High Level Expression and Purification of the Clinically Active Antimicrobial Peptide P-113 in Escherichia coli


Abstract
1. Introduction
2. Results
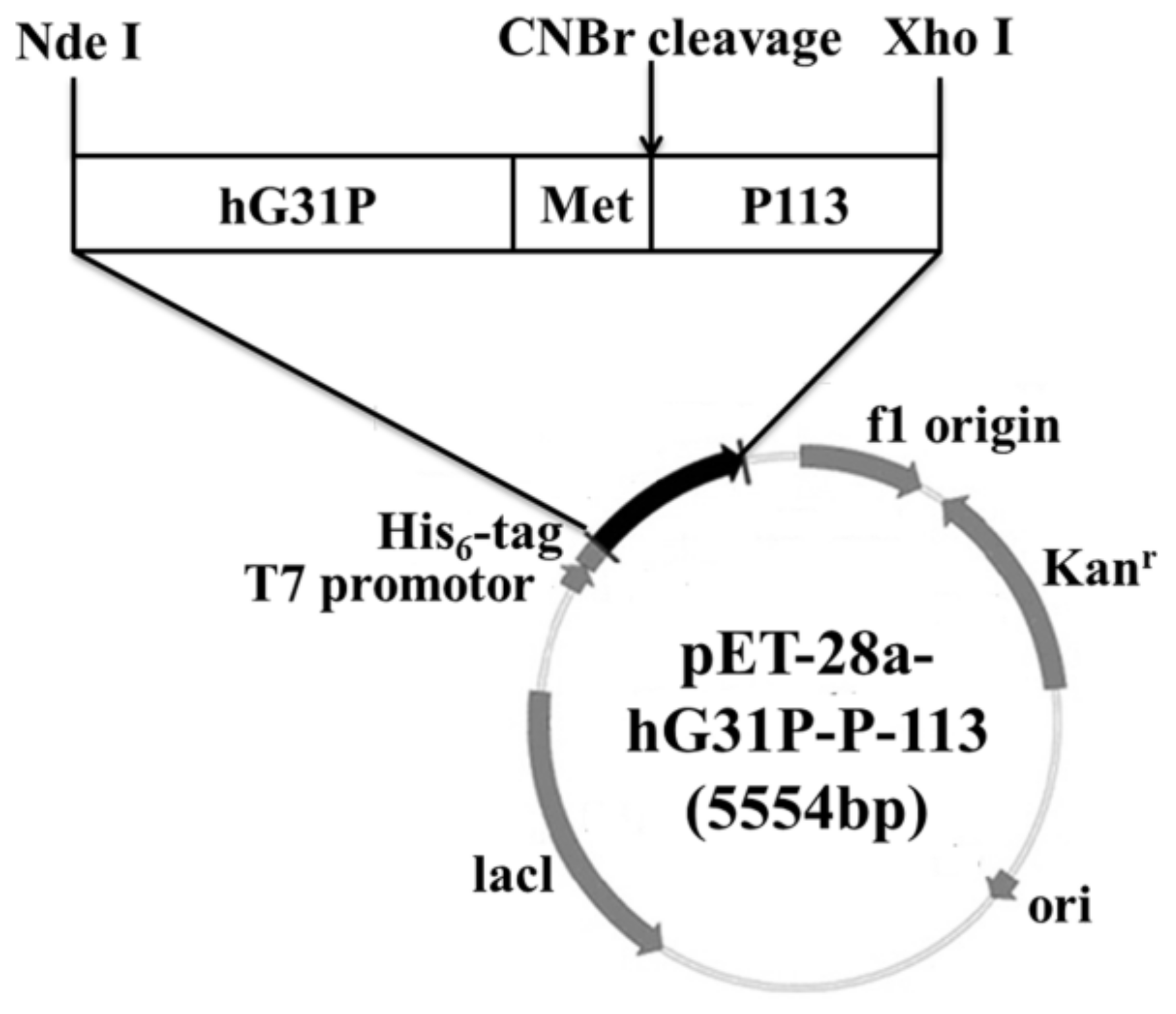
2.1. Construction of the Recombinant Plasmid
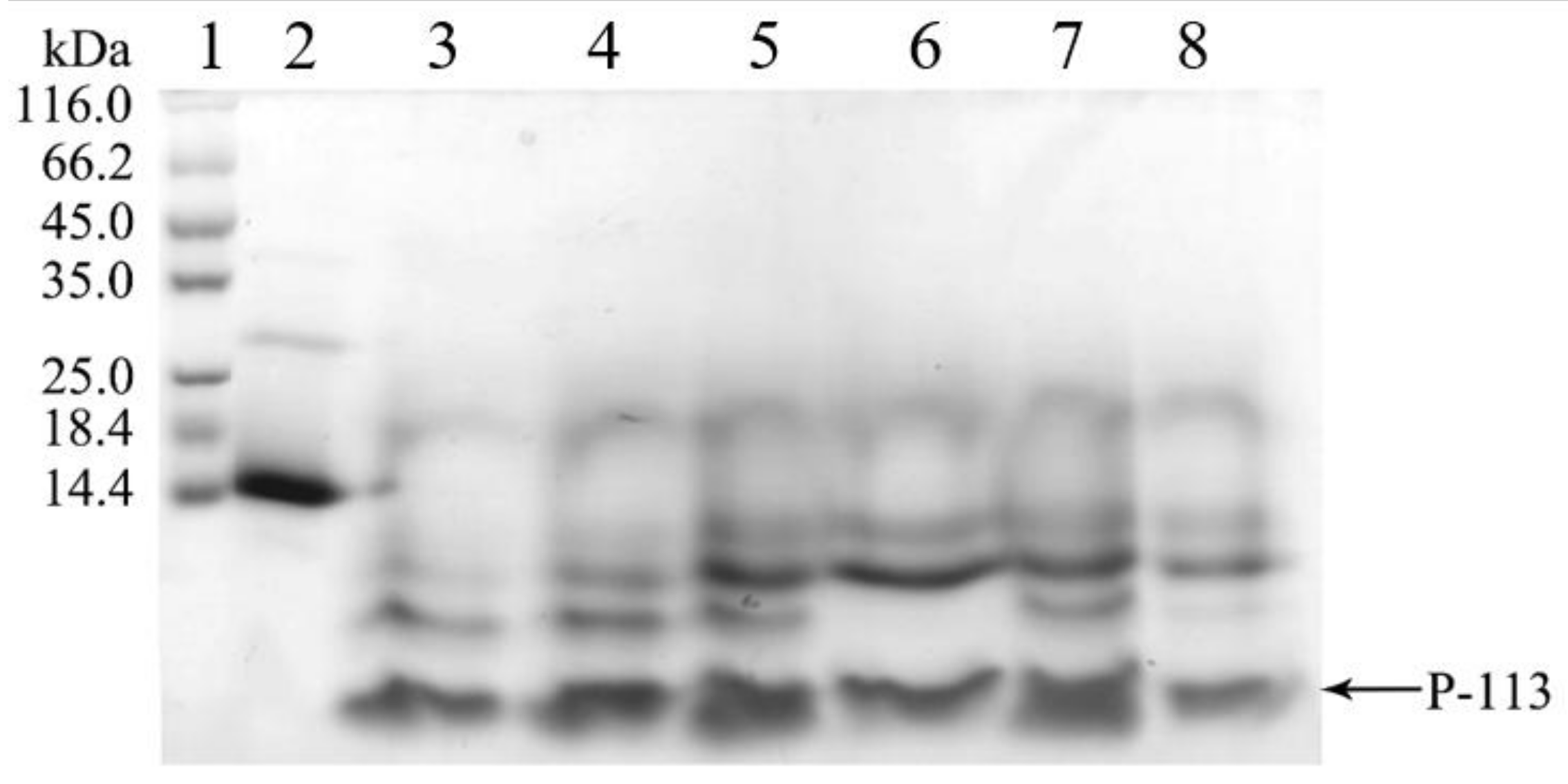
2.2. Expression, Extraction and Purification of hG31P-P-113
2.3. Quantification and Characterization of P-113
2.4. Activity Assays
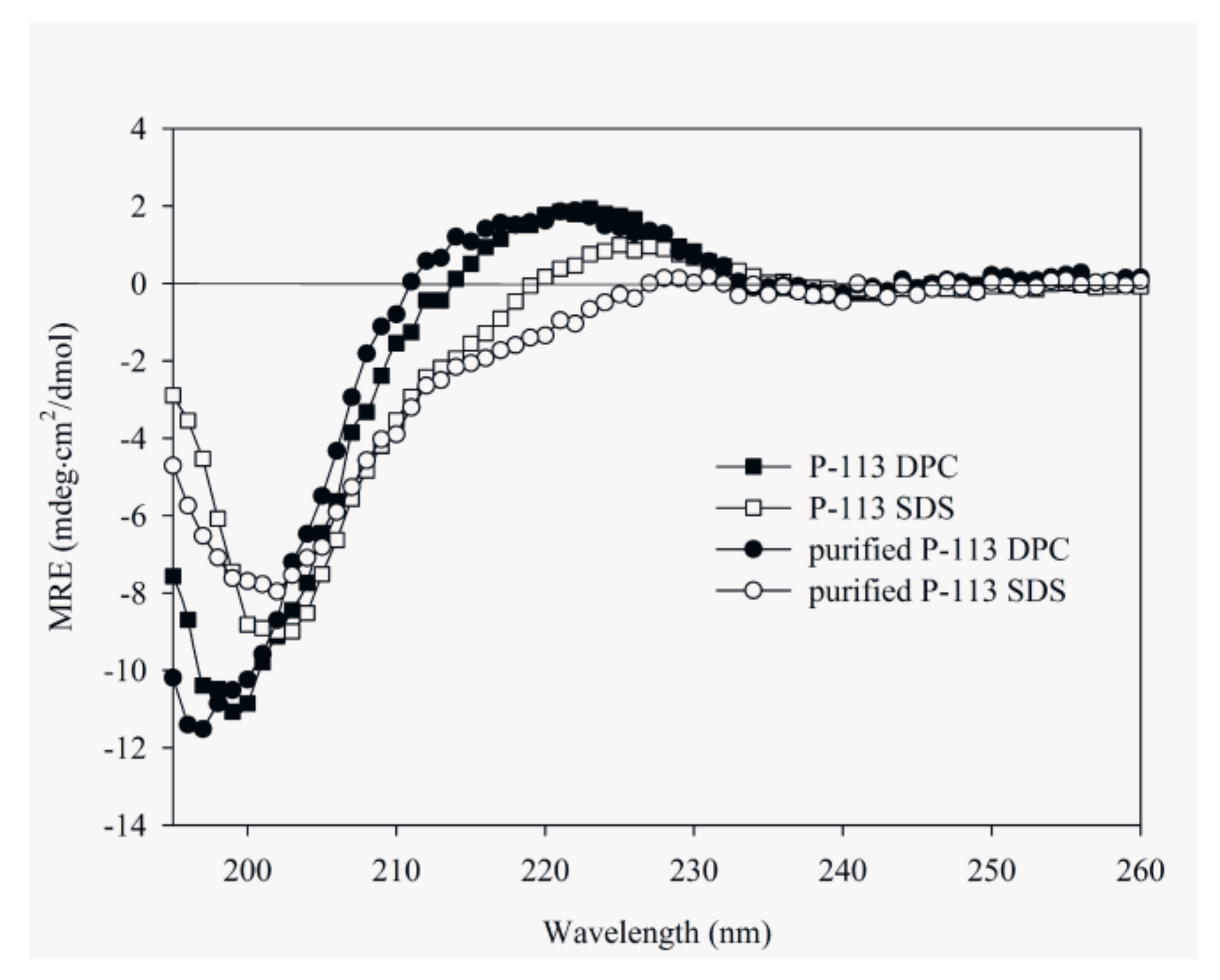
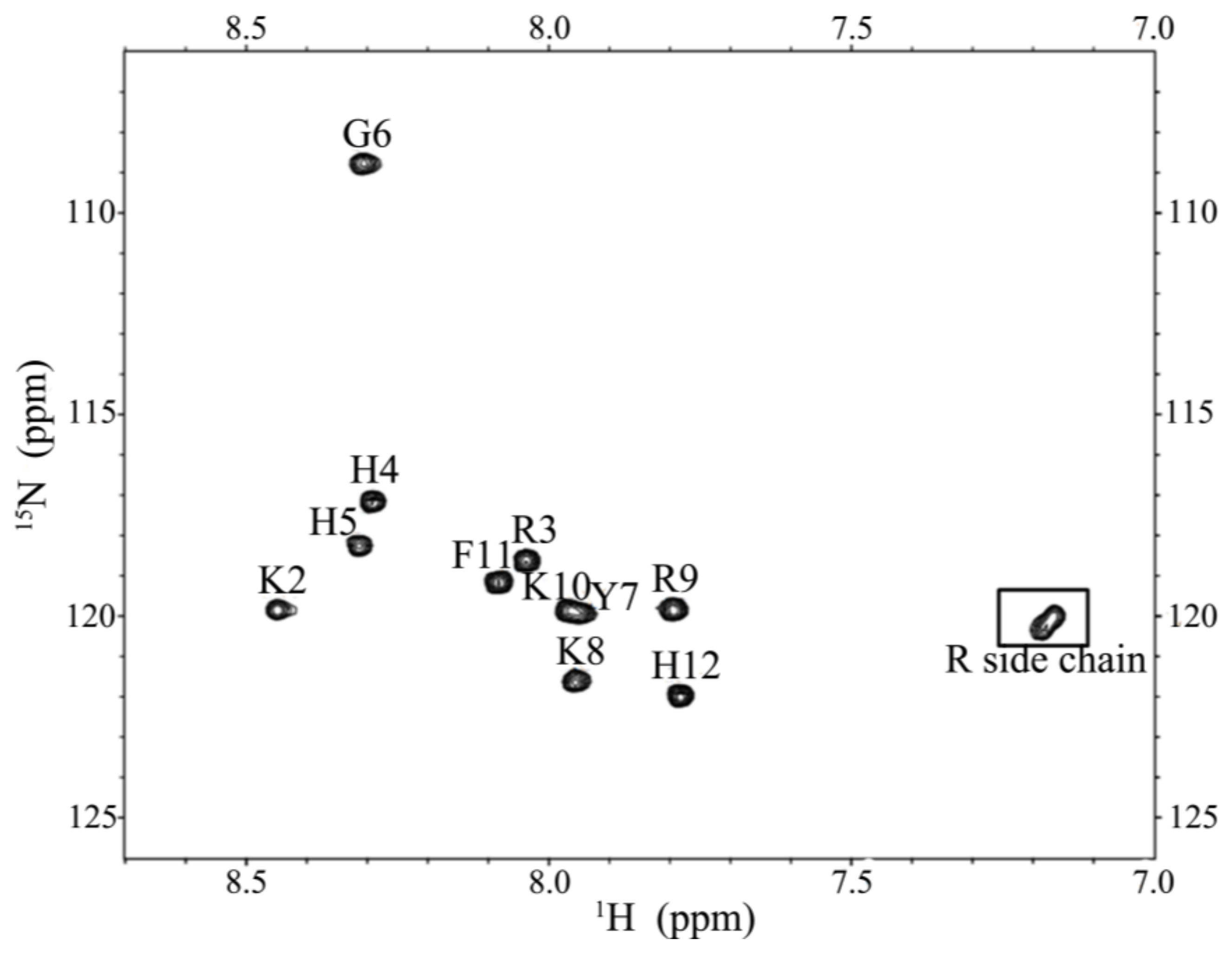
2.5. Characterization of the Expressed P-113 by CD and NMR
3. Discussion
4. Materials and Methods
4.1. Construction of the Expression Plasmid
4.2. Expression of the Recombinant hG31P-P-113
4.3. Lysis of Cells and Purification of Recombinant hG31P-P-113
4.4. Cleavage of hG31P-P-113
4.5. Purification of P-113 by HPLC
4.6. Antimicrobial Activity Assays
4.7. Circular Dichroism Spectroscopy (CD)
4.8. Nuclear Magnetic Resonance Spectroscopy (NMR)
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Andreu, D.; Rivas, L. Animal antimicrobial peptides: An overview. Biopolymers 1998, 47, 415–433. [Google Scholar] [CrossRef]
- Tossi, A.; Sandri, L.; Giangaspero, A. Amphipathic, a-helical antimicrobial peptides. Biopolymers 2000, 55, 4–30. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, D.M.; Spacciapoli, P.; Tran, L.T.; Xu, T.; Roberts, F.D.; Dalla Serra, M.; Buxton, D.K.; Oppenheim, F.G.; Friden, P. Anticandida activity is retained in P-113, a 12-amino-acid fragment of histatin 5. Antimcrob. Agents Chemother. 2001, 45, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Helmerhorst, E.J.; Oppenheim, F.G.; Choi, L.; Cheng, J.W.; Reiner, N.E. Evaluation of a new host-derived synthetic antifungal peptide (PAC-113) in the treatment of oral candidiasis. In Proceedings of the International Meeting on Antimicrobial Chemotherapy in Clinical Practice (ACCP), Poster, Portofino, Italy, 15–17 November 2007. [Google Scholar]
- Mickels, N.; McMamus, C.; Massaro, J.; Friden, P.; Braman, V.; D’Agostino, R.; Oppenheim, F.G.; Warbington, M.; Dibart, S.; Van Dyke, T. Clinical and microbial evaluation of a histatin-containing mouthrinse in humans with experimental gingivitis. J. Clin. Periodontol. 2001, 28, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Paquette, D.W.; Simpson, D.M.; Friden, P.; Braman, V.; Williams, R.C. Safety and clinical effects of topical histatin gels in humans with experimental gingivitis. J. Clin. Periodontol. 2002, 29, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Van Dyke, T.; Paquette, D.; Grossi, S.; Braman, V.; Massaro, J.; D’Agostino, R.; Dibart, S.; Friden, P. Clinical and microbial evaluation of a histatin-containing mouthrinse in humans with experimental gingivitis: A phase-2 multi-center study. J. Clin. Periodontol. 2002, 29, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Andersson, L.; Blomberg, L.; Flegel, M.; Lepsa, L.; Nilsson, B.; Verlander, M. Large-Scale Synthesis of Peptides. Biopolym. Pept. Sci. 2000, 55, 227–250. [Google Scholar] [CrossRef]
- Li, Y. Recombinant production of antimicrobial peptides in Escherichia coli: A review. Protein Expr. Purif. 2011, 80, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Li, J.F.; Zhang, J.; Song, R.; Zhang, J.X.; Shen, Y.; Zhang, S.Q. Production of a cytotoxic cationic antibacterial peptide in Escherichia coli using SUMO fusion partner. Appl. Microbiol. Biotechnol. 2009, 84, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Aleinein, R.A.; Hamoud, R.; Schafer, H.; Wink, M. Molecular cloning and expression of ranalexin, a bioactive antimicrobial peptide from Rana catesbeiana in Escherichia coli and assessments of its biological activities. Appl. Microbiol. Biotechnol. 2013, 97, 3535–3543. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Kim, Y.S.; Seo, J.H.; Jang, W.S.; Lee, I.H.; Cha, H.J. Facilitation of expression and purification of an antimicrobial peptide by fusion with baculoviral polyhedrin in Escherichia coli. Appl. Environ. Microbiol. 2005, 71, 5038–5043. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hara, S.; Yamakawa, M. Production in Escherichia coli of moricin, a novel type antibacterial peptide from the silkworm Bombyx mori. Biochem. Biophys. Res. Commun. 1996, 220, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Skosyrev, V.S.; Rudenko, V.; Yakhnin, A.V.; Zagranichny, V.E.; Popova, L.I.; Zakharov, M.V.; Gorokhovatsky, A.Y.; Vinokurov, L.M. EGFP as a fusion partner for the expression and organic extraction of small polypeptides. Protein Expr. Purif. 2003, 27, 55–62. [Google Scholar] [CrossRef]
- Zhang, L.; Falla, T.J.; Wu, M.; Fidai, S.; Burian, J.; Kay, W.; Hancock, R.E.W. Determinants of recombinant production of antimicrobial cationic peptides and creation of peptide variants in bacteria. Biochem. Biophys. Res. Commun. 1998, 247, 674–680. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pyo, S.H.; Lee, J.H.; Park, H.B.; Cho, J.S.; Kim, H.R.; Han, B.H.; Park, Y.S. Expression and purification of a recombinant buforin derivative from Escherichia coli. Process Biochem. 2004, 39, 1731–1736. [Google Scholar] [CrossRef]
- Wang, H.; Meng, X.L.; Xu, J.P.; Wang, J.; Wang, H.; Ma, C.W. Production, purification, and characterization of the cecropin from Plutella xylostella, pxCECA1, using an intein-induced self-cleavable system in Escherichia coli. Appl. Microbiol. Biotechnol. 2012, 94, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, A.; Moon, J.Y.; Henzler-Wildman, K.A. Expression and purification of a recombinant LL-37 from Escherichia coli. Biochim. Biophys. Acta 2006, 1758, 1351–1358. [Google Scholar]
- Rao, X.C.; Li, S.; Hu, J.C.; Jin, X.L.; Hu, X.M.; Huang, J.J.; Chen, Z.J.; Zhu, J.M.; Hu, F.Q. A novel carrier molecule for high-level expression of peptide antibiotics in Escherichia coli. Protein Expr. Purif. 2004, 36, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Chun, D.S.; Kim, J.S.; Yun, C.H.; Lee, J.H.; Hong, S.K.; Kang, D.K. Expression of the cationic antimicrobial peptide lactoferricin fused with the anionic peptide in Escherichia coli. Appl. Microbiol. Biotechnol. 2006, 72, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Ishida, H.; Nguyen, L.T.; Gopal, R.; Aizawa, T.; Vogel, H.J. Overexpression of antimicrobial, anticancer, and transmembrane peptides in Escherichia coli through a calmodulin-peptide fusion system. J. Am. Chem. Soc. 2016, 138, 11318–11326. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.T.; Huang, K.C.; Yu, H.Y.; Gao, K.J.; Zhao, X.; Li, F.; Town, J.R.; Gordon, J.R.; Cheng, J.W. A new protocol for high-yield purification of recombinant human CXCL8(3-72)K11R/G31P expressed in Escherichia coli. Protein Expr. Purif. 2008, 61, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Mura, M.; Wang, J.; Zhou, Y.; Pinna, M.; Zvelindovsky, A.V.; Dennison, S.R.; Phoenix, D.A. The effect of amidation on the behaviour of antimicrobial peptides. Eur. Biophys. J. 2016, 45, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Yip, B.S.; Chen, H.L.; Cheng, H.T.; Wu, J.M.; Cheng, J.W. Solution structure and model membrane interactions of P-113, a clinically active antimicrobial peptide derived from human saliva. J. Chin. Chem. Soc. 2009, 56, 961–966. [Google Scholar] [CrossRef]
- Fox, J.L. Antimicrobial peptides stage a comeback. Nat. Biotechnol. 2013, 31, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Liu, C.; Guo, J.; Song, X.; Li, J.; Xu, W.; Li, Z. Recombinant expression, purification, and antimicrobial activity of a novel hybrid antimicrobial peptide LFT33. Appl. Microbiol. Biotechnol. 2012, 95, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Marques, L.; Oomen, R.J.F.J.; Aumelas, A.; Jean, M.L.; Berthomieu, P. Production of an Arobidopsis halleri foliar defensin in Esherichia coli. J. Applied Microbiol. 2009, 106, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhu, F.; Xin, Y.; Liu, J.; Luo, L.; Yin, Z. Expression and purification of antimicrobial peptide fuforin IIb in Escherichia coli. Biotechnol. Lett. 2011, 33, 2121–2126. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.W.; Yip, B.S.; Cheng, H.T.; Wang, A.H.; Chen, H.L.; Cheng, J. W.; Lo, H.J. Increased potency of a novel d-b-naphthylalanine-substituted antimicrobial peptide against fluconazole-resistant fungal pathogens. FEMS Yeast Res. 2009, 9, 967–970. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Candida sp. | Source | Strain * | Purified P-113 | Synthetic P-113 * |
|---|---|---|---|---|
| C. krusei | ATCC 6258 | YLO6 | 6.25 | 1.56 |
| C. albicans | ATCC 90028 | YLO12 | 6.25 | 1.56 |
| C. tropicalis | ATCC 13803 | YLO86 | 3.13 | 0.78 |
| C. albicans | HIV patient | YH050001 | 6.25 | 1.56 |
| C. dubliniensis | HIV patient | YH050092 | 6.25 | 1.56 |
| C. glabrata | HIV patient | YH050105 | 12.5 | 3.13 |
| C. krusei | HIV patient | YH050075 | 6.25 | 1.56 |
| C. tropicalis | HIV patient | YH050007 | 3.13 | 1.56 |
| C. tropicalis | HIV patient | YH050013 | 3.13 | 0.78 |
| C. tropicalis | HIV patient | YH050114 | 3.13 | 1.56 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, K.-T.; Wu, C.-L.; Yip, B.-S.; Yu, H.-Y.; Cheng, H.-T.; Chih, Y.-H.; Cheng, J.-W. High Level Expression and Purification of the Clinically Active Antimicrobial Peptide P-113 in Escherichia coli. Molecules 2018, 23, 800. https://doi.org/10.3390/molecules23040800
Cheng K-T, Wu C-L, Yip B-S, Yu H-Y, Cheng H-T, Chih Y-H, Cheng J-W. High Level Expression and Purification of the Clinically Active Antimicrobial Peptide P-113 in Escherichia coli. Molecules. 2018; 23(4):800. https://doi.org/10.3390/molecules23040800
Chicago/Turabian StyleCheng, Kuang-Ting, Chih-Lung Wu, Bak-Sau Yip, Hui-Yuan Yu, Hsi-Tsung Cheng, Ya-Han Chih, and Jya-Wei Cheng. 2018. "High Level Expression and Purification of the Clinically Active Antimicrobial Peptide P-113 in Escherichia coli" Molecules 23, no. 4: 800. https://doi.org/10.3390/molecules23040800
APA StyleCheng, K.-T., Wu, C.-L., Yip, B.-S., Yu, H.-Y., Cheng, H.-T., Chih, Y.-H., & Cheng, J.-W. (2018). High Level Expression and Purification of the Clinically Active Antimicrobial Peptide P-113 in Escherichia coli. Molecules, 23(4), 800. https://doi.org/10.3390/molecules23040800