An In Silico Study of the Antioxidant Ability for Two Caffeine Analogs Using Molecular Docking and Quantum Chemical Methods

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Results and Discussion
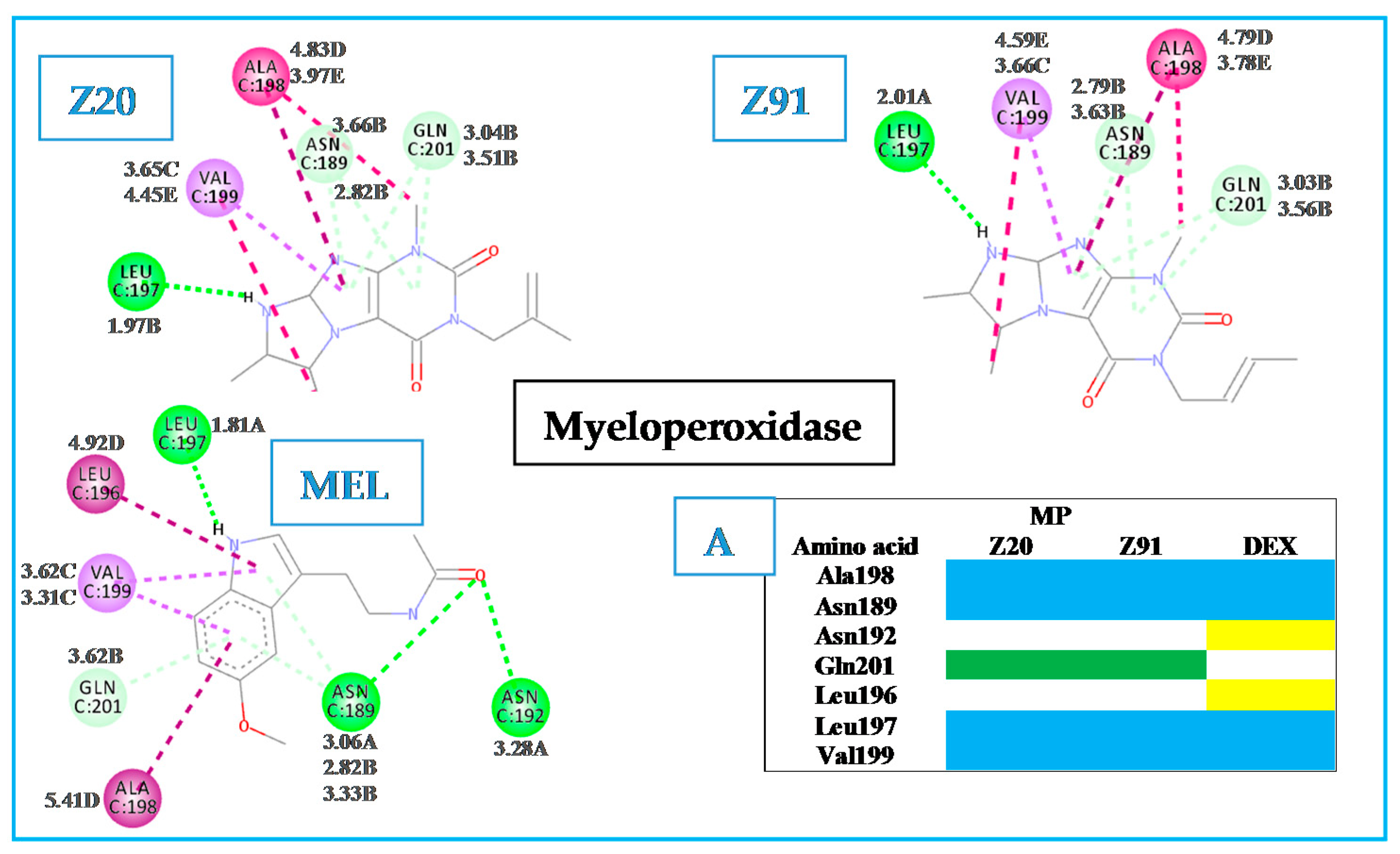
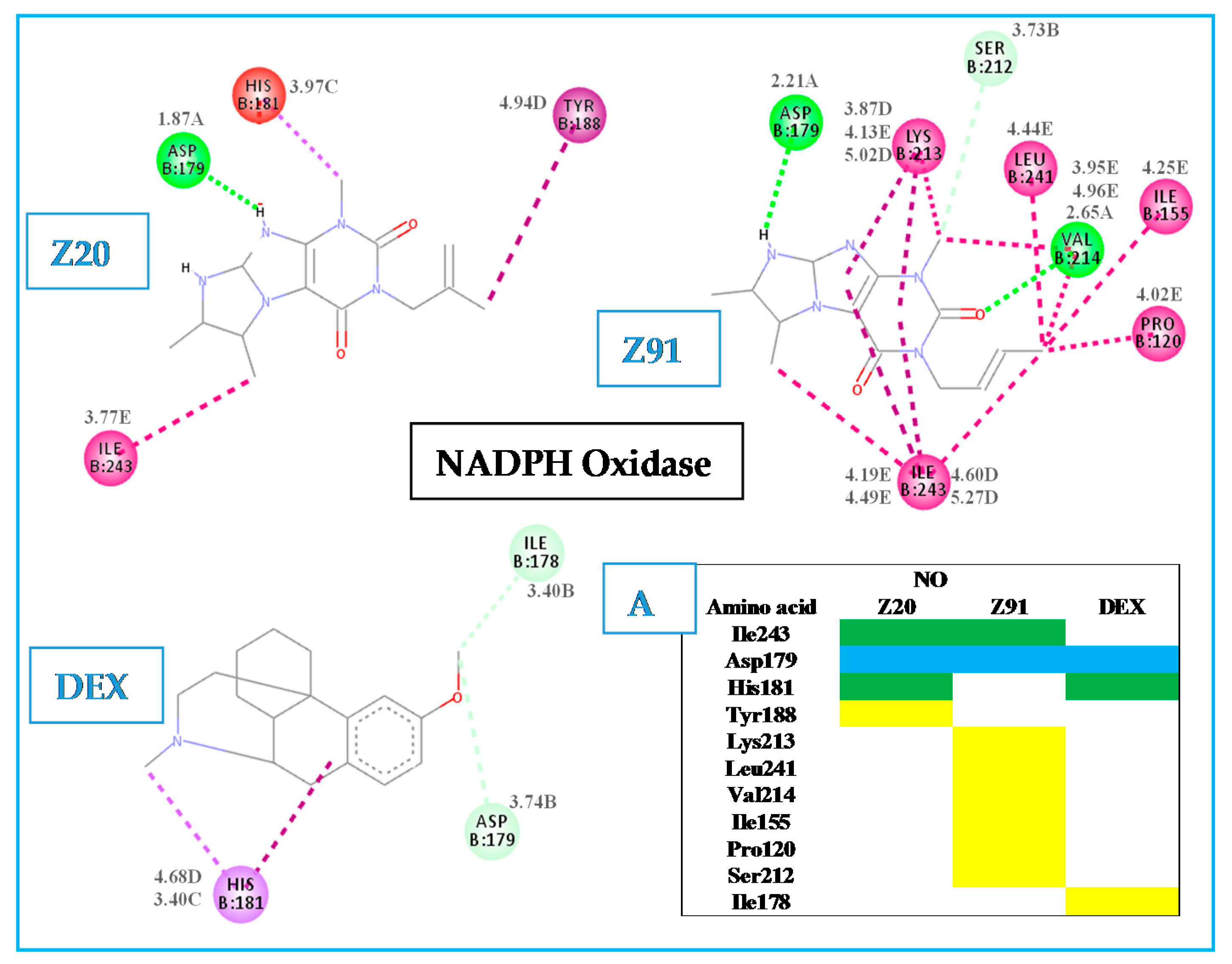
2.1. Evaluation of Molecular Docking
2.2. Molecular Descriptors and Pearson Correlations
2.3. Molecular Orbitals and Characteristic of Antioxidant Ability
3. Materials and Methods
3.1. Calculation of Receptor-Ligand Interaction for Evaluation of Antioxidant Potential
3.2. Quantum Chemical Calculations
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Silva, A.A.; Gonçalves, R.C. Reactive oxygen species and the respiratory tract diseases of large animals. Ciência Rural 2010, 40, 994–1002. [Google Scholar] [CrossRef]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- White, P.A.; Oliveira, R.C.; Oliveira, A.P.; Serafini, M.R.; Araújo, A.A.; Gelain, D.P.; Moreira, J.C.; Almeida, J.R.; Quintans, J.S.; Quintans-Junior, L.J.; et al. Antioxidant activity and mechanisms of action of natural compounds isolated from lichens: A systematic review. Molecules 2014, 19, 14496–14527. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E. Basic mechanisms of antioxidant activity. BioFactors 1997, 6, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Dharmaraja, A.T. Role of reactive oxygen species (ROS) in therapeutics and drug resistance in cancer and bacteria. J. Med. Chem. 2017, 60, 3221–3240. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Sharma, R.; Kumar, A. Docking techniques in pharmacology: How much promising? Comp. Biol. Chem. 2018, 76, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Bandari, S.K.; Kammari, B.R.; Madda, J.; Kommu, N.; Lakkadi, A.; Vuppala, S.; Tigulla, P. Synthesis of new chromeno-carbamodithioate derivatives and preliminary evaluation of their antioxidant activity and molecular docking studies. Bioorg. Med. Chem. Lett. 2017, 27, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Niu, H.; Wang, W.; Li, J.; Lei, Y.; Zhao, Y.; Yang, W.; Zhao, C.; Lin, B.; Song, S.; Wang, S. A novel structural class of coumarin-chalcone fibrates as PPARα/γ agonists with potent antioxidant activities: Design, synthesis, biological evaluation and molecular docking studies. Eur. J. Med. Chem. 2017, 138, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, A.; Kujawski, J.; Czaja, K.; Szeląg, M. Antioxidant properties of several caffeic acid derivatives: A theoretical study. C. R. Chim. 2017, 20, 1072–1082. [Google Scholar] [CrossRef]
- Akbas, E.; Ekin, S.; Ergan, E.; Karakus, Y. Synthesis, DFT calculations, spectroscopy and in vitro antioxidant activity studies on 4-hydroxyphenyl substituted thiopyrimidine derivatives. J. Mol. Struct. 2018, 1174, 177–183. [Google Scholar] [CrossRef]
- Gür, M.; Muğlu, H.; Çavuş, M.S.; Güder, A.; Sayıner, H.S.; Kandemirli, F. Synthesis, characterization, quantum chemical calculations and evaluation of antioxidant properties of 1,3,4-thiadiazole derivatives including 2- and 3-methoxy cinnamic acids. J. Mol. Struct. 2017, 1134, 40–50. [Google Scholar] [CrossRef]
- Teles Fujishima, M.A.; Silva, N.S.R.; Ramos, R.S.; Batista Ferreira, E.F.; Santos, K.L.B.; Silva, C.H.T.P.; Silva, J.O.; Campos Rosa, J.M.; Santos, C.B.R. An Antioxidant potential, quantum-chemical and molecular docking study of the major chemical constituents present in the leaves of curatella americana linn. Pharmaceuticals 2018, 11, 72. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.S.; Costa, K.S.L.; Cruz, J.V.; Ramos, R.S.; Silva, L.B.; Brasil, D.S.B.; Silva, C.H.T.P.; Santos, C.B.R.; Macêdo, W.J.C. Virtual screening and statistical analysis in the design of new caffeine analogues molecules with potential epithelial anticancer activity. Curr. Pharm. Des. 2018, 24, 576–594. [Google Scholar] [CrossRef] [PubMed]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J. Chem. Inf. Model. 2009, 49, 444–460. [Google Scholar] [CrossRef] [PubMed]
- Gowthaman, U.; Jayakanthan, M.; Sundar, D. Molecular docking studies of dithionitrobenzoic acid and its related compounds to protein disulfide isomerase: Computational screening of inhibitors to HIV-1 entry. BMC Bioinform. 2008, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Cosme, J.; Ward, A.; Angove, H.C.; Vinkovic´, D.M.; Jhoti, H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature 2003, 424, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Blair-Johnson, M.; Fiedler, T.; Fenna, R. Human myeloperoxidase: Structure of a cyanide complex and its interaction with bromide and thiocyanate substrates at 1.9 A resolution. Biochemistry 2001, 40, 13990–13997. [Google Scholar] [CrossRef] [PubMed]
- Lountos, G.T.; Jiang, R.; Wellborn, W.B.; Thaler, T.L.; Bommarius, A.S.; Orville, A.M. The crystal structure of NAD(P)H oxidase from lactobacillus sanfranciscensis: Insights into the conversion of O2 into two water molecules by the flavoenzyme. Biochemistry 2006, 2006 45, 9648–9659. [Google Scholar] [CrossRef]
- Mohan, C.; Long, K.D.; Mutneja, M. An Introduction to Inhibitors and Their Biological Applications, 1st ed.; EMD Millipore: Darmstadt, Germany, 2013; pp. 1–48. [Google Scholar]
- Vieira, J.B.; Braga, F.S.; Lobato, C.C.; Santos, C.F.; Costa, J.S.; Bittencourt, J.A.; Brasil, D.S.; Silva, J.O.; Hage-Melim, L.I.; Macêdo, W.J.; et al. A QSAR, pharmacokinetic and toxicological study of new artemisinin compounds with anticancer activity. Molecules 2014, 19, 10670–10697. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.B.R.; Vieira, J.B.; Lobato, C.C.; Hage-Melim, L.I.S.; Souto, R.N.P.; Lima, C.S.; Costa, E.V.M.; Brasil, D.S.B.; Macêdo, W.J.C.; Carvalho, J.C.T. A SAR and QSAR study of new artemisinin compounds with antimalarial activity. Molecules 2014, 19, 367–399. [Google Scholar] [CrossRef] [PubMed]
- Yalkowsky, S.H.; Valvani, S.C. Solubilities and partitioning. 2. Relationships between aqueous solubilities, partition coefficients, and molecular surface areas of rigid aromatic hydrocarbons. J. Chem. Eng. Data 1979, 24, 127–129. [Google Scholar] [CrossRef]
- Panayiotou, C. Redefining solubility parameters: Bulk and surface properties from unified molecular descriptors. J. Chem. Thermodyn. 2017, 111, 207–220. [Google Scholar] [CrossRef]
- Gubskaya, A.V.; Kusalik, P.G. The total molecular dipole moment for liquid water. J. Chem. Phys. 2002, 117, 5290–5302. [Google Scholar] [CrossRef]
- Farasat, M.; Shojaei, S.H.R.; Golzan, M.M.; Farhadi, K. Theoretical study of the potential energy surface and electric dipole moment of aniline. J. Mol. Struct. 2016, 1108, 341–346. [Google Scholar] [CrossRef]
- Tognetti, V.; Morell, C.; Joubert, L. Atomic electronegativities in molecules. Chem. Phys. Lett. 2015, 635, 111–115. [Google Scholar] [CrossRef]
- Cunha, E.L.; Santos, C.F.; Braga, F.S.; Costa, J.S.; Silva, R.C.; Favacho, H.A.S.; Hage-Melim, L.I.S.; Carvalho, J.C.T.; Silva, C.H.T.P.; Santos, C.B.R. Computational investigation of antifungal compounds using molecular modeling and prediction of ADME/Tox properties. J. Comput. Theor. Nanosci. 2015, 12, 3682–3691. [Google Scholar] [CrossRef]
- Yang, W.; Lee, C.; Ghosh, S.K. Molecular softness as the average of atomic softnesses: Companion principle to the geometric mean principle for electronegativity equalization. J. Phys. Chem. 1985, 89, 5412–5414. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Korchowiec, J.; Zhou, Z. Molecular hardness and softness parameters and their use in chemistry. Int. J. Quantum Chem. 1988, 34, 349–366. [Google Scholar] [CrossRef]
- Liu, P.H.; Hunt, K.L.C. Molecular softness, hypersoftness, infrared absorption, and vibrational Raman scattering: New relations derived from nonlocal polarizability densities. J. Chem. Phys. 1995, 103, 10597–10604. [Google Scholar] [CrossRef]
- Ghanty, T.K.; Ghosh, S.K. Molecular Hardness, polarizability and valency variation of formamide and thioformamide on internal rotation: A density functional study. J. Phys. Chem. A 2000, 104, 2975–2979. [Google Scholar] [CrossRef]
- Kaya, S.; Kaya, C. A new method for calculation of molecular hardness: A theoretical study. Comput. Theor. Chem. 2015, 1060, 66–70. [Google Scholar] [CrossRef]
- Tarazona, M.P.; Sáiz, E. Understanding chemical potential. J. Chem. Educ. 1995, 72, 882–883. [Google Scholar] [CrossRef]
- Cook, G.; Dickerson, R.H. Understanding the chemical potential. Am. J. Phys. 1995, 63, 737–742. [Google Scholar] [CrossRef]
- Job, G.; Herrmann, F. Chemical potential—A quantity in search of recognition. Eur. J. Phys. 2006, 27, 353–371. [Google Scholar] [CrossRef]
- Grant, G.H.; Richards, W.G. Computational Chemistry; Oxford Science Publications: Oxford, UK, 1996; pp. 1–96. [Google Scholar]
- Anh, N.T. Regio- and stereo-selectivities in some nucleophilic reactions. In Organic Chemistry Syntheses and Reactivity; Springer: Berlin/Heidelberg, Germany, 1980; Volume 88, pp. 145–162. [Google Scholar]
- Bassindale, A.R.; Glynn, S.J.; Taylor, P.G. Reaction mechanisms of nucleophilic attack at silicon. In The Chemistry of Organic Silicon Compounds; Patai, S., Rappoport, Z., Eds.; Wiley Online Library: New York, NY, USA, 1898; Volume 2, pp. 495–511. [Google Scholar]
- Mihçiokur, O.; Özpozan, T. Molecular structure, vibrational spectroscopic analysis (IR & Raman), HOMO-LUMO and NBO analysis of anti-cancer drug sunitinib using DFT method. J. Mol. Struct. 2017, 1149, 27–41. [Google Scholar] [CrossRef]
- Zhang, G.; Musgrave, C.B. Comparison of DFT methods for molecular orbital eigenvalue calculations. J. Phys. Chem. A 2007, 111, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- PDB—Protein Data Bank. Available online: http://www.ncbi.nlm.nih.gov/protein/ (accessed on 20 January 2018).
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Cruz, J.V.; Serafim, R.B.; da Silva, G.M.; Giuliatti, S.; Rosa, J.M.C.; Neto, M.F.A.; Leite, F.H.A.; Taft, C.A.; da Silva, C.H.T.P.; Santos, C.B.R. Computational design of new protein kinase 2 inhibitors for the treatment of inflammatory diseases using QSAR, pharmacophore-structure-based virtual screening, and molecular dynamics. J. Mol. Model. 2018, 24, 225. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.C.; Poiani, J.G.C.; Ramos, R.S.; Costa, J.S.; Silva, C.H.T.P.; Brasil, D.S.B.; Santos, C.B.R. Ligand- and structure- based virtual screening from 16-(N,N-diisobutylaminomethyl)-6α-hydroxyivouacapan-7β,17β-lactone compound with potential anti-prostate cancer activity. J. Serb. Chem. Soc. 2018, 83, 6472. [Google Scholar] [CrossRef]
- Santos, C.B.R.; Ramos, R.S.; Sánchez Ortiz, B.L.; Silva, G.M.; Giuliatti, S.; Balderas-Lopez, J.L.; Navarrete, A.; Carvalho, J.C.T. Oil from the fruits of Pterodon emarginatus Vog.: A traditional anti-inflammatory. Study combining in vivo and in silico. J. Ethnopharmacol. 2018, 222, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.V.; Neto, M.F.A.; Silva, L.B.; da Ramos, R.; da Costa, J.; Brasil, D.S.B.; Lobato, C.C.; da Costa, G.V.; Bittencourt, J.A.H.M.; da Silva, C.H.T.P.; et al. Identification of novel protein kinase receptor type 2 inhibitors using pharmacophore and structure-based virtual screening. Molecules 2018, 23, 453. [Google Scholar] [CrossRef] [PubMed]
- Viloria, J.S.; Allega, M.F.; Lambrughi, M.; Papaleo, E. An optimal distance cutoff for contact-based Protein Structure Networks using side-chain centers of mass. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Pereira, A.L.; Santos, G.B.; Franco, M.S.; Federico, L.B.; Silva, C.H.; Santos, C.B. Molecular modeling and statistical analysis in the design of derivatives of human dipeptidyl peptidase IV. J. Biomol. Struct. Dyn. 2018, 36, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Padilha, E.C.; Serafim, R.B.; Sarmiento, D.Y.R.; Santos, C.F.; Santos, C.B.; Silva, C.H. New PPARα/γ/δ optimal activator rationally designed by computational methods. J. Braz. Chem. Soc. 2016, 27, 1636–1647. [Google Scholar] [CrossRef]
- Rogozin, E.A.; Lee, K.W.; Kang, N.J.; Yu, H.; Nomura, M.; Miyamoto, K.I.; Conney, A.H.; Bode, A.M.; Dong, Z. Inhibitory effects of caffeine analogues on neoplastic transformation: Structure–activity relationship. Carcinogenesis 2008, 29, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; Galano, J.; Durand, T.; Guennec, J.; Lee, J.C.Y. Physiological role of reactive oxygen species as promoters of natural defenses. FASEB J. 2017, 31, 3729–3745. [Google Scholar] [CrossRef] [PubMed]
- Działo, M.; Mierziak, J.; Korzun, U.; Preisner, M.; Szopa, J.; Kulma, A. The potential of plant phenolics in prevention and therapy of skin disorders. Int. J. Mol. Sci. 2016, 17, 160. [Google Scholar] [CrossRef] [PubMed]
- Reinisalo, M.; Kårlund, A.; Koskela, A.; Kaarniranta, K.; Karjalainen, R.O. Polyphenol stilbenes: Molecular mechanisms of defence against oxidative stress and aging-related diseases. Oxid. Med. Cell. Longev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gunes, A.; Coskun, U.; Boruban, C.; Gunel, N.; Babaoglu, M.O.; Sencan, O.; Bozkurt, A.; Rane, A.; Hassan, M.; Zengil, H.; et al. Inhibitory effect of 5-fluorouracil on cytochrome P450 2C9 activity in cancer patients. Basic Clin. Pharmacol. Toxicol. 2006, 98, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Drugbank. Available online: https://www.drugbank.ca/unearth/ (accessed on 7 July 2018).
- Saul, D.; Gleitz, S.; Nguyen, H.H.; Kosinsky, R.L.; Sehmisch, S.; Hoffmann, D.B.; Wassmann, M.; Menger, B.; Komrakova, M. Effect of the lipoxygenase-inhibitors baicalein and zileuton on the vertebra in ovariectomized rats. Bone 2017, 101, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Carter, G.W.; Young, P.R.; Albert, D.H.; Bouska, J.; Dyer, R.; Bell, R.L.; Summers, J.B.; Brooks, D.W. 5-Lipoxygenase inhibitory activity of zileuton. J. Pharmacol. Exp. Ther. 1991, 256, 929–937. [Google Scholar] [PubMed]
- Allegra, M.; Furtmüller, P.G.; Regelsberger, G.; Turco-Liveri, M.L.; Tesoriere, L.; Perretti, M.; Livrea, M.A.; Obinger, C. Mechanism of reaction of melatonin with human myeloperoxidase. Biochem. Biophys. Res. Commun. 2001, 282, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Galijasevic, S.; Abdulhamid, I.; Abu-Soud, H.M. Melatonin is a potent inhibitor for myeloperoxidase. Biochemistry 2008, 47, 2668–2677. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.L.; Li, Y.H.; Shi, G.Y.; Tang, S.H.; Jiang, S.J.; Huang, C.W.; Liu, P.Y.; Hong, J.S.; Wu, H.L. Dextromethorphan reduces oxidative stress and inhibits atherosclerosis and neointima formation in mice. Cardiovasc. Res. 2009, 82, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.C.; Chao, C.Y.; Lin, S.J.; Chen, J.W. Low-dose dextromethorphan, a NADPH oxidase inhibitor, reduces blood pressure and enhances vascular protection in experimental hypertension. PLoS ONE 2012, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Malik, U.Z.; Hundley, N.J.; Romero, G.; Radi, R.; Freeman, B.A.; Tarpey, M.M.; Kelley, E.E. Febuxostat inhibition of endothelial-bound XO: Implications for targeting vascular ROS production. Free Radic. Biol. Med. 2011, 51, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.S.; Santos, C.B.R.; Costa, K.S.L.; Ramos, R.S.; Silva, C.H.T.P.; Macêdo, W.J.C. Validation of computational methods applied in molecular modeling of caffeine with epithelial anticancer activity: Theoretical study of geometric, thermochemical and spectrometric data. Quím. Nova 2018, 7, 732–742. [Google Scholar] [CrossRef]
- STATISTICA (Data Analysis Software System), Version 6.1; StatSoft Inc.: Tulsa, OK, USA, 2004.
Sample Availability: Samples of the compounds are not available from the authors. |



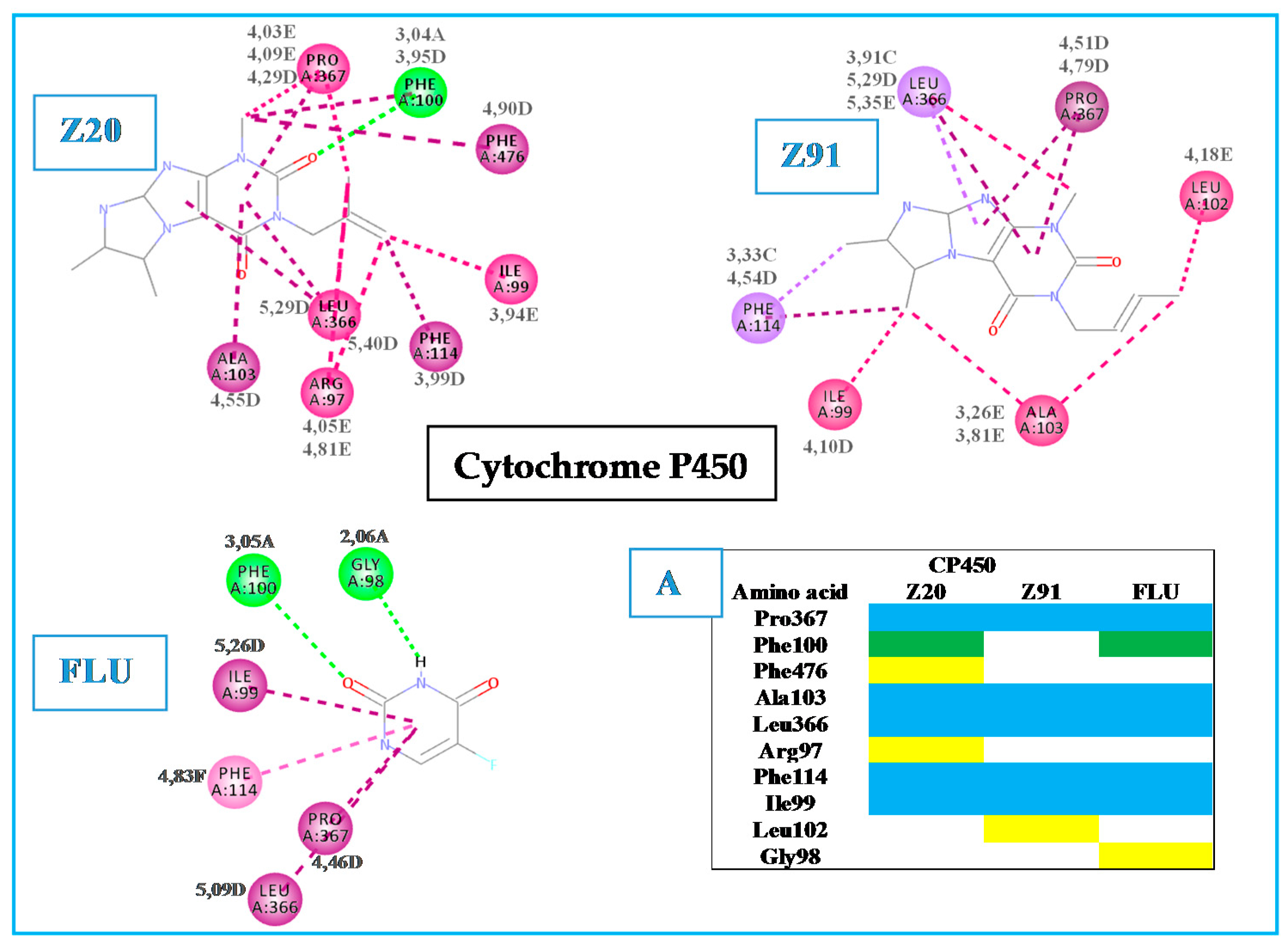
 Hydrogen bond (A);
Hydrogen bond (A);  carbon-hydrogen bond (B);
carbon-hydrogen bond (B);  pi-sigma (C);
pi-sigma (C);  pi-alkyl (D);
pi-alkyl (D);  alkyl (E).
Hydrogen bond (A); carbon-hydrogen bond (B); pi-sigma (C); pi-alkyl (D); alkyl (E).
alkyl (E).
Hydrogen bond (A); carbon-hydrogen bond (B); pi-sigma (C); pi-alkyl (D); alkyl (E). Hydrogen bond (A); carbon-hydrogen bond (B); pi-sigma (C); pi-alkyl (D); alkyl (E).
Hydrogen bond (A); carbon-hydrogen bond (B); pi-sigma (C); pi-alkyl (D); alkyl (E).
Hydrogen bond (A); carbon-hydrogen bond (B); pi-sigma (C); pi-alkyl (D); alkyl (E).
Hydrogen bond (A); carbon-hydrogen bond (B); pi-sigma (C); pi-alkyl (D); alkyl (E). Hydrogen bond (A); carbon-hydrogen bond (B); pi-sigma (C); pi-alkyl (D); akyl (E).
Hydrogen bond (A); carbon-hydrogen bond (B); pi-sigma (C); pi-alkyl (D); akyl (E).
Hydrogen bond (A); carbon-hydrogen bond (B); pi-sigma (C); pi-alkyl (D); akyl (E).
Hydrogen bond (A); carbon-hydrogen bond (B); pi-sigma (C); pi-alkyl (D); akyl (E).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Assignment |
|---|---|
 | Tested molecule |
 | Tested molecule |
| Descriptors | Z91 | Z20 | FLU | MEL | DEX |
| AST (Å2) | 457.6300 | 432.0800 | 229.5500 | 438.0500 | 371.1200 |
| MV (Å3) | 831.4000 | 817.9600 | 348.7100 | 740.5100 | 821.6600 |
| χ (eV) | 2.9553 | 2.9776 | 3.5946 | 1.9003 | 1.9594 |
| η (eV) | 5.2982 | 5.3058 | 6.3539 | 5.4811 | 5.9292 |
| 1/η (eV) | 0.1887 | 0.1885 | 0.1574 | 0.1824 | 0.1687 |
| μ (eV) | −2.9553 | −2.9776 | −3.5946 | −1.9003 | −1.9594 |
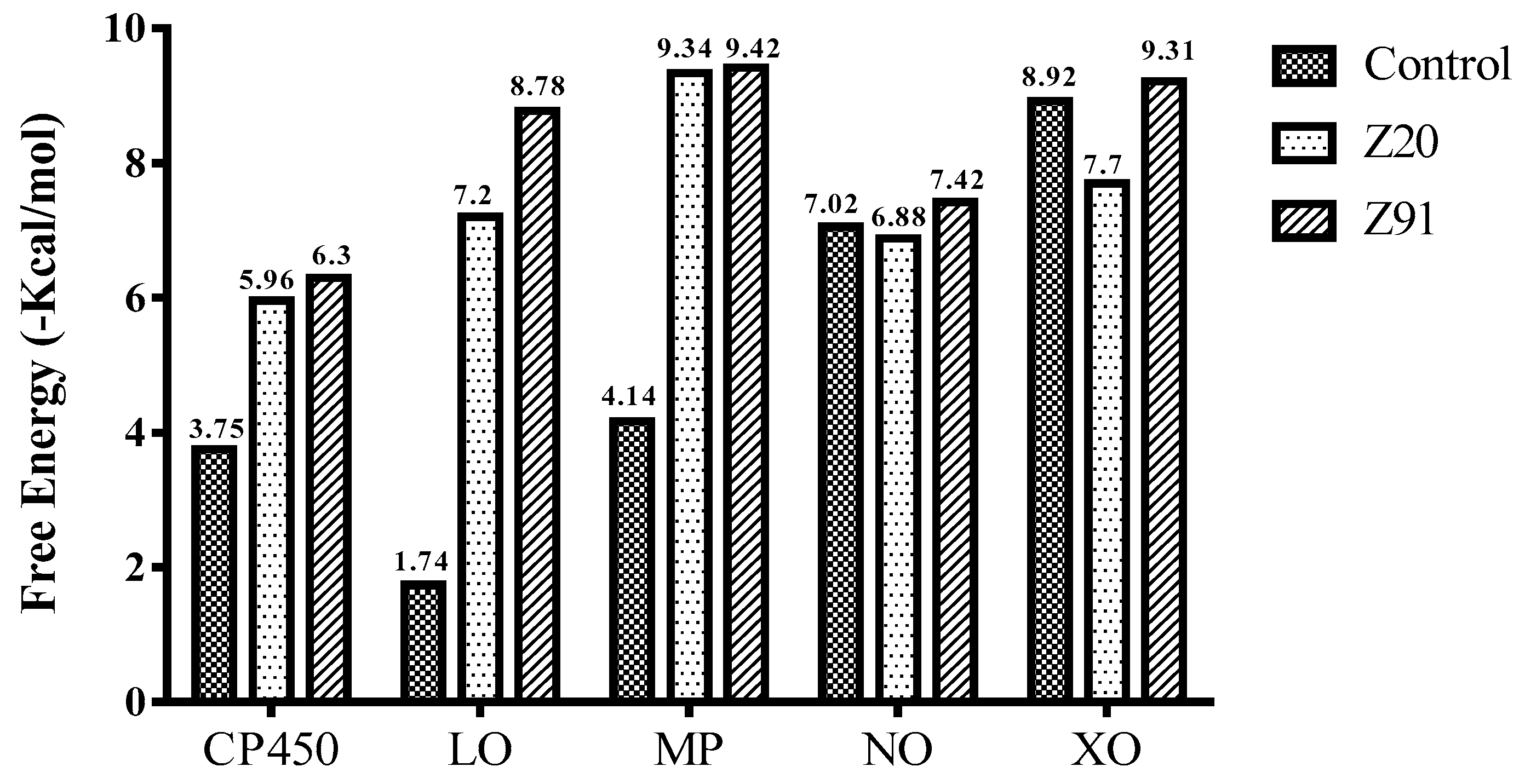
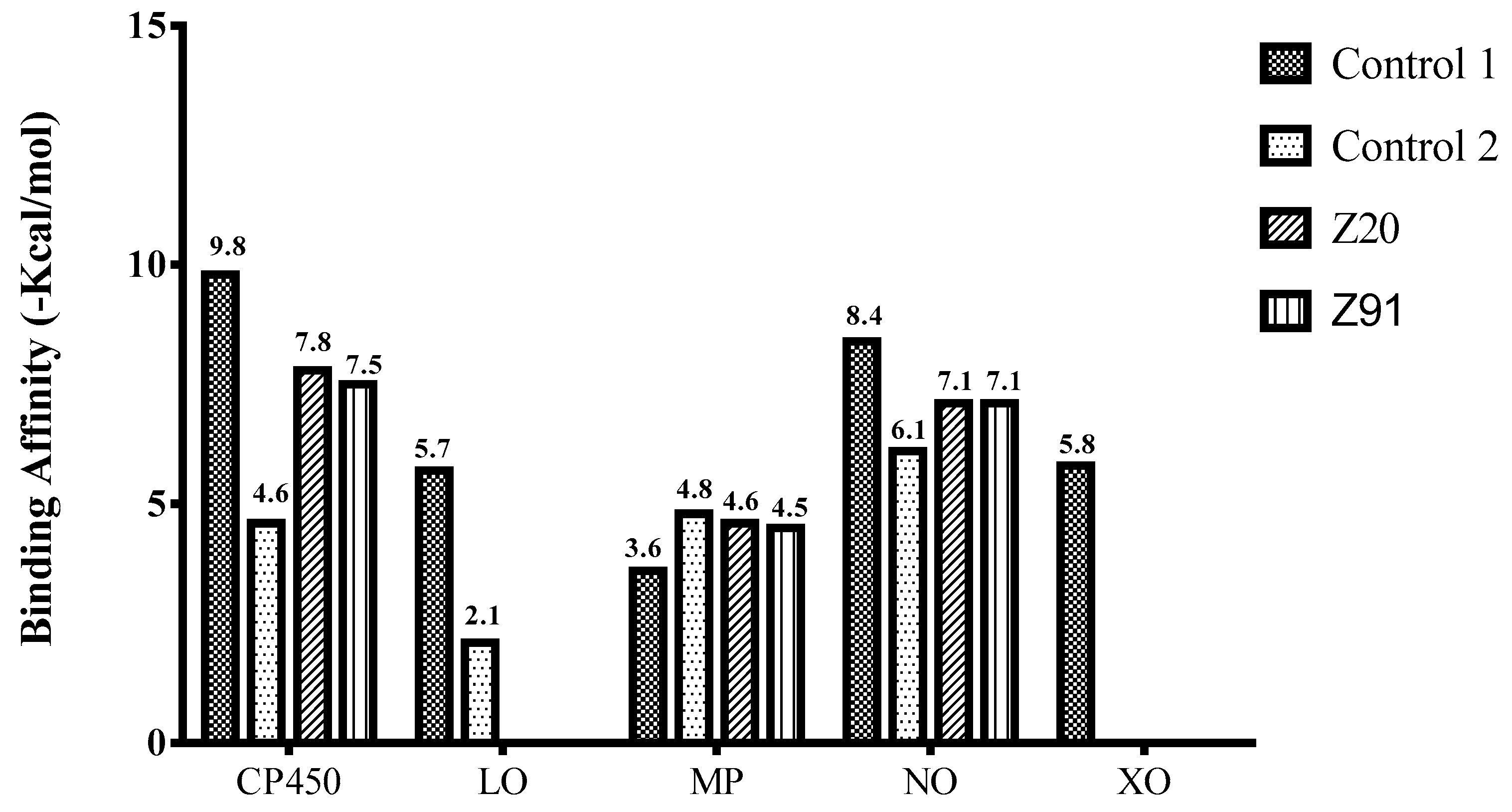
| Ki-CP450 (µM) | 24.30 | 17.89 | 182.0 | - | - |
| Ki-MP (µM) | 22.58 | 19.97 | - | 185.0 | - |
| Ki-NO (µM) | 9.83 | 3.36 | - | - | 7.11 |
| Descriptors | CKi-CP450 | CKi-MP | CKi-NO | - | - |
| AST | −0.95 | −0.28 | −0.51 | - | - |
| VM | −0.99 | −0.05 | −0.74 | - | - |
| χ | −0.67 | −0.60 | −0.08 | - | - |
| η | 0.85 | 0.19 | 0.49 | - | - |
| 1/η | −0.83 | −0.17 | −0.48 | - | - |
| μ | −0.67 | −0.60 | −0.08 | - | - |
| # | LUMO+1 | LUMO | HOMO | HOMO-1 |
| Z20 |  |  |  |  |
| GAP1 = 10.6116 eV | GAP2 = 12.6035 eV | GAP3 = 11.7711 eV | GAP4 = 13.7630 eV | |
| Z91 |  |  |  |  |
| GAP1 = 10.5964 eV | GAP2 = 12.5932 eV | GAP3 = 11.7572 eV | GAP4 = 13.7540 eV | |
| FLU |  |  |  |  |
| GAP1 = 12.7077 eV | GAP2 = 14.8923 eV | GAP3 = 14.7945 eV | GAP4 = 16.9791 eV | |
| MEL |  |  |  |  |
| GAP1 = 10.9621 eV | GAP2 = 12.6495 eV | GAP3 = 11.2799 eV | GAP4 = 12.9853 eV | |
| DEX |  |  |  |  |
| GAP1 = 11.8585 eV | GAP2 = 12.4686 eV | GAP3 = 12.4479 eV | GAP4 = 13.3285 eV |
| Descriptors | Enzymes | ||
|---|---|---|---|
| CP450 | MP | NO | |
| HOMO-1 (eV) | −0.38 | −0.81 | −0.92 |
| HOMO (eV) | −0.13 | −0.47 | −0.22 |
| LUMO (eV) | 0.91 | 0.54 | 0.30 |
| LUMO+1 (eV) | 0.88 | 0.60 | 0.23 |
| ΔGAP | GAP1 * | GAP2 * | GAP3 * | GAP4 * |
|---|---|---|---|---|
| ΔGAP (Z20-FLU) | −2.0961 | −2.2888 | −3.0234 | −3.2161 |
| ΔGAP (Z91-FLU) | −2.1113 | −2.2991 | −3.0373 | −3.2251 |
| ΔGAP (Z20-MEL) | −0.3505 | −0.3586 | 0.4912 | 0.7777 |
| ΔGAP (Z91-MEL) | −0.3657 | −0.3689 | 0.4773 | 0.7687 |
| ΔGAP (Z20-DEX) | −1.2469 | 0.1349 | −0.6768 | 0.4345 |
| ΔGAP (Z91-DEX) | −1.2621 | 0.1246 | −0.6907 | 0.4255 |
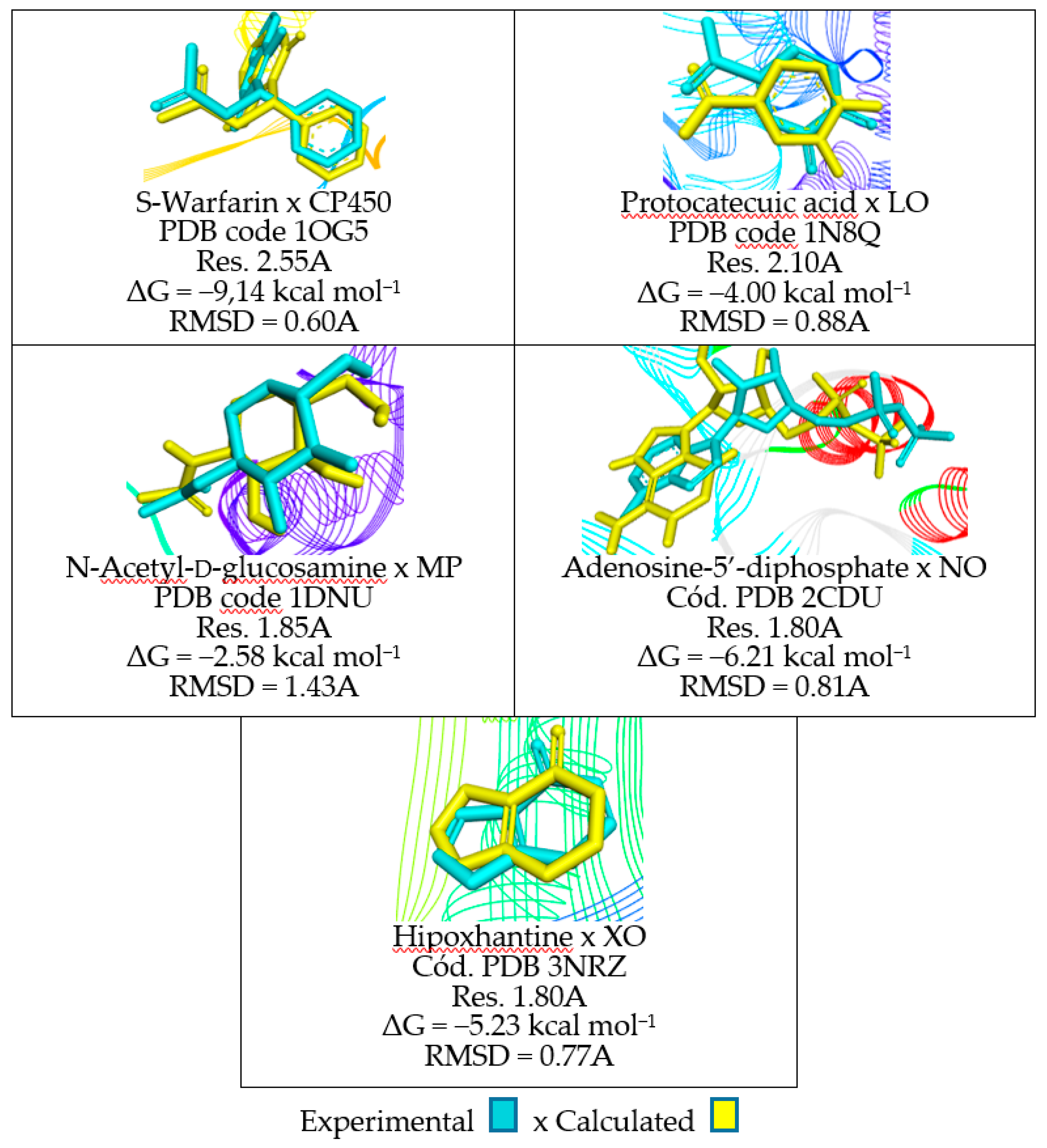
| Receptor | Ligand | Coordinates of the Grid Center | Grid Size (points) |
|---|---|---|---|
| CP450 (PDB code: 1OG5) | S-Warfarin | −20.257x 86.991y 38.581z | 22 x 20 y 24 z |
| LO (PDB code: 1N8Q) | Protocatecuic acid | 21.864x 2.184y 18.909z | 24 x 18 y 12 z |
| MP (PDB code: 1DNU) | N-Acetyl-D-glucosamine | 39.817x −38.635y −5.308z | 22 x 30 y 18 z |
| NO (PDB code: 2CDU) | Adenosine-5’-diphosphate | 1.687x 9.885y 54.962z | 30 x 14 y 32 z |
| XO (PDB code: 3NRZ) | Hypoxanthine | 89.018x 9.4501y 18.290z | 37 x 37 y 37 z |
| Molecule | Assignment |
|---|---|
 | Molecule (control) present in commercially available drug with inhibitory activity at the CP450 receptor [55,56]. |
 | Molecule (control) present in commercially available drug with inhibitory activity at the LO receptor [56,57,58,59]. |
 | Molecule (control) present in commercially available drug with inhibitory activity at the MP receptor [56,59,60]. |
 | Molecule (control) present in commercially available drug with inhibitory activity at the NO receptor [56,61,62]. |
 | Molecule (control) present in commercially available drug with inhibitory activity at the XO receptor [13,56,63]. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, J.d.S.; Ramos, R.d.S.; Costa, K.d.S.L.; Brasil, D.d.S.B.; Silva, C.H.T.d.P.d.; Ferreira, E.F.B.; Borges, R.d.S.; Campos, J.M.; Macêdo, W.J.d.C.; Santos, C.B.R.d. An In Silico Study of the Antioxidant Ability for Two Caffeine Analogs Using Molecular Docking and Quantum Chemical Methods. Molecules 2018, 23, 2801. https://doi.org/10.3390/molecules23112801
Costa JdS, Ramos RdS, Costa KdSL, Brasil DdSB, Silva CHTdPd, Ferreira EFB, Borges RdS, Campos JM, Macêdo WJdC, Santos CBRd. An In Silico Study of the Antioxidant Ability for Two Caffeine Analogs Using Molecular Docking and Quantum Chemical Methods. Molecules. 2018; 23(11):2801. https://doi.org/10.3390/molecules23112801
Chicago/Turabian StyleCosta, Josivan da Silva, Ryan da Silva Ramos, Karina da Silva Lopes Costa, Davi do Socorro Barros Brasil, Carlos Henrique Tomich de Paula da Silva, Elenilze Figueiredo Batista Ferreira, Rosivaldo dos Santos Borges, Joaquín María Campos, Williams Jorge da Cruz Macêdo, and Cleydson Breno Rodrigues dos Santos. 2018. "An In Silico Study of the Antioxidant Ability for Two Caffeine Analogs Using Molecular Docking and Quantum Chemical Methods" Molecules 23, no. 11: 2801. https://doi.org/10.3390/molecules23112801
APA StyleCosta, J. d. S., Ramos, R. d. S., Costa, K. d. S. L., Brasil, D. d. S. B., Silva, C. H. T. d. P. d., Ferreira, E. F. B., Borges, R. d. S., Campos, J. M., Macêdo, W. J. d. C., & Santos, C. B. R. d. (2018). An In Silico Study of the Antioxidant Ability for Two Caffeine Analogs Using Molecular Docking and Quantum Chemical Methods. Molecules, 23(11), 2801. https://doi.org/10.3390/molecules23112801