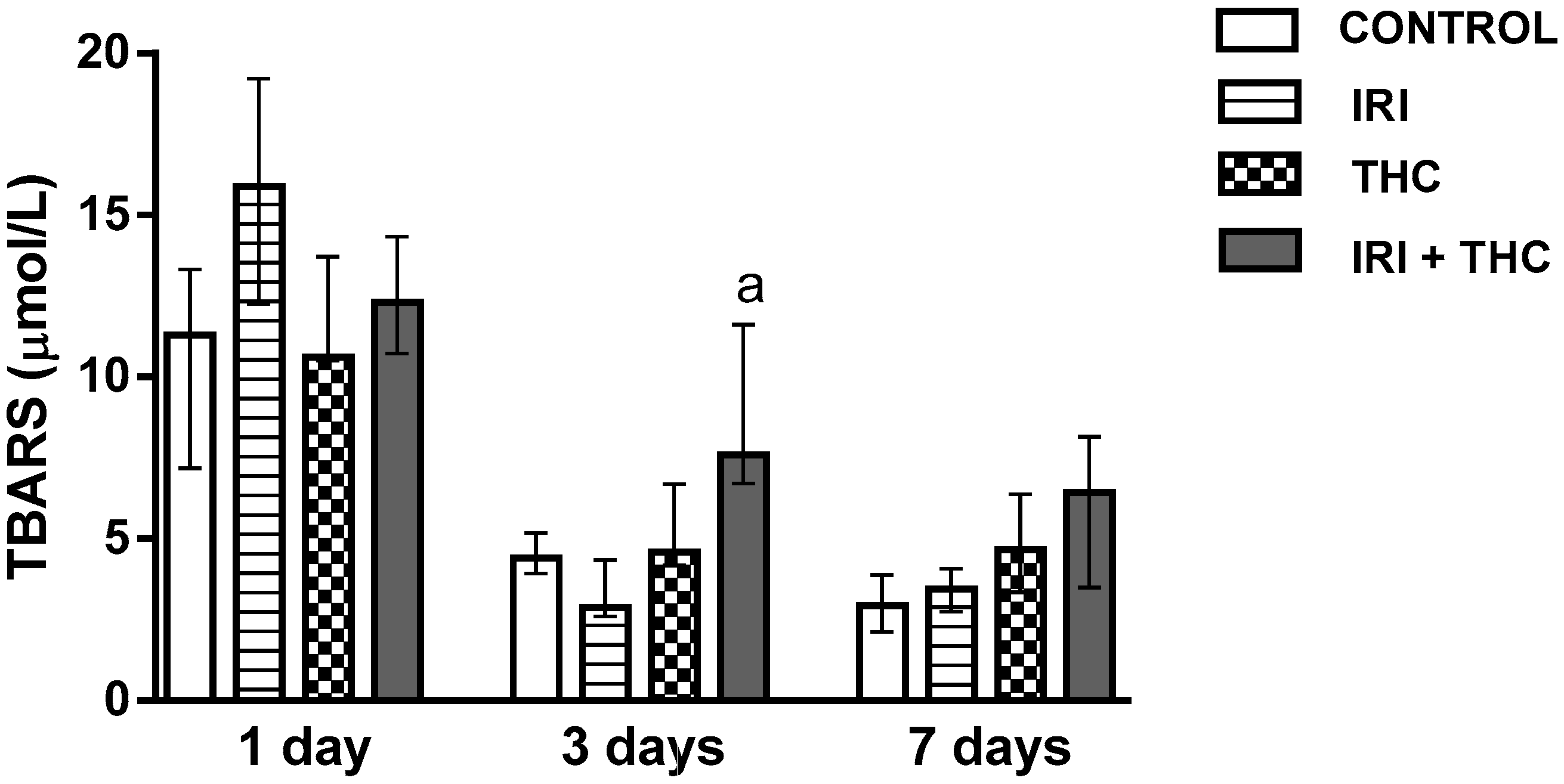
2.2.1. Lipid Peroxidation
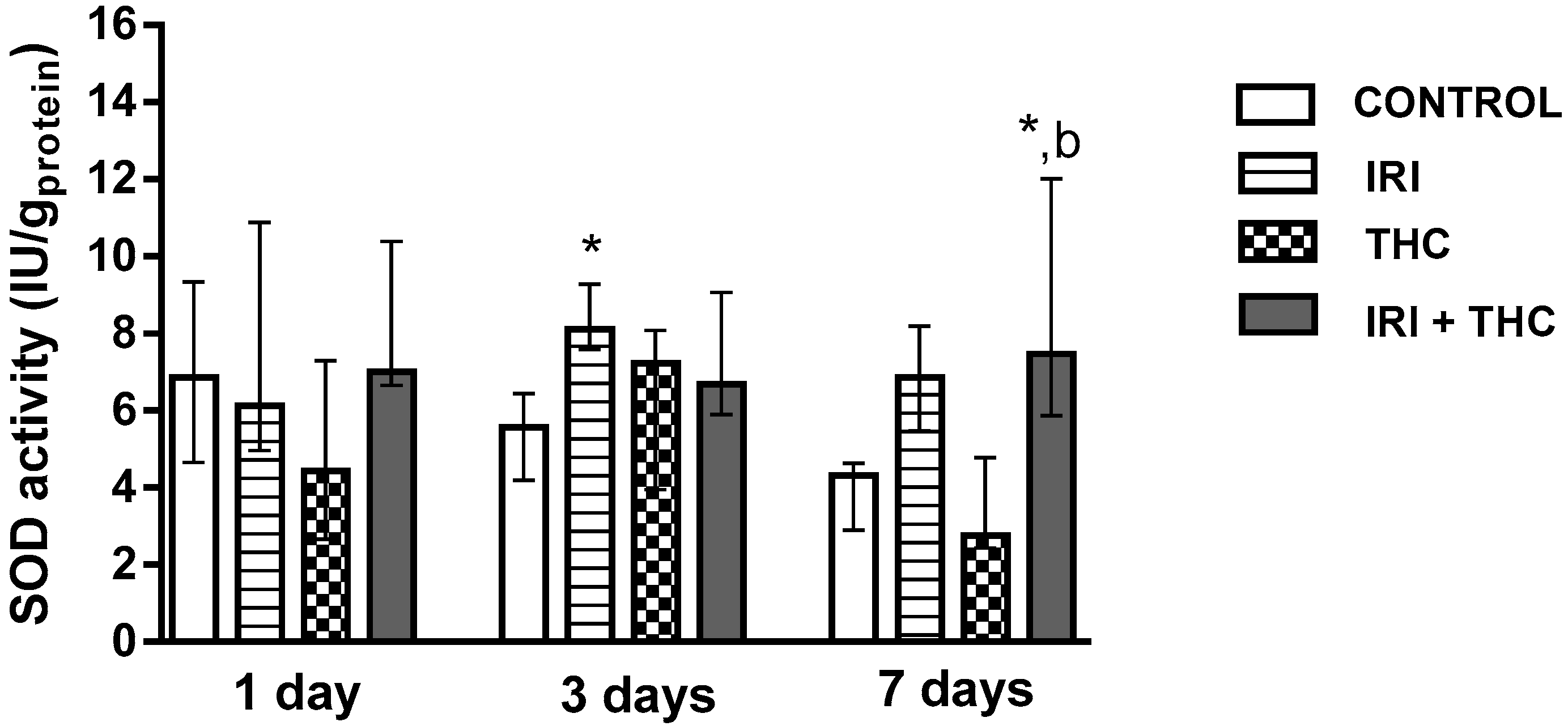
2.2.2. Superoxide Dismutase (SOD)
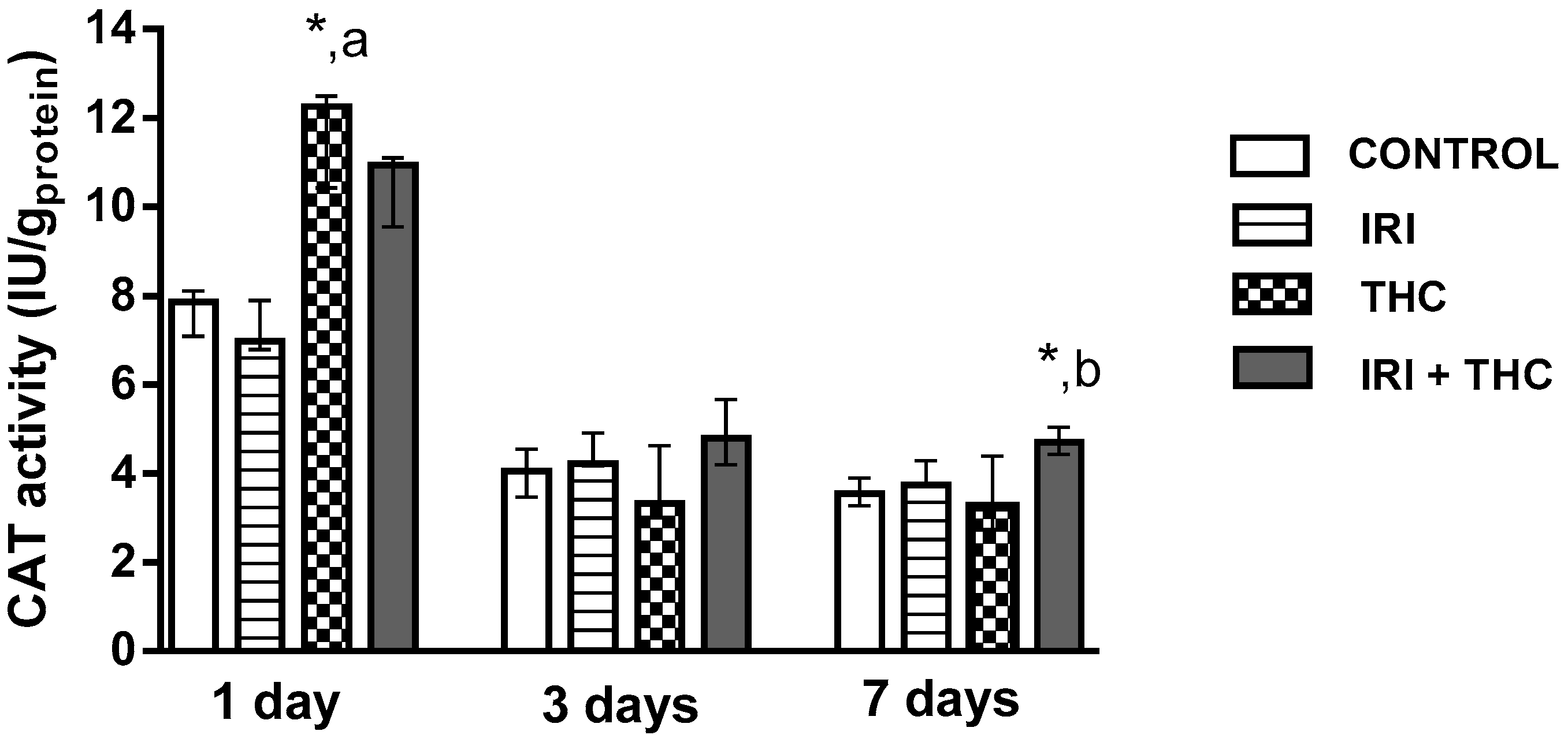
2.2.3. Catalase (CAT)
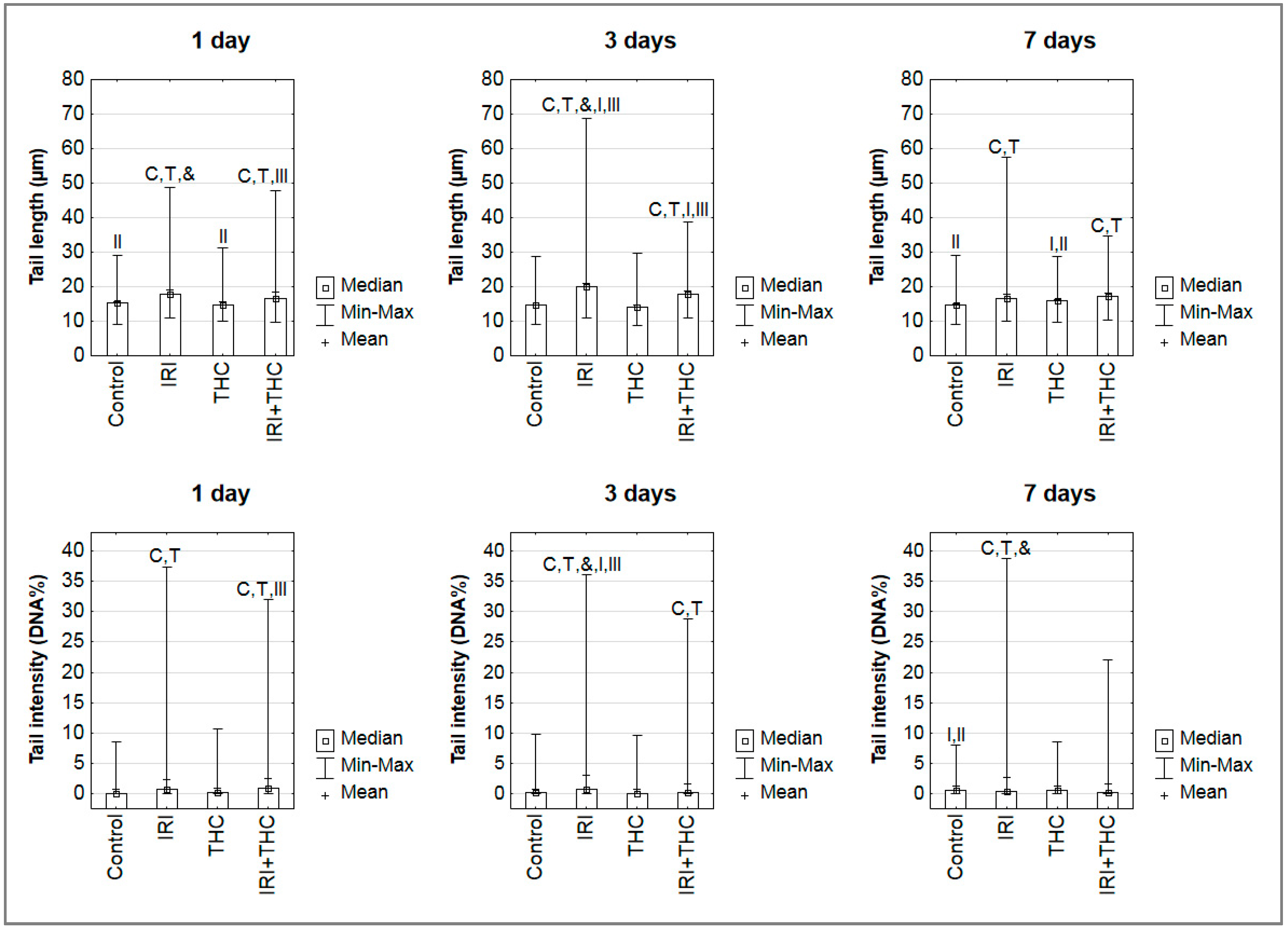
2.3. Primary DNA Damage in Rat Hepatocytes
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Animals
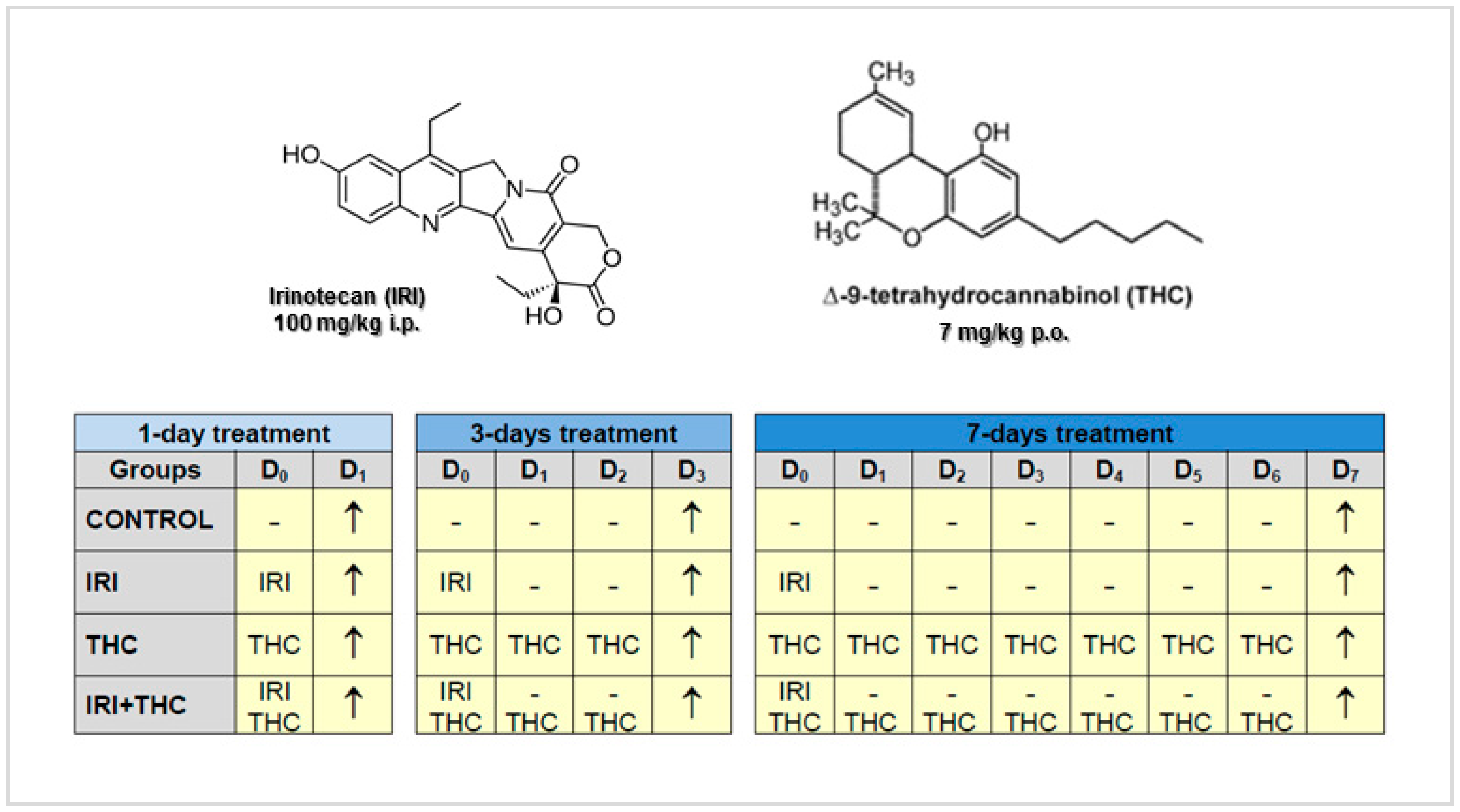
3.3. Preparation and Dosing of the Tested Compounds
3.4. Experimental Design
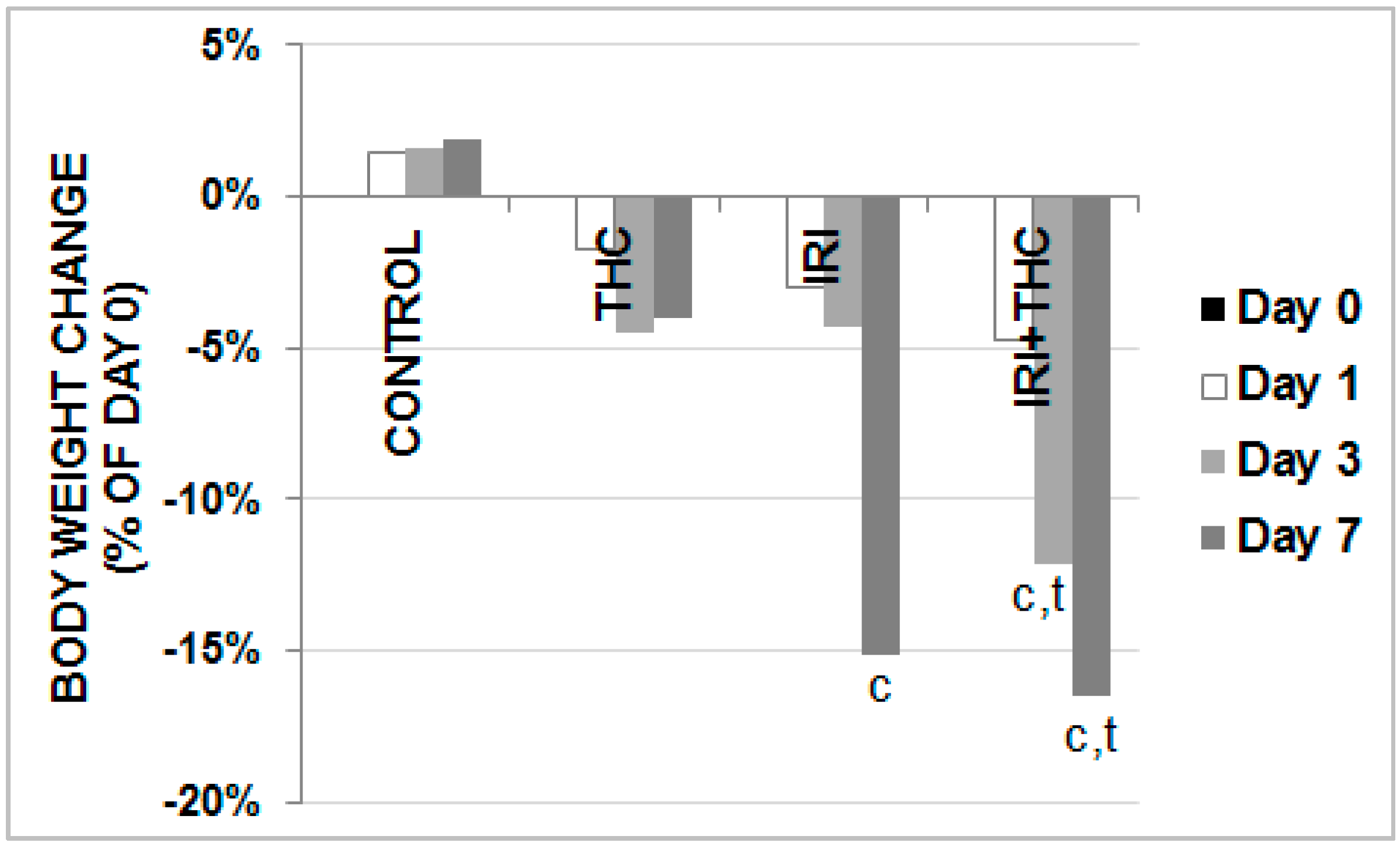
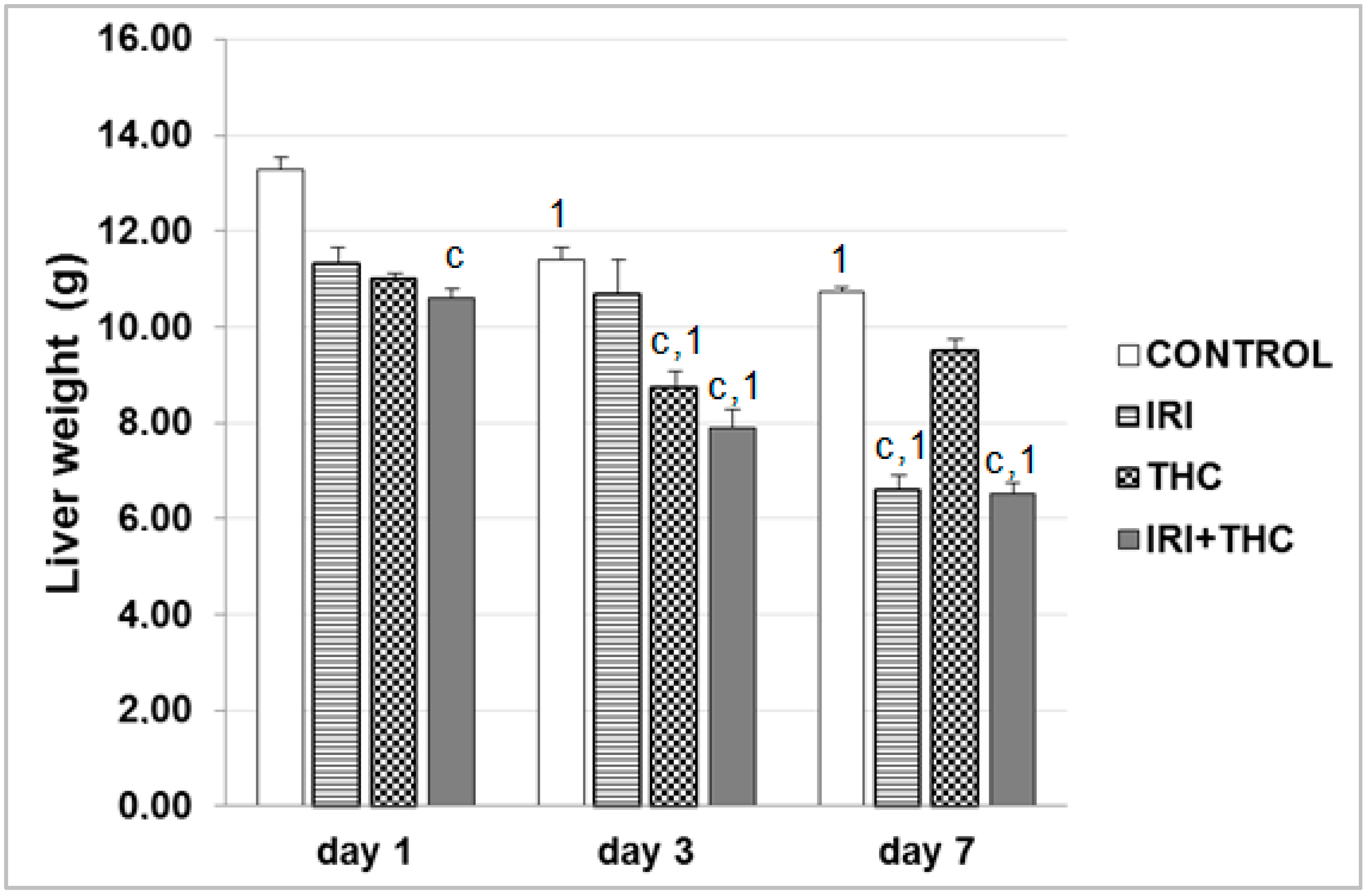
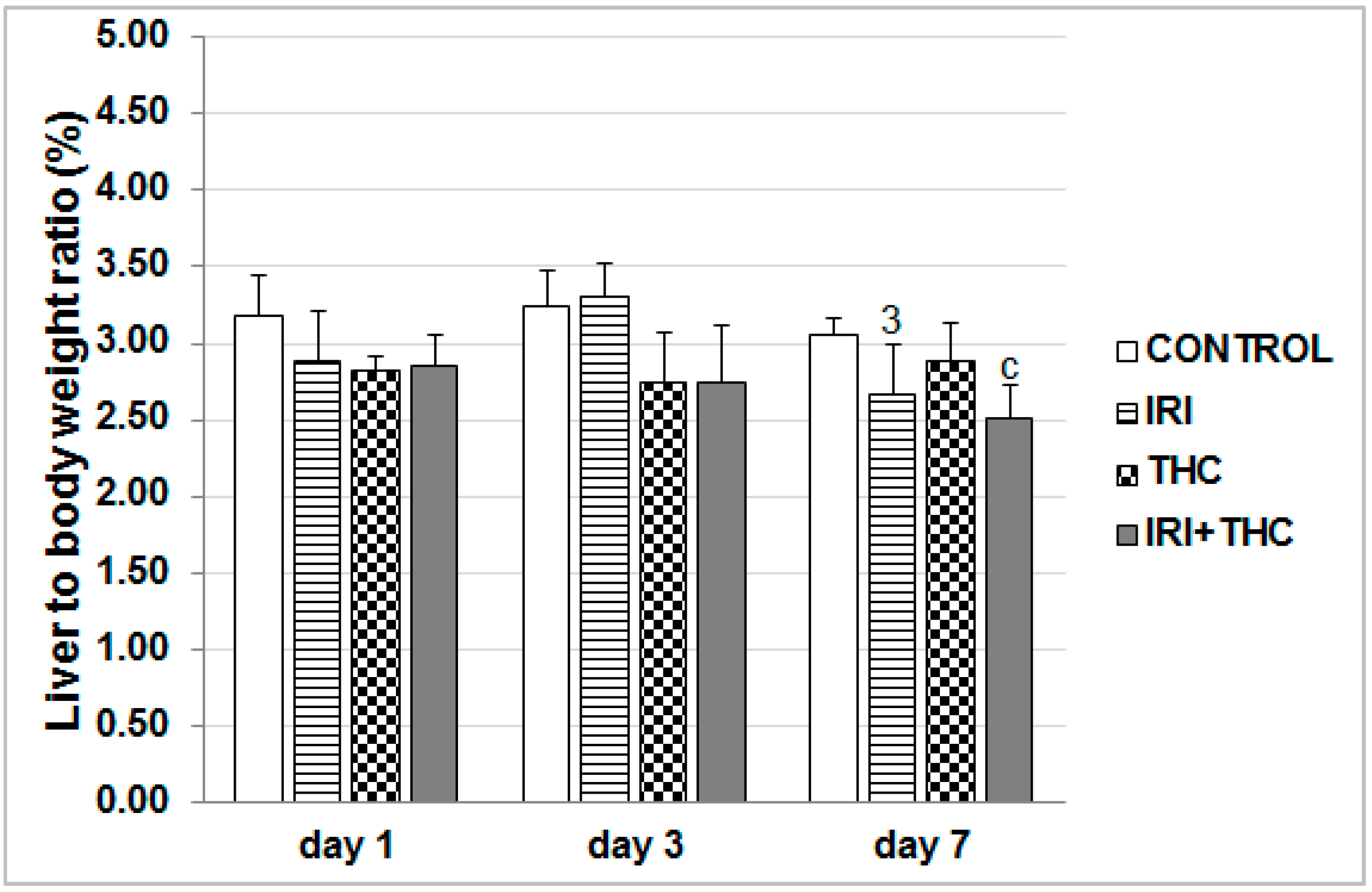
3.5. Body Weight, Liver Weight, and Calculation of Liver to Body Weight Ratio
3.6. Biochemical Serum Analysis
3.7. Lipid Peroxidation
3.8. Superoxide Dismutase Activity
3.9. Catalase Activity
3.10. Protein Quantification
3.11. The Alkaline Comet Assay
3.12. Statistical Analysis
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- ElSohly, M.A. Chemical Constituents of Cannabis. In Cannabis and Cannabinoids: Pharmacology,Toxicology, and Therapeutic Potential; Grotenhermen, F., Russo, E., Eds.; The Haworth Press Inc.: Binghamton, NY, USA, 2002; Volume 1, pp. 27–35. [Google Scholar]
- Grotenhermen, F. Clinical Pharmacokinetics of Cannabinoids. J. Cannabis Ther. 2003, 3, 3–51. [Google Scholar] [CrossRef]
- Badowski, M.E. A review of oral cannabinoids and medical marijuana for the treatment of chemotherapy-induced nausea and vomiting: A focus on pharmacokinetic variability and pharmacodynamics. Cancer Chemother. Pharmacol. 2017, 80, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Mathijssen, R.H.J.; Van Alphen, R.J.; Verweij, J.; Loos, W.J.; Nooter, K.; Stoter, G.; Sparreboom, A. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11). Clin. Cancer Res. 2001, 7, 2182–2194. [Google Scholar] [PubMed]
- Smith, N.F.; Figg, W.D.; Sparreboom, A. Pharmacogenetics of irinotecan metabolism and transport: An update. Toxicol. In Vitro 2006, 20, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.; Mitchell, E.P.; Hoff, P.M. Irinotecan in the treatment of colorectal cancer. Cancer Treat. Rev. 2006, 32, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Alimonti, A.; Gelibter, A.; Pavese, I.; Satta, F.; Cognetti, F.; Ferretti, G.; Rasio, D.; Vecchione, A.; Di Palma, M. New approaches to prevent intestinal toxicity of irinotecan-based regimens. Cancer Treat. Rev. 2004, 30, 555–562. [Google Scholar] [CrossRef] [PubMed]
- De Jong, F.A. A Roadmap to Individualized Irinotecan Dosing; Erasmus University Rotterdam: Rotterdam, The Netherlands, 2006; ISBN 978-90-90-21065-0. [Google Scholar]
- Xu, Y.; Villalona-Calero, M.A. Irinotecan: Mechanisms of tumor resistance and novel strategies for modulating its activity. Ann. Oncol. 2002, 13, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Sanghani, S.P.; Quinney, S.K.; Fredenburg, T.B.; Davis, W.I.; Murry, D.J.; Bosron, W.F. Hydrolysis of irinotecan and its oxidative metabolites, 7-ethyl-10-[4-N-(5-aminopentanoic acid)-1-piperidino] carbonyloxycamptothecin and 7-ethyl-10-[4-(1-piperidino)-1-amino]-carbonyloxycamptothecin, by human carboxylesterases CES1A1, CES2, and a newly expressed carboxylesterase isoenzyme. Drug Metab. Dispos. 2004, 32, 505–511. [Google Scholar] [PubMed]
- Ribeiro, R.A.; Wanderley, C.W.S.; Wong, D.V.T.; Mota, J.M.S.C.; Leite, C.A.V.G.; Souza, M.H.L.P.; Cunha, F.Q.; Lima-Júnior, R.C.P. Irinotecan- and 5-fluorouracil-induced intestinal mucositis: insights into pathogenesis and therapeutic perspectives. Cancer Chemother. Pharmacol. 2016, 78, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, C.H.; Wright, J.; Arbuck, S.G. Clinical applications of the camptothecins. Biochim. Biophys. Acta Gene Struct. Expr. 1998, 1400, 107–119. [Google Scholar] [CrossRef]
- Dodds, H.M.; Haaz, M.C.; Riou, J.F.; Robert, J.; Rivory, L.P. Identification of a new metabolite of CPT-11 (irinotecan): pharmacological properties and activation to SN-38. J. Pharmacol. Exp. Ther. 1998, 286, 578–583. [Google Scholar] [PubMed]
- Santos, A.; Zanetta, S.; Cresteil, T.; Deroussent, A.; Pein, F.; Raymond, E.; Vernillet, L.; Risse, M.L.; Boige, V.; Gouyette, A.; et al. Metabolism of irinotecan (CPT-11) by CYP3A4 and CYP3A5 in humans. Clin. Cancer Res. 2000, 6, 2012–2020. [Google Scholar] [PubMed]
- Takasuna, K.; Hagiwara, T.; Hirohashi, M.; Kato, M.; Nomura, M.; Nagai, E.; Yokoi, T.; Kamataki, T. Involvement of β-glucuronidase in intestinal microflora in the intestinal toxicity of the antitumor camptothecin derivative irinotecan hydrochloride (CPT-11) in rats. Cancer Res. 1996, 56, 3752–3757. [Google Scholar] [PubMed]
- Cai, Z.; Yang, J.; Shu, X.; Xiong, X. Chemotherapy-associated hepatotoxicity in colorectal cancer. Vascular 2014, 14, 15. [Google Scholar]
- Grigorian, A.; O’Brien, C.B. Hepatotoxicity secondary to chemotherapy. J. Clin. Transl. Hepatol. 2014, 2, 95–102. [Google Scholar] [PubMed]
- Sawano, T.; Shimizu, T.; Yamada, T.; Nanashima, N.; Miura, T.; Morohashi, S.; Kudo, D.; Hui, F.M.; Kijima, H.; Hakamada, K.; et al. Fatty acid synthase-positive hepatocytes and subsequent steatosis in rat livers by irinotecan. Oncol. Rep. 2015, 33, 2151–2160. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, J.; Guo, G. Mechanistic review of drug-induced steatohepatitis. Toxicol. Appl. Pharmacol. 2015, 289, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Samuel, T.; Olasore, H.; Nanaghan, E.; Turner, I.; Owoyale, A.; Magbagbeola, O. Neuroendocrinological, metabolic and toxicological effects of chronic cannabis use among male in Lagos. Asian J. Biochem. 2012, 7, 226–231. [Google Scholar]
- Purohit, V.; Rapaka, R.; Shurtleff, D. Role of cannabinoids in the development of fatty liver (steatosis). AAPS J. 2010, 12, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Borini, P.; Cardoso Guimarães, R.; Bicalho Borini, S. Possible hepatotoxicity of chronic marijuana usage. Sao Paulo Med. J. 2004, 122, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Stout, S.M.; Cimino, N.M. Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: A systematic review. Drug Metab. Rev. 2014, 46, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Mazur, A.; Lichti, C.F.; Prather, P.L.; Zielinska, A.K.; Bratton, S.M.; Gallus-Zawada, A.; Finel, M.; Miller, G.P.; Radomińska-Pandya, A.; Moran, J.H. Characterization of human hepatic and extrahepatic UDP-glucuronosyltransferase enzymes involved in the metabolism of classic cannabinoids. Drug Metab. Dispos. 2009, 37, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Grotenhermen, F. Pharmacokinetics and pharmacodynamics of cannabinoids. Clin. Pharm. 2003, 42, 327–359. [Google Scholar] [CrossRef] [PubMed]
- Bland, T.M.; Haining, R.L.; Tracy, T.S.; Callery, P.S. CYP2C-catalyzed delta(9)-tetrahydrocannabinol metabolism: Kinetics, pharmacogenetics and interaction with phenytoin. Biochem. Pharmacol. 2005, 70, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Hryhorowicz, S.; Walczak, M.; Zakerska-Banaszak, O.; Słomski, R.; Skrzypczak-Zielińska, M. Pharmacogenetics of cannabinoids. Eur. J. Drug Metab. Pharmacokinet. 2018, 43, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Murthy, P.; Bharath, M.M.S. Chemistry, metabolism, and toxicology of cannabis: Clinical implications. Iran. J. Psychiatry 2012, 7, 149–156. [Google Scholar] [PubMed]
- Huestis, M.A. Human cannabinoid pharmacokinetics. Chem. Biodivers. 2007, 4, 1770–1804. [Google Scholar] [CrossRef] [PubMed]
- McGilveray, I.J. Pharmacokinetics of cannabinoids. Pain Res. Manag. 2005, 10, 15A–22A. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.Y.; Kato, Y.; Sugiyama, Y. Multiplicity of biliary excretion mechanisms for irinotecan, CPT-11, and its metabolites in rats. Cancer Res. 1997, 57, 1934–1938. [Google Scholar] [PubMed]
- Sugiyama, Y.; Kato, Y.; Chu, X. Multiplicity of biliary excretion mechanisms for the camptothecin derivative irinotecan (CPT-11), its metabolite SN-38, and its glucuronide: role of canalicular multispecific organic anion transporter and P-glycoprotein. Cancer Chemother. Pharm. 1998, 42, S44–S49. [Google Scholar] [CrossRef]
- Horikawa, M.; Kato, Y.; Sugiyama, Y. Reduced gastrointestinal toxicity following inhibition of the biliary excretion of irinotecan and its metabolites by probenecid in rats. Pharm. Res. 2002, 19, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Takemoto, I.; Itagaki, S.; Sasaki, K.; Hirano, T.; Iseki, K. Biliary excretion of irinotecan and its metabolites. J. Pharm. Pharm. Sci. 2004, 7, 13–18. [Google Scholar] [PubMed]
- Bansal, T.; Awasthi, A.; Jaggi, M.; Khar, R.K.; Talegaonkar, S. Pre-clinical evidence for altered absorption and biliary excretion of irinotecan (CPT-11) in combination with quercetin: Possible contribution of P-glycoprotein. Life Sci. 2008, 83, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Celik, S.; Kartal, K.; Ozseker, H.; Hayran, M.; Hamaloglu, E. Hepatoprotective effect of pioglitazone in cases of chemotherapy induced steatohepatitis. Chirurgia 2015, 110, 49–55. [Google Scholar] [PubMed]
- Guan, H.; Li, P.F.; Wang, X.M.; Yue, J.J.; He, Y.; Luo, X.M.; Su, M.F.; Liao, S.G.; Shi, Y. Shengjiang Xiexin decoction alters pharmacokinetics of irinotecan by regulating metabolic enzymes and transporters: A multi-target therapy for Alleviating the gastrointestinal toxicity. Front. Pharm. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Salam, O.M.E.; Metwaly, S.; Sleem, A.A.; Morsy, F.A.; Sharaf, H.A. Cannabis sativa exacerbates hepatic injury caused by acetaminophen or carbon tetrachloride in rats. Comp. Clin. Pathol. 2013, 22, 209–218. [Google Scholar] [CrossRef]
- Coşkun, Z.M.; Bolkent, S. The changes of oxidative stress and Δ9-tetrahydrocannabinol accumulation in liver of type-2 diabetic rats. IUFS J. Biol. 2015, 74, 1–8. [Google Scholar]
- Musa, E.M.; EL Badwi, S.M.; Jah Elnabi, M.A.; Osman, E.A.; Dahab, M.M. Hepatoprotective and toxicity assessment of Cannabis sativa seed oil in Albino rat. Int. J. Chem. Biochem. Sci. 2012, 1, 69–76. [Google Scholar]
- Okwari, O.O.; Emerole, C.G.; Dasofunjo, K.; Alagwu, E.A.; Olatunji, T.L.; Osim, E.E. Impact of repeated administration of Cannabis sativa on some biochemical parameters in albino rats. J. Pharm. Biol. Sci. 2014, 9, 51–57. [Google Scholar]
- Cao, S.; Rustum, Y.M. Synergistic antitumor activity of irinotecan in combination with 5-fluorouracil in rats bearing advanced colorectal cancer: Role of drug sequence and dose. Cancer Res. 2000, 60, 3717–3721. [Google Scholar] [PubMed]
- Gibson, R.J.; Bowen, J.M.; Inglis, M.R.B.; Cummins, A.G.; Keefe, D.M.K. Irinotecan causes severe small intestinal damage, as well as colonic damage, in the rat with implanted breast cancer. J. Gastroenterol. Hepatol. 2003, 18, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Barth, S.W.; Briviba, K.; Watzl, B.; Jäger, N.; Marko, D.; Esselen, M. In vivo bioassay to detect irinotecan-stabilized DNA/topoisomerase I complexes in rats. Biotechnol. J. 2010, 5, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Esselen, M.; Barth, S.W.; Winkler, S.; Baechler, S.; Briviba, K.; Watzl, B.; Skrbek, S.; Marko, D. Anthocyanins suppress the cleavable complex formation by irinotecan and diminish its DNA-strand-breaking activity in the colon of wistar rats. Carcinogenesis 2013, 34, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, N.; Kurita, A.; Hosokawa, Y.; Yokokura, T.; Awazu, S. Intravenous administration of irinotecan elevates the blood β-glucuronidase activity in rats. Cancer Res. 1997, 57, 5305–5308. [Google Scholar] [PubMed]
- Gibson, R.J.; Bowen, J.M.; Alvarez, E.; Finnie, J.; Keefe, D.M.K. Establishment of a single-dose irinotecan model of gastrointestinal mucositis. Chemotherapy 2007, 53, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Logan, R.M.; Gibson, R.J.; Bowen, J.M.; Stringer, A.M.; Sonis, S.T.; Keefe, D.M.K. Characterisation of mucosal changes in the alimentary tract following administration of irinotecan: Implications for the pathobiology of mucositis. Cancer Chemother. Pharmacol. 2008, 62, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Stringer, A.M.; Gibson, R.J.; Logan, R.M.; Bowen, J.M.; Yeoh, A.S.J.; Keefe, D.M.K. Faecal microflora and β-glucuronidase expression are altered in an irinotecan-induced diarrhoea model in rats. Cancer Biol. Ther. 2008, 7, 1919–1925. [Google Scholar] [CrossRef] [PubMed]
- Rtibi, K.; Selmi, S.; Grami, D.; Sebai, H.; Amri, M.; Marzouki, L. Irinotecan chemotherapy-induced intestinal oxidative stress: Underlying causes of disturbed mucosal water and electrolyte transport. Pathophysiology 2017, 24, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Al-Dasooqi, N.; Bowen, J.M.; Gibson, R.J.; Logan, R.M.; Stringer, A.M.; Keefe, D.M. Irinotecan-induced alterations in intestinal cell kinetics and extracellular matrix component expression in the dark agouti rat. Int. J. Exp. Pathol. 2011, 92, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.I.; Kubota, Y.; Ishida, H.; Sasaki, Y. Irinotecan, a key chemotherapeutic drug for metastatic colorectal cancer. World J. Gastroenterol. 2015, 21, 12234–12248. [Google Scholar] [CrossRef] [PubMed]
- Guichard, S.; Chatelut, E.; Lochon, I.; Bugat, R.; Mahjoubi, M.; Canal, P. Comparison of the pharmacokinetics and eficacy of irinotecan after administration by the intravenous versus intraperitoneal route in mice. Cancer Chemother. Pharmacol. 1998, 42, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Nebendahl, K. Routes of Administration. In The Laboratory Rat; Krinke, G., Ed.; Academic Press: London, UK, 2000; pp. 463–483. [Google Scholar]
- Turner, P.V; Brabb, T.; Pekow, C.; Vasbinder, M.A. Administration of substances to laboratory animals: Routes of administration and factors to consider. J. Am. Assoc. Lab. Anim. Sci. 2011, 50, 600–613. [Google Scholar] [PubMed]
- Bansal, T.; Mishra, G.; Jaggi, M.; Khar, R.K.; Talegaonkar, S. Effect of P-glycoprotein inhibitor, verapamil, on oral bioavailability and pharmacokinetics of irinotecan in rats. Eur. J. Pharm. Sci. 2009, 36, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Garg, T.; Bhandari, S.; Rath, G.; Goyal, A.K. Current strategies for targeted delivery of bio-active drug molecules in the treatment of brain tumor. J. Drug Target. 2015, 23, 865–887. [Google Scholar] [CrossRef] [PubMed]
- Hatefi, A.; Amsden, B. Camptothecin delivery methods. Pharm. Res. 2002, 19, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, D.N.; Sutherland, B.W.; Santos, N.D.; Masin, D.; Osooly, M.; Strutt, D.; Ostlund, C.; Anantha, M.; Harasym, N.; Manisali, I.; et al. Irinophore C™, a lipid nanoparticle formulation of irinotecan, abrogates the gastrointestinal effects of irinotecan in a rat model of clinical toxicities. Investig. New Drugs 2014, 32, 1071–1082. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ligresti, A. Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J. Pharm. Exp. Ther. 2006, 318, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Loflin, M.; Earleywine, M. A new method of cannabis ingestion: The dangers of dabs? Addict. Behav. 2014, 39, 1430–1433. [Google Scholar] [CrossRef] [PubMed]
- National Toxicology Program. NTP 1-Trans-delta9-tetrahydrocannabinol (CAS No. 1972-08-3) in F344/N rats and B6C3F1 mice (gavage studies). Natl. Toxicol. Program Tech. Rep. Ser. 1996, 446, 1–317. [Google Scholar]
- Dasofunjo, K.; Okwari, O.; Obembe, A.; Olantuji, T.; Ezugwu, H.; Osim, E. Assessment of the effect of Cannabis sativa and Nicotiana tobacum leaves on some haematological and liver function indices of albino rats. IOSR J. Pharm. Biol. Sci. 2014, 9, 33–40. [Google Scholar]
- Mukhtar, A.H.; Elbagir, N.M. Effect of Cannabis sativa on hematological indices in rats and men. Pakistan J. Nutr. 2011, 10, 313–316. [Google Scholar] [CrossRef][Green Version]
- Harvey, D.J. Metabolism and pharmacokinetics of the cannabinoids. In Biochemistry and Physiology of Substance Abuse; Watson, R., Ed.; CRC Press: Boca Raton, FL, USA, 1991; Volume III, pp. 279–365. [Google Scholar]
- Bridgeman, M.B.; Abazia, D.T. Medicinal cannabis: History, pharmacology, and implications for the acute care setting. Pharm. Ther. 2017, 42, 180–188. [Google Scholar]
- Engels, F.K.; de Jong, F.A.; Sparreboom, A.; Mathot, R.A.A.; Loos, W.J.; Kitzen, J.J.E.M.; de Bruijn, P.; Verweij, J.; Mathijssen, R.H.J. Medicinal cannabis does not influence the clinical pharmacokinetics of irinotecan and docetaxel. Oncologist 2007, 12, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Kurita, A.; Kado, S.; Kaneda, N.; Onoue, M.; Hashimoto, S.; Yokokura, T. Modified irinotecan hydrochloride (CPT-11) administration schedule improves induction of delayed-onset diarrhea in rats. Cancer Chemother. Pharmacol. 2000, 46, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Takakura, A.; Kurita, A.; Asahara, T.; Yokoba, M.; Yamamoto, M.; Ryuge, S.; Igawa, S.; Yasuzawa, Y.; Sasaki, J.; Kobayashi, H.; et al. Rapid deconjugation of SN-38 glucuronide and adsorption of released free SN-38 by intestinal microorganisms in rat. Oncol. Lett. 2012, 3, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kurita, A.; Asahara, T.; Takakura, A.; Katono, K.; Iwasaki, M.; Ryuge, S.; Wada, M.; Onoda, S.; Yanaihara, T.; et al. Metabolism of irinotecan and its active metabolite SN-38 by intestinal microflora in rats. Oncol. Rep. 2008, 20, 727–730. [Google Scholar] [PubMed]
- Hayes, W. Principles and Methods of Toxicology, 5th ed.; Taylor and Francis: London, UK, 2007; pp. 1465–1508. ISBN 9781420005424. [Google Scholar]
- Sturgill, M.G.; Lambert, G.H. Xenobiotic-induced hepatotoxicity: Mechanisms of liver injury and methods of monitoring hepatic function. Clin. Chem. 1997, 43, 1512–1526. [Google Scholar] [PubMed]
- Thapa, B.R.; Walia, A. Liver function tests and their interpretation. Indian J. Paediatr. 2007, 74, 67–75. [Google Scholar] [CrossRef]
- Takeba, Y.; Kumai, T.; Matsumoto, N.; Nakaya, S.; Tsuzuki, Y.; Yanagida, Y.; Kobayashi, S. Irinotecan activates p53 with its active metabolite, resulting in human hepatocellular carcinoma apoptosis. J. Pharm. Sci. 2007, 104, 232–242. [Google Scholar] [CrossRef]
- Fulco, R.A.; Costa, C.; Germano, M.P.; Torre, E.A.; Viscomi, M.G.; Salimbeni, V.; Maisano, R.; Giudice, A.; Costa, G. Hepatotoxicity of camptothecin derivatives in a primary culture system of rat hepatocytes. J. Chemother. 2000, 12, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.; Roberts, L.R.; Jones, B.A.; Gores, G.J. Dysregulation of apoptosis as a mechanism of liver disease: an overview. Semin. Liver Dis. 1998, 18, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Bas, H.; Pandir, D.; Kalender, S. Furan-induced hepatotoxic and hematologic changes in diabetic rats: the protective role of lycopene. Arh. Hig. Rada Toksikol. 2016, 67, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Limdi, J.K.; Hyde, G.M. Evaluation of abnormal liver function tests. Postgrad. Med. J. 2003, 79, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Navarro, V.J.; Senior, J.R. Drug-related hepatotoxicity. N. Engl. J. Med. 2006, 354, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Field, K.M.; Dow, C.; Michael, M. Part I: Liver function in oncology: biochemistry and beyond. Lancet Oncol. 2008, 9, 1092–1101. [Google Scholar] [CrossRef]
- Yokooji, T.; Kawabe, Y.; Mori, N.; Murakami, T. Effect of genistein, a natural soy isoflavone, on the pharmacokinetics and intestinal toxicity of irinotecan hydrochloride in rats. J. Pharm. Pharm. 2013, 65, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Marcolino Assis-Júnior, E.; Melo, A.T.; Pereira, V.B.M.; Wong, D.V.T.; Sousa, N.R.P.; Oliveira, C.M.G.; Malveira, L.R.C.; Moreira, L.S.; Souza, M.H.L.P.; Almeida, P.R.C.; et al. Dual effect of silymarin on experimental non-alcoholic steatohepatitis induced by irinotecan. Toxicol. Appl. Pharm. 2017, 327, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.L.V.; Lima, R.C.P.; Aragão, K.S.; Medeiros, R.P.; Marques-Neto, R.D.; De Sá Grassi, L.; Leite, L.L.; Nunes, L.G.; De Mesquita Neto, J.W.B.; De Castro Brito, G.A.; et al. Chemotherapy-associated steatohepatitis induced by irinotecan: A novel animal model. Cancer Chemother. Pharmacol. 2014, 74, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Isaq, H.; Khan, Z.S.; Noureen, H. Effect of Cannabis sativa extract on the liver and cardic enzymes of normal healthy mice. KJMS 2016, 9, 117–119. [Google Scholar]
- Tothova, C.; Nagy, O.; Kovac, G. Serum proteins and their diagnostic utility in veterinary medicine: A review. Vet. Med. 2016, 61, 475–496. [Google Scholar] [CrossRef]
- Fanali, G.; Cao, Y.; Ascenzi, P.; Trezza, V.; Rubino, T.; Parolaro, D.; Fasano, M. Binding of Δ9-tetrahydrocannabinol and diazepam to human serum albumin. IUBMB Life 2011, 63, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Gupta, E.; Safa, A.R.; Wang, X.; Ratain, M.J. Pharmacokinetic modulation of irinotecan and metabolites by cyclosporin A. Cancer Res. 1996, 56, 1309–1314. [Google Scholar] [PubMed]
- Dinis-Oliveira, R.J. Metabolomics of Δ9-tetrahydrocannabinol: Implications in toxicity. Drug Metab. Rev. 2016, 48, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Yamaori, S.; Funahashi, T.; Kimura, T.; Yamamoto, I. Cytochrome P450 enzymes involved in the metabolism of tetrahydrocannabinols and cannabinol by human hepatic microsomes. Life Sci. 2007, 80, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- Kapitulnik, J. Bilirubin: An endogenous product of heme degradation with both cytotoxic and cytoprotective properties. Mol. Pharm. 2004, 66, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Kamisako, T.; Kobayashi, Y.; Takeuchi, K.; Ishihara, T.; Higuchi, K.; Tanaka, Y.; Gabazza, E.C.; Adachi, Y. Recent advances in bilirubin metabolism research: The molecular mechanism of hepatocyte bilirubin transport and its clinical relevance. J. Gastroenterol. 2000, 35, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Repetto, M.; Semprine, J.; Boveris, A. Lipid peroxidation: Chemical mechanism, biological implications and analytical determination. In Lipid Peroxidation; Catala, A., Ed.; IntechOpen Limited: London, UK, 2012; Volume I, pp. 3–30. ISBN 978-953-51-0716-3. [Google Scholar]
- Matés, J.M.; Pérez-Gómez, C.; De Castro, I.N. Antioxidant enzymes and human diseases. Clin. Biochem. 1999, 32, 595–603. [Google Scholar] [CrossRef]
- Alfonso-Prieto, M.; Vidossich, P.; Rovira, C. The reaction mechanisms of heme catalases: An atomistic view by ab initio molecular dynamics. Arch. Biochem. Biophys. 2012, 525, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Sadzuka, Y.; Hirota, S. Effect of CPT-11 on lipid peroxide level in mouse tissue. Jpn. J. Cancer Res. 1997, 88, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Vauthey, J.N.; Pawlik, T.M.; Ribero, D.; Wu, T.T.; Zorzi, D.; Hoff, P.M.; Xiong, H.Q.; Eng, C.; Lauwers, G.Y.; Mino-Kenudson, M.; et al. Chemotherapy regimen predicts steatohepatitis and an increase in 90-day mortality after surgery for hepatic colorectal metastases. J. Clin. Oncol. 2006, 24, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Van Houten, B.; Woshner, V.; Santos, J.H. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair 2006, 5, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Sarafian, T.A.; Kouyoumjian, S.; Khoshaghideh, F.; Tashkin, D.P.; Roth, M.D. Δ9-Tetrahydrocannabinol disrupts mitochondrial function and cell energetics. Am. J. Physiol. Cell. Mol. Physiol. 2003, 284, L298–L306. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wolff, V.; Schlagowski, A.-I.; Rouyer, O.; Charles, A.-L.; Singh, F.; Auger, C.; Schini-Kerth, V.; Marescaux, C.; Raul, J.-S.; Zoll, J.; et al. Tetrahydrocannabinol induces brain mitochondrial respiratory chain dysfunction and increases oxidative stress: A potential mechanism involved in cannabis-related stroke. Biomed. Res. Int. 2015, 2015, 323706. [Google Scholar] [CrossRef] [PubMed]
- Hézode, C.; Zafrani, E.S.; Roudot-Thoraval, F.; Costentin, C.; Hessami, A.; Bouvier-Alias, M.; Medkour, F.; Pawlostky, J.M.; Lotersztajn, S.; Mallat, A. Daily cannabis use: A novel risk factor of steatosis severity in patients with chronic hepatitis C. Gastroenterology 2008, 134, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Jurica, K.; Brčić Karačonji, I.; Benković, V.; Kopjar, N. In vitro assessment of the cytotoxic, DNA damaging, and cytogenetic effects of hydroquinone in human lymphocytes. Arh. Hig. Rada Toksikol. 2017, 68, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Azqueta, A.; Shaposhnikov, S.; Collins, A. DNA repair measured by the comet assay. In DNA Repair; Kruman, I., Ed.; Intech Open Limited: London, UK, 2011; Volume 1, pp. 615–636. ISBN 978-953-307-697-3. [Google Scholar]
- Collins, A.R. The comet assay for DNA damage and repair: principles, applications, and limitations. Mol. Biotechnol. 2004, 26, 249–261. [Google Scholar] [CrossRef]
- Tice, R.R.; Agurell, E.; Anderson, D.; Burlinson, B.; Hartmann, A.; Kobayashi, H.; Miyamae, Y.; Rojas, E.; Ryu, J.C.; Sasaki, Y.F. Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ. Mol. Mutagen. 2000, 35, 206–221. [Google Scholar] [CrossRef]
- Lisičić, D.; Benković, V.; Đikić, D.; Blažević, A.S.; Mihaljević, J.; Oršolić, N.; Knežević, A.H. Addition of propolis to irinotecan therapy prolongs survival in Ehrlich ascites tumor-bearing mice. Cancer Biother. Radiopharm. 2014, 29, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Horvat Knežević, A.; Đikić, D.; Lisičić, D.; Kopjar, N.; Oršolić, N.; Karabeg, S.; Benković, V. Synergistic effects of irinotecan and flavonoids on Ehrlich ascites tumour-bearing mice. Basic Clin. Pharm. Toxicol. 2011, 109, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Attia, S. Modulation of irinotecan-induced genomic DNA damage by theanine. Food Chem. Toxicol. 2012, 50, 1749–1754. [Google Scholar] [CrossRef] [PubMed]
- Lucić Vrdoljak, A.; Berend, S.; Želježić, D.; Piljac-Žegarac, J.; Pleština, S.; Kuča, K.; Radić, B.; Mladinić, M.; Kopjar, N. Irinotecan side effects relieved by the use of HI-6 oxime: in vivo experimental approach. Basic Clin. Pharm. Toxicol. 2009, 105, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Lin, L.F. Genetic toxicology of abused drugs: A brief review. Mutagenesis 1998, 13, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Marselos, M.; Karamanakos, P. Mutagenicity, developmental toxicity and carcinogenicity of cannabis. Addict. Biol. 1999, 4, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, S.; Zimmerman, A.M. Genetic effects of marijuana. Int. J. Addict. 1990, 25, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Reece, A.S.; Hulse, G.K. Chromothripsis and epigenomics complete causality criteria for cannabis- and addiction-connected carcinogenicity, congenital toxicity and heritable genotoxicity. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2016, 789, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Son, B.H.; Lee, S.Y.; Chung, K.H.; Oh, S.M. The role of p53 in marijuana smoke condensates-induced genotoxicity and apoptosis. Environ. Health Toxicol. 2012, 27, e2012017. [Google Scholar] [CrossRef] [PubMed]
- Schwilke, E.W.; Schwope, D.M.; Karschner, E.L.; Lowe, R.H.; Darwin, W.D.; Kelly, D.L.; Goodwin, R.S.; Gorelick, D.A.; Huestis, M.A. Δ9-tetrahydrocannabinol (THC), 11-hydroxy-THC, and 11-nor-9-carboxy-THC plasma pharmacokinetics during and after continuous high-dose oral THC. Clin. Chem. 2009, 55, 2180–2189. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, N.; Long, L.E.; Dawson, B.L.; Hansen, G.H.; Richardson, D.P.; Li, K.M.; Arnold, J.C.; McGregor, I.S. Reintoxication: The release of fat-stored Δ9-tetrahydrocannabinol (THC) into blood is enhanced by food deprivation or ACTH exposure. Br. J. Pharm. 2009, 158, 1330–1337. [Google Scholar] [CrossRef] [PubMed]
- Jamontt, J.M.; Molleman, A.; Pertwee, R.G.; Parsons, M.E. The effects of Δ9-tetrahydrocannabinol and cannabidiol alone and in combination on damage, inflammation and in vitro motility disturbances in rat colitis. Br. J. Pharm. 2010, 160, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Kumaravel, T.S.; Vilhar, B.; Faux, S.P.; Jha, A.N. Comet Assay measurements: A perspective. Cell Biol. Toxicol. 2009, 25, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Choucroun, P.; Gillet, D.; Dorange, G.; Sawicki, B.; Dewitte, J.D. Comet assay and early apoptosis. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2001, 478, 89–96. [Google Scholar] [CrossRef]
- Roser, S.; Pool-Zobel, B.L.; Rechkemmer, G. Contribution of apoptosis to responses in the comet assay. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2001, 497, 169–175. [Google Scholar] [CrossRef]
- Drury, J.A.; Nycyk, J.A.; Cooke, R.W.I. Comparison of urinary and plasma malondialdehyde in preterm infants. Clin. Chim. Acta 1997, 263, 177–185. [Google Scholar] [CrossRef]
- Flohé, L.; Ötting, F. Superoxide dismutase assays. Method Enzymol. 1984, 105, 93–104. [Google Scholar]
- Aebi, H. Oxygen radicals in biological systems. Method Enzymol. 1984, 105, 121–126. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Singh, N.P.; McCoy, M.T.; Tice, R.R.; Schneider, E.L. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res. 1988, 175, 184–191. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |













© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucić Vrdoljak, A.; Fuchs, N.; Mikolić, A.; Žunec, S.; Brčić Karačonji, I.; Jurič, A.; Prester, L.; Micek, V.; Neuberg, M.; Čanović, S.; et al. Irinotecan and Δ9-Tetrahydrocannabinol Interactions in Rat Liver: A Preliminary Evaluation Using Biochemical and Genotoxicity Markers. Molecules 2018, 23, 1332. https://doi.org/10.3390/molecules23061332
Lucić Vrdoljak A, Fuchs N, Mikolić A, Žunec S, Brčić Karačonji I, Jurič A, Prester L, Micek V, Neuberg M, Čanović S, et al. Irinotecan and Δ9-Tetrahydrocannabinol Interactions in Rat Liver: A Preliminary Evaluation Using Biochemical and Genotoxicity Markers. Molecules. 2018; 23(6):1332. https://doi.org/10.3390/molecules23061332
Chicago/Turabian StyleLucić Vrdoljak, Ana, Nino Fuchs, Anja Mikolić, Suzana Žunec, Irena Brčić Karačonji, Andreja Jurič, Ljerka Prester, Vedran Micek, Marijana Neuberg, Samir Čanović, and et al. 2018. "Irinotecan and Δ9-Tetrahydrocannabinol Interactions in Rat Liver: A Preliminary Evaluation Using Biochemical and Genotoxicity Markers" Molecules 23, no. 6: 1332. https://doi.org/10.3390/molecules23061332
APA StyleLucić Vrdoljak, A., Fuchs, N., Mikolić, A., Žunec, S., Brčić Karačonji, I., Jurič, A., Prester, L., Micek, V., Neuberg, M., Čanović, S., Mršić, G., & Kopjar, N. (2018). Irinotecan and Δ9-Tetrahydrocannabinol Interactions in Rat Liver: A Preliminary Evaluation Using Biochemical and Genotoxicity Markers. Molecules, 23(6), 1332. https://doi.org/10.3390/molecules23061332