
A QM/MM Study of Nitrite Binding Modes in a Three-Domain Heme-Cu Nitrite Reductase


Abstract

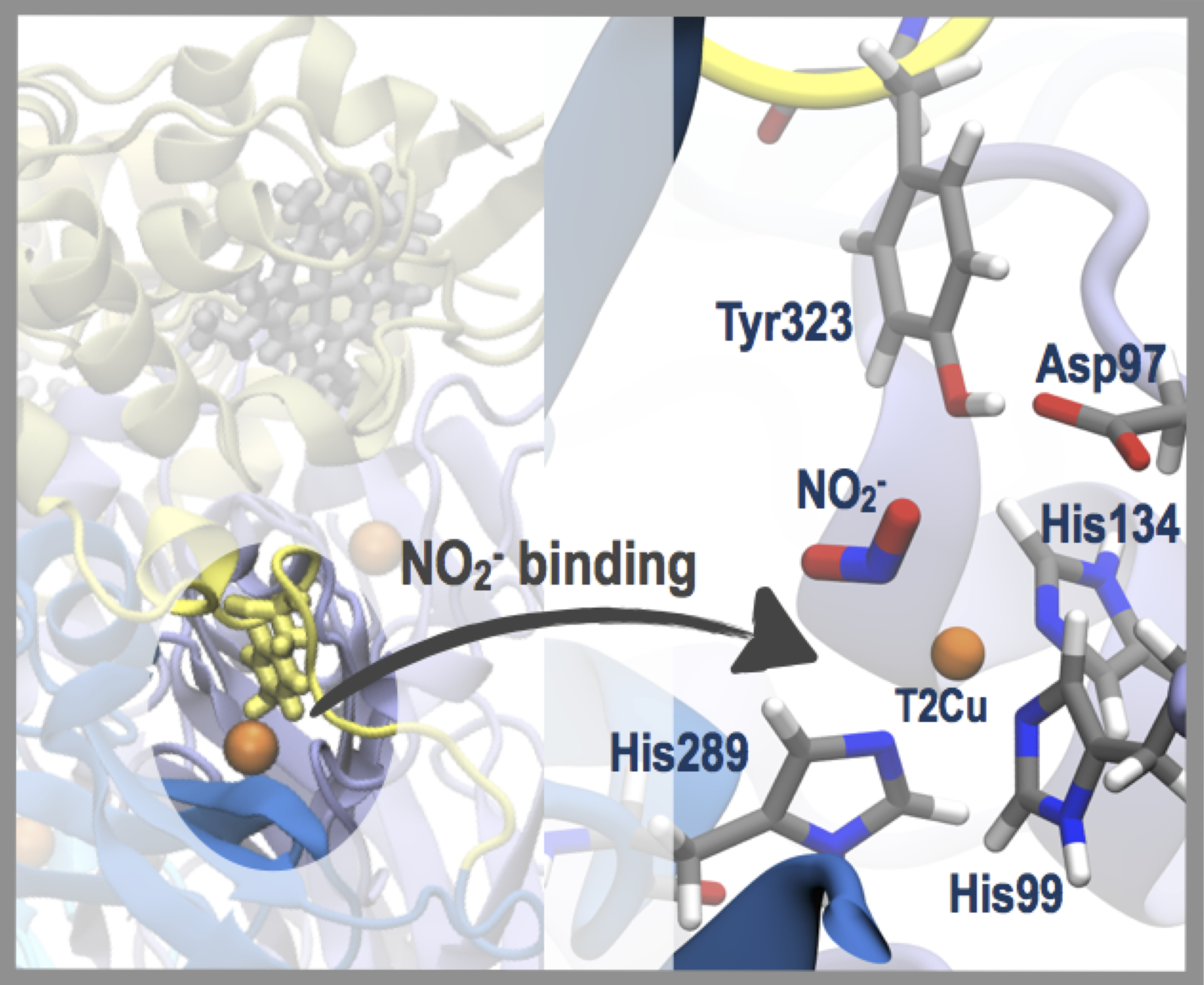
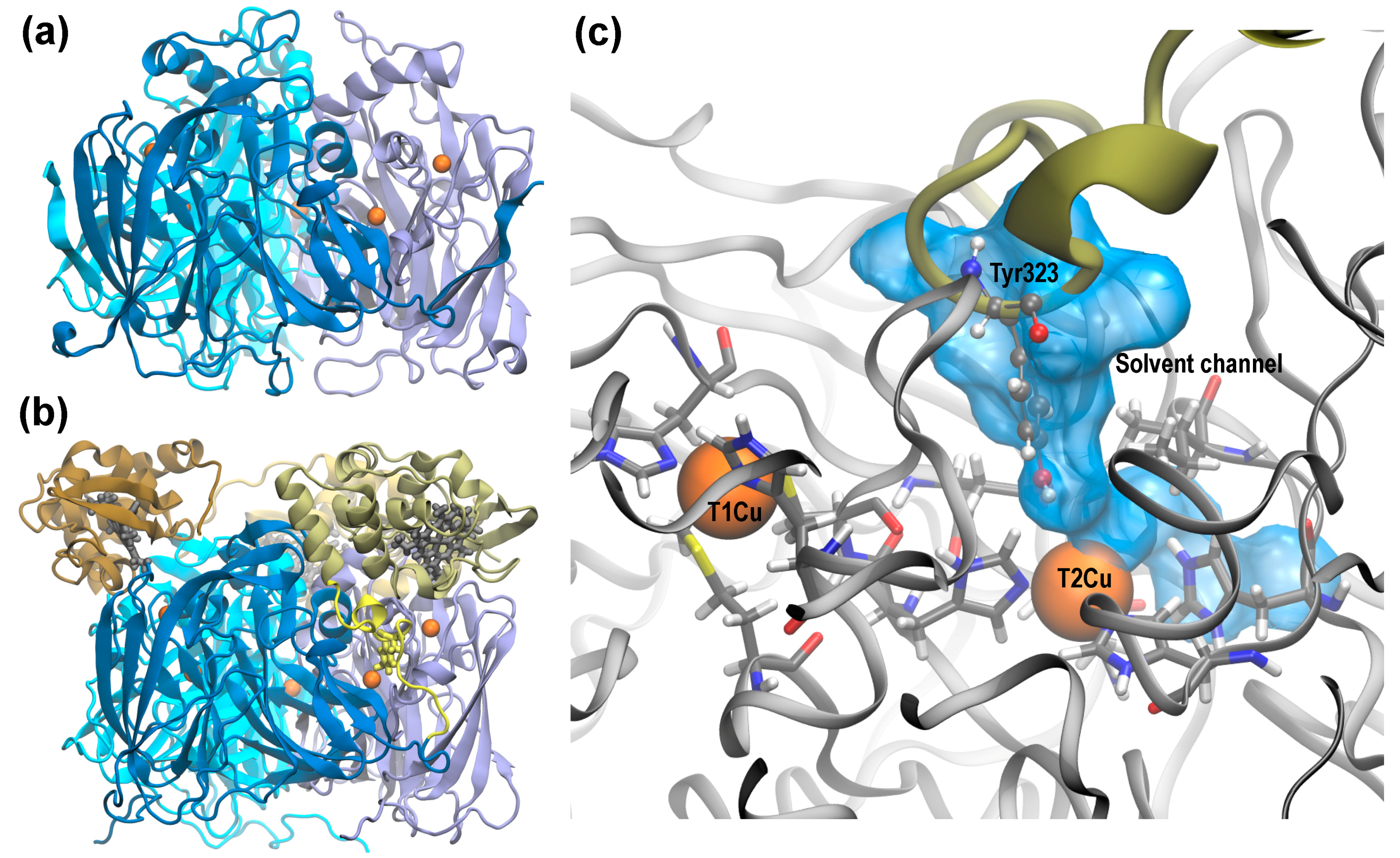
1. Introduction
2. Results
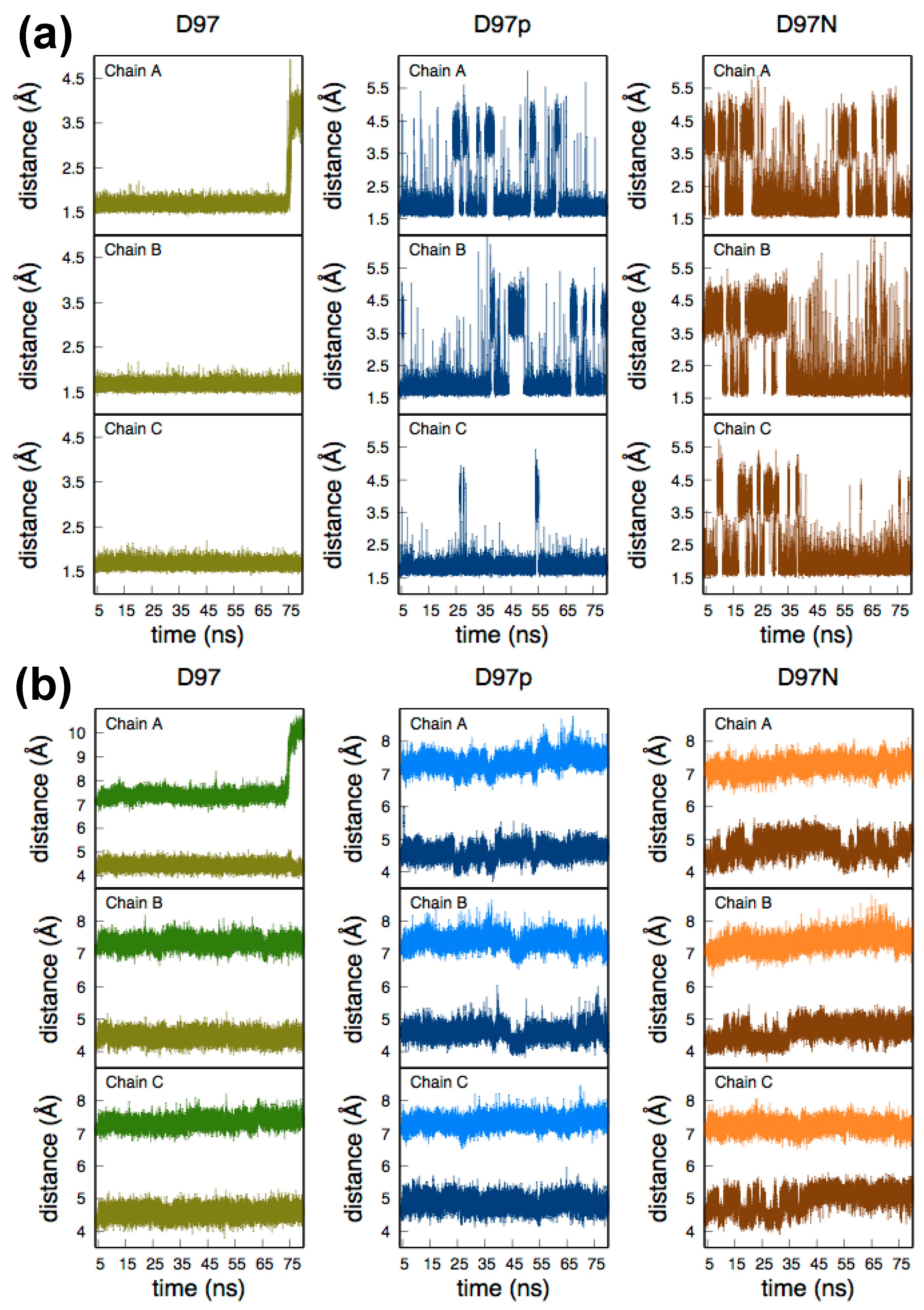
2.1. Molecular Dynamics of Native and D97N RpNiR in the Resting State
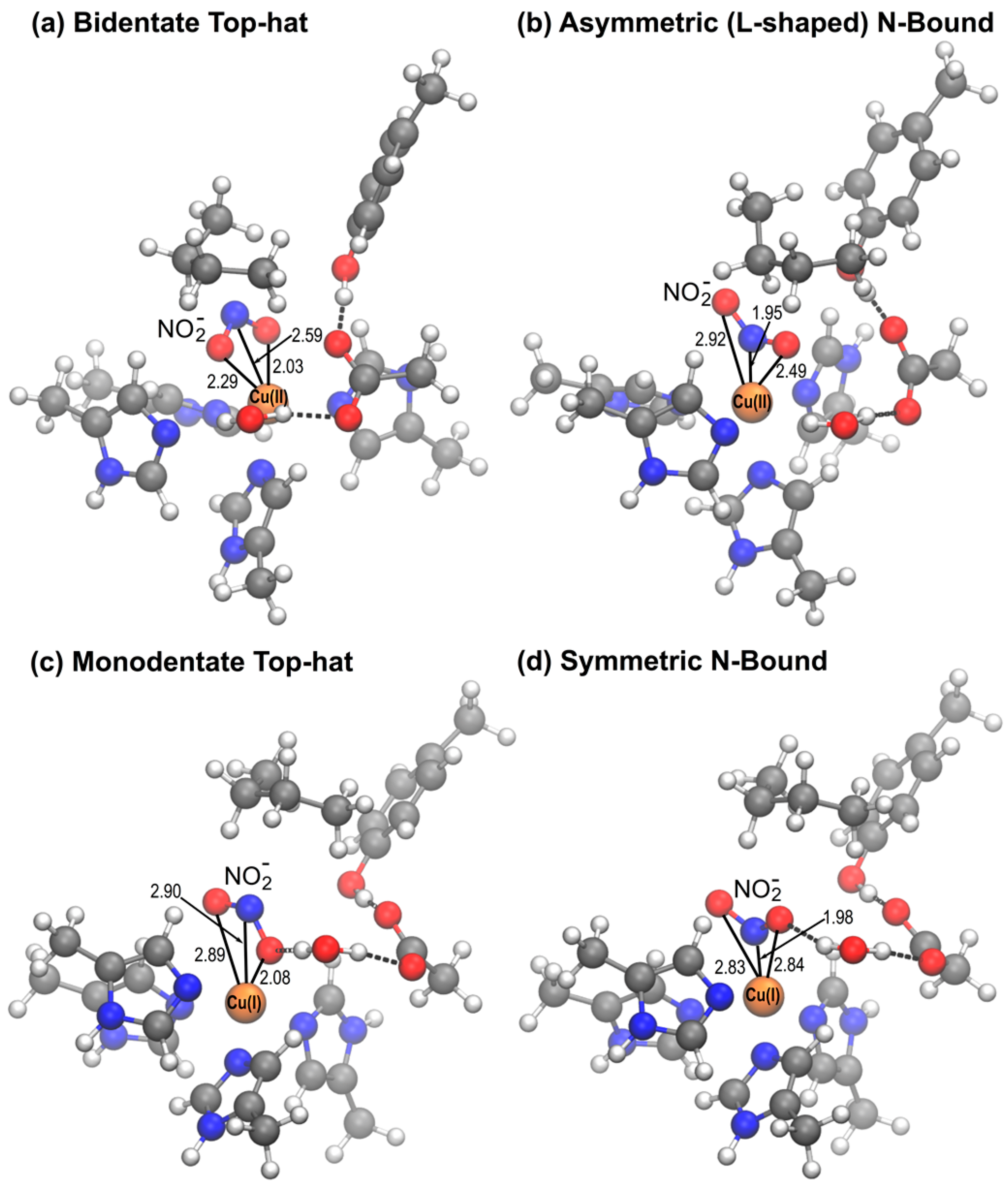
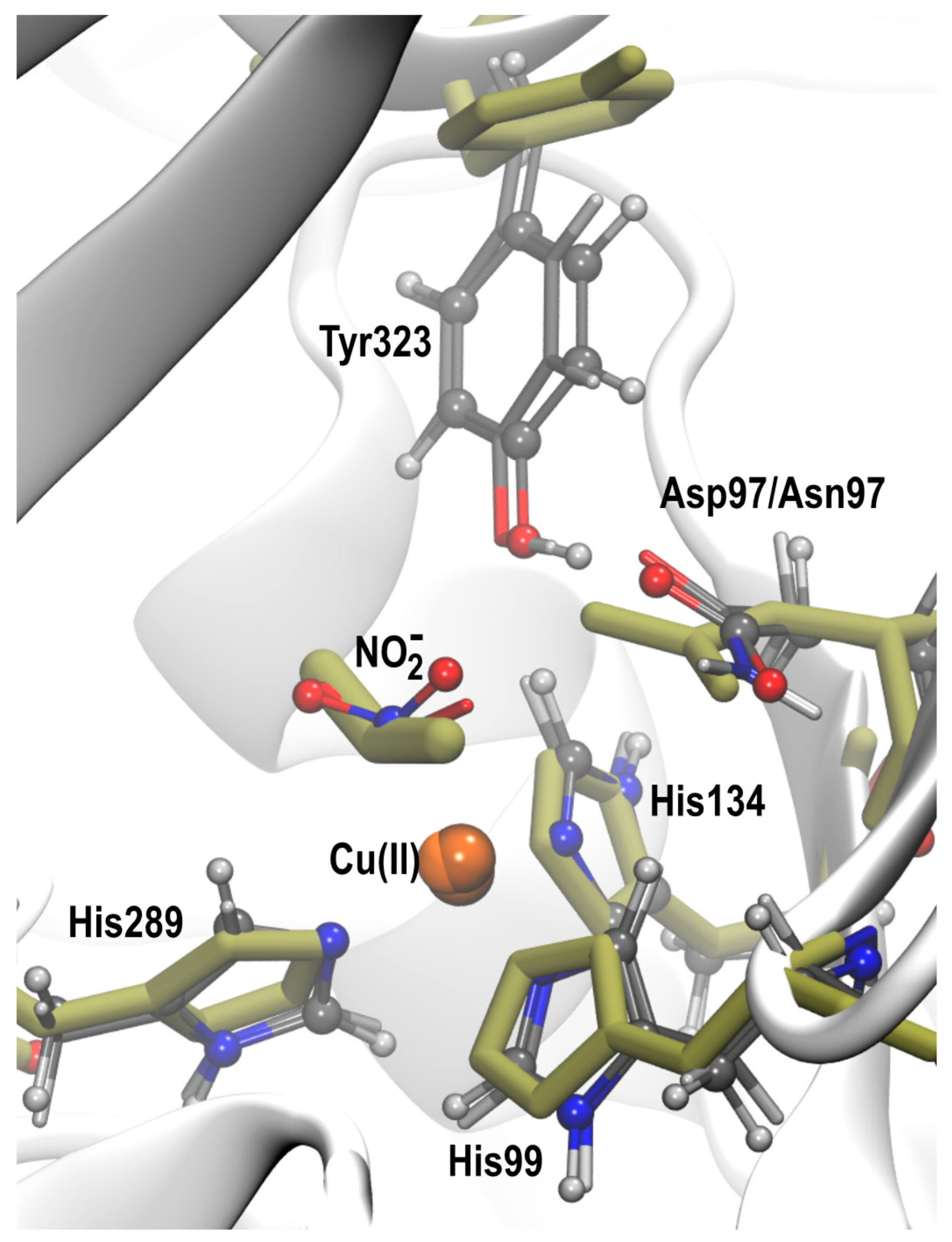
2.2. QM/MM Optimization of NO2− Bound Structures
2.2.1. D97 RpNiR
2.2.2. D97p RpNiR
2.2.3. D97N Mutant RpNiR
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maia, L.B.; Moura, J.J. How biology handles nitrite. Chem. Rev. 2014, 114, 5273–5357. [Google Scholar] [CrossRef] [PubMed]
- Zumft, W.G. Cell biology and the molecular basis of denitrification. Microbiol. Mol. Biol. Rev. 1997, 61, 533–616. [Google Scholar] [PubMed]
- Eady, R.R.; Hasnain, S.S. Comprehensive Coordination Chemistry II: Bio-Coordination Chemistry; Que, L.T.W., Tolman, W., Eds.; Elsevier: Oxford, UK, 2003; Volume 8, pp. 759–786. [Google Scholar]
- Nojiri, M.; Xie, Y.; Inoue, T.; Yamamoto, T.; Matsumura, H.; Kataoka, K.; Deligeer; Yamaguchi, K.; Kai, Y.; Suzuki, S. Structure and function of a hexameric copper-containing nitrite reductase. Proc. Natl. Acad. Sci. USA 2007, 104, 4315–4320. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Kataoka, K.; Kobayashi, M.; Itoh, K.; Fukui, A.; Suzuki, S. Characterization of two type 1 Cu sites of Hyphomicrobium denitrificans nitrite reductase: A new class of copper-containing nitrite reductases. Biochemistry 2004, 43, 14180–14188. [Google Scholar] [CrossRef] [PubMed]
- Bertini, I.; Cavallaro, G.; Rosato, A. Cytochrome c: Occurrence and functions. Chem. Rev. 2006, 106, 90–115. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Wright, G.S.; Fisher, K.; Rigby, S.E.; Eady, R.R.; Hasnain, S.S. Characterization of a novel copper-haem c dissimilatory nitrite reductase from Ralstonia pickettii. Biochem. J. 2012, 444, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.J.; Grossmann, J.G.; Eady, R.R.; Hasnain, S.S. Genomic analysis reveals widespread occurrence of new classes of copper nitrite reductases. J. Biol. Inorg. Chem. 2007, 12, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Antonyuk, S.V.; Han, C.; Eady, R.R.; Hasnain, S.S. Structures of protein-protein complexes involved in electron transfer. Nature 2013, 496, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Antonyuk, S.V.; Strange, R.W.; Sawers, G.; Eady, R.R.; Hasnain, S.S. Atomic resolution structures of resting-state, substrate- and product-complexed Cu-nitrite reductase provide insight into catalytic mechanism. Proc. Natl. Acad. Sci. USA 2005, 102, 12041–12046. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.J.; Dodd, F.E.; Sawers, G.; Eady, R.R.; Hasnain, S.S. Atomic resolution structures of native copper nitrite reductase from Alcaligenes xylosoxidans and the active site mutant Asp92Glu. J. Mol. Biol. 2003, 328, 429–438. [Google Scholar] [CrossRef]
- Leferink, N.G.; Antonyuk, S.V.; Houwman, J.A.; Scrutton, N.S.; Eady, R.R.; Hasnain, S.S. Impact of residues remote from the catalytic centre on enzyme catalysis of copper nitrite reductase. Nat. Commun. 2014, 5, 4395. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, Y.; Tse, K.M.; Lintuluoto, M.; Fukunishi, Y.; Mizohata, E.; Matsumura, H.; Takami, H.; Nojiri, M.; Inoue, T. Structural insights into the function of a thermostable copper-containing nitrite reductase. J. Biochem. 2014, 155, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Sen, K.; Horrell, S.; Kekilli, D.; Yong, C.W.; Keal, T.W.; Atakisi, H.; Moreau, D.W.; Thorne, R.E.; Hough, M.A.; Strange, R.W. Active-site protein dynamics and solvent accessibility in native Achromobacter cycloclastes copper nitrite reductase. IUCrJ 2017, 4, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Tocheva, E.I.; Eltis, L.D.; Murphy, M.E. Conserved active site residues limit inhibition of a copper-containing nitrite reductase by small molecules. Biochemistry 2008, 47, 4452–4460. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, M.J.; Kukimoto, M.; Nishiyama, M.; Horinouchi, S.; Murphy, M.E. Catalytic roles for two water bridged residues (Asp-98 and His-255) in the active site of copper-containing nitrite reductase. J. Biol. Chem. 2000, 275, 23957–23964. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.J.; Prudencio, M.; Dodd, F.E.; Strange, R.W.; Sawers, G.; Eady, R.R.; Hasnain, S.S. Biochemical and crystallographic studies of the Met144Ala, Asp92Asn and His254Phe mutants of the nitrite reductase from Alcaligenes xylosoxidans provide insight into the enzyme mechanism. J. Mol. Biol. 2002, 316, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Furusawa, H.; Takagi, K.; Yamaguchi, K.; Suzuki, S. Functional Analysis of Conserved Aspartate and Histidine Residues Located Around the Type 2 Copper Site of Copper-Containing Nitri Reductase. J. Biochem. 2000, 127, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Prudencio, M.; Eady, R.R.; Sawers, G. Catalytic and spectroscopic analysis of blue copper-containing nitrite reductase mutants altered in the environment of the type 2 copper centre: Implications for substrate interaction. Biochem. J. 2001, 353, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Sasaki, D.; Eady, R.R.; Antonyuk, S.V.; Hasnain, S.S. Identification of a tyrosine switch in copper-haem nitrite reductases. IUCrJ 2018, 5, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Horrell, S.; Kekilli, D.; Sen, K.; Owen, R.L.; Dworkowski, F.S.N.; Antonyuk, S.V.; Keal, T.W.; Yong, C.W.; Eady, R.R.; Hasnain, S.S.; et al. Enzyme catalysis captured using multiple structures from one crystal at varying temperatures. IUCrJ 2018, 5, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, M.J.; Murphy, M.E. Directing the mode of nitrite binding to a copper-containing nitrite reductase from Alcaligenes faecalis S-6: Characterization of an active site isoleucine. Protein Sci. 2003, 12, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Dodd, F.E.; Hasnain, S.S.; Abraham, Z.H.; Eady, R.R.; Smith, B.E. Structures of a blue-copper nitrite reductase and its substrate-bound complex. Acta Crystallogr. D Biol. Crystallogr. 1997, 53, 406–418. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S.; Heyes, D.J.; Hay, S.; Hough, M.A.; Eady, R.R.; Hasnain, S.S.; Scrutton, N.S. Demonstration of proton-coupled electron transfer in the copper-containing nitrite reductases. J. Biol. Chem. 2009, 284, 25973–25983. [Google Scholar] [CrossRef] [PubMed]
- Hough, M.A.; Antonyuk, S.V.; Strange, R.W.; Eady, R.R.; Hasnain, S.S. Crystallography with online optical and X-ray absorption spectroscopies demonstrates an ordered mechanism in copper nitrite reductase. J. Mol. Biol. 2008, 378, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Dey, A.; Sun, Y.; Scholes, C.P.; Solomon, E.I. Spectroscopic and computational studies of nitrite reductase: Proton induced electron transfer and backbonding contributions to reactivity. J. Am. Chem. Soc. 2009, 131, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Wijma, H.J.; Jeuken, L.J.; Verbeet, M.P.; Armstrong, F.A.; Canters, G.W. A random-sequential mechanism for nitrite binding and active site reduction in copper-containing nitrite reductase. J. Biol. Chem. 2006, 281, 16340–16346. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, R.H.; Tabares, L.C.; Kostrz, D.; Dennison, C.; Aartsma, T.J.; Canters, G.W.; Moerner, W.E. Redox cycling and kinetic analysis of single molecules of solution-phase nitrite reductase. Proc. Natl. Acad. Sci. USA 2011, 108, 17269–17274. [Google Scholar] [CrossRef] [PubMed]
- Källrot, N.; Nilsson, K.; Rasmussen, T.; Ryde, U. Theoretical study of structure of catalytic copper site in nitrite reductase. Int. J. Quantum Chem. 2005, 102, 520–541. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Ryde, U.; Roos, B.O.; Pierloot, K. On the relative stability of tetragonal and trigonal Cu(II) complexes with relevance to the blue copper proteins. J. Biol. Inorg. Chem. 1998, 3, 109–125. [Google Scholar] [CrossRef]
- Sundararajan, M.; Hillier, I.H.; Burton, N.A. Mechanism of nitrite reduction at T2Cu centers: Electronic structure calculations of catalysis by copper nitrite reductase and by synthetic model compounds. J. Phys. Chem. B 2007, 111, 5511–5517. [Google Scholar] [CrossRef] [PubMed]
- Silaghi-Dumitrescu, R. Copper-containing nitrite reductase: A DFT study of nitrite and nitric oxide adducts. J. Inorg. Biochem. 2006, 100, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Horrell, S.; Antonyuk, S.V.; Eady, R.R.; Hasnain, S.S.; Hough, M.A.; Strange, R.W. Serial crystallography captures enzyme catalysis in copper nitrite reductase at atomic resolution from one crystal. IUCrJ 2016, 3, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, P.; de Vries, A.H.; Guest, M.F.; Schreckenbach, G.; Catlow, C.R.A.; French, S.A.; Sokol, A.A.; Bromley, S.T.; Thiel, W.; Turner, A.J.; et al. QUASI: A general purpose implementation of the QM/MM approach and its application to problems in catalysis. J. Mol. Struct. Theochem. 2003, 632, 1–28. [Google Scholar] [CrossRef]
- Metz, S.; Kästner, J.; Sokol, A.A.; Keal, T.W.; Sherwood, P. ChemShell—A modular software package for QM/MM simulations. WIREs Comput. Mol. Sci. 2014, 4, 101–110. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Smith, W.; Forester, T.R. DL_POLY_2.0: A general-purpose parallel molecular dynamics simulation package. J. Mol. Graph. 1996, 14, 136–141. [Google Scholar] [CrossRef]
- Kästner, J.; Carr, J.M.; Keal, T.W.; Thiel, W.; Wander, A.; Sherwood, P. DL-FIND: An open-source geometry optimizer for atomistic simulations. J. Phys. Chem. A 2009, 113, 11856–11865. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D97 | D97p | D97N | Crystal | ||
|---|---|---|---|---|---|
| Asp97/Asn97-Cu | Chain A | 4.45 ± 0.18 | 4.61 ± 0.23 | 4.76 ± 0.32 | 4.48 (D97) [4.30] a |
| Chain B | 4.46 ± 0.18 | 4.58 ± 0.24 | 4.60 ± 0.29 | ||
| Chain C | 4.57 ± 0.19 | 4.88 ± 0.23 | 4.96 ± 0.34 | ||
| Tyr323-Cu | Chain A | 7.37 ± 0.19 9.83 ± 0.49 b | 7.38 ± 0.24 | 7.22 ± 0.22 | 7.16 [9.83] a |
| Chain B | 7.33 ± 0.18 | 7.38 ± 0.23 | 7.35 ± 0.26 | ||
| Chain C | 7.35 ± 018 | 7.35 ± 0.20 | 7.21 ± 0.20 |
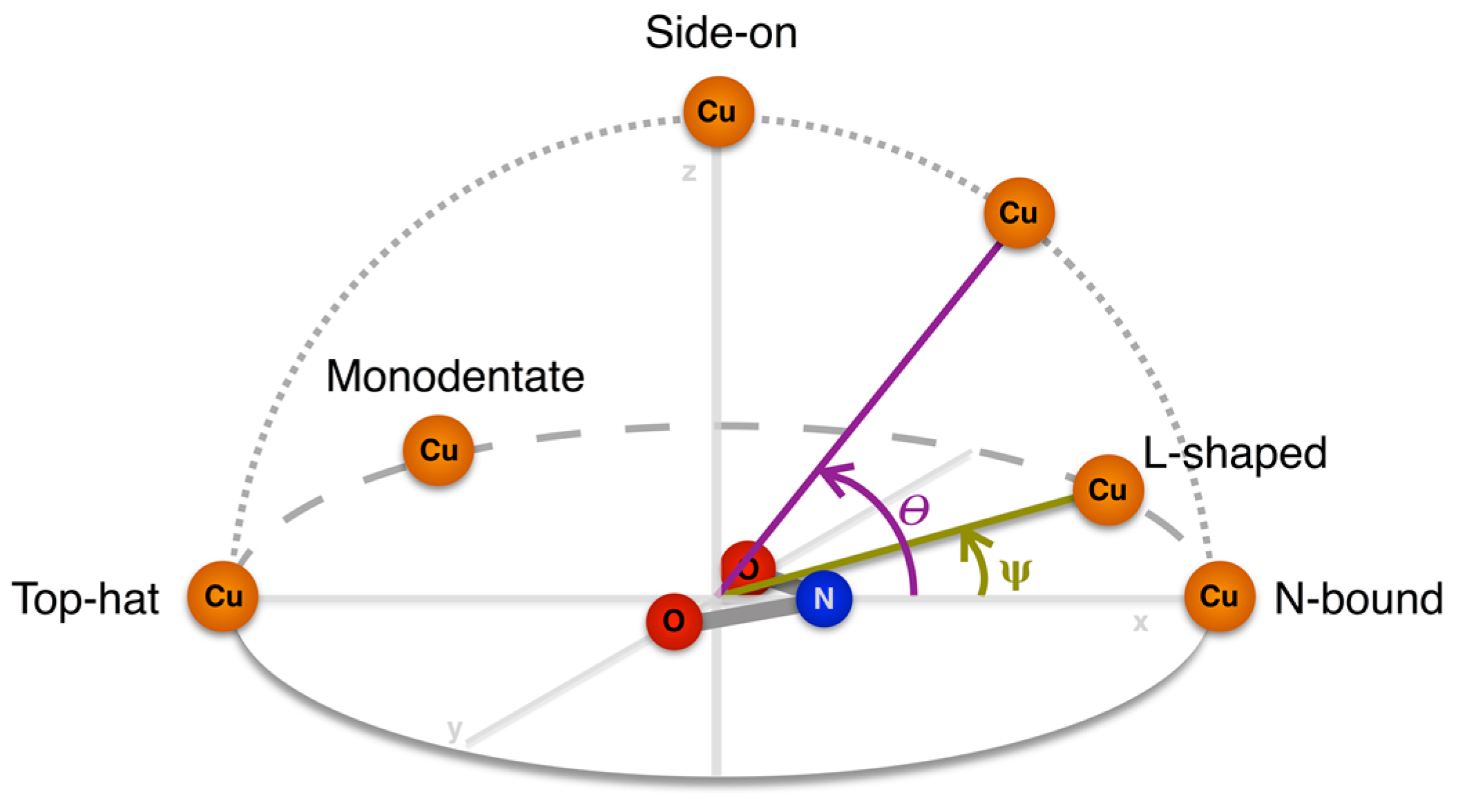
| Altitudinal Geometry | θ | Azimuthal Geometry | ψ |
|---|---|---|---|
| N-bound | 0–20° | l-shaped | 10–30° |
| Symmetrical | 0–10° | ||
| Top-Hat | 160–180° | Bidentate | 170–180° |
| Monodentate | 140–170° | ||
| Side-on | 70–110° | n/a |
| Geometrical Parameters and Distribution | Cu(II) | Cu(I) | |||||
|---|---|---|---|---|---|---|---|
| D97 | D97p | D97N | D97 | D97p | D97N | ||
| Bidentate top-hat | θ/° | 175.8 (1.1) | 178.1 (1.6) | 177.7 (1.7) | - | - | - |
| ψ/° | 174.2 (0.6) | 175.0 (1.1) | 174.1 (2.7) | - | - | - | |
| % structures b | 100 | 100 | 100 | - | - | - | |
| Monodentate top-hat | θ/° | - | - | - | 175.5 (3.9) | 174.5 (4.8) | 175.3 (5.4) |
| ψ/° | - | - | - | 156.7 (2.4) | 160.4 (5.6) | 158.8 (5.3) | |
| % structures b | - | - | - | 83.3 a | 71.4 a | 66.7 a | |
| l-shaped N-bound | θ/° | 5.8 (1.1) | 6.1 (1.5) | 5.1 (2.4) | - | - | - |
| ψ/° | 12.6 (1.5) | 12.2 (1.5) | 15.8 (1.0) | - | - | - | |
| % structures b | 66.7 | 71.4 | 33.3 | - | - | - | |
| Symmetrical N-bound | θ/° | 5.4 c | 5.0 c | 5.8 (3.1) | 5.8 (1.3) | 7.1 (3.5) | 4.3 (3.1) |
| ψ/° | 6.6 c | 4.9 c | 6.4 (1.9) | 1.0 (0.7) | 3.3 (1.9) | 2.4 (1.8) | |
| % structures b | 33.3 | 28.6 | 66.7 | 100 | 100 | 100 | |
| System | Number of Starting Structures | Oxidation State | Average ΔE Energy in kcal/mol (std) |
|---|---|---|---|
| D97 | 6 | Cu(II) | −0.27 (2.96) |
| Cu(I) | 8.66 (6.39) | ||
| D97p | 7 | Cu(II) | 0.24 (2.73) |
| Cu(I) | 4.64 (7.66) | ||
| D97N | 9 | Cu(II) | −1.22 (4.29) |
| Cu(I) | 5.17 (4.87) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sen, K.; Hough, M.A.; Strange, R.W.; Yong, C.W.; Keal, T.W. A QM/MM Study of Nitrite Binding Modes in a Three-Domain Heme-Cu Nitrite Reductase. Molecules 2018, 23, 2997. https://doi.org/10.3390/molecules23112997
Sen K, Hough MA, Strange RW, Yong CW, Keal TW. A QM/MM Study of Nitrite Binding Modes in a Three-Domain Heme-Cu Nitrite Reductase. Molecules. 2018; 23(11):2997. https://doi.org/10.3390/molecules23112997
Chicago/Turabian StyleSen, Kakali, Michael A. Hough, Richard W. Strange, Chin W. Yong, and Thomas W. Keal. 2018. "A QM/MM Study of Nitrite Binding Modes in a Three-Domain Heme-Cu Nitrite Reductase" Molecules 23, no. 11: 2997. https://doi.org/10.3390/molecules23112997
APA StyleSen, K., Hough, M. A., Strange, R. W., Yong, C. W., & Keal, T. W. (2018). A QM/MM Study of Nitrite Binding Modes in a Three-Domain Heme-Cu Nitrite Reductase. Molecules, 23(11), 2997. https://doi.org/10.3390/molecules23112997