Can Adsorption on Graphene be Used for Isotopic Enrichment? A DFT Perspective



Abstract
1. Introduction
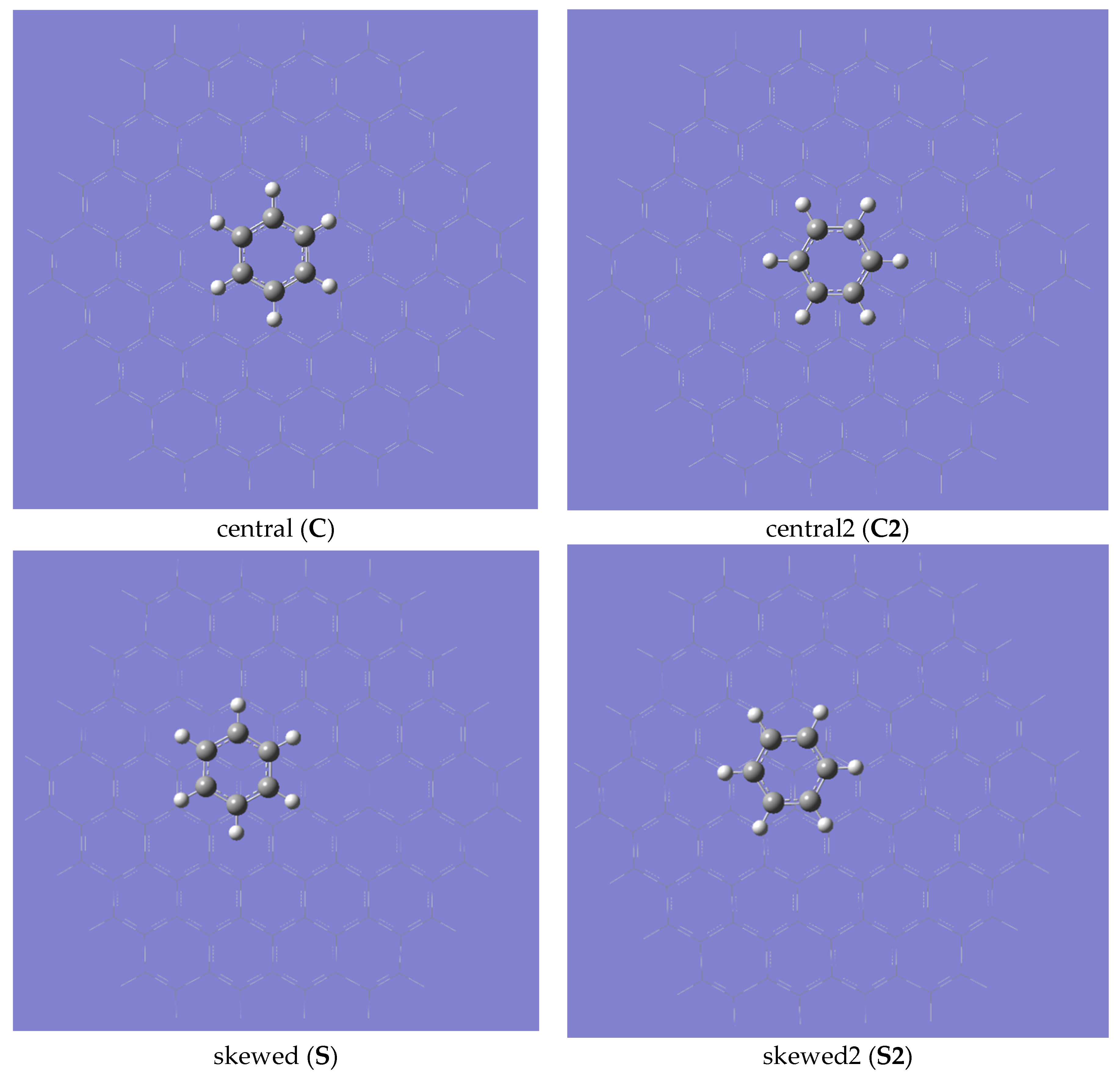
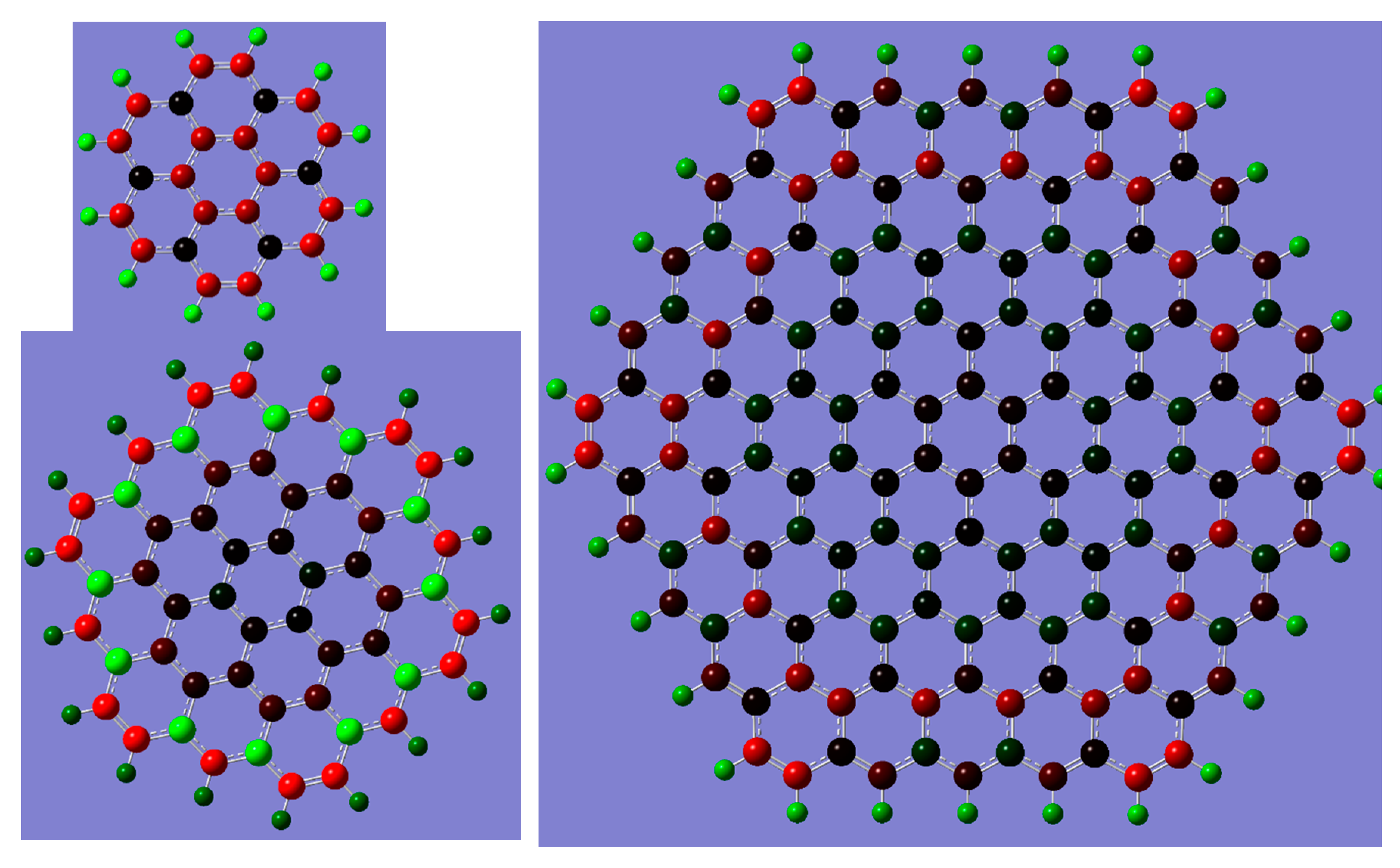
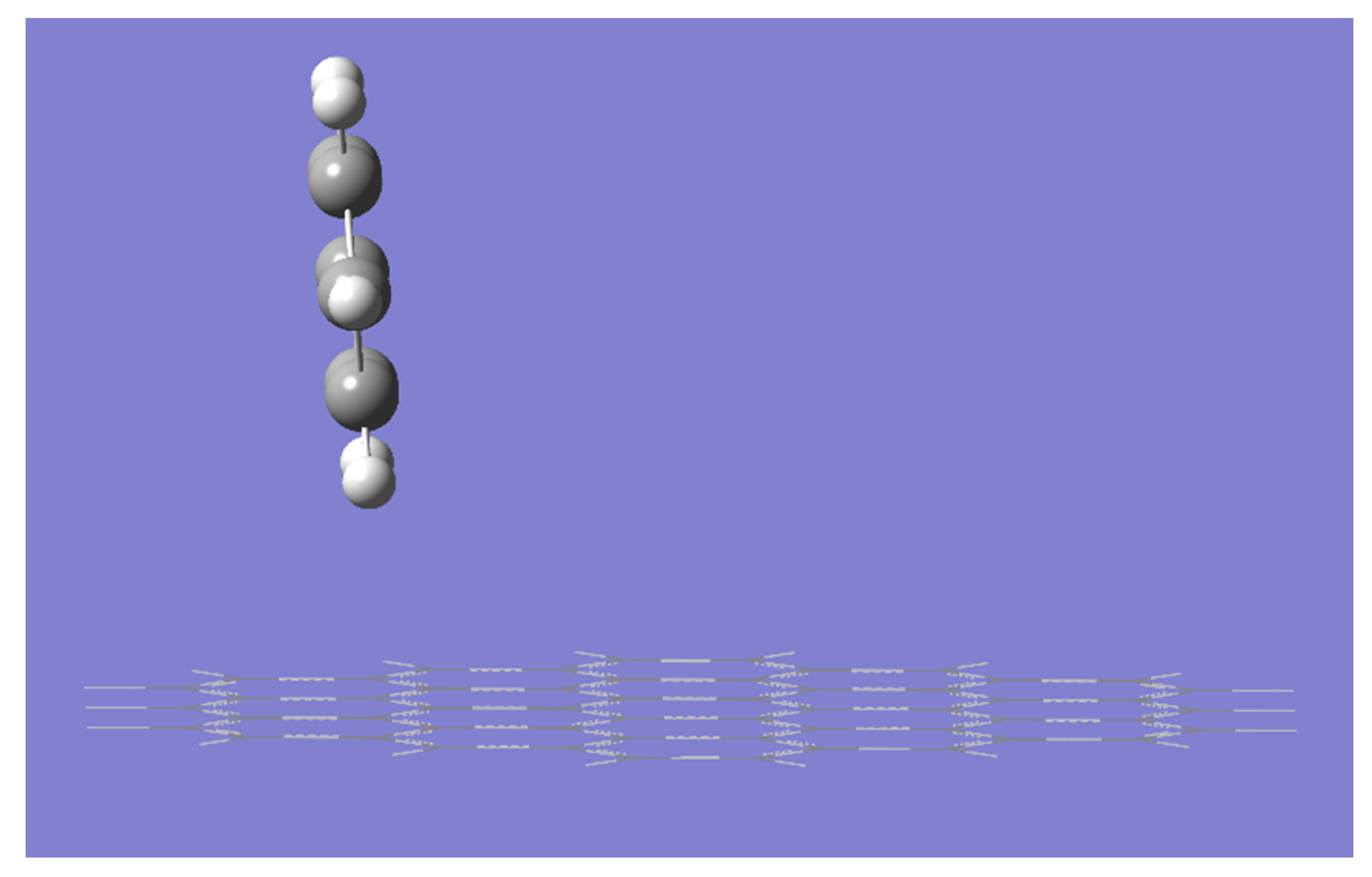
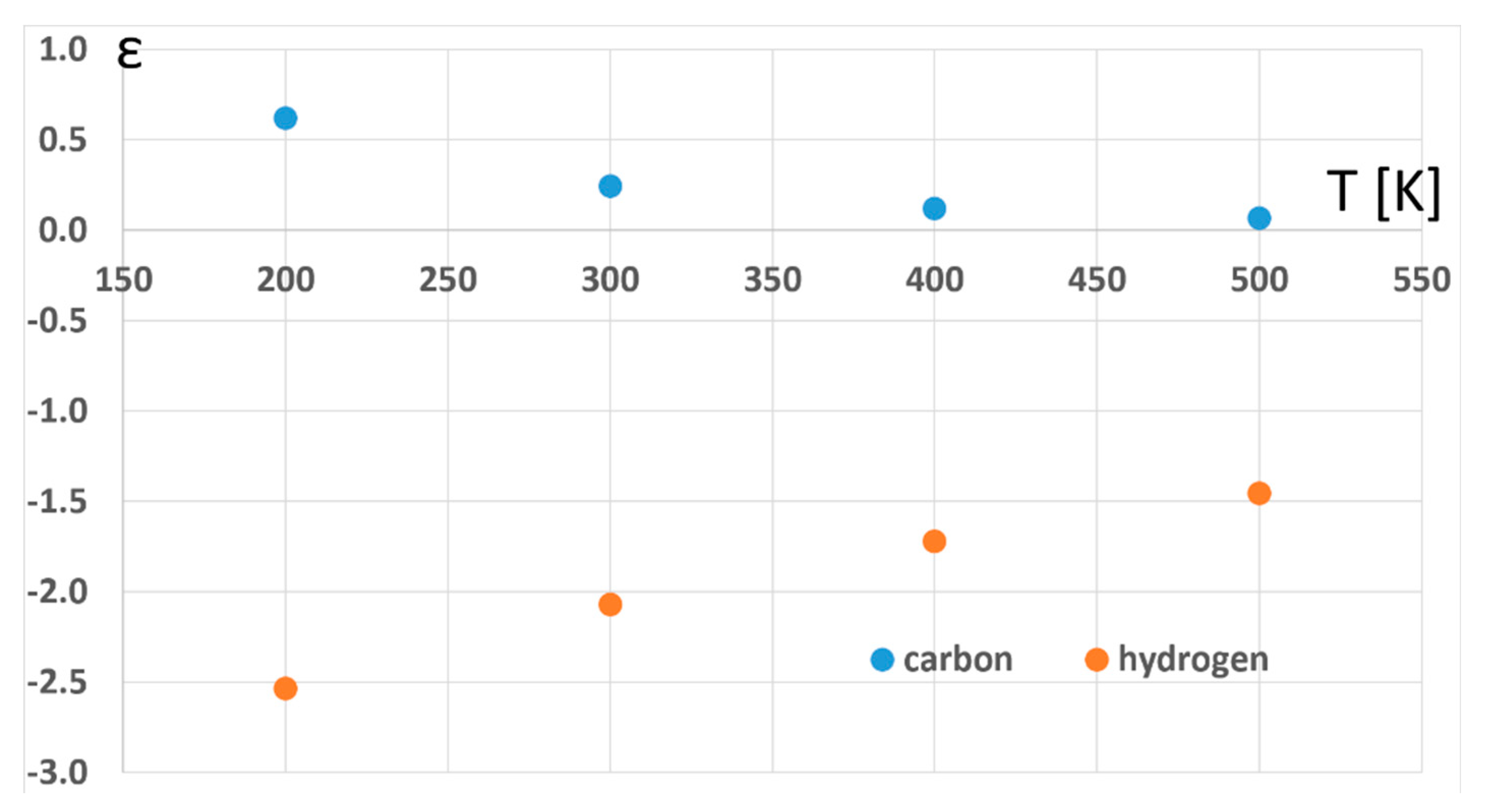
2. Results and Discussion
3. Computation Methods
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of interest
References
- Clancy, A.J.; Bayazit, M.K.; Hodge, S.A.; Skipper, N.T.; Howard, C.A.; Shafffer, M.S.P. Charged Carbon Nanomaterials: Redox Chemistries of Fullerenes, Carbon Nanotubes, and Graphenes. Chem. Rev. 2018, 118, 7363–7408. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Meric, I.; Huang, P.Y.; Gao, Q.; Gao, Y.; Tran, H.; Taniguchi, T.; Watanabe, K.; Campos, L.M.; Muller, D.A.; et al. One-Dimensional Electrical Contact to a Two-Dimensional Material. Science 2013, 342, 614–617. [Google Scholar] [CrossRef] [PubMed]
- Balandin, A.A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior Thermal Conductivity of Single-layer Graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Sato, S. Application of graphene to electronic devices. In Proceedings of the 24th International Workshop on Active-Matrix Flatpanel Displays and Devices (AM-FPD), Kyoto, Japan, 4–7 July 2017; pp. 90–93. [Google Scholar]
- Pérez-Ramírez, E.E.; Luz-Asunción, M.; Martínez-Hernández, A.L.; Velasco-Santos, C. Graphene Materials to Remove Organic Pollutants and Heavy Metals from Water: Photocatalysis and Adsorption, Semiconductor Photocatalysis. Semicond. Photocatal. Mater. Mech. Appl. 2016. [Google Scholar] [CrossRef]
- Ji, L.; Chen, W.; Xu, Z.; Zheng, S.; Zhu, D. Graphene Nanosheets and Graphite Oxide as Promising Adsorbents for Removal of Organic Contaminants from Aqueous Solution. J. Environ. Qual. 2013, 42, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Paixão, M.M.; Vianna, M.T.G.; Marques, M. Graphene and Graphene Nanocomposites for the Removal of Aromatic Organic Compounds from the Water: Systematic Review. Mater. Res. Express 2018, 5. [Google Scholar] [CrossRef]
- Yang, G.C.C.; Tang, P.L. Removal of Phthalates and Pharmaceuticals from Municipal Wastewater by Graphene Adsorption Process. Water Sci. Technol. 2016, 73, 2268–2274. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Gu, X.; Wu, J.; Zhong, S.; Chen, J. Advances in Graphene for Adsorption of Heavy Metals in Wastewater. Adv. Mat. Res. 2012, 550, 2121–2124. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, Y.; Liu, S.; Zeng, G.; Hu, X.; Hu, X.; Guo, Z.; Tan, X.; Wang, L.; Wu, Z. Adsorption of Estrogen Contaminants by Graphene Nanomaterials under Natural Organic Matter Preloading: Comparison to Carbon Nanotube, Biochar, and Activated Carbon. Environ. Sci. Technol. 2017, 51, 6352–6359. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Ramaprabhu, S. Carbon Dioxide Adsorption in Graphene Sheets. AIP Adv. 2011, 1. [Google Scholar] [CrossRef]
- Saha, D.; Nelson, K.; Chen, J.; Lu, Y.; Ozcan, S. Adsorption of CO2, CH4, and N2 in Micro-Mesoporous Nanographene: A Comparative Study. J. Chem. Eng. Data 2015, 60, 2636–2645. [Google Scholar] [CrossRef]
- Szczęśniak, B.; Choma, J.; Jaroniec, M. Gas adsorption properties of graphene-based materials. Adv. Colloid Interface Sci. 2017, 243, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Yamamoto, S.; Hamamoto, Y.; Shiozawa, Y.; Tashima, K.; Fukidome, H.; Koitaya, T.; Mukai, K.; Yoshimoto, S.; Suemitsu, M.; et al. Adsorption of CO2 on Graphene: A Combined TPD, XPS, and vdW-DF Study. J. Phys. Chem. C 2017, 121, 2807–2814. [Google Scholar] [CrossRef]
- Kong, L.; Enders, A.; Rahman, T.S.; Dowben, P.A. Molecular Adsorption on Graphene. J. Phys. Condens. Matter 2014, 26. [Google Scholar] [CrossRef] [PubMed]
- Spasenovic, M. The Price of Graphene. Graphenea. Available online: http://www.graphenea.com/pages/graphene-price (accessed 4 September 2018).
- Sütay, B.; Yurtsever, M. Adsorption of Dihalogen Molecules on Pristine Graphene Surface: Monte Carlo and Molecular Dynamics Simulation Studies. J. Mol. Model. 2017, 23, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Comer, J.; Chen, Z.; Chen, J.; Gumbart, J.C. Exploring Adsorption of Neutral Aromatic Pollutants onto Graphene Nanomaterials via Molecular Dynamics Simulations and Theoretical Linear Solvation Energy Relationships. Environ. Sci. Nano 2018, 5, 2117–2128. [Google Scholar] [CrossRef]
- Li, K.; Li, H.; Yan, N.; Wang, T.; Zhao, Z. Adsorption and Dissociation of CH4 on Graphene: A Density Functional Theory Study. Appl. Surf. Sci. 2018, 459, 693–699. [Google Scholar] [CrossRef]
- Lee, G.; Lee, B.; Kim, J.; Cho, K. Ozone Adsorption on Graphene: Ab Initio Study and Experimental Validation. J. Phys. Chem. C 2009, 113, 14225–14229. [Google Scholar] [CrossRef]
- Ni, J.; Yang, N.; Liang, Q.; Jiang, J.; Meng, R.; Huang, Y.; Chen, X.; Chen, X. Gas Adsorption on Graphene with Different Layers: A First-Principles Study. In Proceedings of the 16th International Conference on Electronic Packaging Technology (ICEPT), Shanghai, China, 11–14 August 2015; pp. 989–993. [Google Scholar]
- Pramanik, A.; Kang, H.S. Density Functional Theory Study of O2 and NO Adsorption on Heteroatom-Doped Graphenes Including the van der Waals Interaction. J. Phys. Chem. C 2011, 115, 10971–10978. [Google Scholar] [CrossRef]
- Lazar, P.; Karlický, F.; Jurecka, P.; Kocman, M.; Otyepková, E.; Šafárová, K.; Otyepka, M. Adsorption of Small Organic Molecules on Graphene. J. Am. Chem. Soc. 2013, 135, 6372–6377. [Google Scholar] [CrossRef] [PubMed]
- Karlický, F.; Otyepková, E.; Lo, R.; Pitoňak, M.; Jurečka, P.; Pykal, M.; Hobza, P.; Otyepka, M. Adsorption of Organic Molecules to van der Waals Materials: Comparison of Fluorographene and Fluorographite with Graphene and Graphite. J. Chem. Theory Comput. 2017, 13, 1328–1340. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ou, P.; Wei, Y.; Zhang, X.; Song, J. Polycyclic Aromatic Hydrocarbons Adsorption onto Graphene: A DFT and AIMD Study. Materials 2018, 11, 726. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Cheng, X.; Wu, W. Al-doped Graphene as an Effective Adsorber for Some Toxic Derivatives of Aromatic Hydrocarbons. J. Theor. Comput. Chem. 2017, 16. [Google Scholar] [CrossRef]
- Yu, S.; Wang, X.; Ai, Y.; Hayat, T.; Hu, W.; Wang, X. Experimental and Theoretical Studies on Competitive Adsorption of Aromatic Compounds on Reduced Graphene Oxides. J. Mater. Chem. A 2016, 4, 5654–5662. [Google Scholar] [CrossRef]
- Michalkova, A.; Gorb, L.; Hill, F.; Leszczynski, J. Can the Gibss Free Energy of Adsorption Be Predicted Efficiently and Accurately: An M05-2X DFT Study. J. Phys. Chem. A 2011, 115, 2423–2430. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Sun, T.; Zhang, Y.; Wang, Y.-B. Benchmark Calculations of the Adsorption of Aromatic Molecules on Graphene. J. Comput. Chem. 2015, 36, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, Y.; Wang, Y.-B. Noncovalent Interaction Between Graphene and Aromatic Molecule: Structure, Energy, and Nature. J. Chem. Phys. 2014, 140. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Sun, T.; Zhang, Y.; Wang, Y.-B. Substituent Effects in the π-π Interaction Between Graphene and Benzene: An Indication for the Noncovalent Functionalization of Graphene. Comput. Theor. Chem. 2014, 1046, 64–69. [Google Scholar] [CrossRef]
- Raad, T.M.; Behnejad, H.; El Jamal, M. Equilibrium and Kinetic Studies for the Adsorption of Benzene and Toluene by Graphene Nanosheets: A Comparison with Carbon Nanotubes. Surf. Interface Anal. 2016, 48, 117–125. [Google Scholar] [CrossRef]
- Wolfsberg, M.; Van Hook, W.A.; Paneth, P. Isotope Effects: In the Chemical, Geological, and Bio Sciences; Springer: London, UK, 2009. [Google Scholar]
- Vasquez, L.; Rostkowski, M.; Gelman, F.; Dybała-Defratyka, A. Can PIMD Make a Good Approximation for Vapor Pressure Isotope Effects Prediction for Organic Solvents? A Comparison to ONIOM QM/MM and QM Cluster Calculation. J. Phys. Chem. B 2018, 122, 7353–7364. [Google Scholar] [CrossRef] [PubMed]
- Świderek, K.; Paneth, P. Binding Isotope Effects. Chem. Rev. 2013, 113, 7851–7879. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Gordon-Head, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Melander, L.C.S.; Saunders, W.H. Isotope Effect on Reaction Rates; The Ronald Press Company: New York, NY, USA, 1960. [Google Scholar]
- Paneth, P. Isotopic Fractionation—Chemical v. Environmental Perspective. Environ. Chem. 2012, 9, 67–68. [Google Scholar]
- Gold, V. Rule of the Geometric Mean: Its Role in the Treatment of Thermodynamic and Kinetic Deuterium Solvent Isotope Effects. Trans. Faraday Soc. 1968, 64, 2770–2779. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian, Version 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Svensson, M.; Humbel, S.; Froese, R.D.J.; Matsubara, T.; Sieber, S.; Morokuma, K. ONIOM: A Multilayered Integrated MO + MM Method for Geometry Optimizations and Single Point Energy Predictions. A Test for Diels−Alder Reactions and Pt(P(t-Bu)3)2 + H2 Oxidative Addition. J. Phys. Chem. 1996, 100, 19357–19363. [Google Scholar] [CrossRef]
- Field, M.J.; Albe, M.; Bret, C.; Proust-De Martin, F.; Thomas, A. The dynamo library for molecular simulations using hybrid quantum mechanical and molecular mechanical potentials. J. Comput. Chem. 2000, 21, 1088–1100. [Google Scholar] [CrossRef]
- Williams, I.H. Kinetic Isotope Effects from QM/MM Subset Hessians: “Cut-Off” Analysis for SN2 Methyl Transfer in Solution. J. Chem. Theory Comput. 2012, 8, 542–553. [Google Scholar] [CrossRef] [PubMed]
- HyperChem Professional, Version 8.0.1; Hypercube, Inc.: Gainesville, FL, USA, 2011.
- GaussView, version 6; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comp. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. Long-Range Correction Scheme for Generalized-Gradient-Approximation Exchange Functionals. J. Chem. Phys. 2001, 115, 3540–3544. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C.; New, A. Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin-density calculations—A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Hehre, W.J. Ab Initio Molecular Orbital Theory. Acc. Chem. Res. 1976, 9, 399–406. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III.* The 3-21 + G Basis Set for First-Row Elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-Consistent Molecular Orbital Methods 25. Supplementary Functions for Gaussian Basis Sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian-Basis Sets for Molecular Calculations. 1. 2nd Row Atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Raghavachari, K.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. 20. Basis set for correlated wave-functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar]
- Stewart, J.J.P. Optimization of Parameters for Semi-Empirical Methods. V. Modification of NDDO Approximations and Application to 70 Elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Porezag, D.; Frauenheim, T.; Köhler, T.; Seifert, G.; Kaschner, R. Construction of Tight-Binding-Like Potentials on the Basis of Density-Functional Theory: Application to Carbon. Phys. Rev. B 1995, 51, 12947–12957. [Google Scholar] [CrossRef]
- Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.; Frauenheim, T.; Suhai, S.; Seifert, G. Self-Consistent-Charge Density-Functional Tight-Binding Method for Simulations of Complex Materials Properties. Phys. Rev. B 1998, 58, 7260–7268. [Google Scholar] [CrossRef]
- Anisimov, V.; Paneth, P. ISOEFF98. A Program for Studies of Isotope Effects Using Hessian Modifications. J. Mathem. Chem. 1999, 26, 75–86. [Google Scholar] [CrossRef]
- Świderek, K.; Paneth, P. Importance of the Lactate Dehydrogenase Quaternary Structure in Theoretical Calculations. J. Phys. Chem. B 2010, 114, 3393–3397. [Google Scholar] [CrossRef] [PubMed]
- Leenaerts, O.; Partoens, B.; Peeters, F.M. Adsorption of small molecules on graphene. Microelectr. J. 2009, 40, 860–862. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gaussian Version | Convergence Criteria | “IE” | “ε” |
|---|---|---|---|
| G09 rev. E01 | default ⇄ tight | 1.000374 | −0.37 |
| tight ⇄ vtight | 1.000007 | −0.01 | |
| G16 rev. B01 | default ⇄ tight | 1.000559 | −0.56 |
| tight ⇄ vtight | 1.000007 | −0.01 | |
| G09 ⇄ G16 | default | 1.001907 | −1.90 |
| tight | 1.002092 | −2.09 | |
| vtight | 1.002056 | −2.05 |
| Functional | Basis Set | D6-IE | εperD | 13C6-IE | εperC |
|---|---|---|---|---|---|
| ωB97xd | def2-TZVPP | 1.01674 | −2.76 | 0.99801 | 0.33 |
| ωB97xd | 6-311 + G(d,p) | 1.00138 | −0.23 | 0.99901 | 0.16 |
| ωB97xd a | def2-SVPP | 1.01762 | −2.91 | 0.99864 | 0.23 |
| ωB97xd | def2-SVPP | 1.01252 | −2.07 | 0.99853 | 0.25 |
| B97d3 | def2-SVPP | 0.97265 | 4.63 | 0.99725 | 0.46 |
| B97d | def2-SVPP | 0.97778 | 3.75 | 0.99675 | 0.54 |
| am-B3LYP | def2-SVPP | 1.00366 | −0.61 | 1.00094 | −0.16 |
| LC-BLYP | def2-SVPP | 1.02588 | −4.25 | 1.00091 | −0.15 |
| M06-2X | def2-SVPP | 1.06471 | −10.40 | 1.00163 | −0.27 |
| ωB97xd | 6-31 + G(d,p) | 0.99595 | 0.68 | 0.99944 | 0.09 |
| B97d3 | 6-31 + G(d,p) | 0.98191 | 3.05 | 0.99750 | 0.42 |
| B97d | 6-31 + G(d,p) | 0.98998 | 1.68 | 0.99770 | 0.38 |
| cam-B3LYP | 6-31 + G(d,p) | 1.00441 | −0.73 | 1.00115 | −0.19 |
| LC-BLYP | 6-31 + G(d,p) | 1.02935 | −4.81 | 1.00109 | −0.18 |
| M06-2X | 6-31 + G(d,p) | 1.07768 | −12.39 | 1.00151 | −0.25 |
| B3LYP | 6-31 + G(d,p) | 0.99871 | 0.21 | 1.00098 | −0.16 |
| :QM | Structure | D6-IE | εperD | 13C6-IE | εperC |
|---|---|---|---|---|---|
| - | S | 1.01674 | −2.76 | 0.99801 | 0.33 |
| PM7 | C2 | 0.97754 | 3.79 | 0.99918 | 0.14 |
| PM7 | S | 0.99944 | 0.09 | 0.98089 | 3.22 |
| PM7 | S2 | 0.97889 | 3.56 | 0.99964 | 0.06 |
| DFTB | V | 1.00969 | −1.61 | 1.00032 | −0.05 |
| DFTB | S2 | 1.00011 | −0.02 | 1.00038 | −0.06 |
| PM6 | C2 | 1.00146 | −0.24 | 0.99954 | 0.08 |
| PM6 | S | 1.00112 | −0.19 | 0.99987 | 0.02 |
| PM6 | S2 | 1.00412 | −0.69 | 0.99997 | 0.01 |
| Graphene Model | Degrees of Freedom | Point Charges | D6-IE | εperD | 13C6-IE | εperC |
|---|---|---|---|---|---|---|
| C54 | 3nBG-6 | - | 1.01674 | −2.76 | 0.99801 | 0.33 |
| C54 | 3nB-6 | Mülliken | 1.01533 | −2.53 | 0.99993 | 0.01 |
| C54 | 3nB-6 | APT | 1.00681 | −1.13 | 1.00025 | −0.04 |
| C150 | 3nB-6 | Mülliken | 1.01088 | −1.80 | 1.00019 | −0.03 |
| C150 | 3nB | Mülliken | 1.01074 | −1.78 | 1.00013 | −0.02 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pokora, M.; Paneth, P. Can Adsorption on Graphene be Used for Isotopic Enrichment? A DFT Perspective. Molecules 2018, 23, 2981. https://doi.org/10.3390/molecules23112981
Pokora M, Paneth P. Can Adsorption on Graphene be Used for Isotopic Enrichment? A DFT Perspective. Molecules. 2018; 23(11):2981. https://doi.org/10.3390/molecules23112981
Chicago/Turabian StylePokora, Mateusz, and Piotr Paneth. 2018. "Can Adsorption on Graphene be Used for Isotopic Enrichment? A DFT Perspective" Molecules 23, no. 11: 2981. https://doi.org/10.3390/molecules23112981
APA StylePokora, M., & Paneth, P. (2018). Can Adsorption on Graphene be Used for Isotopic Enrichment? A DFT Perspective. Molecules, 23(11), 2981. https://doi.org/10.3390/molecules23112981