
Synthesis, Biological Activity and Preliminary in Silico ADMET Screening of Polyamine Conjugates with Bicyclic Systems

 ,
, 

Abstract
:
1. Introduction
2. Results and Discussion
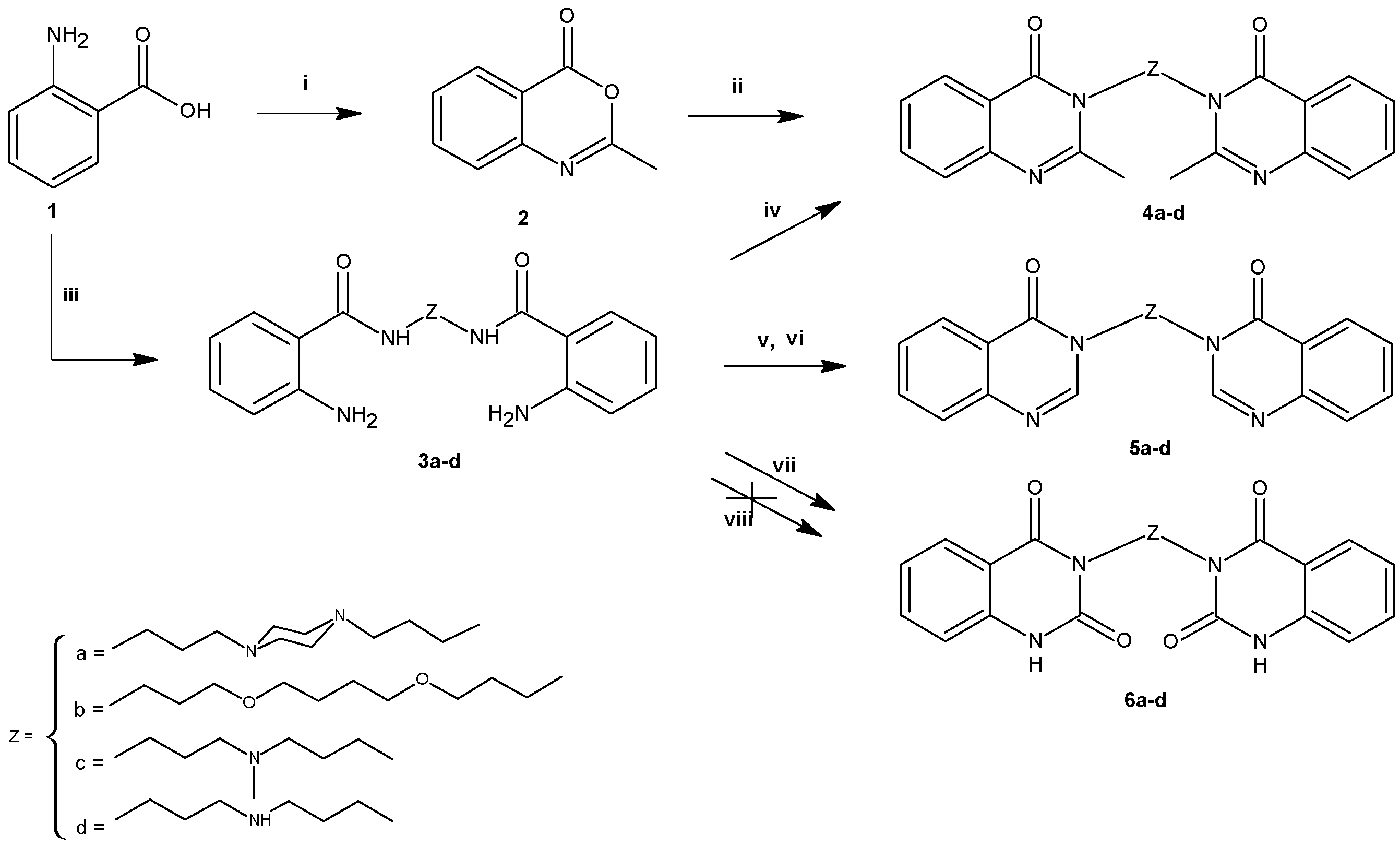
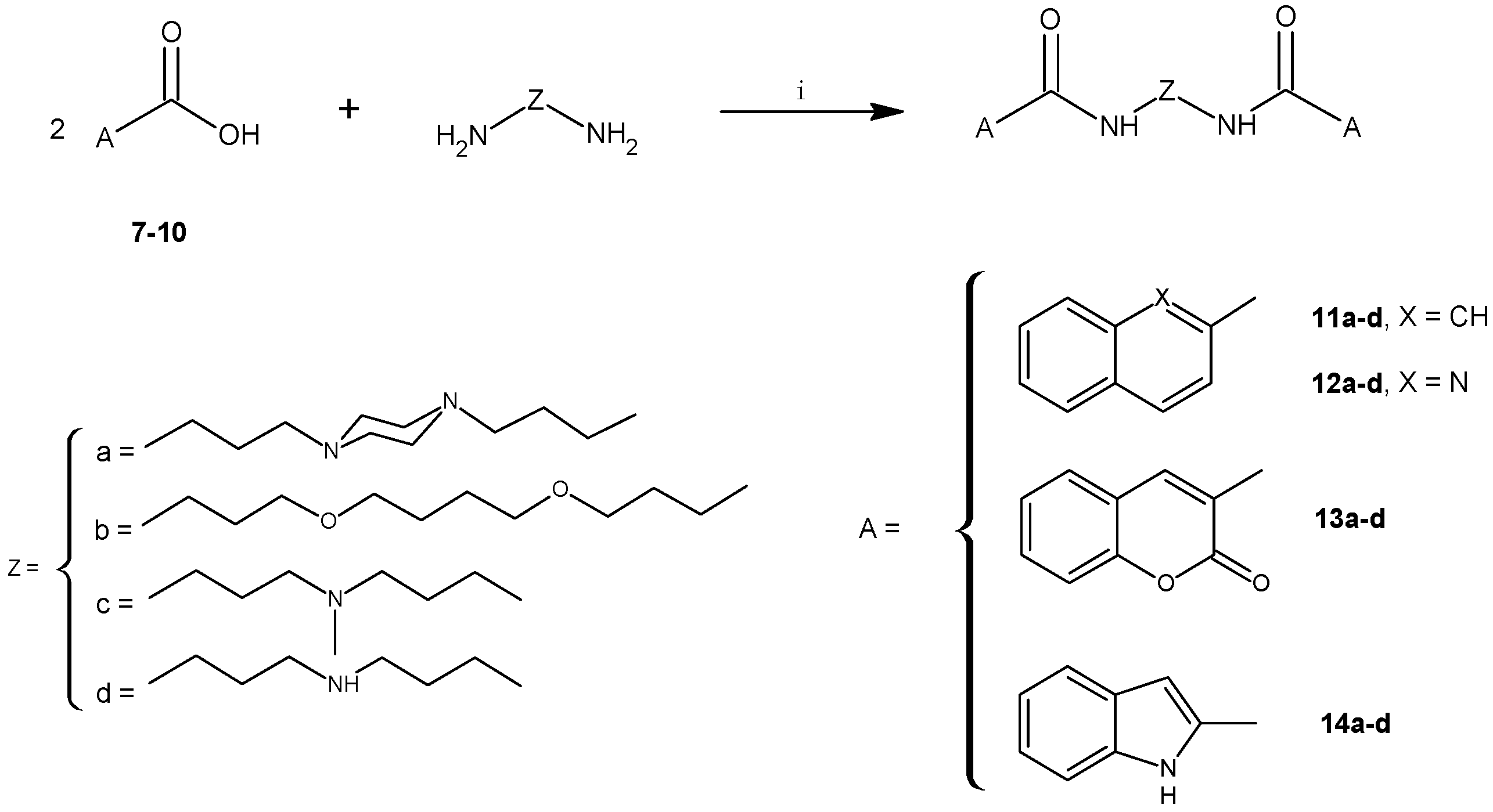
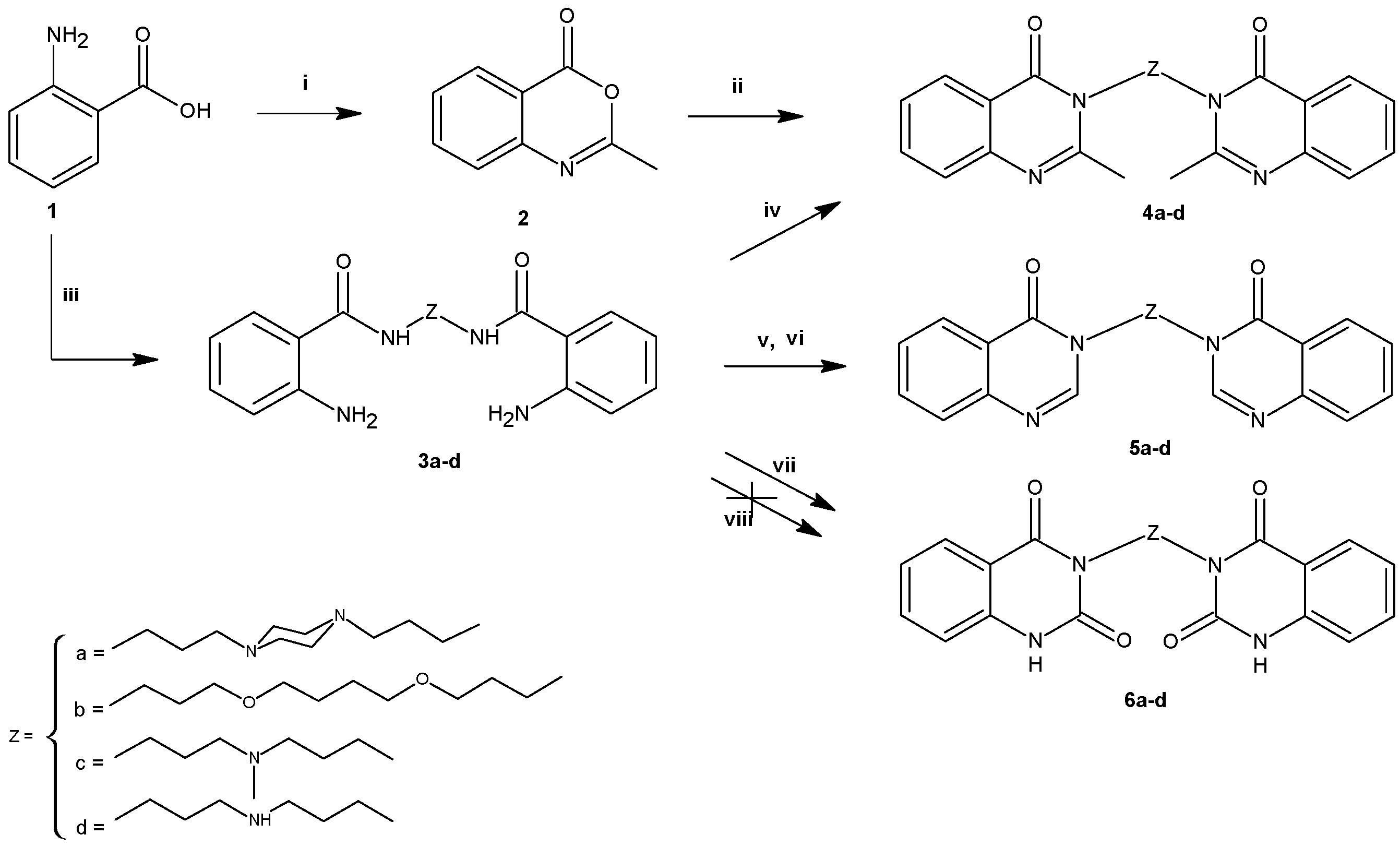
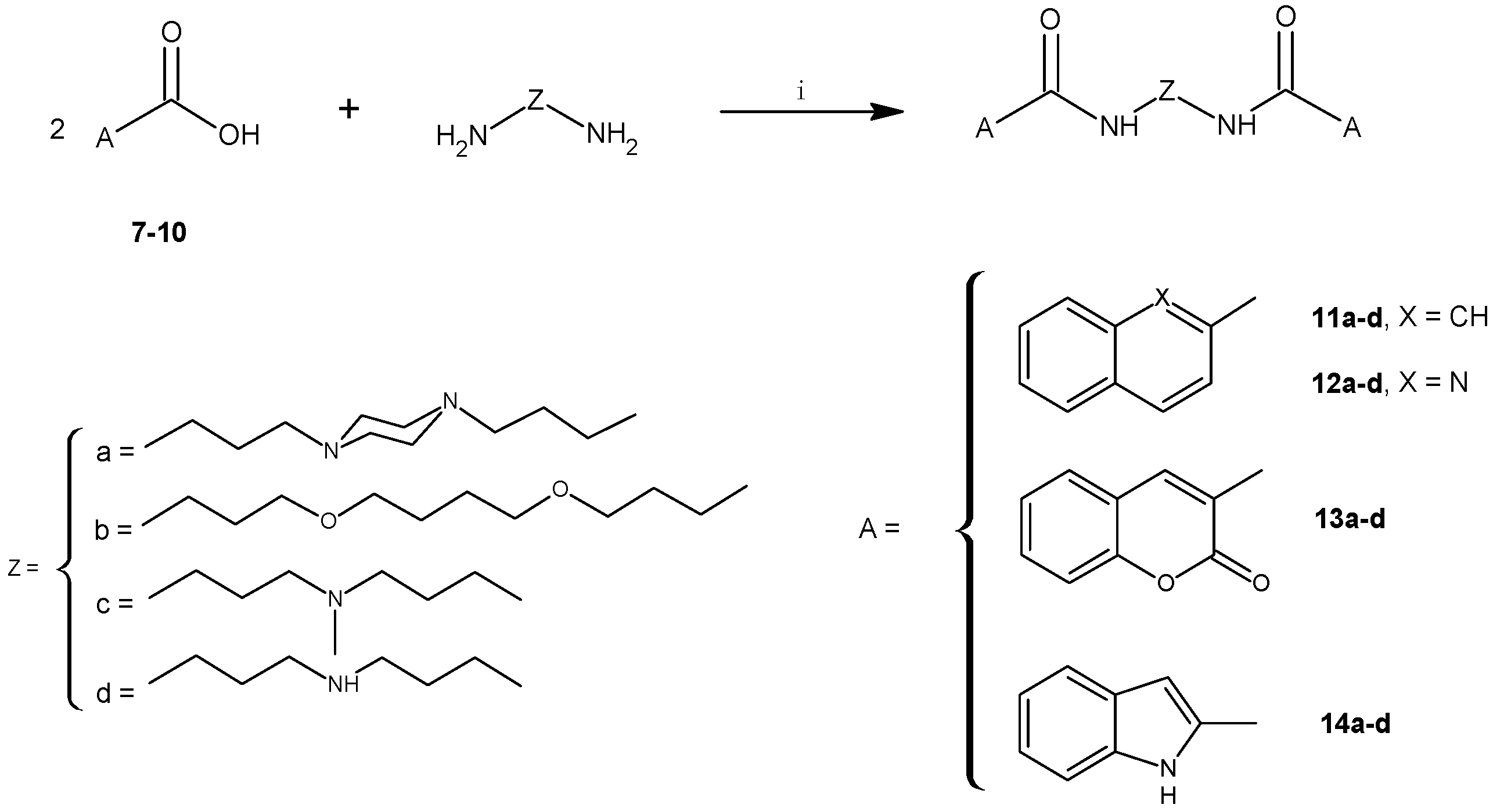
2.1. Chemistry
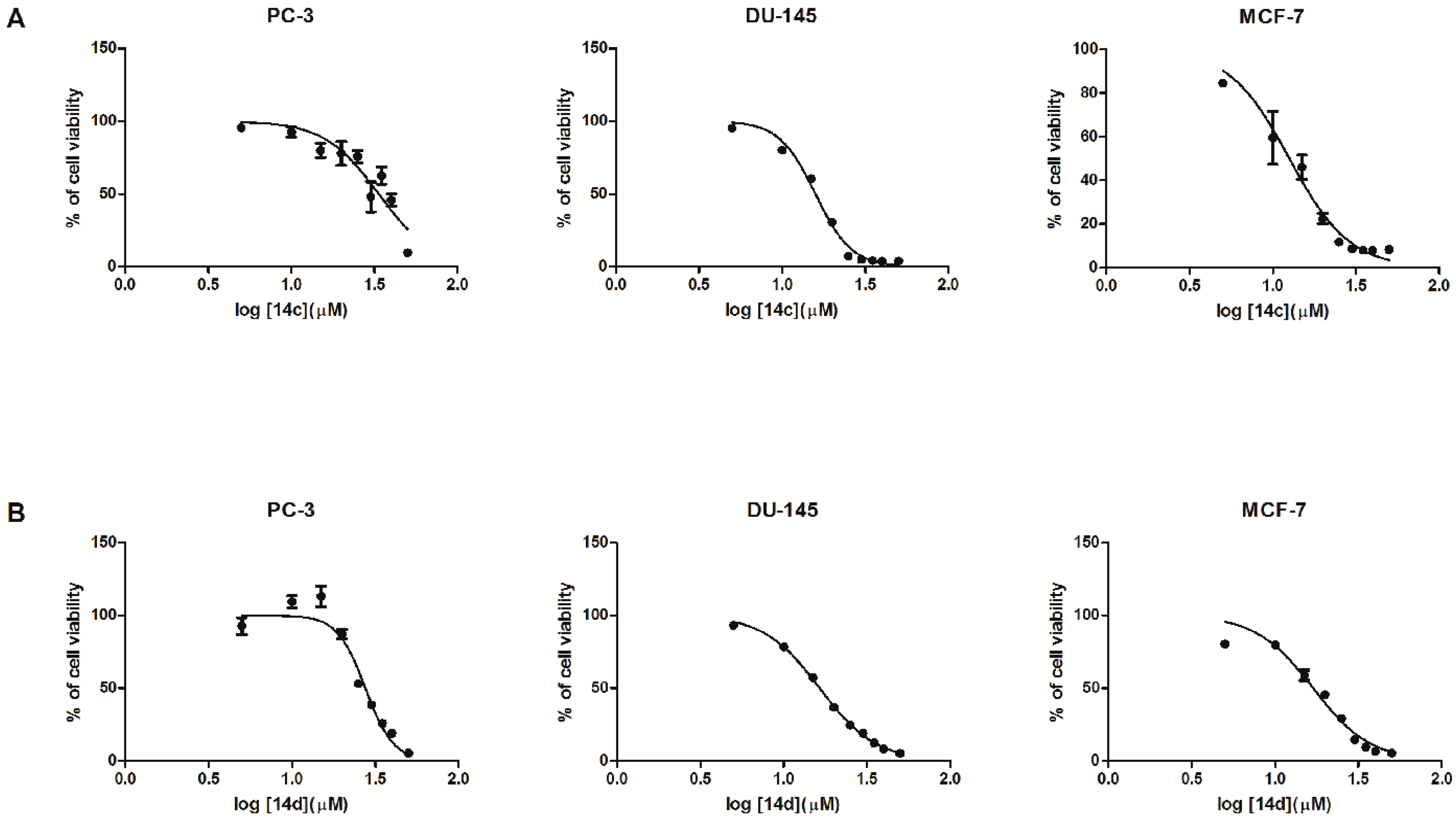
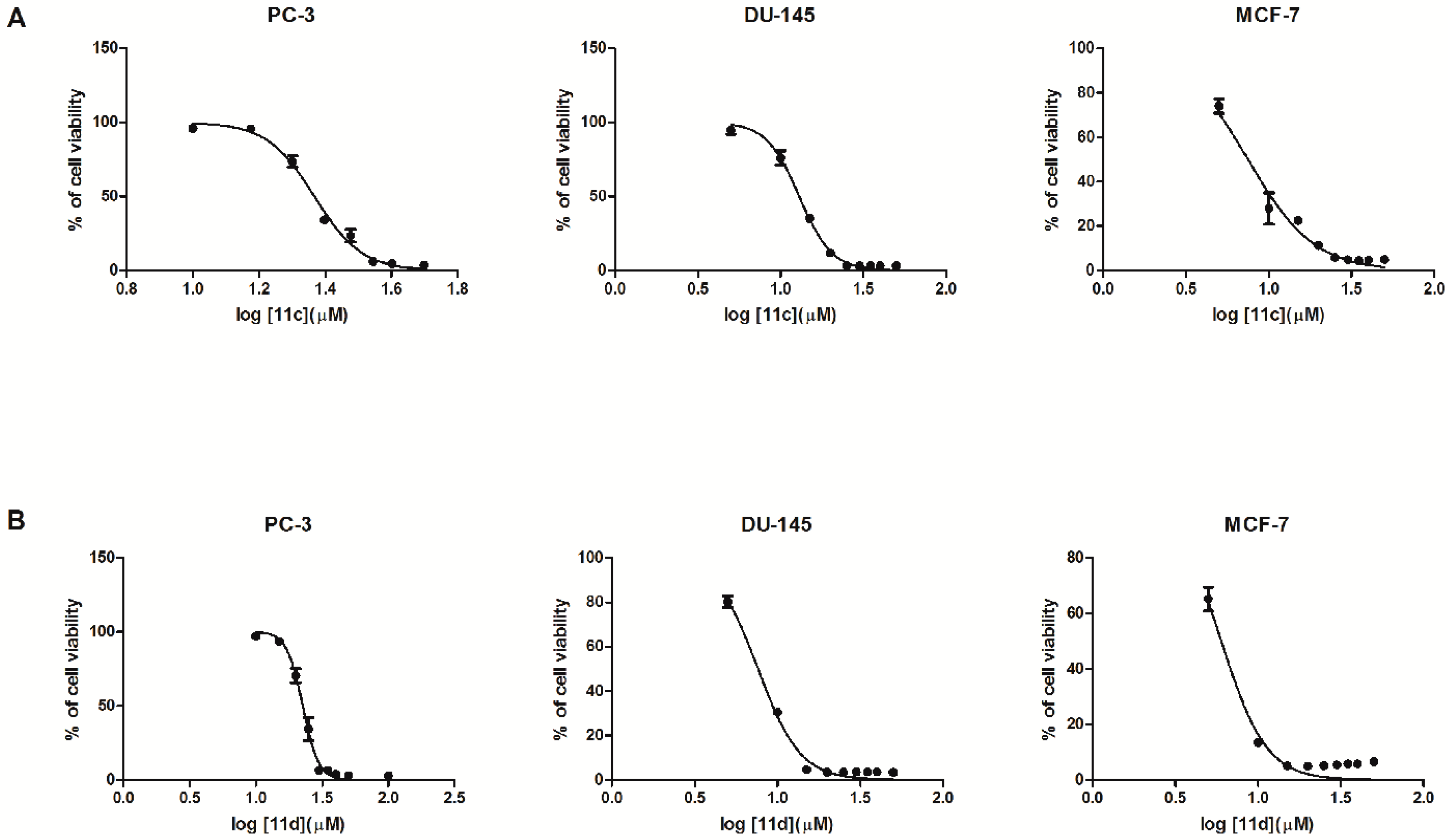
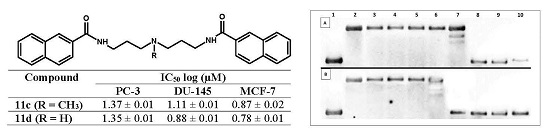
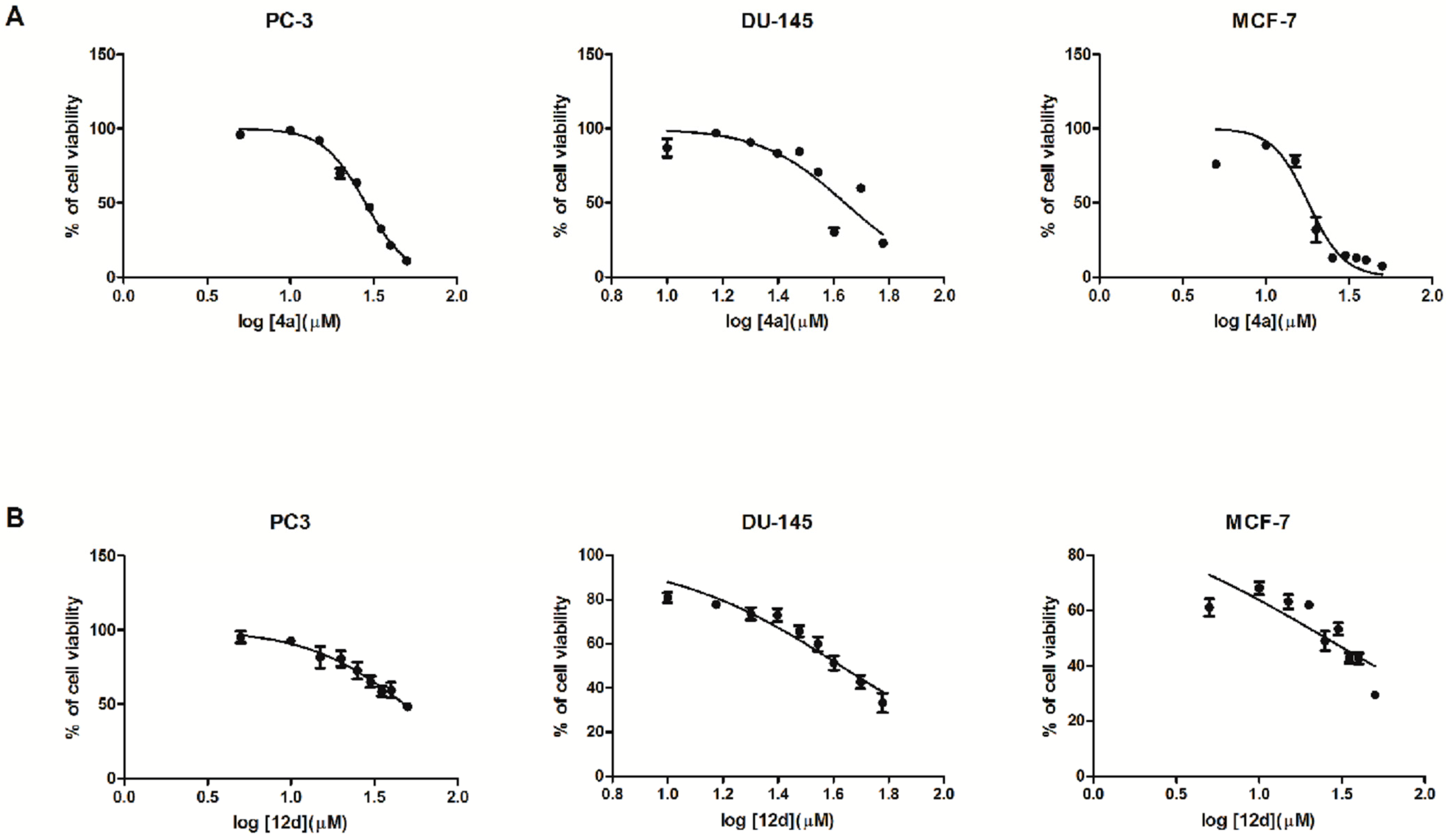
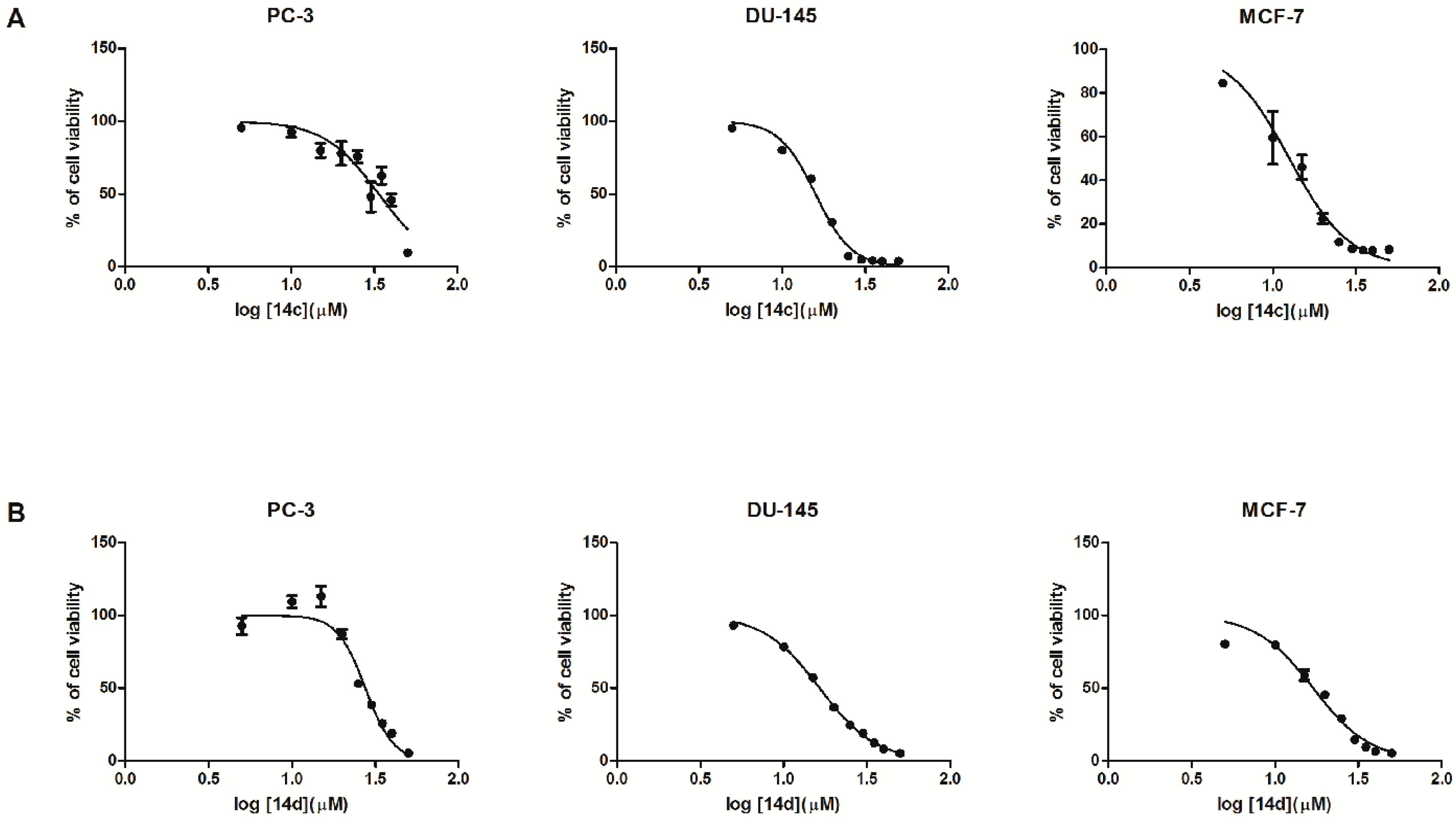
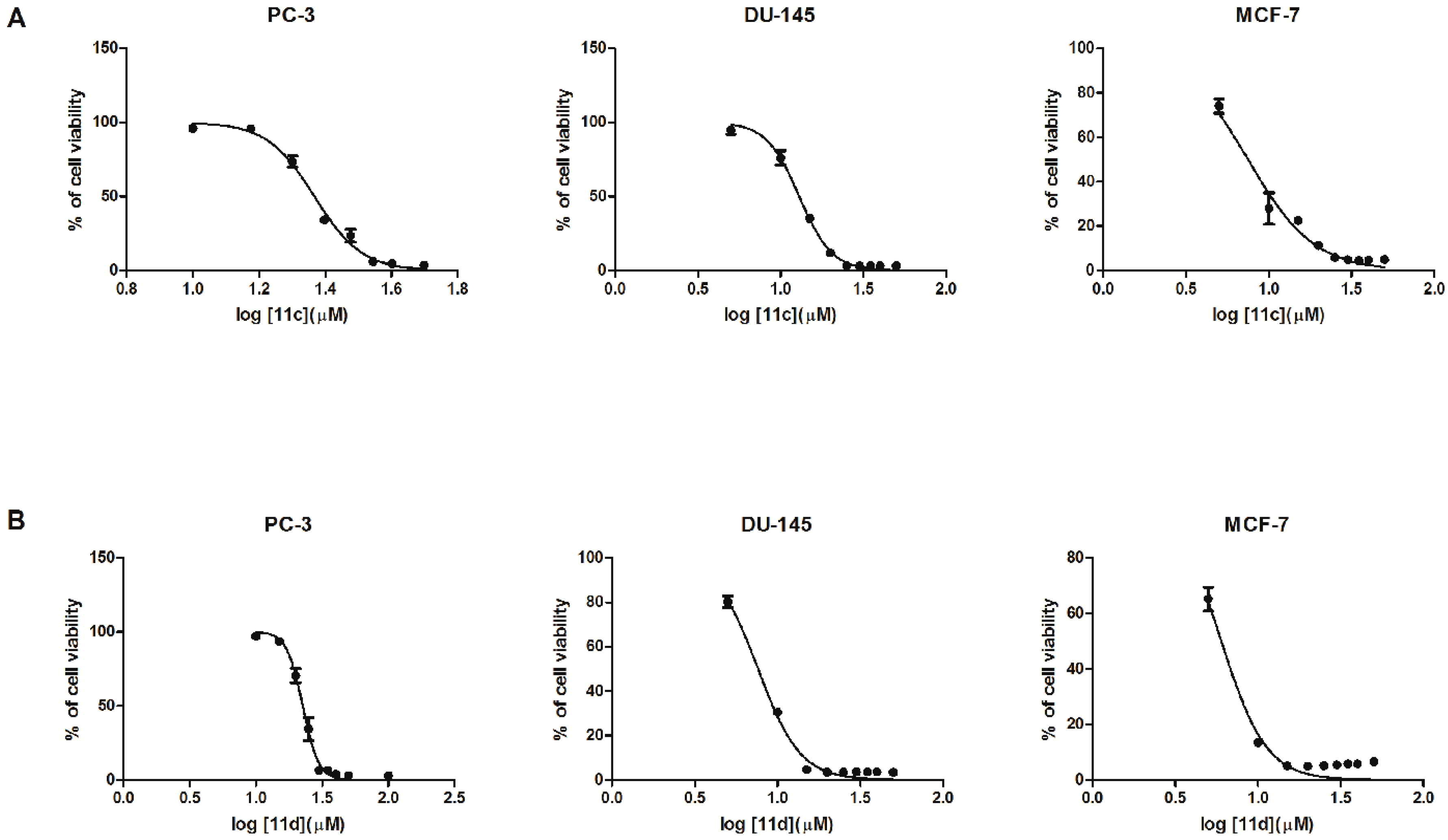
2.2. Biological In Vitro Evaluation
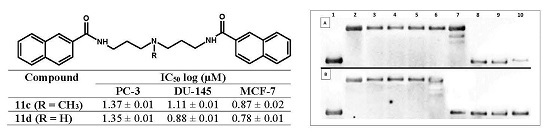
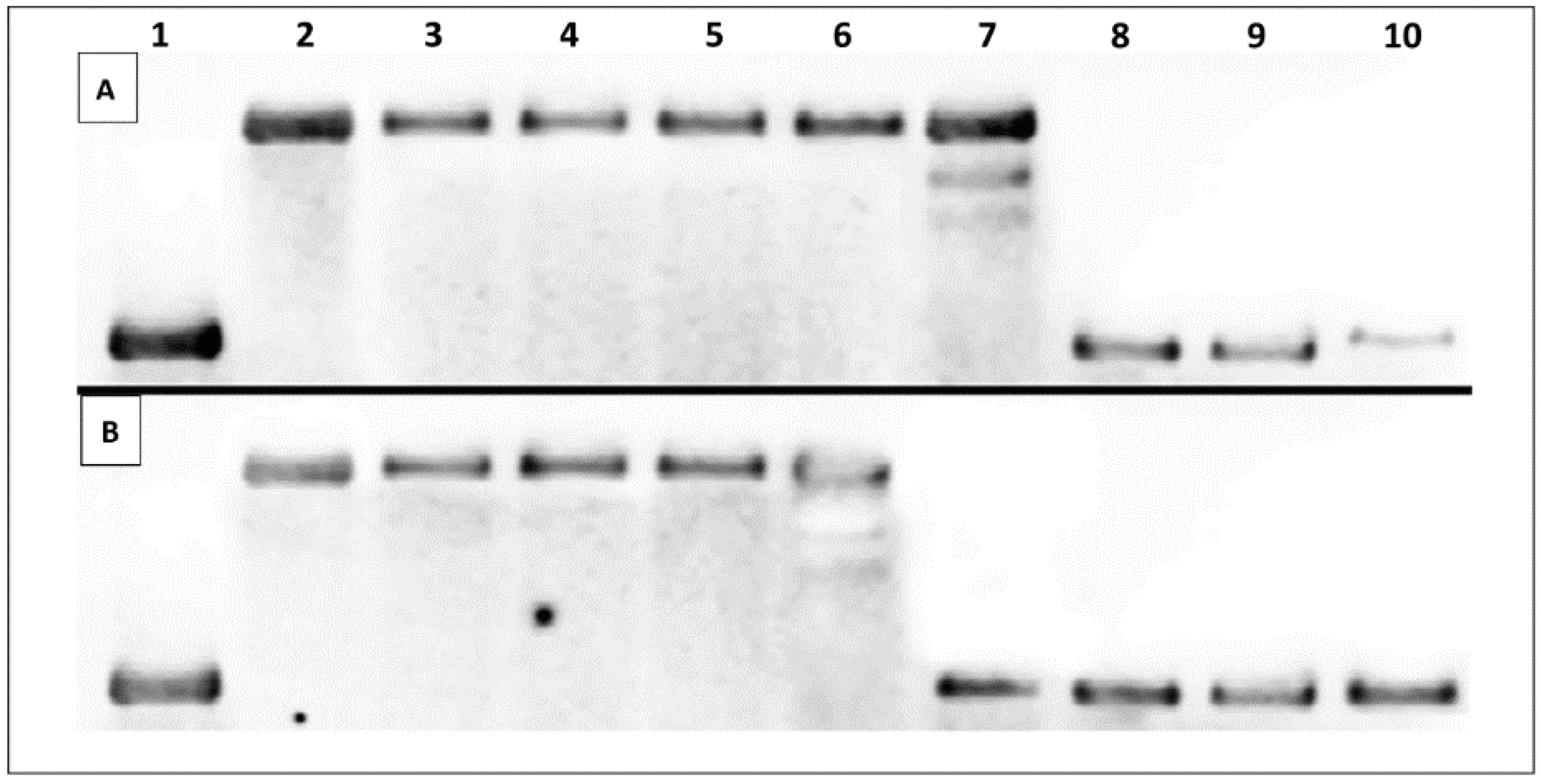
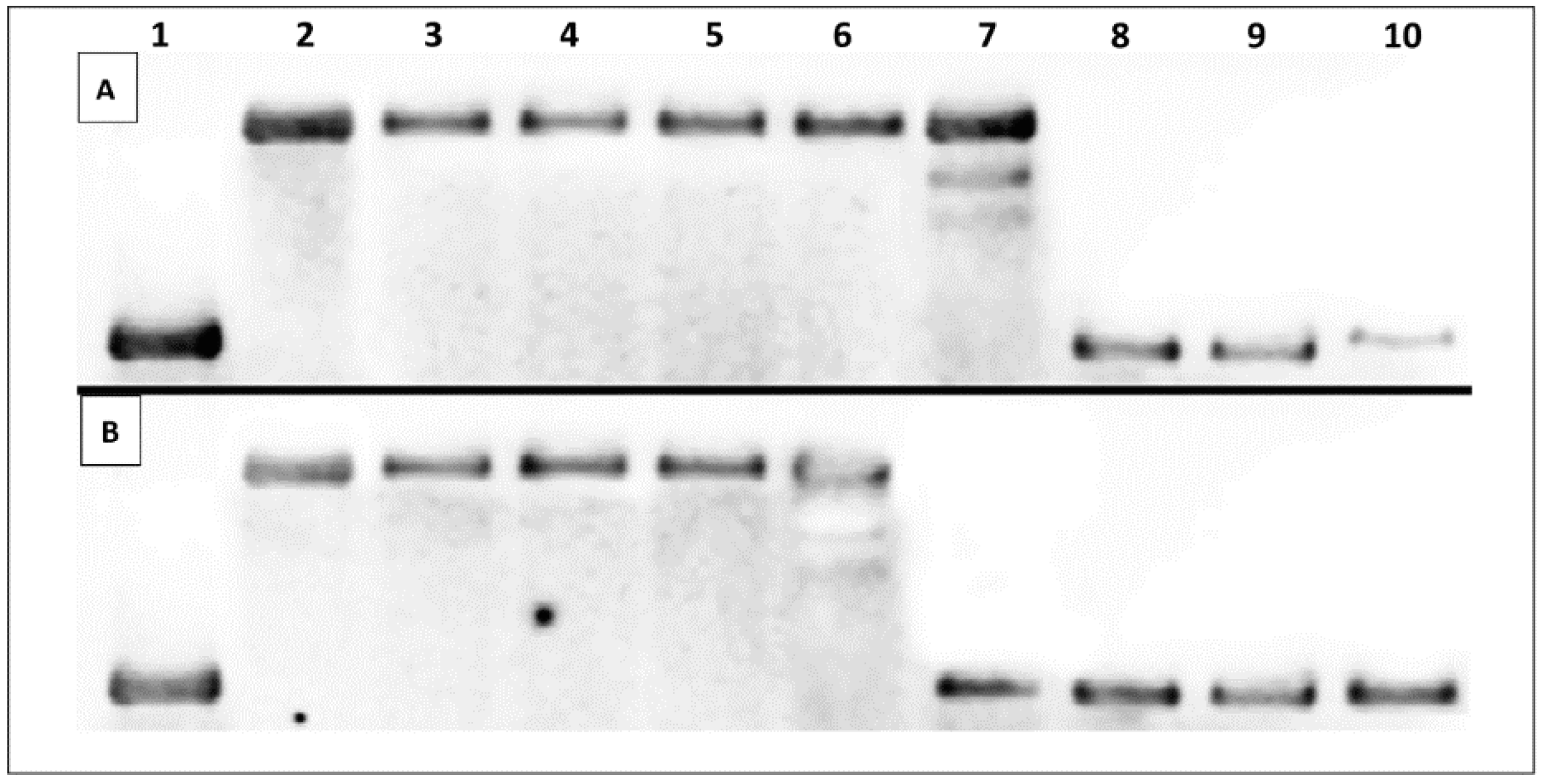
2.3. DNA Interaction Studies
2.3.1. Thermal Melting Studies
2.3.2. Topoisomerase I Activity Assay
2.4. Preliminary In Silico ADME Screening
3. Materials and Methods
3.1. General Information
3.2. Chemistry
3.2.1. General Procedure for the Synthesis of 3a–d
3.2.2. General Procedure for the Synthesis of 4a–d
3.2.3. General Procedure for the Synthesis of 5a–d
3.2.4. General Procedure for the Synthesis of 6a–d
3.2.5. General Procedure for the Synthesis of 11a–d to 14a–d
3.3. Biological In Vitro Evaluation
3.3.1. Preparation of Drug Stock and Working Solutions
3.3.2. Cell Culture
3.3.3. Drug Treatment
3.3.4. WST–1 Cell Viability Assay
3.4. DNA Interaction Studies
3.4.1. Thermal Melting Studies
3.4.2. Strains and Media
3.4.3. Bacterial Culture and Plasmid Isolation
3.4.4. Topoisomerase I Activity Assay
3.5. Preliminary in Silico ADME Screening
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhang, R.; Wu, X.; Yalowich, J.C.; Hasinoff, B.B. Design, synthesis, and biological evaluation of a novel series of bisintercalating DNA-binding piperazine-linked bisanthrapyrazole compounds as anticancer agents. Bioorg. Med. Chem. 2011, 19, 7023–7032. [Google Scholar] [CrossRef] [PubMed]
- Avendano, C.; Carlos, M.J. DNA intercalators and topiosomerase inhibitors. In Medicinal Chemistry of Anticancer Drugs; Elsevier Inc.: Madrid, Spain, 2008; pp. 199–228. [Google Scholar]
- Lorente, A.; Vázquez, Y.; Fernández, M.J.; Ferrández, A. Bisacridines with aromatic linking chains. Synthesis, DNA interaction, and antitumor activity. Bioorg. Med. Chem. 2004, 12, 4307–4312. [Google Scholar] [CrossRef] [PubMed]
- Rescifina, A.; Zagni, C.; Varrica, M.G.; Pistara, V.; Corsaro, A. Recent advances in small organic molecules as DNA intercalating agents: Synthesis, activity, and modeling. Eur. J. Med. Chem. 2014, 74, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Brana, M.F.; Cacho, M.; Gradillas, A.; de Pascual-Teresa, B.; Ramos, A. Intercalators as anticancer drugs. Curr. Pharm. Des. 2001, 7, 1745–1780. [Google Scholar] [CrossRef] [PubMed]
- Rong, R.X.; Sun, Q.; Ma, C.L.; Chen, B.; Wang, W.Y.; Wang, Z.A.; Wang, K.R.; Cao, Z.R.; Li, X.L. Development of novel bis-naphthalimide derivatives and their anticancer properties. Med. Chem. Commun. 2016, 7, 679–685. [Google Scholar] [CrossRef]
- Bestwick, C.S.; Ralton, L.D.; Milne, L.; Kong Thoo Lin, P.; Duthie, S.J. The influence of bisnaphthalimidopropyl polyamines on DNA instability and repair in Caco-2 colon epithelial cells. Cell Biol. Toxicol. 2011, 27, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Brana, M.F.; Cacho, M.; Ramos, A.; Dominguez, M.T.; Pozuelo, J.M.; Abradelo, C.; Rey-Stolle, M.F.; Yuste, M.; Carrasco, C.; Bailly, C. Synthesis, biological evaluation and DNA binding properties of novel mono and bisnaphthalimides. Org. Biomol. Chem. 2003, 1, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Gamage, S.A.; Spicer, J.A.; Atwell, G.J.; Finlay, G.J.; Baguley, B.C.; Denny, W.A. Structure-activity relationships for substituted bis(acridine-4-carboxamides): A new class of anticancer agents. J. Med. Chem. 1999, 42, 2383–2393. [Google Scholar] [CrossRef] [PubMed]
- Demeunynck, M.; Charmantray, F.; Martelli, A. Interest of acridine derivatives in the anticancer chemotherapy. Curr. Pharm. Des. 2001, 7, 1703–1724. [Google Scholar] [CrossRef] [PubMed]
- Antonini, I. Intriguing classes of acridine derivatives as DNA-binding antitumour agents: From pyrimido[5,6,1-de]acridines to bis(acridine-4-carboxamides). Med. Chem. Rev. Online 2004, 1, 267–290. [Google Scholar] [CrossRef]
- Spicer, J.A.; Gamage, S.A.; Finlay, G.J.; Baguley, B.C.; Denny, W.A. Dimeric analogues of non-cationic tricyclic aromatic carboxamides are a new class of cytotoxic agents. Anti-Cancer Drug Des. 1999, 14, 281–289. [Google Scholar]
- Spicer, J.A.; Gamage, S.A.; Rewcastle, G.W.; Finlay, G.J.; Bridewell, D.J.A.; Baguley, B.C.; Denny, W.A. Bis(phenazine-1-carboxamides): Structure-activity relationships for a new class of dual topoisomerase I/II-directed anticancer drugs. J. Med. Chem. 2000, 43, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Gamage, S.A.; Spicer, J.A.; Finlay, G.J.; Stewart, A.J.; Charlton, P.; Baguley, B.C.; Denny, W.A. Dicationic bis(9-methylphenazine-1-carboxamides): Relationships between biological activity and linker chain structure for a series of potent topoisomerase targeted anticancer drugs. J. Med. Chem. 2001, 44, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Yang, D. DNA recognition by a novel bis-intercalator, potent anticancer drug XR5944. Curr. Top. Med. Chem. 2015, 15, 1385–1397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.S.; Fang, L.Y.; Zhu, L.Z.; Sun, D.X.; Wang, P.G. Syntheses and biological activity of bisdaunorubicins. Bioorg. Med. Chem. 2006, 14, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Chaires, J.B.; Leng, F.F.; Przewloka, T.; Fokt, I.; Ling, Y.H.; Perez-Soler, R.; Priebe, W. Structure-based design fill of a new bisintercalating anthracycline antibiotic. J. Med. Chem. 1997, 40, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, S.; Vizcaıno, C.; Rodrıguez-Sanchez, M.A.; Priebe, W.; Portugal, J. Autophagy modulates the effects of bis-anthracycline WP631 on p53-deficient prostate cancer cells. J. Cell. Mol. Med. 2015, 19, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.; Marin, L.; Alvarez-Alonso, R.; Redondo, S.; Carvajal, J.; Villamizar, G.; Villar, C.J.; Lombo, F. Biosynthetic modularity rules in the bisintercalator family of antitumor compounds. Mar. Drugs 2014, 12, 2668–2699. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.R.; LaTurner, S.; Ziemer, J.; McVean, M.; Devens, B.; Carlson, C.L.; Graminski, G.F.; Vanderwerf, S.M.; Weeks, R.S.; Carreon, J. Induction of apoptosis by aryl-substituted diamines: Role of aromatic group substituents and distance between nitrogens. Bioorg. Med. Chem. Lett. 2002, 12, 1263–1267. [Google Scholar] [CrossRef]
- Cain, B.F.; Baguley, B.C.; Denny, W.A. Potential antitumor agents. 28. Deoxyribonucleic-acid polyintercalating agents. J. Med. Chem. 1978, 21, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Sartorius, J.; Schneider, H.J. Intercalation mechanisms with ds-DNA: Binding modes and energy contributions with benzene, naphthalene, quinoline and indole derivatives including some antimalarials. J. Chem. Soc. Perkin Trans. 2 1997, 2319–2327. [Google Scholar] [CrossRef]
- Szumilak, M.; Szulawska-Mroczek, A.; Koprowska, K.; Stasiak, M.; Lewgowd, W.; Stanczak, A.; Czyz, M. Synthesis and in vitro biological evaluation of new polyamine conjugates as potential anticancer drugs. Eur. J. Med. Chem. 2010, 45, 5744–5751. [Google Scholar] [CrossRef] [PubMed]
- Szulawska-Mroczek, A.; Szumilak, M.; Szczesio, M.; Olczak, A.; Nazarski, R.B.; Lewgowd, W.; Czyz, M.; Stanczak, A. Synthesis and biological evaluation of new bischromone derivatives with antiproliferative activity. Arch. Pharm. 2013, 346, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Szumilak, M.; Galdyszynska, M.; Dominska, K.; Stanczak, A.; Piastowska-Ciesielska, A. Antitumor activity of polyamine conjugates in human prostate and breast cancer. Acta Biochim. Pol. 2017, 64. [Google Scholar] [CrossRef]
- Szumilak, M.; Merecz, A.; Strek, M.; Stanczak, A.; Inglot, T.W.; Karwowski, B.T. DNA interaction studies of selected polyamine conjugates. Int. J. Mol. Sci. 2016, 17, 1560. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.N. Optimization of metabolic stability as a goal of modern drug design. Med. Res. Rev. 2001, 21, 412–449. [Google Scholar] [CrossRef] [PubMed]
- Errede, L.A.; Oien, H.T.; Yarian, D.R. Acylanthranils 3. Influence of ring substituents on reactivity and selectivity in reaction of acylanthranils with amines. J. Org. Chem. 1977, 42, 12–18. [Google Scholar] [CrossRef]
- Staab, H.A. Synthesen Mit Heterocyclishen Amiden (Azoliden). Angew. Chem. 1962, 74, 407–423. [Google Scholar] [CrossRef]
- Errede, L.A. Acylanthranils 1. The pathway of quinazolone formation in the reaction of acylanthranils with anilines. J. Org. Chem. 1976, 41, 1763–1765. [Google Scholar] [CrossRef]
- Errede, L.A.; McBrady, J.J.; Oien, H.T. Acylanthranils. 2. The problem of selectivity in the reaction of acetylanthranil with anilines. J. Org. Chem. 1976, 41, 1765–1768. [Google Scholar] [CrossRef]
- Stanczak, A.; Lewgowd, W.; Ochocki, Z.; Pakulska, W.; Szadowska, A. Synthesis, structures and biological activity of some 4-amino-3-cinnoline-carboxylic acid derivatives-Part 2. Pharmazie 1997, 52, 91–97. [Google Scholar] [PubMed]
- Clark, J.; Hitiris, G. Heterocyclic studies 43. Thieno[2,3-d:4,5-d′]dipyrimidines. J. Chem. Soc. Perkin Trans. 1 1984, 9, 2005–2008. [Google Scholar] [CrossRef]
- Gravier, D.; Dupin, J.P.; Casadebaig, F.; Hou, G.; Boisseau, M.; Bernard, H. Synthesis and in vitro study of platelet antiaggregant activity of some 4-quinazolinone derivatives. Pharmazie 1992, 47, 91–94. [Google Scholar] [PubMed]
- Malamas, M.S.; Millen, J. Quinazolineacetic acids and related analogs as aldose reductase inhibitors. J. Med. Chem. 1991, 34, 1492–1503. [Google Scholar] [CrossRef] [PubMed]
- Stanczak, A.; Lewgowd, W.; Pakulska, W. Synthesis and biological activity of some 4-amino-3-cinnoline carboxylic acid derivatives-Part 4: 2,4-dioxo-1,2,3,4-tetrahydropyrimido[5,4-c] cinnolines. Pharmazie 1998, 53, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Piastowska-Ciesielska, A.W.; Kozlowski, M.; Wagner, W.; Dominska, K.; Ochedalski, T. Effect of an angiotensin II type 1 receptor blocker on caveolin-1 expression in prostate cancer cells. Arch. Med. Sci. 2013, 9, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Guedin, A.; Lacroix, L.; Mergny, J.-L. Thermal Melting Studies of Ligand DNA Interactions. In Drug-DNA Interaction Protocols; Fox, K.R., Ed.; Humana Press: New York, NY, USA, 2010; pp. 25–35. [Google Scholar] [CrossRef]
- Palchaudhuri, R.; Hergenrother, P.J. DNA as a target for anticancer compounds: Methods to determine the mode of binding and the mechanism of action. Curr. Opin. Biotechnol. 2007, 18, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Jianling, W.; Urban, L. The impact of early ADME profiling on drug discovery and development strategy. Drug Discov. World 2004, 4, 73–86. [Google Scholar]
- ACD/Percepta, Version 14.0.0 (Build 2726); Advanced Chemistry Development Inc.: Toronto, On, Canada, 2015; Available online: www.acdlabs.com (accessed on 16 January 2017).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Lanevskij, K.; Japertas, P.; Didziapetris, R.; Petrauskas, A. Ionization-specific prediction of blood–brain permeability. J. Pharm. Sci. 2009, 98, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Moroy, G.; Martiny, V.Y.; Vayer, P.; Villoutreix, B.O.; Miteva, M.A. Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov. Today 2012, 17, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Kleandrova, V.V.; Luan, F.; Speck-Planche, A.; Cordeiro, M.N. In silico assessment of the acute toxicity of chemicals: Recent advances and new model for multitasking prediction of toxic effect. Mini Rev. Med. Chem. 2015, 15, 677–686. [Google Scholar] [CrossRef] [PubMed]
- The European Parliament and the Council of the European Union. Regulation (EC) No 1272/2008 of The European Parliament and of The Council of 16 December 2008 on Classification, Labelling and Packaging of Substances and Mixtures, Amending and Repealing Directives 67/548/EEC and 1999/45/EC, and Amending Regulation (EC) No 1907/2006. 2006. [Google Scholar]
- Saleh, A.M.; Aljada, A.; El-Abadelah, M.M.; Sabri, S.S.; Zahra, J.A.; Nasr, A.; Aziz, M.A. The pyridone-annelated isoindigo (5′-Cl) induces apoptosis, dysregulation of mitochondria and formation of ROS in leukemic HL-60 cells. Cell. Physiol. Biochem. 2015, 35, 1958–1974. [Google Scholar] [CrossRef] [PubMed]
- Tsakalozou, E.; Adane, E.D.; Kuo, K.L.; Daily, A.; Moscow, J.A.; Leggas, M. The effect of breast cancer resistance protein, multidrug resistant protein 1, and organic anion-transporting polypeptide 1B3 on the antitumor efficacy of the lipophilic camptothecin 7-t-butyldimethylsilyl-10-hydroxycamptothecin (AR-67) in vitro. Drug Metab. Dispos. 2013, 41, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Sappal, D.S.; McClendon, A.K.; Fleming, J.A.; Thoroddsen, V.; Connolly, K.; Reimer, C.; Blackman, R.K.; Bulawa, C.E.; Osheroff, N.; Charlton, P.; et al. Biological characterization of MLN944: A potent DNA binding agent. Mol. Cancer Ther. 2004, 3, 47–58. [Google Scholar] [PubMed]
- Reynolds, D.P.; Lanevskij, K.; Japertas, P.; Didziapetris, R.; Petrauskas, A. Ionization-specific analysis of human intestinal absorption. J. Pharm. Sci. 2009, 98, 4039–4054. [Google Scholar] [CrossRef] [PubMed]
- Sazonovas, A.; Japertas, P.; Didziapetris, R. Estimation of reliability of predictions and model applicability domain evaluation in the analysis of acute toxicity (LD50). SAR QSAR Environ. Res. 2010, 21, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-A.; Yang, Z.; Lee, J.Y.; De, U.; Kim, T.H.; Park, J.Y.; Lee, H.J.; Park, Y.J.; Chun, P.; Kim, H.S.; Jeong, L.S.; Moona, H.R. Design, synthesis and anticancer activity of novel dihydrobenzofuro[4,5-b][1,8]naphthyridin-6-one derivatives. Bioorg. Med. Chem. 2011, 21, 5730–5734. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Viability Rate % | |||||||||
| PC–3 | ||||||||||
| 5 µM | 10 µM | 15 µM | 20 µM | 25 µM | 30 µM | 35 µM | 40 µM | 50 µM | 1 IC50 µM | |
| 4a | 95.94 ± 2.25 | 98.80 ± 1.39 | 91.90 ± 1.93 | 69.92 ± 3.33 | 63.74 ± 2.85 | 46.91 ± 1.61 | 32.62 ± 1.33 | 21.61 ± 1.32 | 11.08 ± 0.57 | 28.24 |
| 11c | … | 96.10 ± 2.18 | 95.82 ± 1.2 | 73.53 ± 3.9 | 34.14 ± 2.5 | 23.60± 4.4 | 6.16 ± 0.60 | 4.73 ± 0.24 | 3.45 ± 0.14 | 23.30 |
| 11d | 96.69 ± 0.77 | 97.05 ± 0.58 | 58.49 ± 6.00 | 10.70 ± 5.02 | 3.44 ± 0.24 | 3.68 ± 0.22 | 3.50 ± 0.24 | 3.04 ± 0.21 | 2.93 ± 0.21 | 22.57 |
| 12d | 95.31 ± 3.97 | 92.83 ± 1.93 | 81.70 ± 7.40 | 80.66 ± 5.28 | 72.56 ± 5.78 | 65.12 ± 3.77 | 58.44 ± 3.44 | 59.54 ± 5.09 | 48.39 ± 1.96 | 48.08 |
| 14c | 95.62 ± 0.91 | 92.34 ± 3.30 | 89.42 ± 5.22 | 99.90 ± 5.57 | 90.33 ± 4.90 | 47.94 ± 10.29 | 62.48 ± 5.82 | 45.69 ± 4.25 | 9.57 ± 2.74 | 35.72 |
| 14d | 92.66 ± 5.82 | 109.44 ± 4.12 | 113.08 ± 7.15 | 87.05 ± 3.24 | 52.95 ± 1.97 | 38.51 ± 1.58 | 25.82 ± 1.61 | 19.12 ± 0.91 | 5.21 ± 1.41 | 27.59 |
| Entry | Viability Rate % | |||||||||
| DU–145 | ||||||||||
| 5 µM | 10 µM | 15 µM | 20 µM | 25 µM | 30 µM | 35 µM | 40 µM | 50 µM | 1 IC50 µM | |
| 4a | 91.23 ± 3.47 | 100.58 ± 3.57 | 93.26 ± 2.16 | 91.12 ± 3.24 | 86.85 ± 1.78 | 78.77 ± 2.71 | 51.02 ± 6.52 | 54.00 ± 2.21 | 28.24 ± 1.85 | 43.63 |
| 11c | 94.90 ± 2.95 | 76.11 ± 4.76 | 35.16 ± 1.53 | 11.99 ± 0.91 | 3.07 ± 0.09 | 3.02 ± 0.16 | 3.16 ± 0.16 | 3.11 ± 0.09 | 3.25 ± 0.14 | 12.96 |
| 11d | 80.23 ± 2.50 | 30.43 ± 1.33 | 4.72 ± 0.10 | 3.45 ± 0.22 | 3.52 ± 0.15 | 3.58 ± 0.13 | 3.63 ± 0.25 | 3.61 ± 0.13 | 3.40 ± 0.20 | 7.63 |
| 12d | 80.81 ± 2.32 | 77.77 ± 1.63 | 73.62 ± 2.78 | 72.97 ± 2.97 | 65.67 ± 2.49 | 59.84 ± 3.21 | 51.32 ± 3.18 | 42.75 ± 3.06 | 33.24 ± 4.39 | 42.63 |
| 14c | 95.04 ± 2.49 | 80.02 ± 1.82 | 60.43 ± 1.77 | 30.57 ± 1.19 | 7.28 ± 0.68 | 4.82 ± 0.38 | 4.19 ± 0.39 | 3.78 ± 0.36 | 3.77 ± 0.39 | 15.86 |
| 14d | 93.25 ± 1.24 | 78.47 ± 1.67 | 57.33 ± 1.36 | 36.78 ± 0.43 | 24.58 ± 0.80 | 18.89 ± 0.57 | 12.37 ± 0.27 | 8.16 ± 0.29 | 5.07 ± 0.12 | 16.46 |
| Entry | Viability Rate % | |||||||||
| MCF–7 | ||||||||||
| 5 µM | 10 µM | 15 µM | 20 µM | 25 µM | 30 µM | 35 M | 40 M | 50 M | 1 IC50 µM | |
| 4a | 75.91 ± 1.39 | 88.72 ± 2.36 | 78.00 ± 3.89 | 31.76 ± 8.36 | 12.79 ± 0.53 | 14.51 ± 0.63 | 12.95 ± 0.58 | 11.32 ± 1.02 | 7.38 ± 0.53 | 17.95 |
| 11c | 74.01 ± 3.18 | 27.88 ± 7.04 | 22.58 ± 1.05 | 11.25 ± 0.63 | 5.88 ± 0.33 | 4.86 ± 0.24 | 4.56 ± 0.14 | 4.73 ± 0.35 | 4.98 ± 0.19 | 7.48 |
| 11d | 65.16 ± 4.29 | 13.49 ± 1.31 | 5.08 ± 0.26 | 4.97 ± 0.32 | 5.14 ± 0.36 | 5.44 ± 0.41 | 5.74 ± 0.20 | 5.74 ± 0.39 | 6.58 ± 0.28 | 6.00 |
| 12d | 61.13 ± 3.10 | 68.18 ± 2.27 | 63.21 ± 2.57 | 62.06 ± 0.95 | 49.05 ± 3.46 | 53.31 ± 2.23 | 42.92 ± 1.78 | 42.54 ± 1.93 | 29.53 ± 1.51 | 25.45 |
| 14c | 149.95 ± 13.45 | 106.03 ± 19.03 | 46.02 ± 5.56 | 22.48 ± 2.32 | 11.71 ± 1.87 | 8.66 ± 1.17 | 7.96 ± 0.99 | 7.83 ± 0.92 | 8.23 ± 0.98 | 15.51 |
| 14d | 80.29 ± 1.26 | 79.63 ± 1.68 | 58.73 ± 3.57 | 45.53 ± 2.32 | 29.16 ± 1.13 | 14.52 ± 1.33 | 9.43 ± 1.07 | 6.61 ± 0.32 | 5.26 ± 0.23 | 16.91 |
| Additive | Oligonucleotide | Melting Temperature, Tm (°C) |
|---|---|---|
| None (negative control) | ds–DNA | 61.69 ± 0.58 |
| 4a | ds–DNA | 61.67 ± 0.56 |
| 11c | ds–DNA | 65.10 ± 0.11 |
| 11d | ds–DNA | 67.52 ± 0.72 |
| 12d | ds–DNA | 61.02 ± 0 |
| 14c | ds–DNA | 61.34 ± 1.18 |
| 14d | ds–DNA | 61.02 ± 1.73 |
| 9-AA (positive control) | ds–DNA | 70.08 ± 1.08 |
| Entry | Drug-likeness | |||
|---|---|---|---|---|
| 1 HBD | 2 HBA | 3 Mw | 4 logP | |
| 4a | 0 | 8 | 486.61 | 2.18 |
| 11c | 2 | 5 | 453.58 | 4.17 |
| 11d | 3 | 5 | 439.55 | 4.37 |
| 12d | 3 | 7 | 441.53 | 2.34 |
| 14c | 4 | 7 | 431.53 | 3.6 |
| 14d | 5 | 7 | 417.5 | 3.8 |
| Computed ADMET Parameters | 4a | 11c | 11d | 12d | 14c | 14d |
|---|---|---|---|---|---|---|
| 1 %HIA | 100 | 100 | 100 | 99.02 | 99.57 | 94.54 |
| 2 Pe, 10−4 cm/s | 6.14 | 6.73 | 6.11 | 1.77 | 2.23 | 0.97 |
| 3 ka, min−1 | 0.04 | 0.05 | 0.04 | 0.01 | 0.02 | 0.01 |
| 4 logPS | −2.17 | −1.65 | −1.93 | −2.93 | −2.73 | −2.99 |
| 5 logBB | 0.1 | −0.22 | −0.31 | −0.93 | −0.08 | −0.08 |
| 6 fu, brain | 0.27 | 0.01 | 0.02 | 0.19 | 0.08 | 0.08 |
| 7 log(PS*fu, brain) | −2.73 | −3.58 | −3.65 | −3.64 | −3.84 | −4.06 |
| 8 %PPB | 65.87 | 99.38 | 98.91 | 97.74 | 93.48 | 92.91 |
| 9 log KaHSA | 3.48 | 5.04 | 4.98 | 4.65 | 4.2 | 4.23 |
| 10 V (L/kg) | 7.51 | 6.53 | 6.78 | 4.63 | 6.13 | 6.00 |
| 11 LD50 | 190 | 1600 | 1600 | 850 | 430 | 420 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szumilak, M.; Galdyszynska, M.; Dominska, K.; Bak-Sypien, I.I.; Merecz-Sadowska, A.; Stanczak, A.; Karwowski, B.T.; Piastowska-Ciesielska, A.W. Synthesis, Biological Activity and Preliminary in Silico ADMET Screening of Polyamine Conjugates with Bicyclic Systems. Molecules 2017, 22, 794. https://doi.org/10.3390/molecules22050794
Szumilak M, Galdyszynska M, Dominska K, Bak-Sypien II, Merecz-Sadowska A, Stanczak A, Karwowski BT, Piastowska-Ciesielska AW. Synthesis, Biological Activity and Preliminary in Silico ADMET Screening of Polyamine Conjugates with Bicyclic Systems. Molecules. 2017; 22(5):794. https://doi.org/10.3390/molecules22050794
Chicago/Turabian StyleSzumilak, Marta, Malgorzata Galdyszynska, Kamila Dominska, Irena I. Bak-Sypien, Anna Merecz-Sadowska, Andrzej Stanczak, Boleslaw T. Karwowski, and Agnieszka W. Piastowska-Ciesielska. 2017. "Synthesis, Biological Activity and Preliminary in Silico ADMET Screening of Polyamine Conjugates with Bicyclic Systems" Molecules 22, no. 5: 794. https://doi.org/10.3390/molecules22050794
APA StyleSzumilak, M., Galdyszynska, M., Dominska, K., Bak-Sypien, I. I., Merecz-Sadowska, A., Stanczak, A., Karwowski, B. T., & Piastowska-Ciesielska, A. W. (2017). Synthesis, Biological Activity and Preliminary in Silico ADMET Screening of Polyamine Conjugates with Bicyclic Systems. Molecules, 22(5), 794. https://doi.org/10.3390/molecules22050794