A Molecular Electron Density Theory Study of the Reactivity of Azomethine Imine in [3+2] Cycloaddition Reactions



Abstract
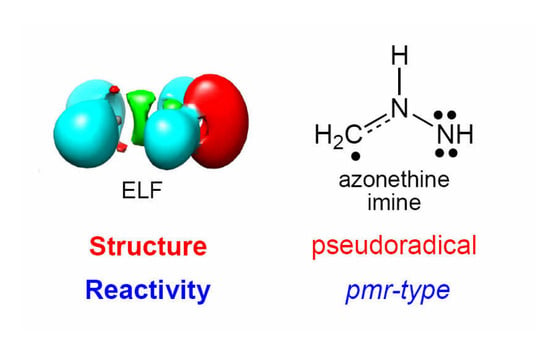
:
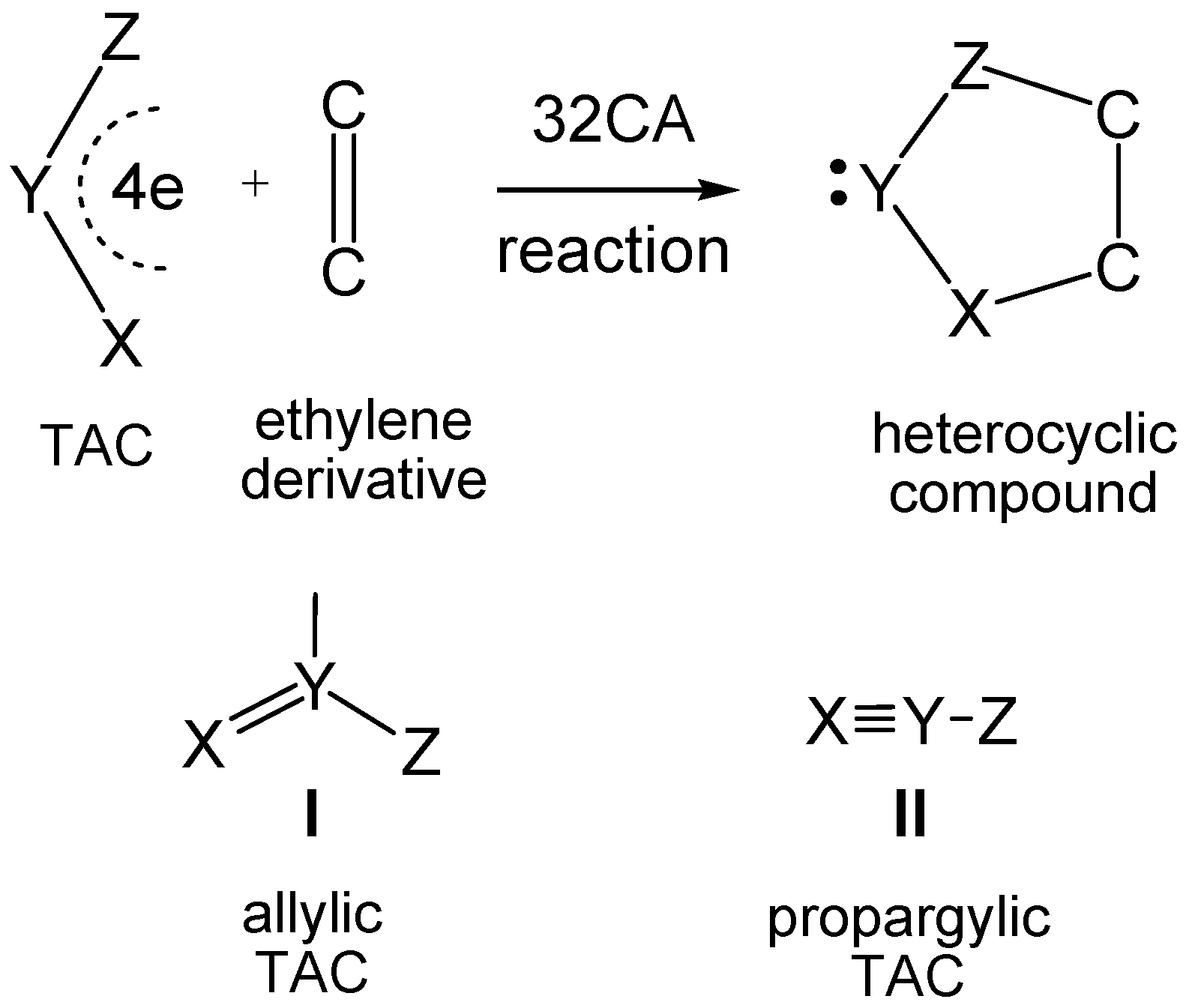
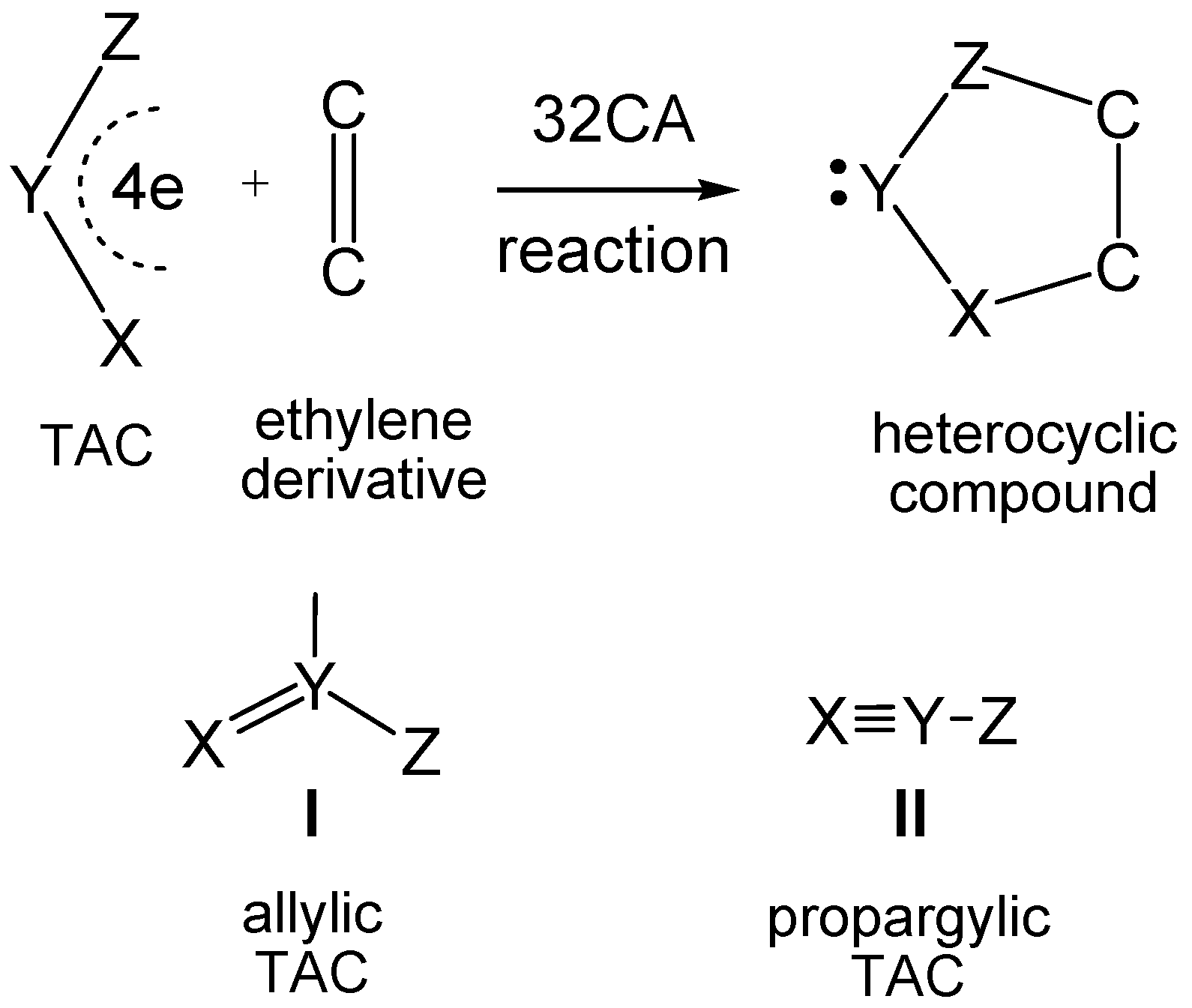
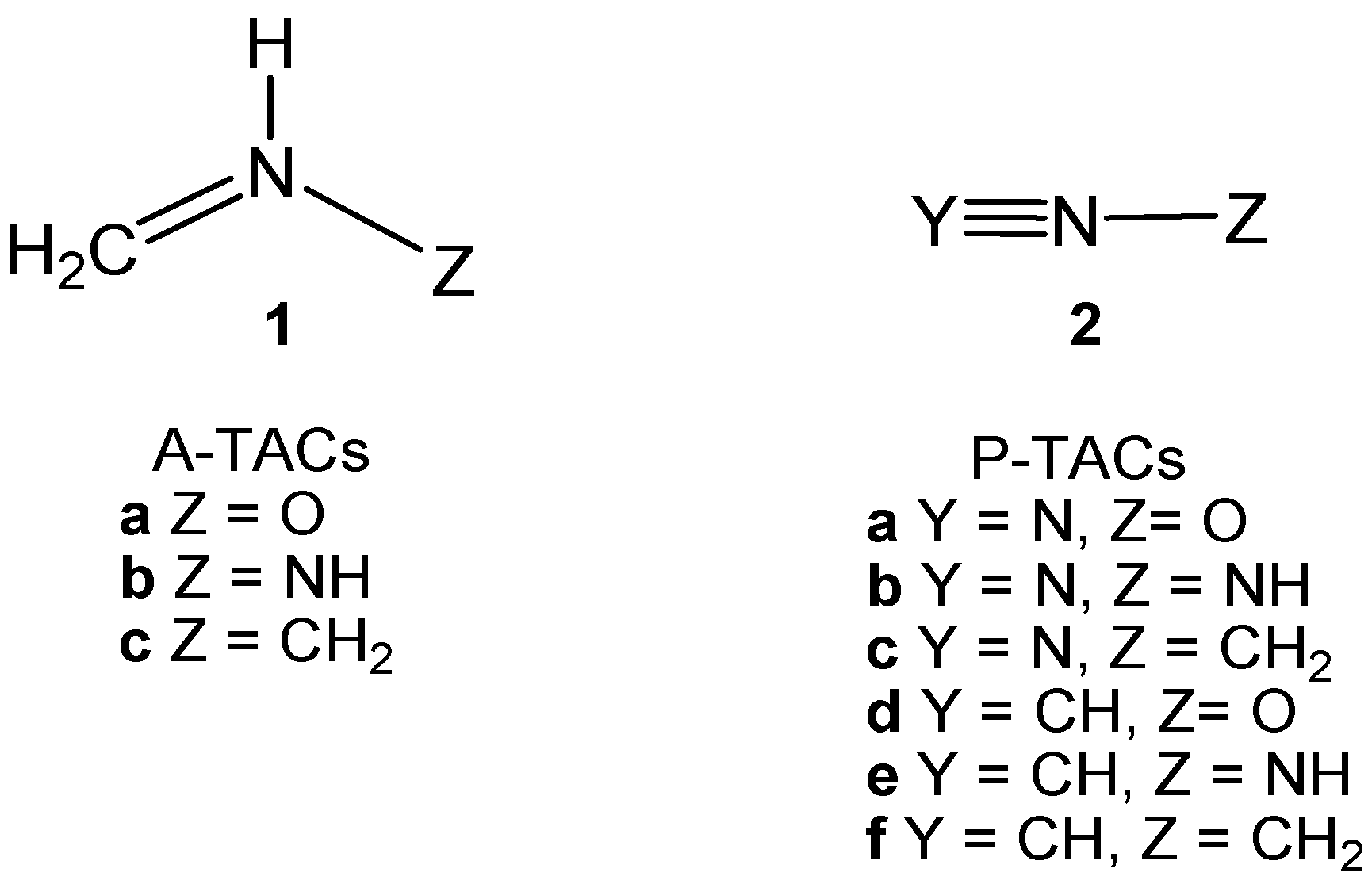
1. Introduction
2. Results and Discussion
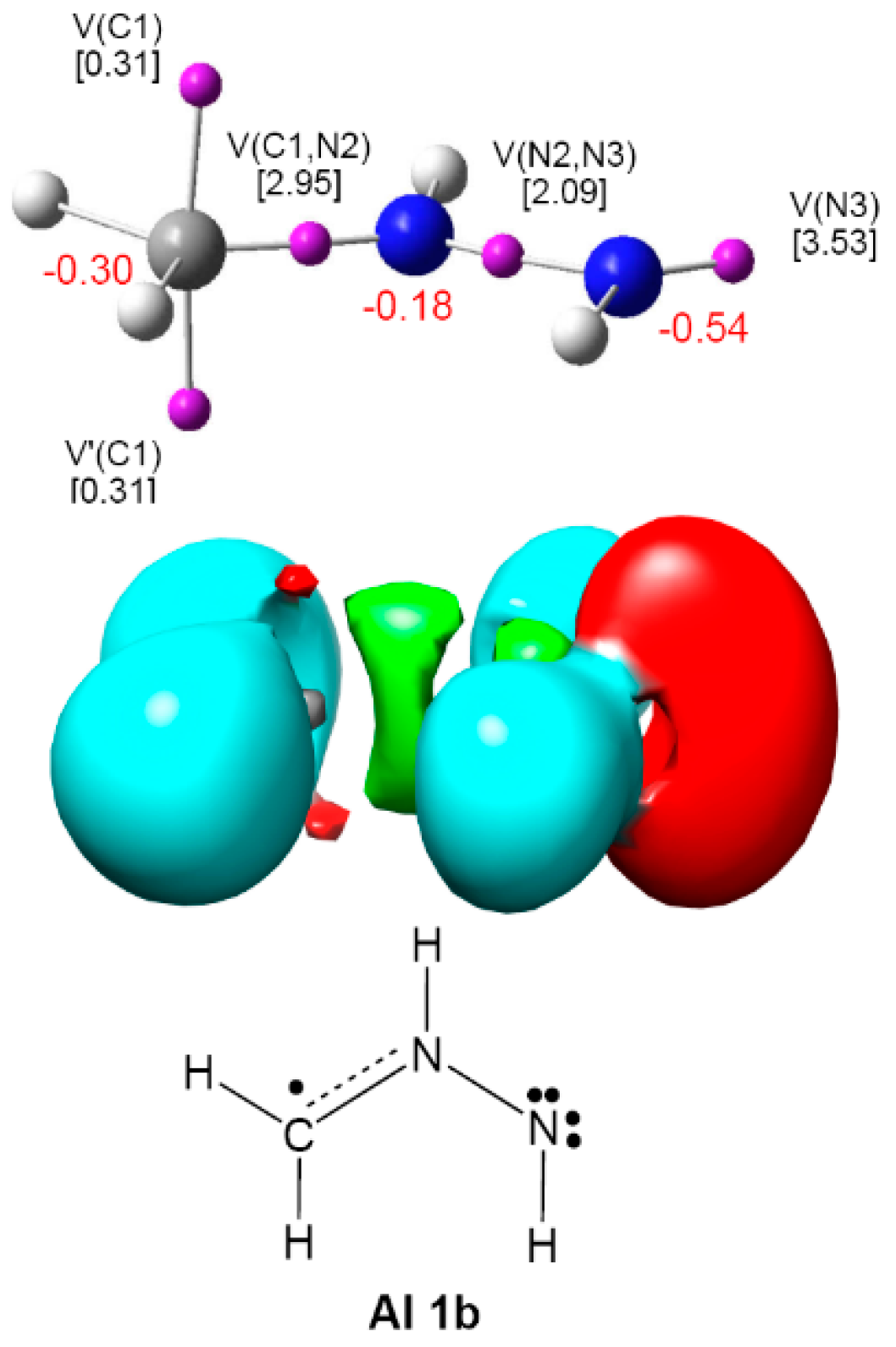
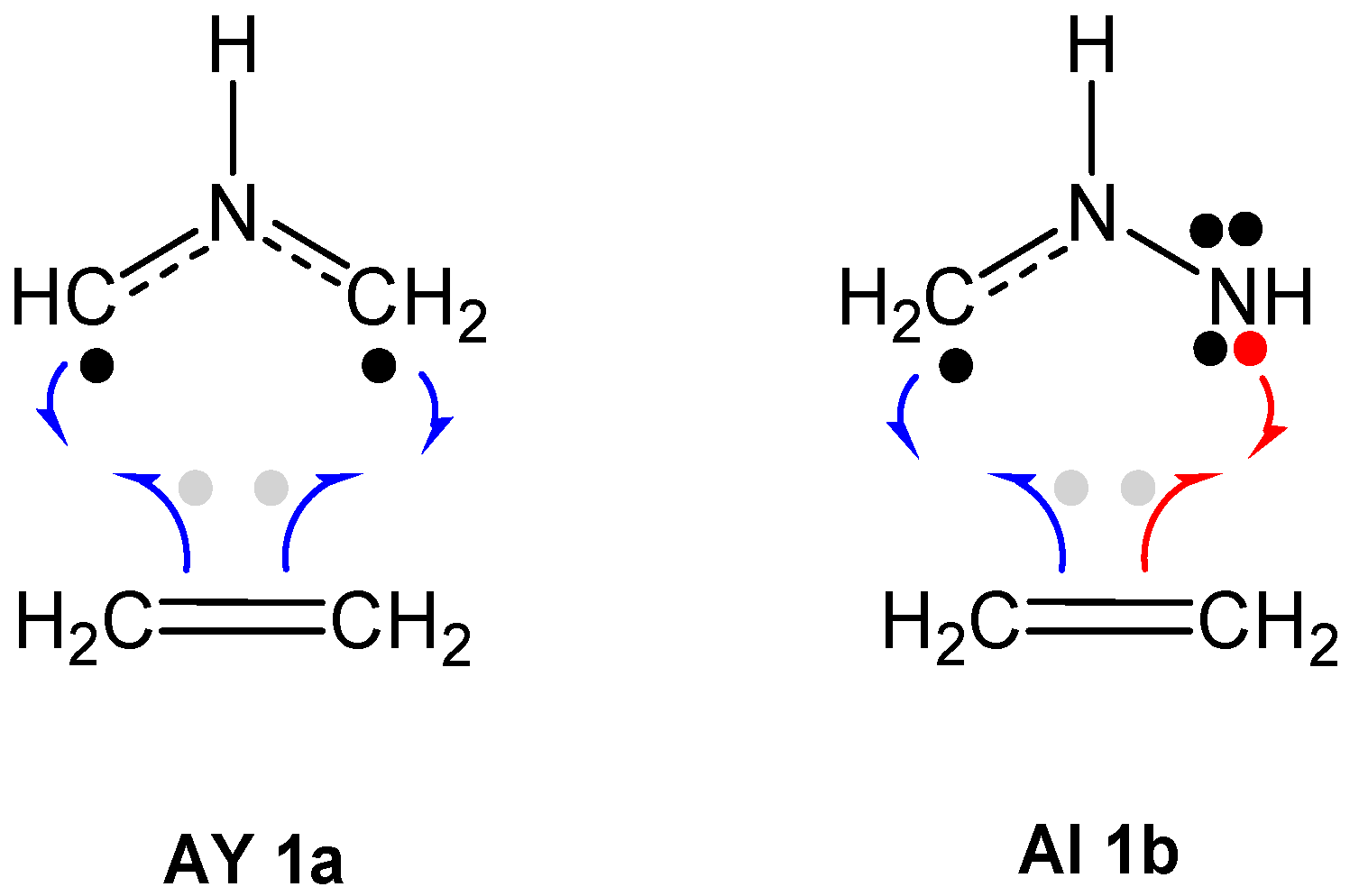
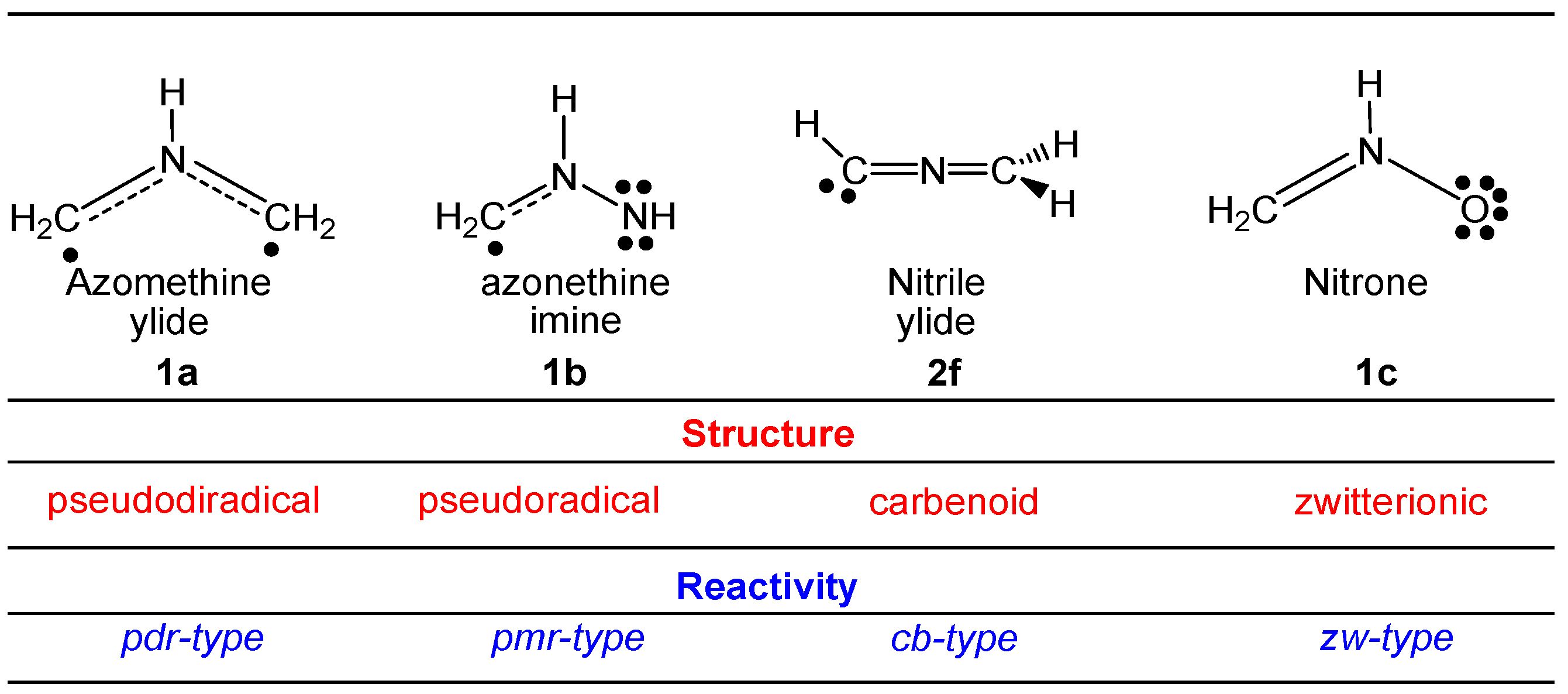
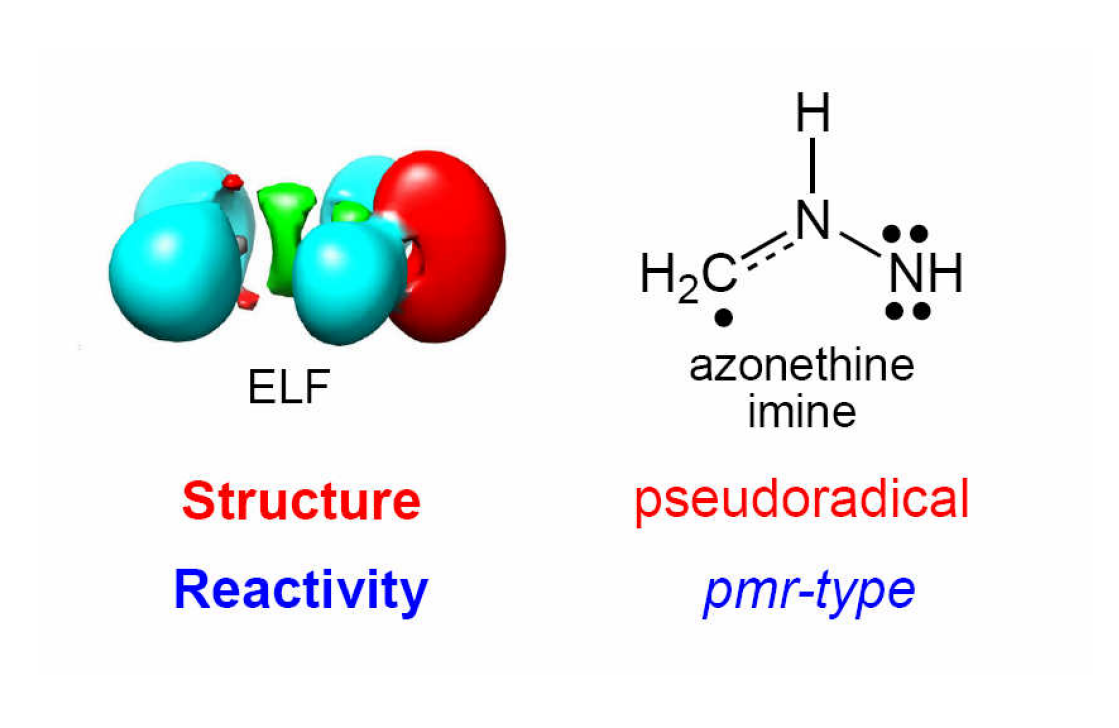
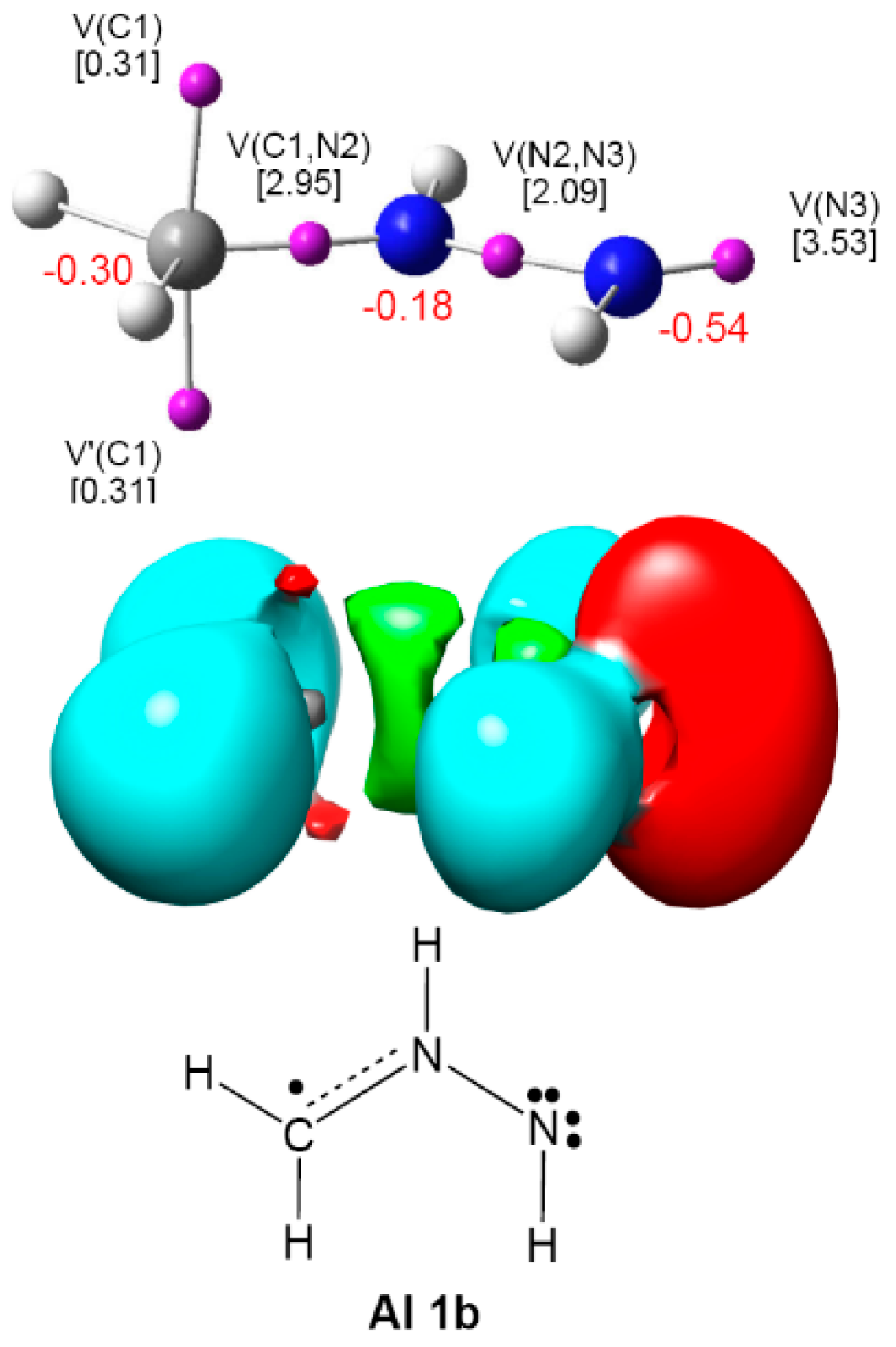
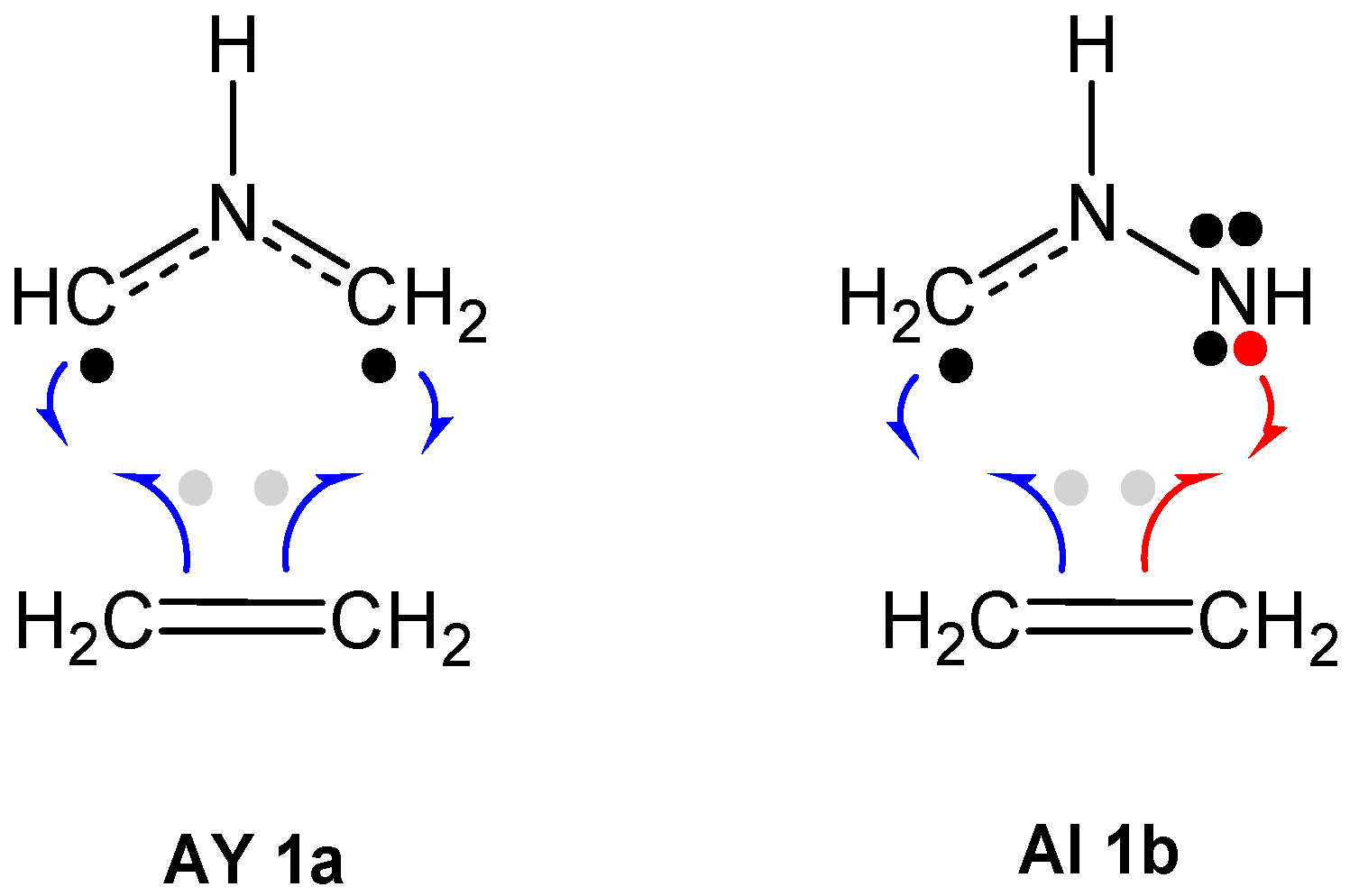
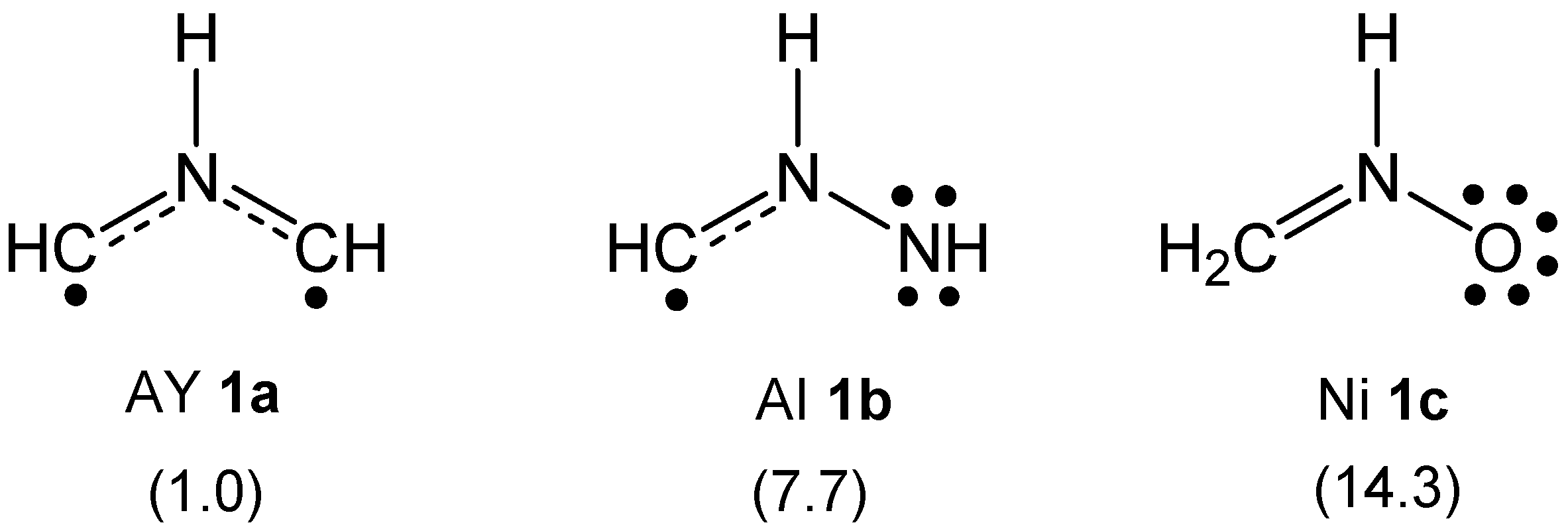
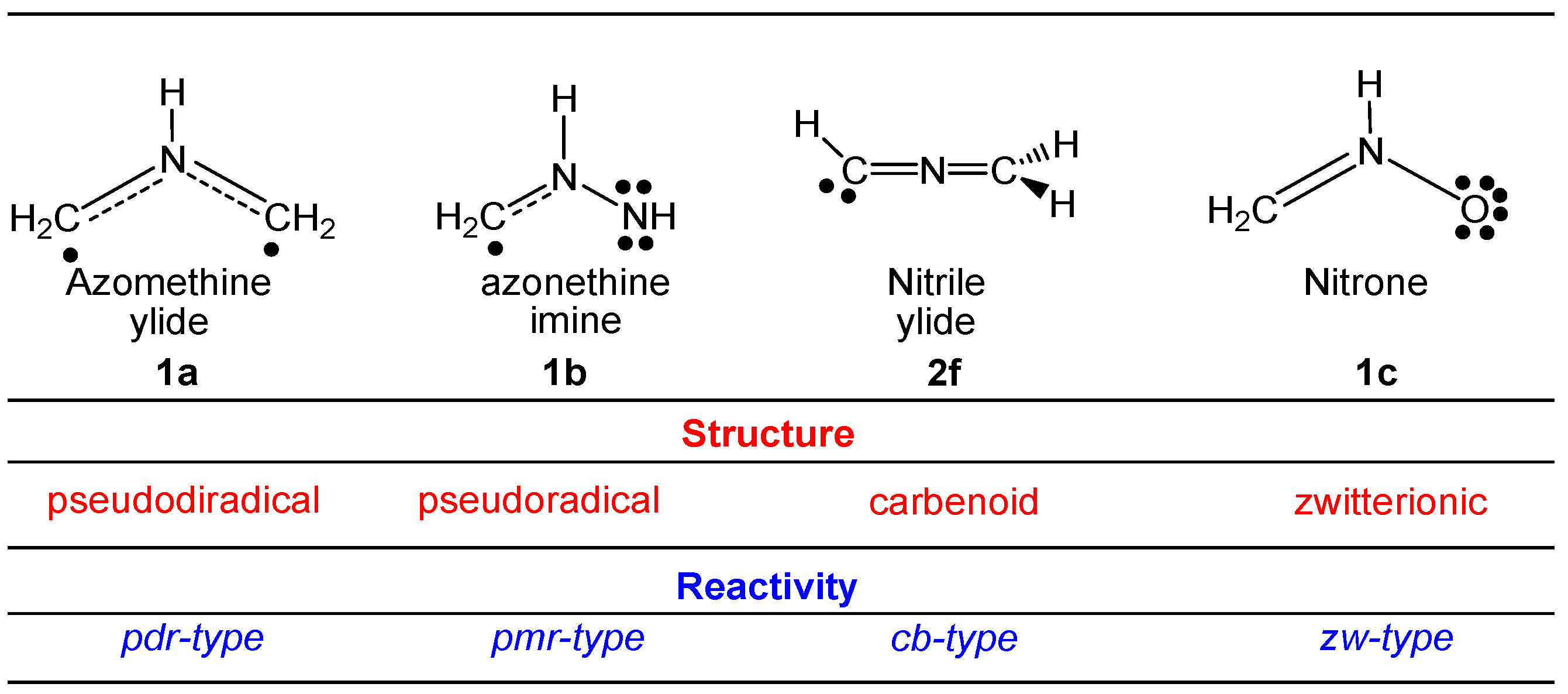
2.1. ELF Topological Analysis and Natural Population Analysis (NPA) of AI 1b
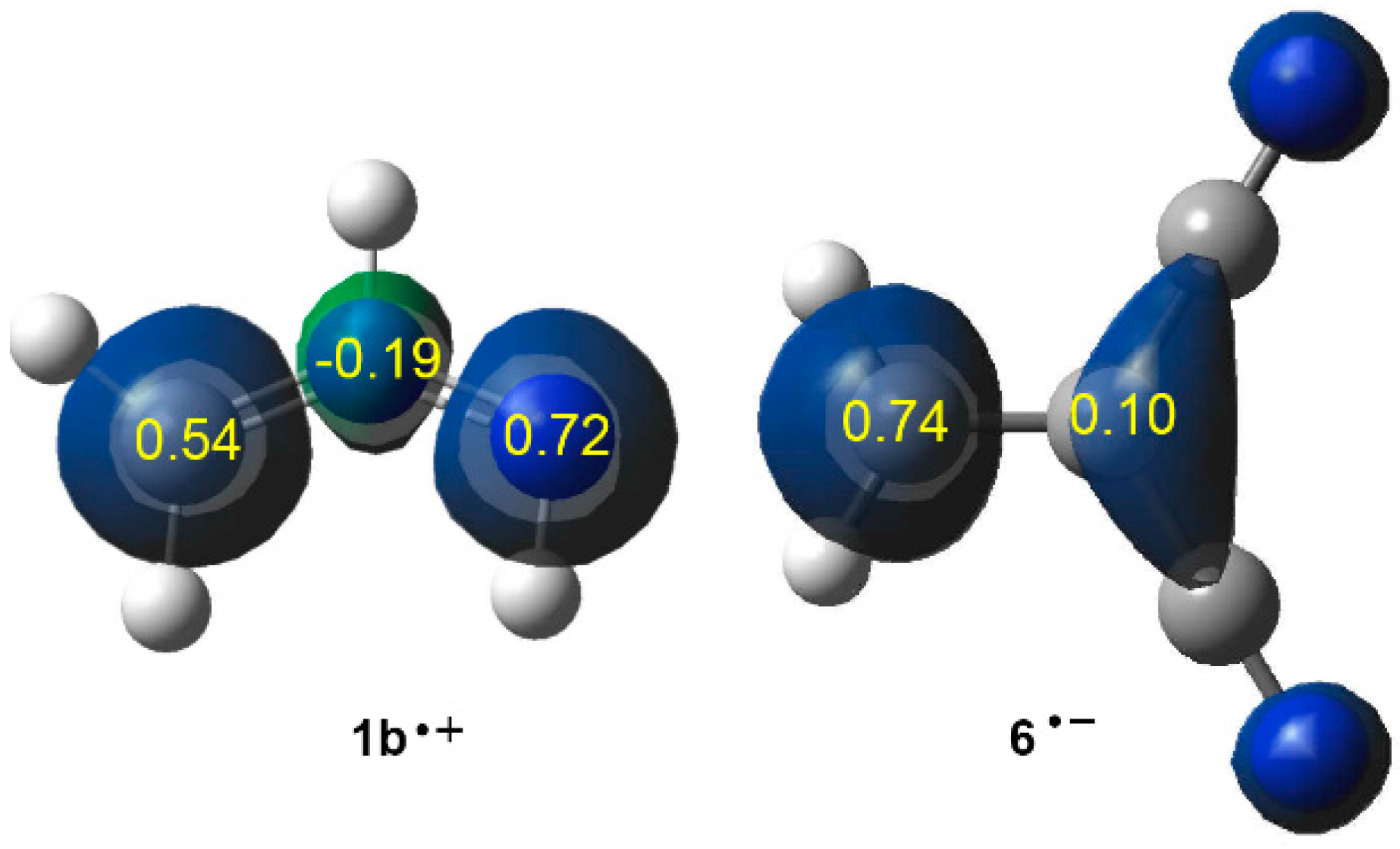
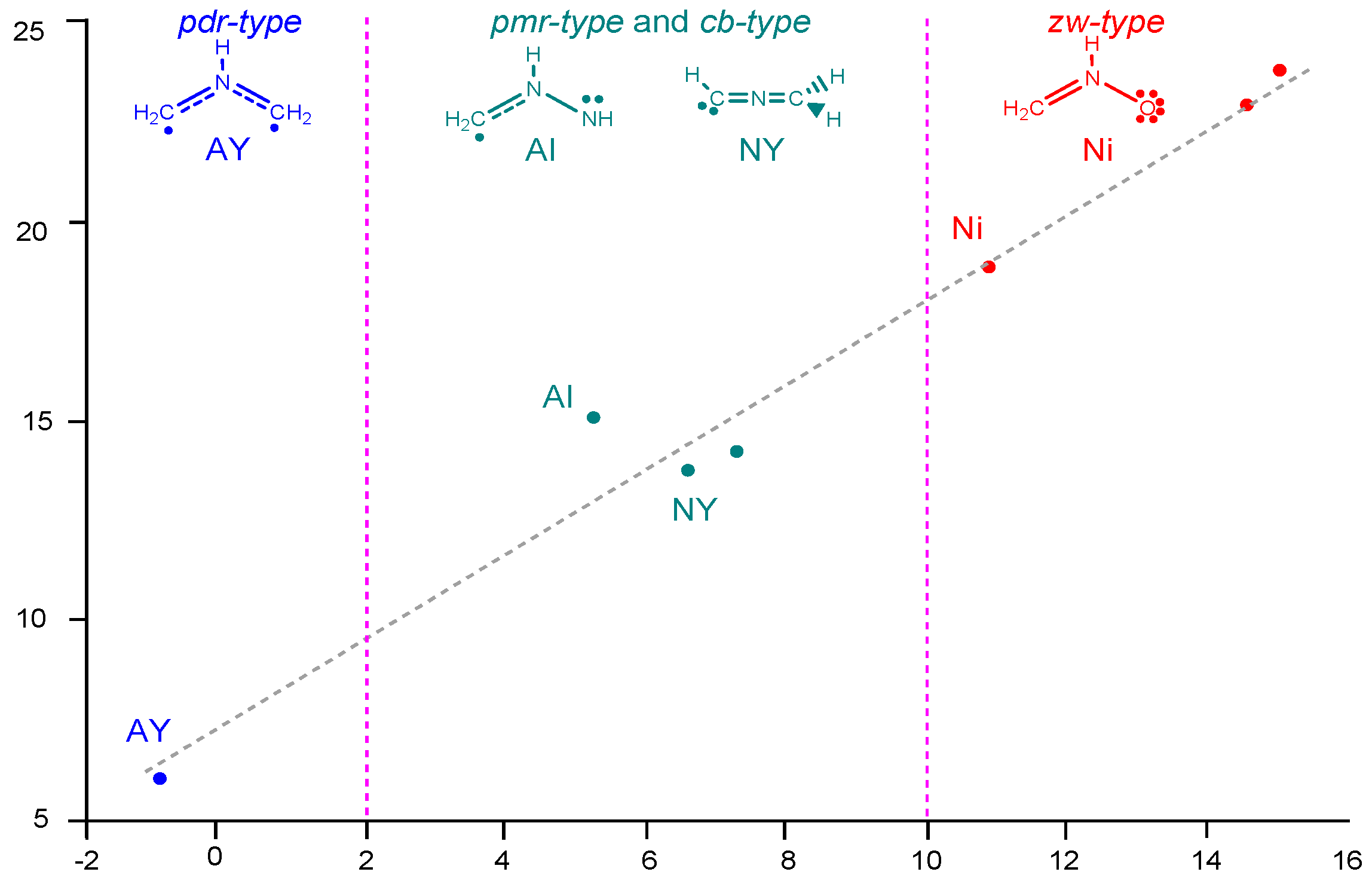
2.2. Analysis of the CDFT Reactivity Indices at the GS of the Reagents
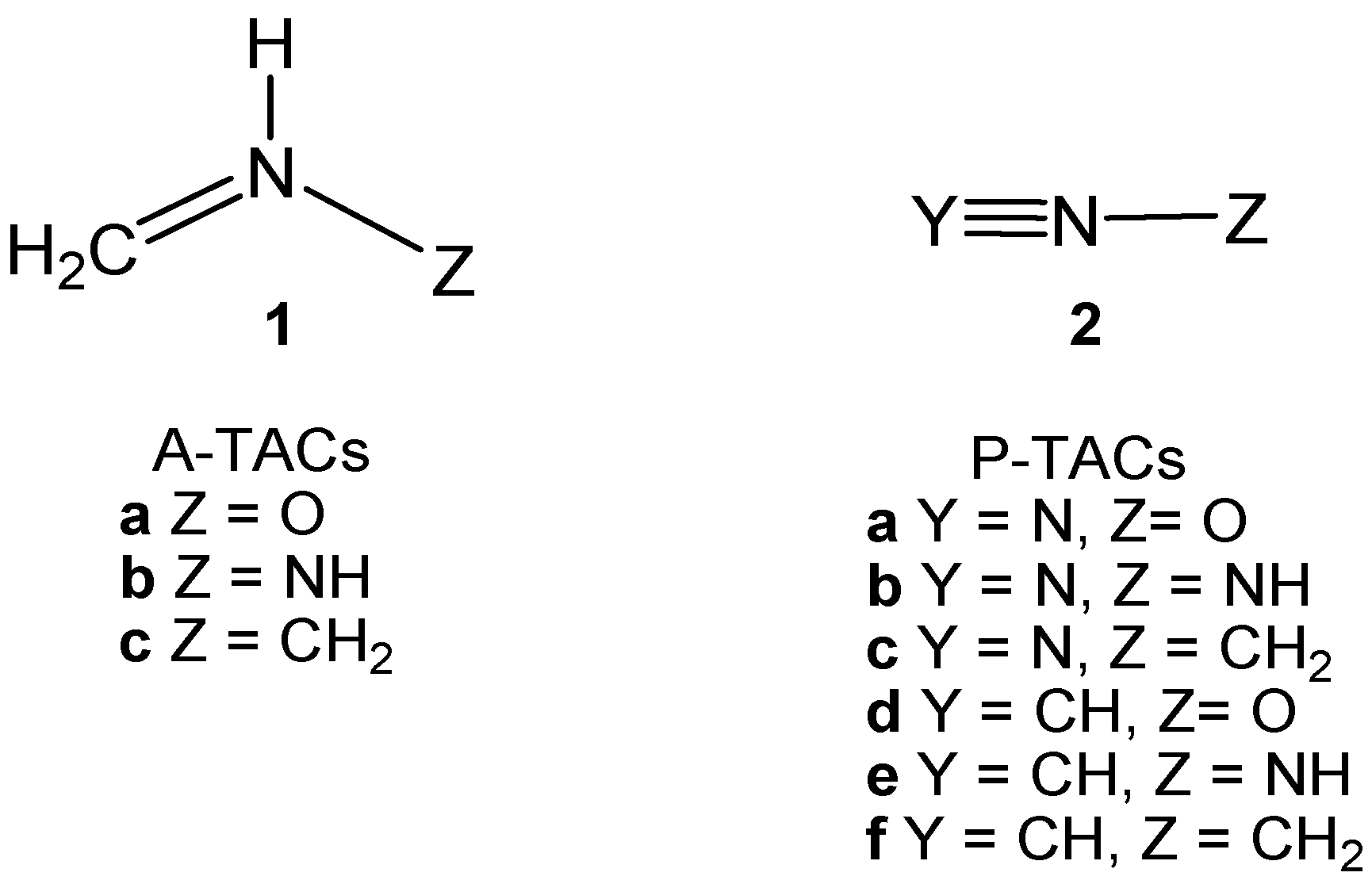
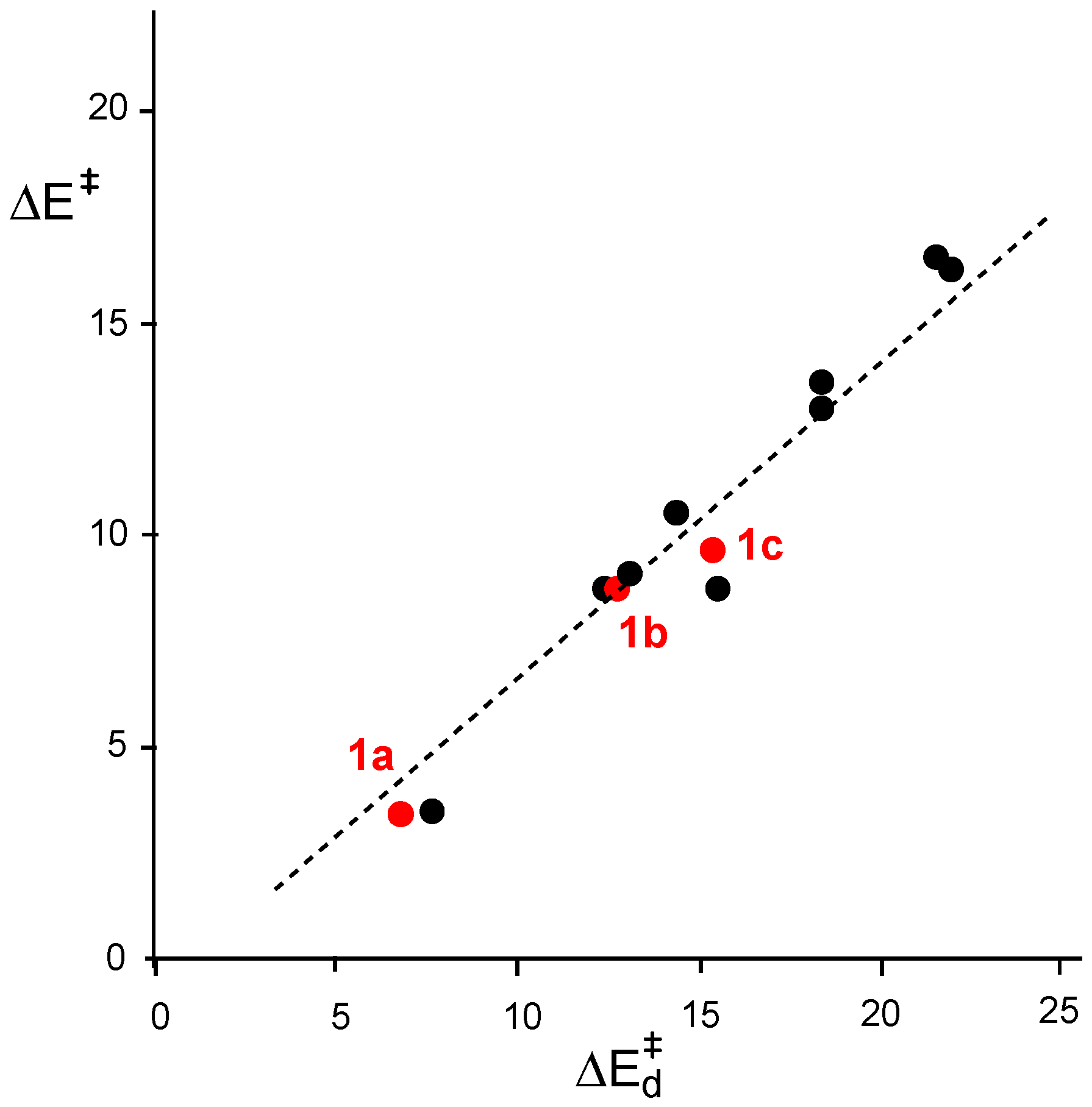
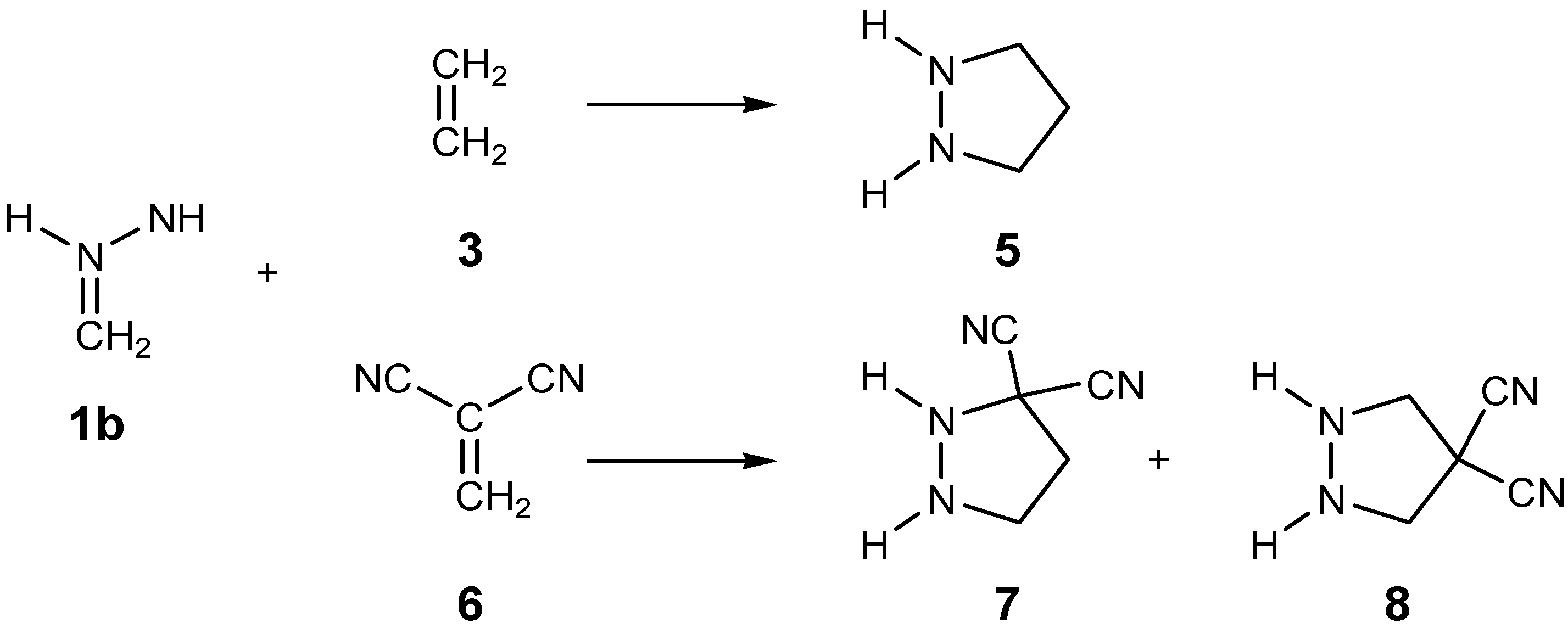
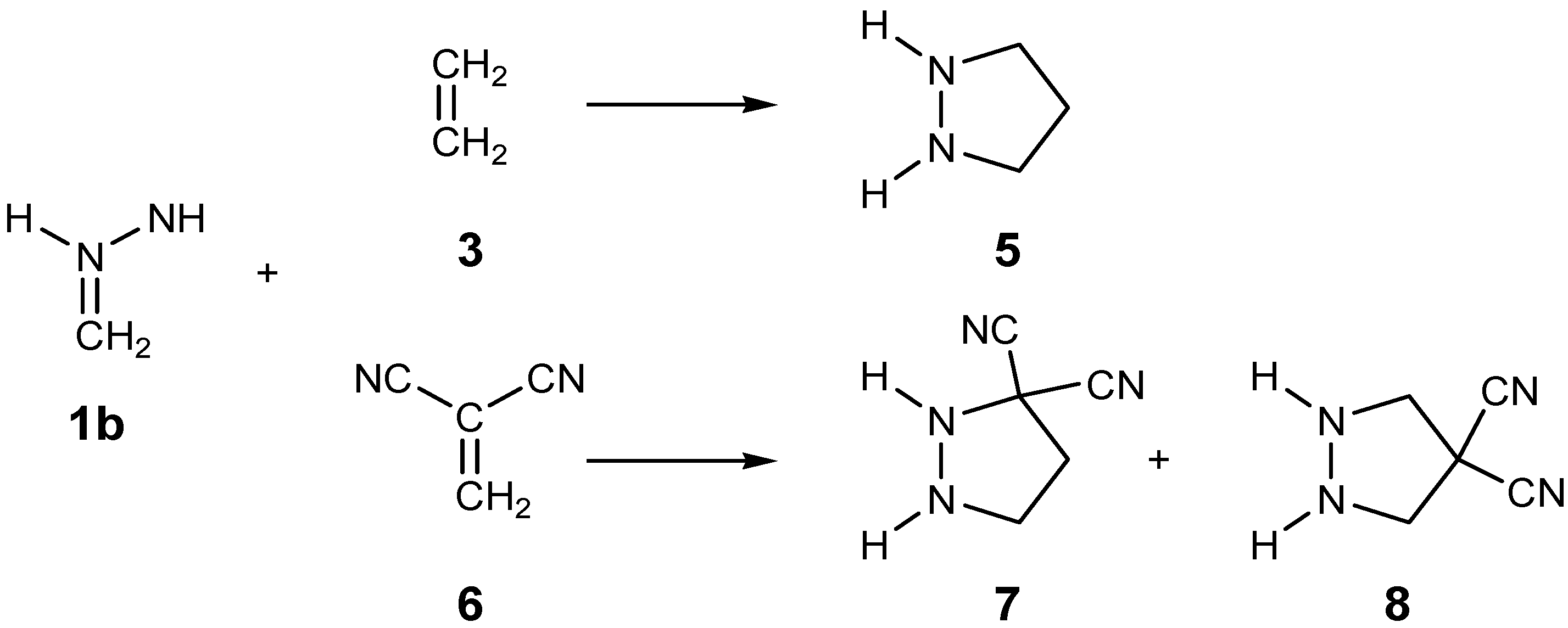
2.3. Study of the Reaction Channels Associated with the 32CA Reactions of AI 1b with Ethylene 3 and DCE 6
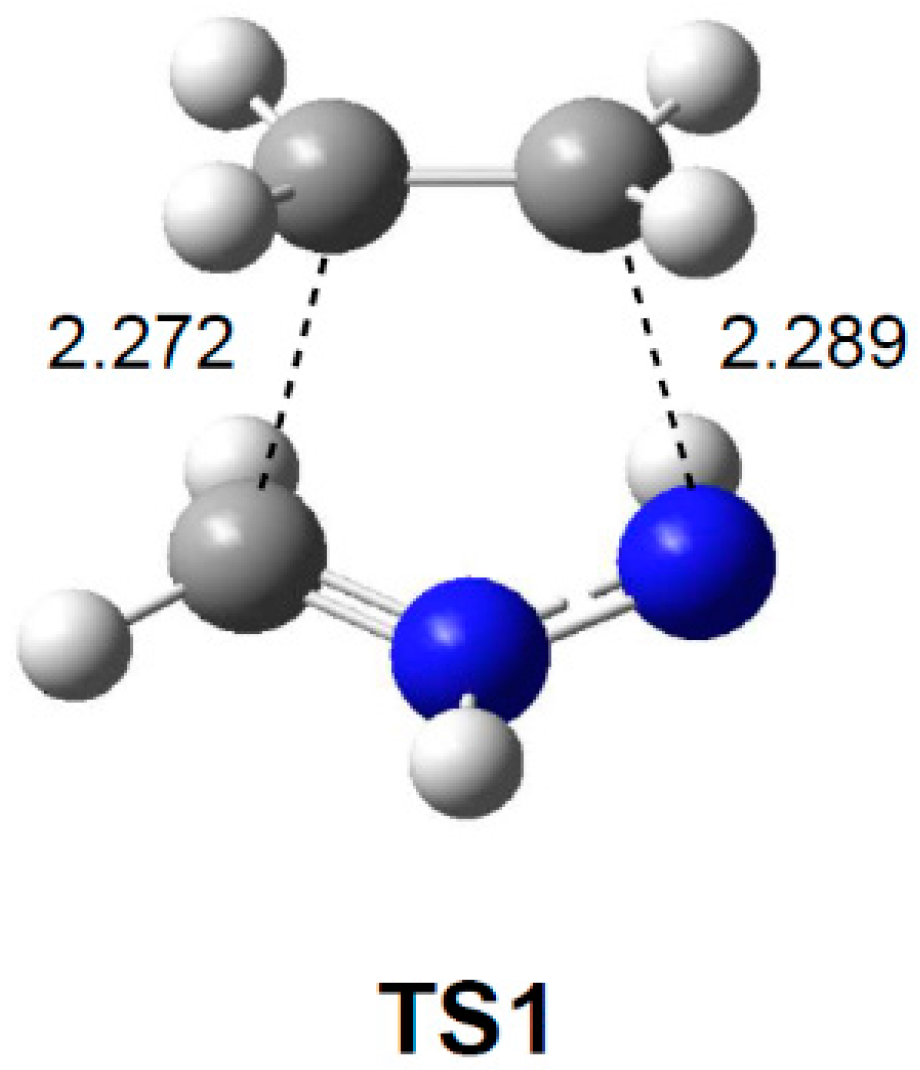
2.3.1. 32CA Reaction Involving Ethylene 3
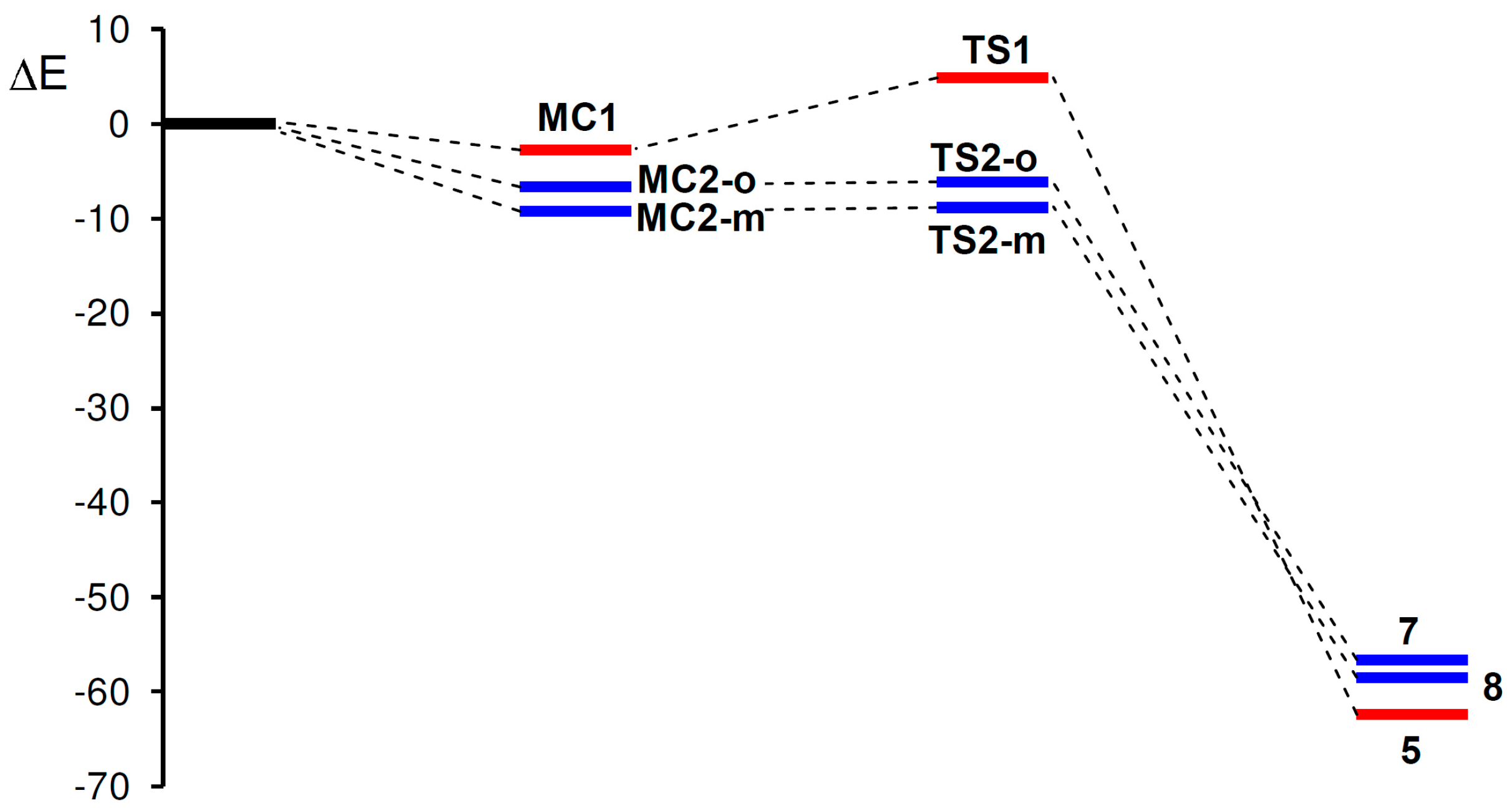
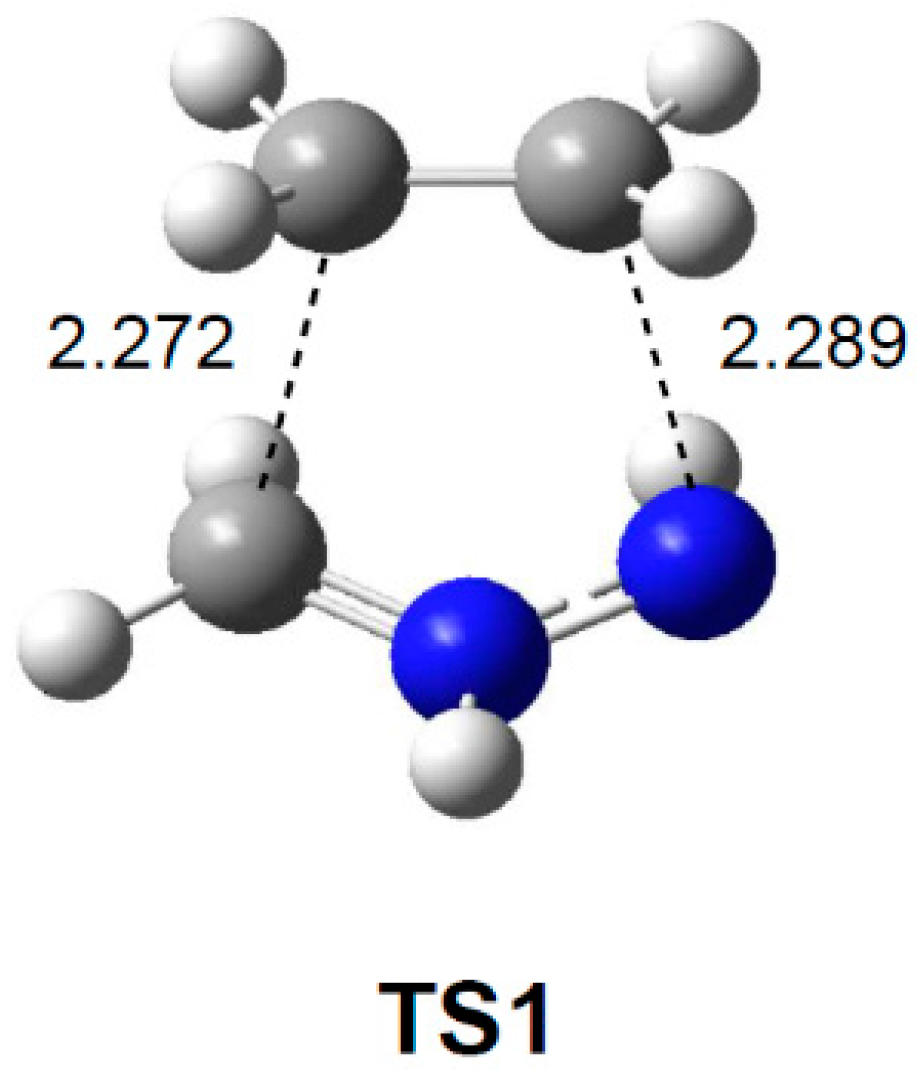
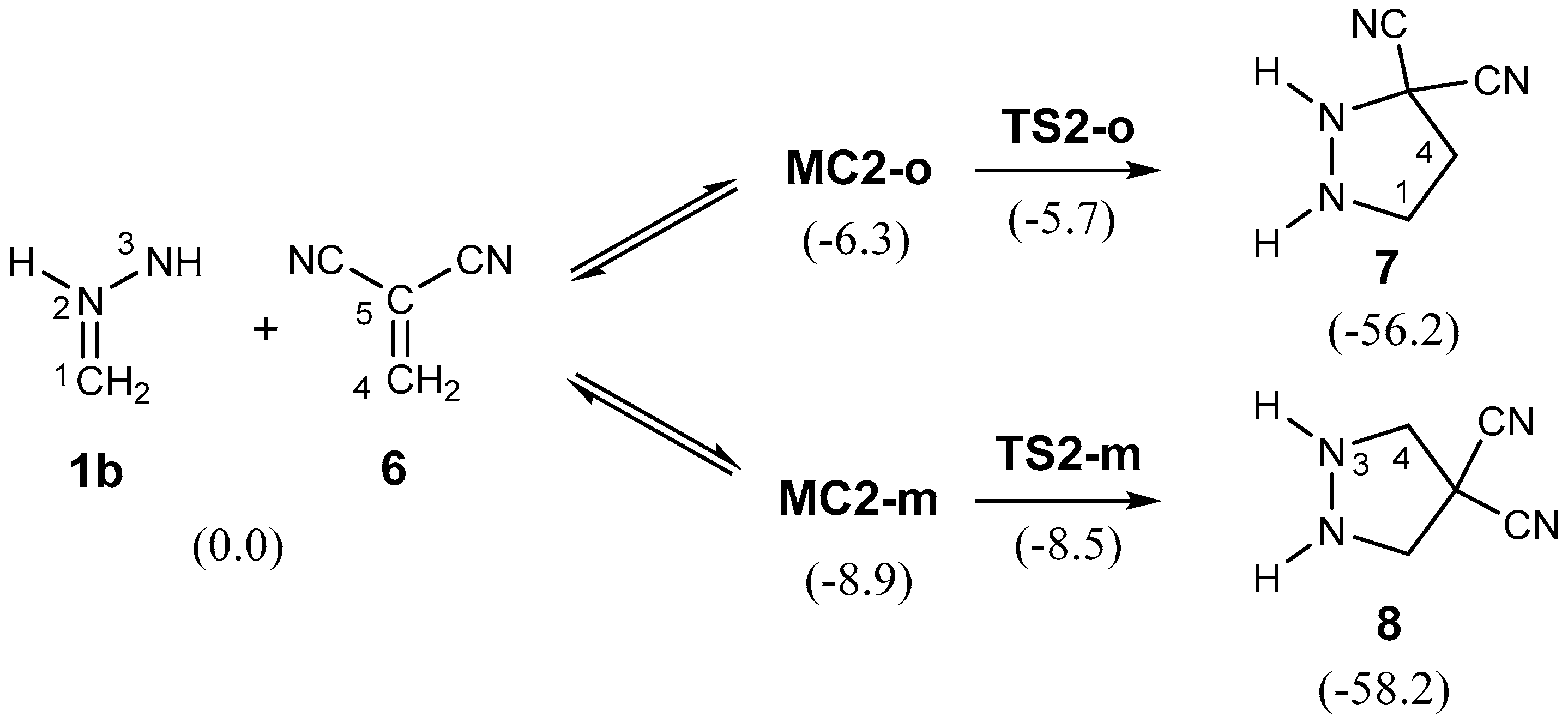
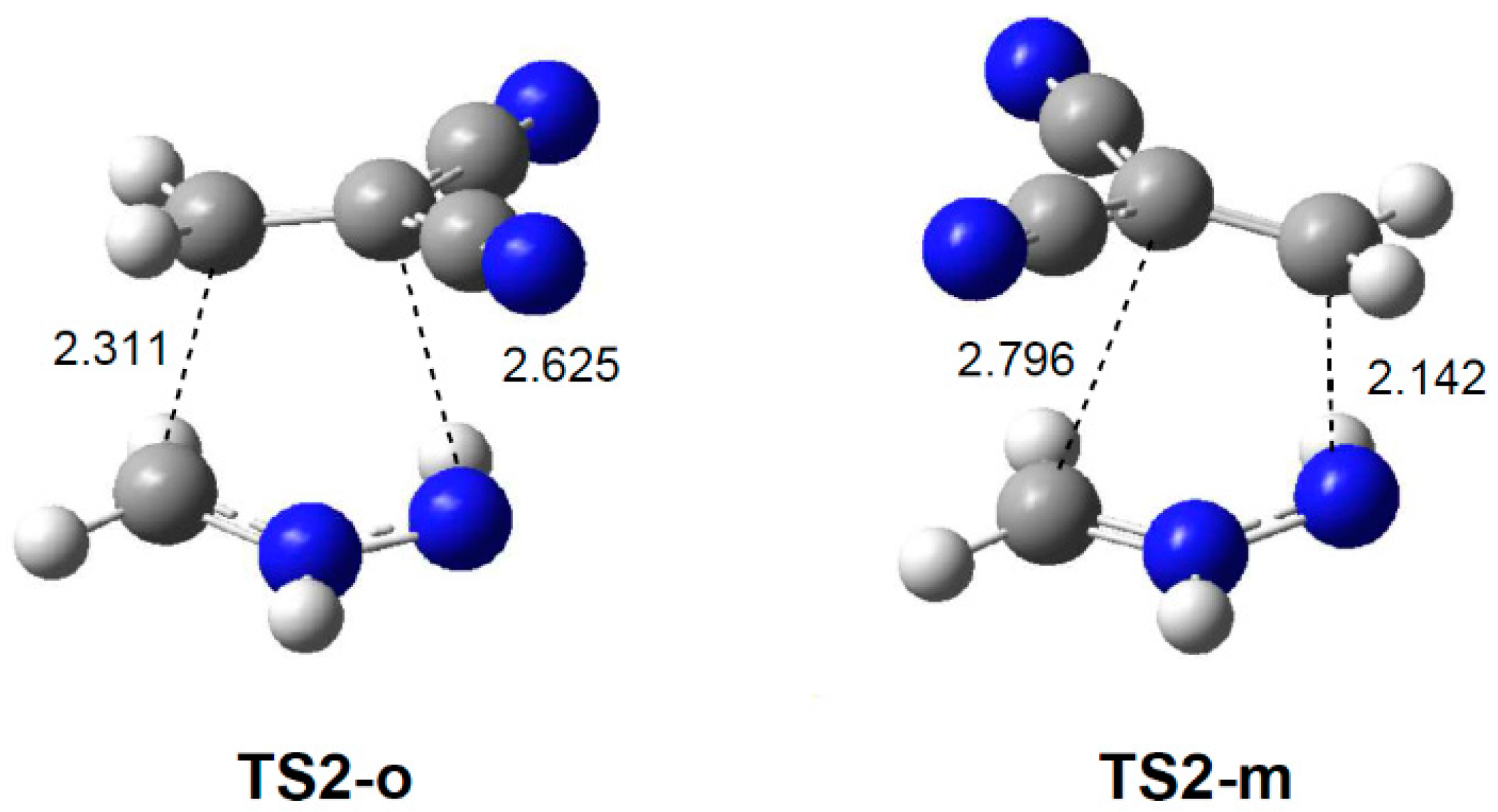
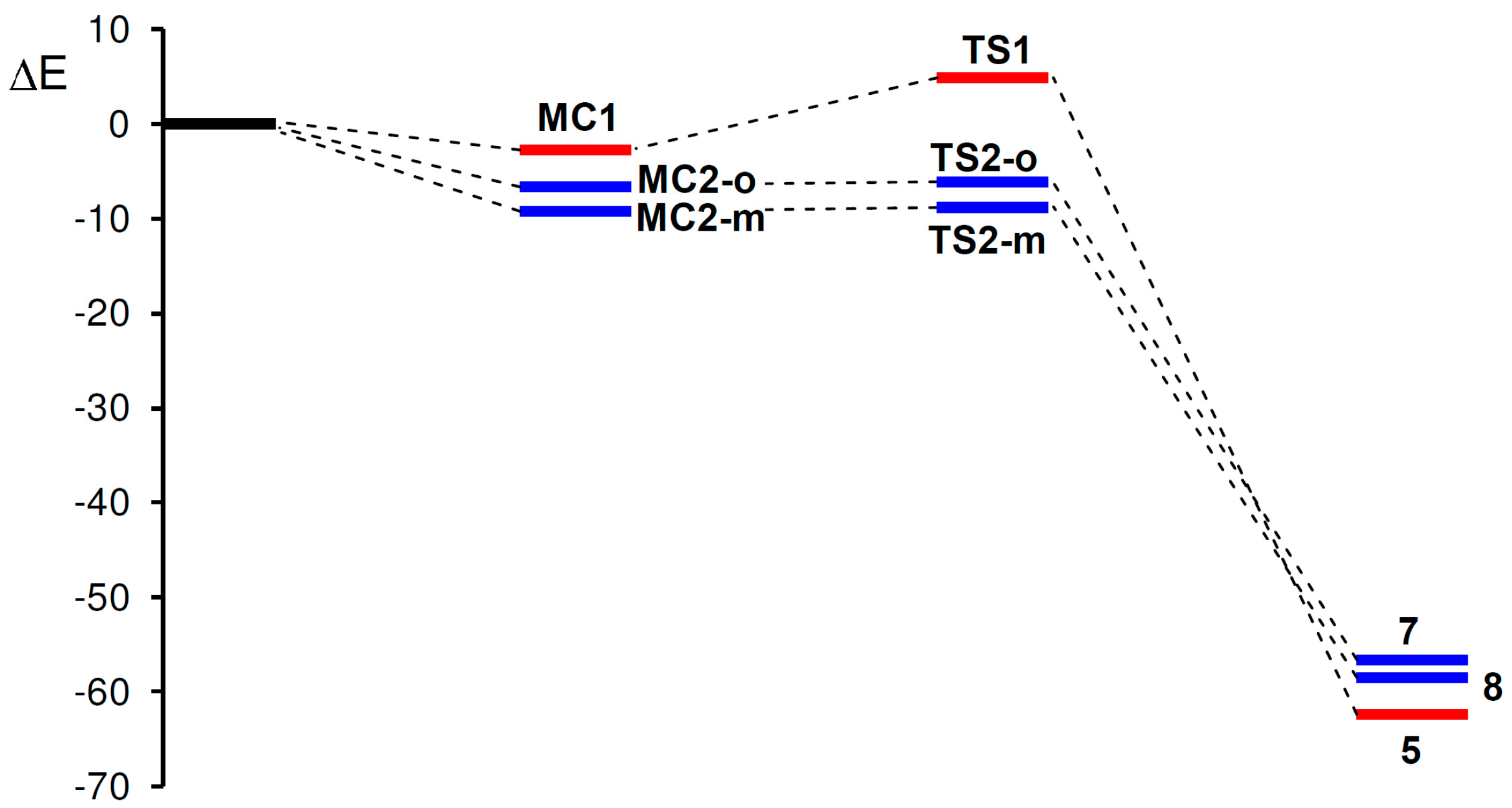
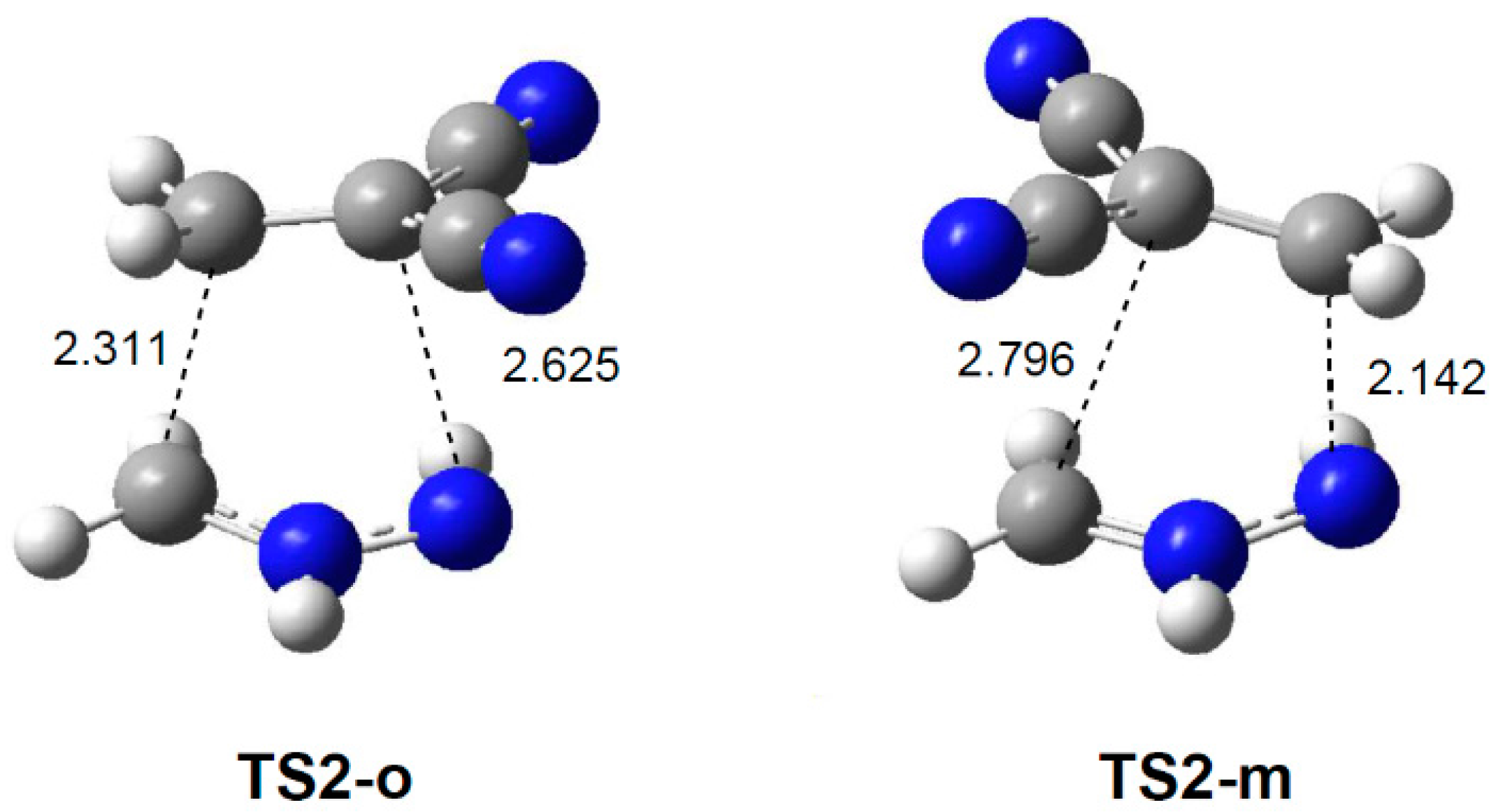
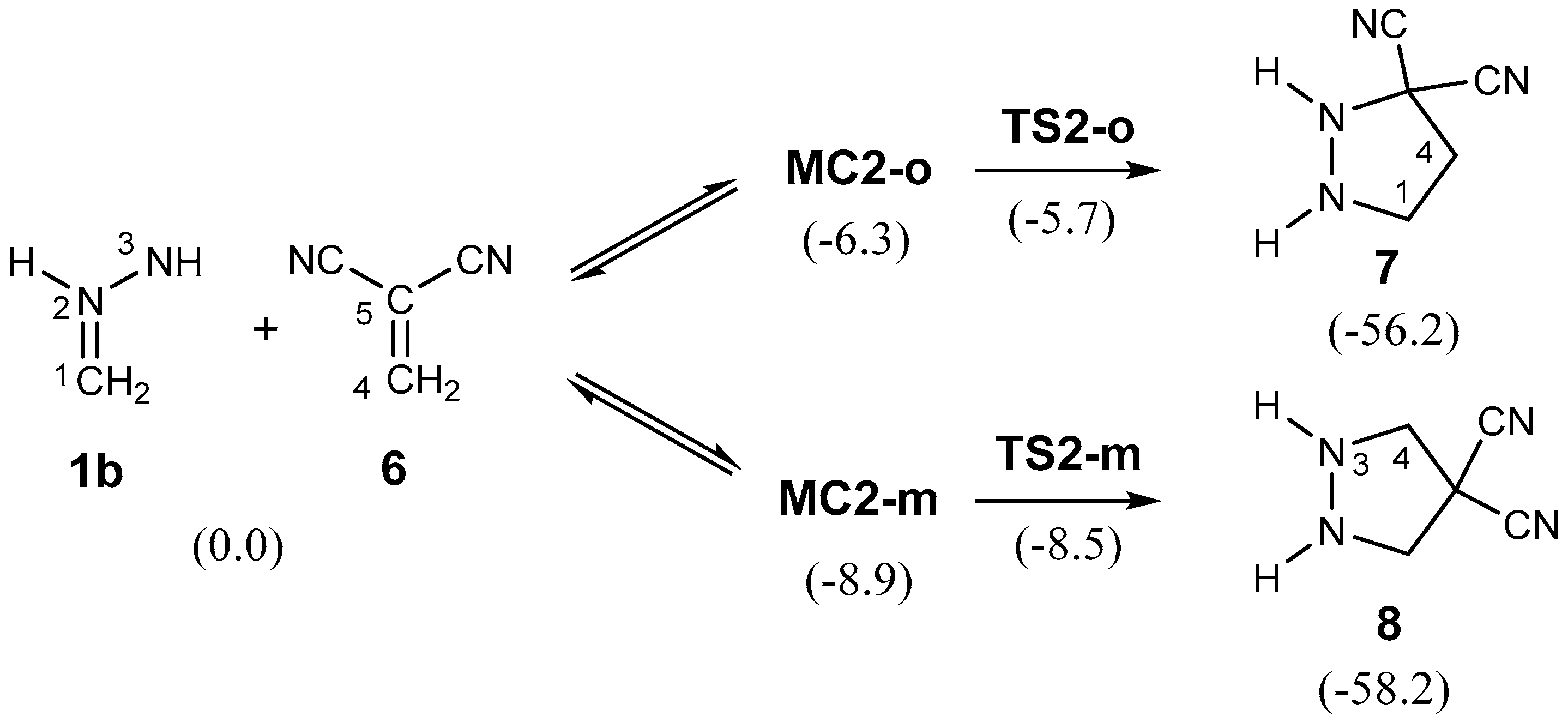
2.3.2. 32CA Reaction Involving DCE 6
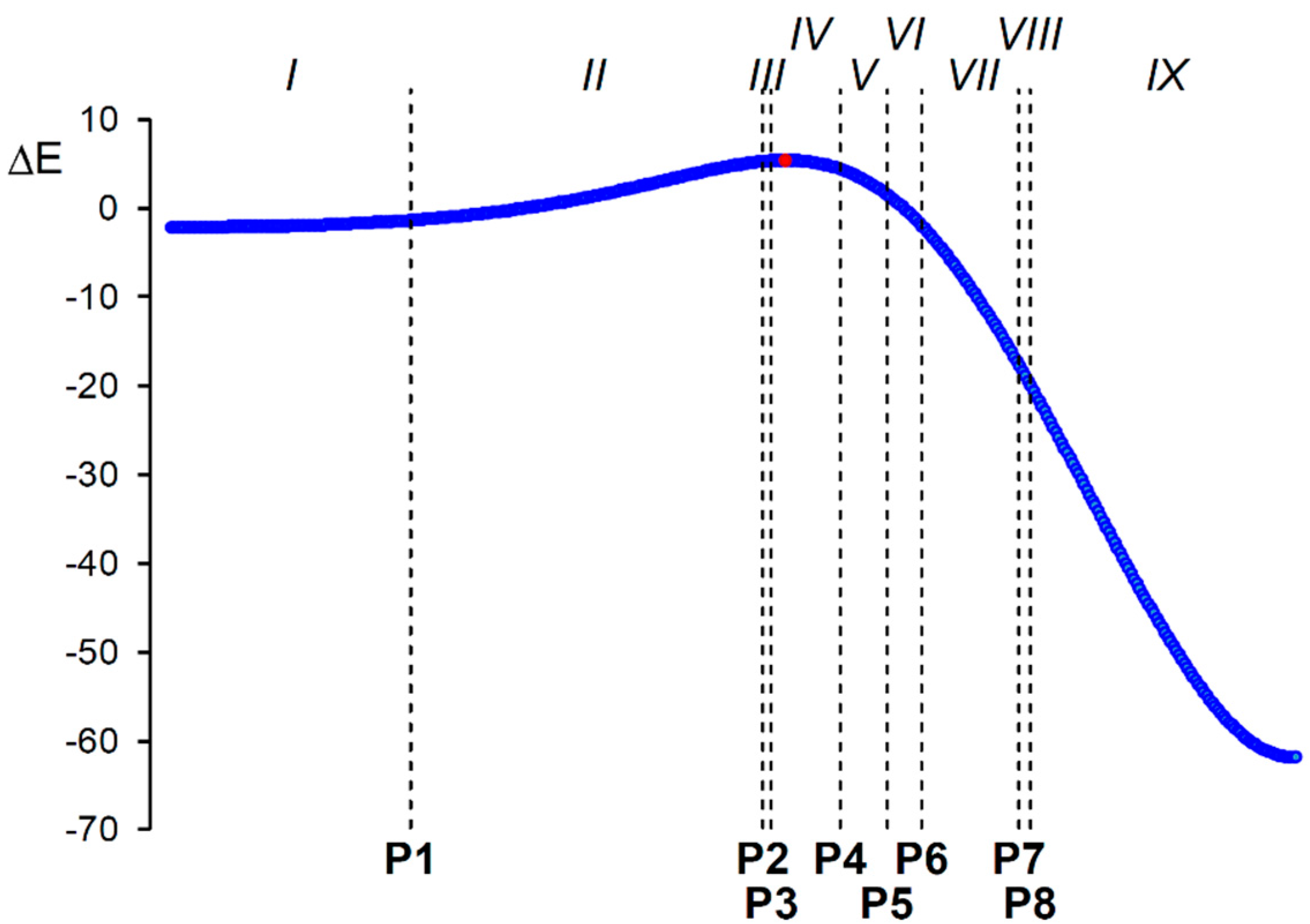
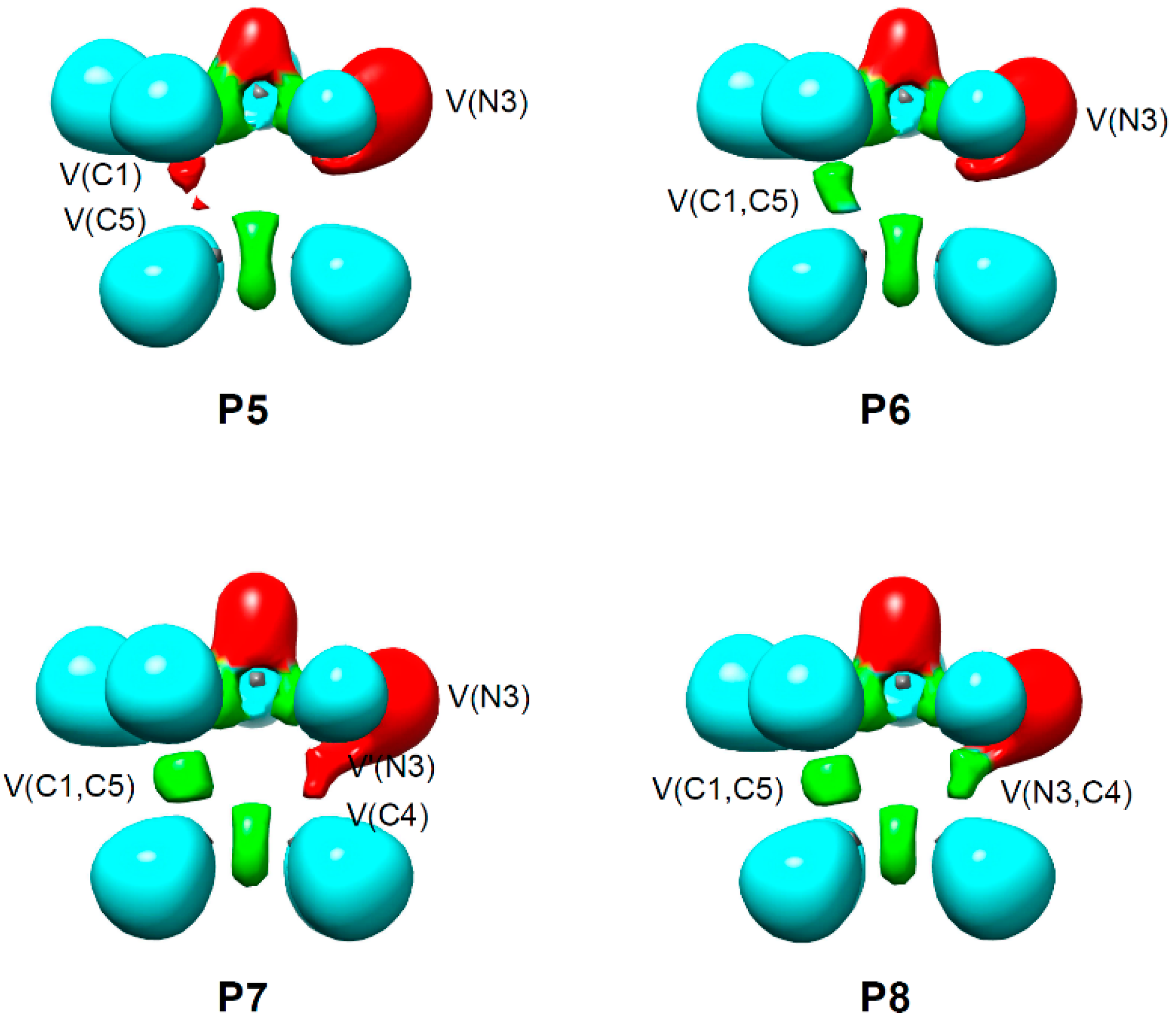
2.4. BET Study of the 32CA Reaction of AI 1b with ethylene 3
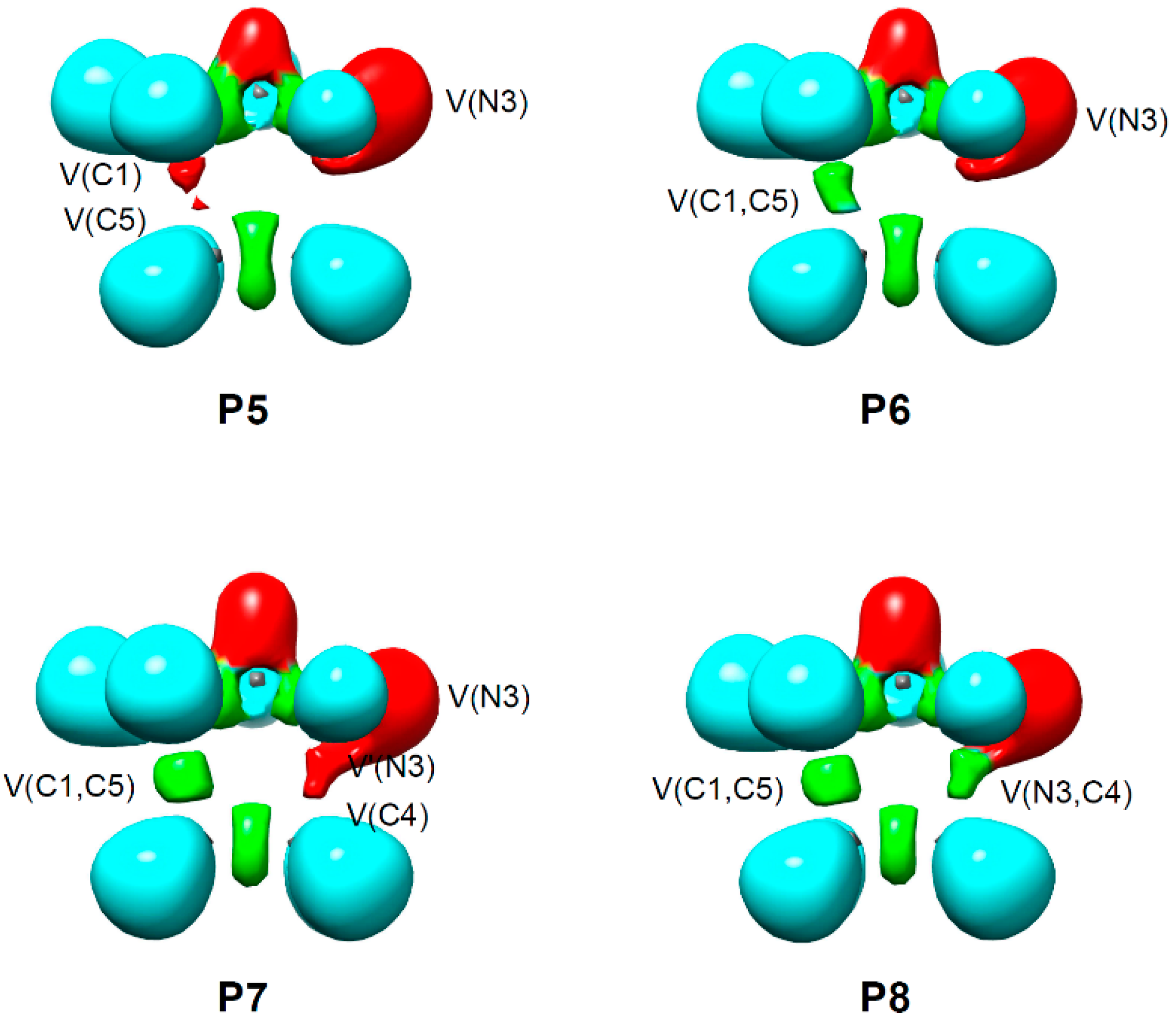
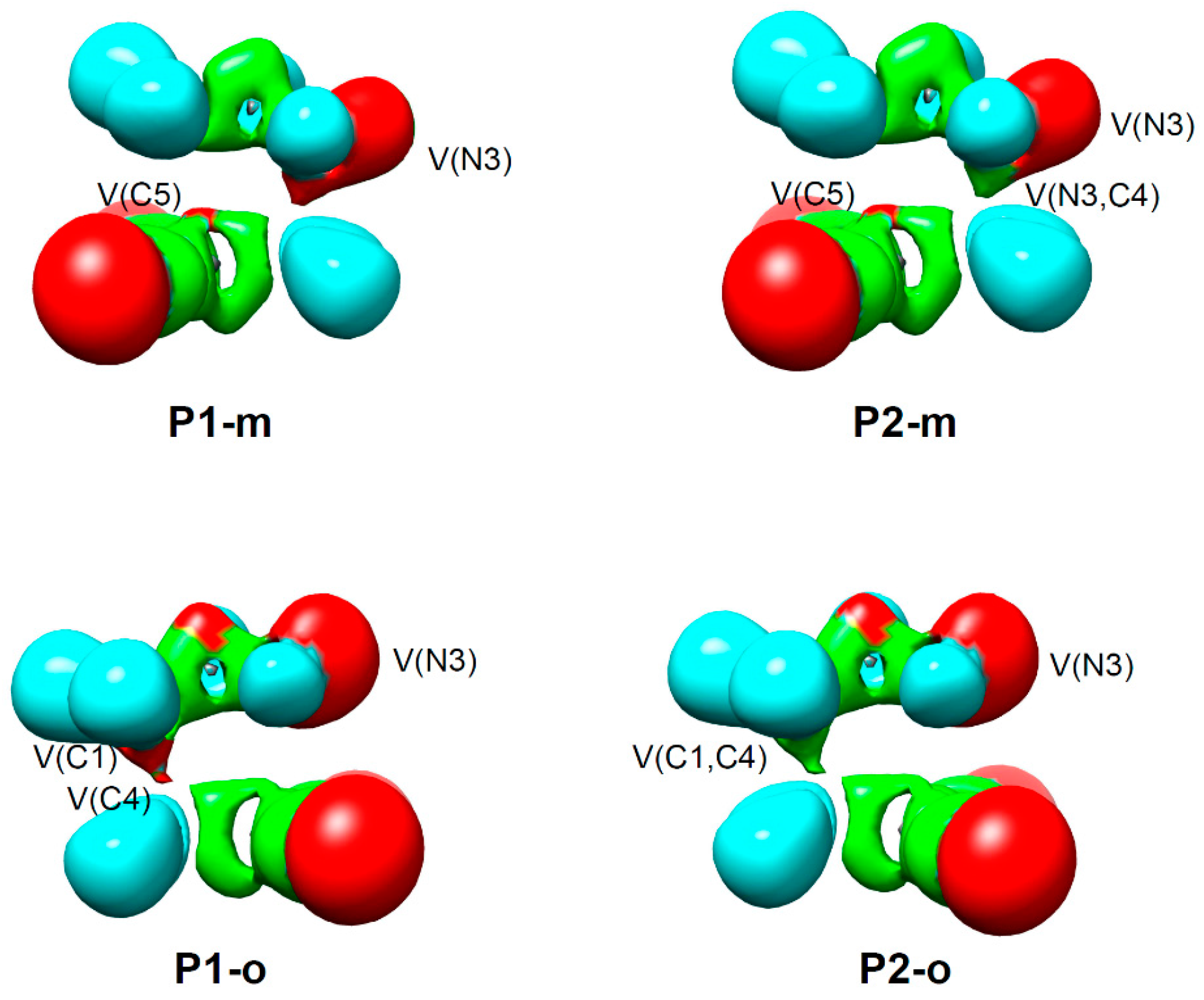
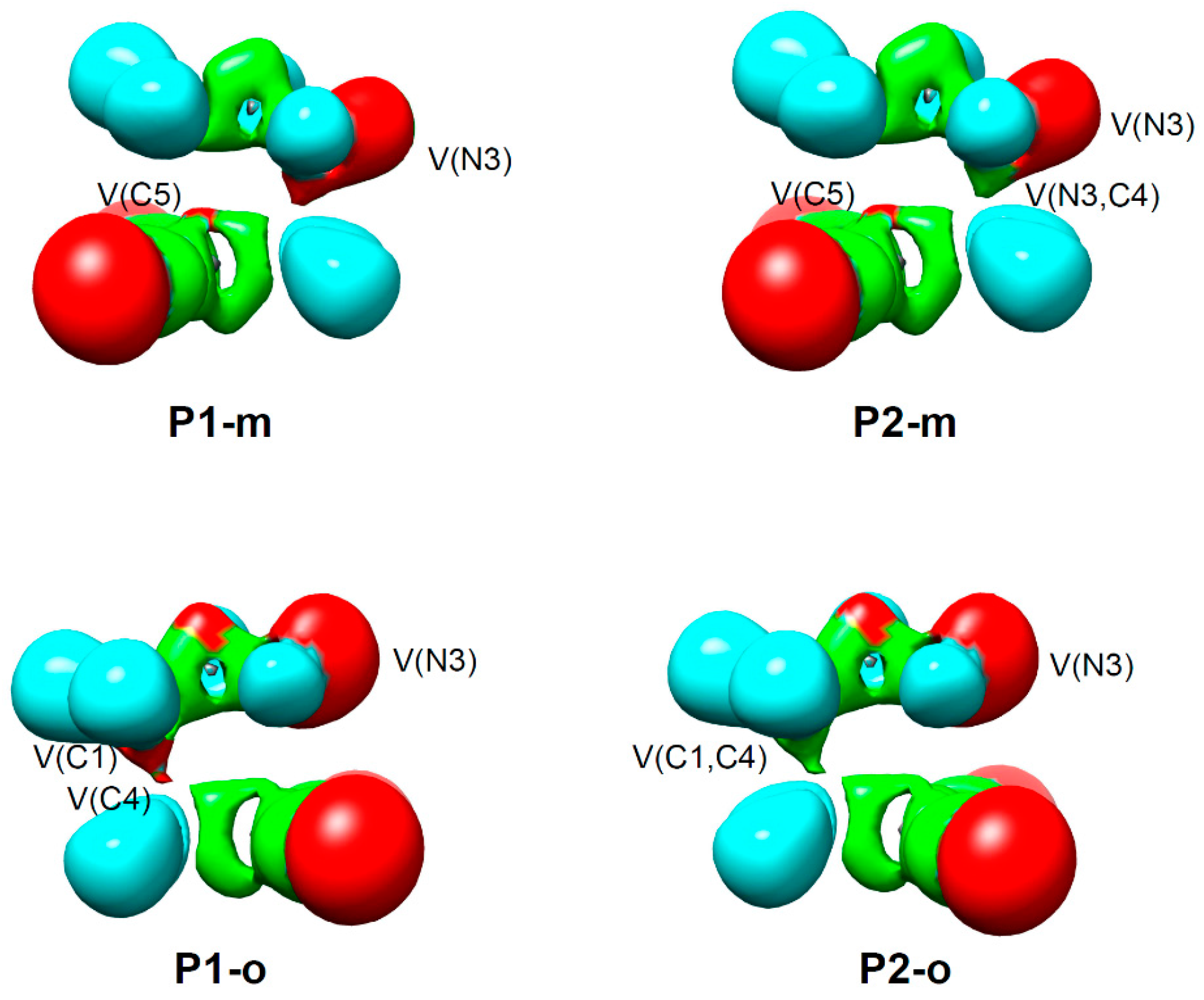
2.5. ELF Topological Analysis of the C–C and N–C Bond Formation Processes along the Polar 32CA Reaction between AI 1b and DCE 6. Understanding the Role of the GEDT
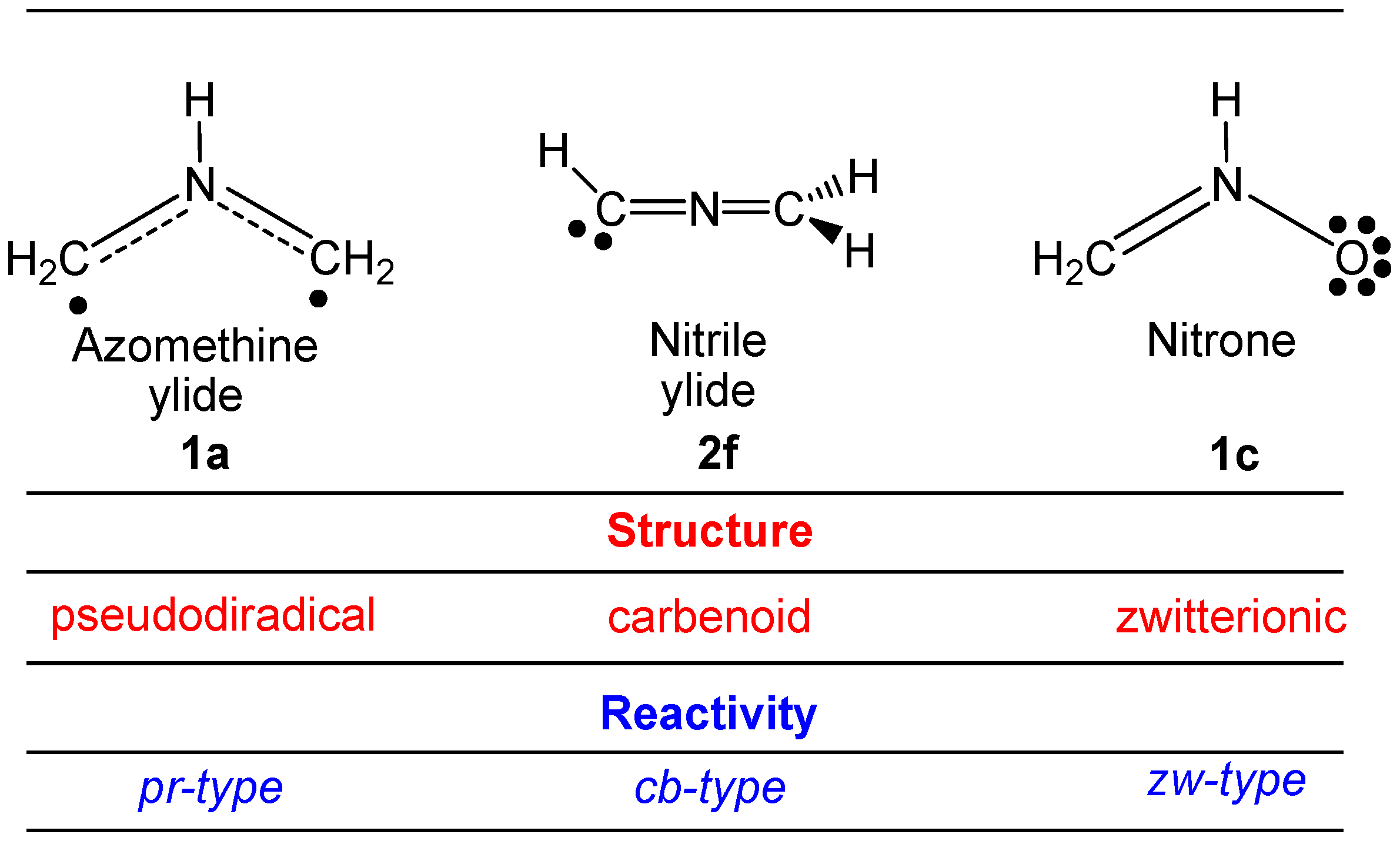
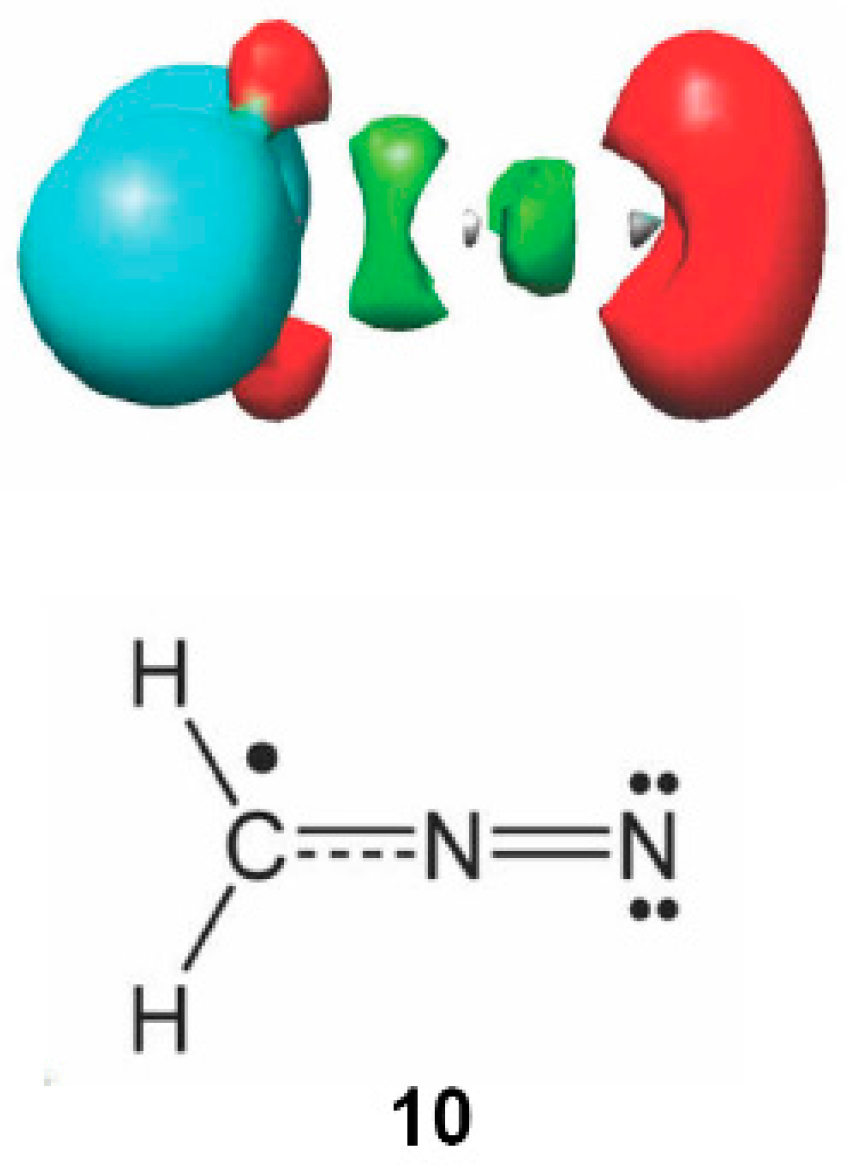
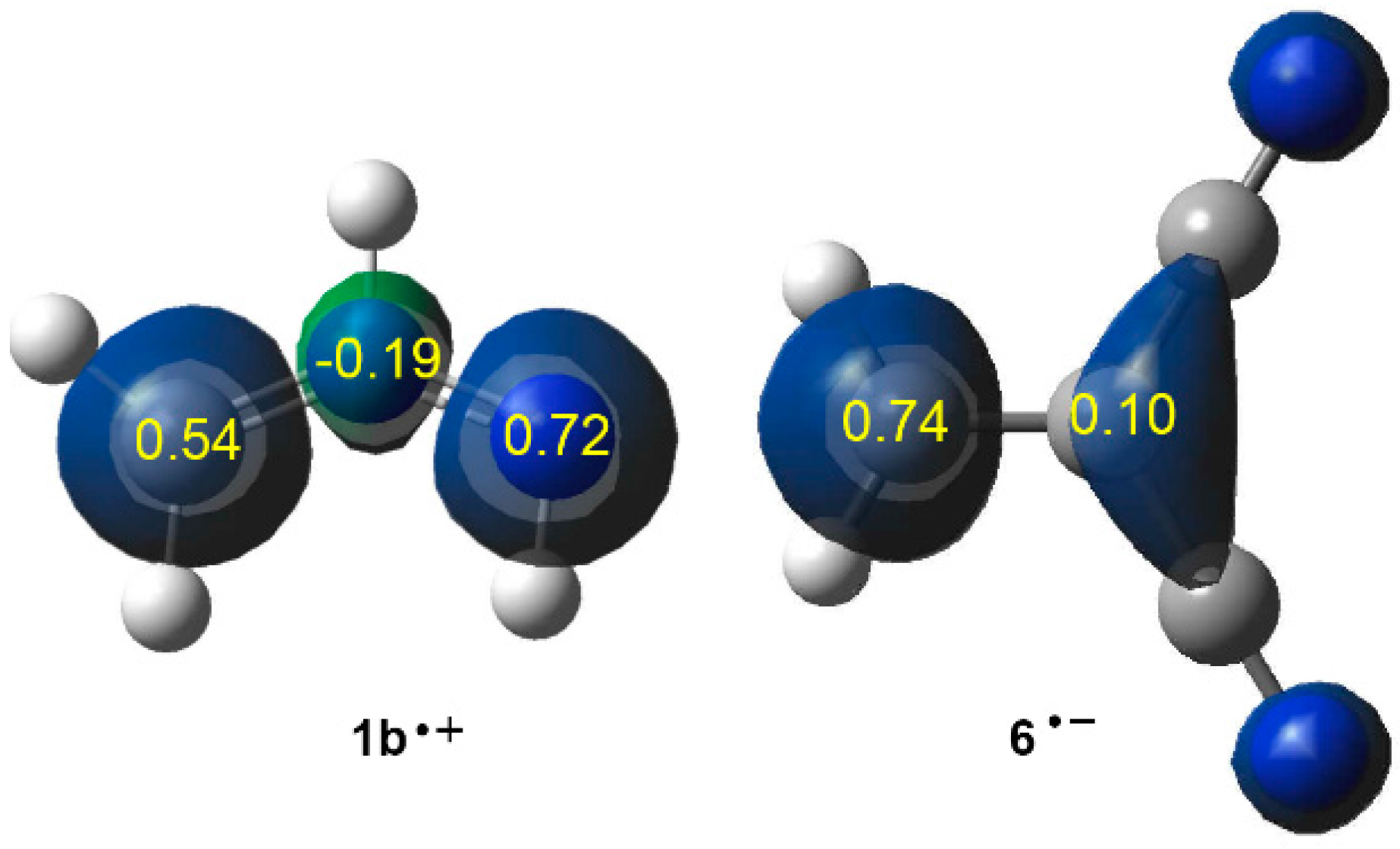
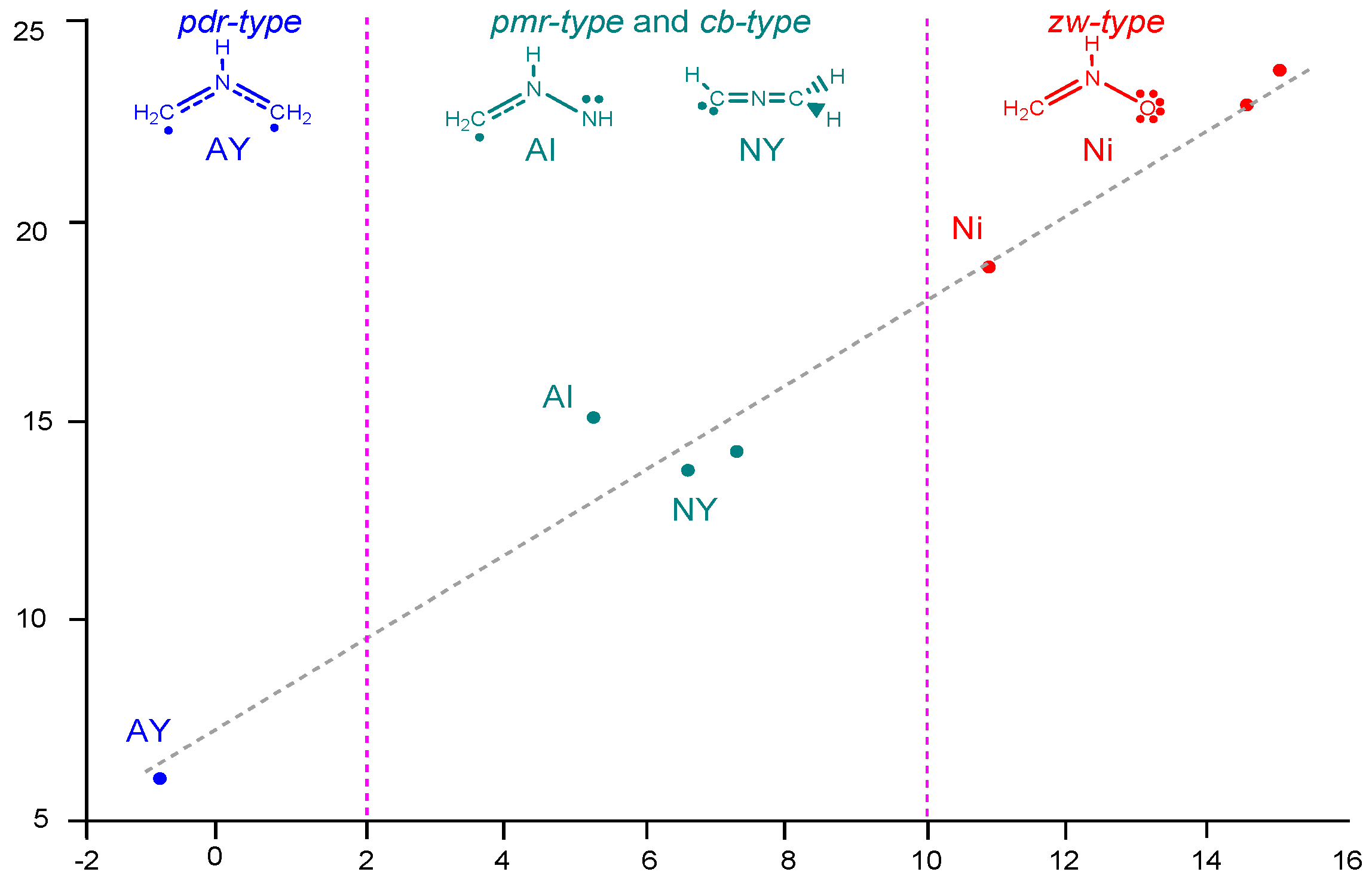
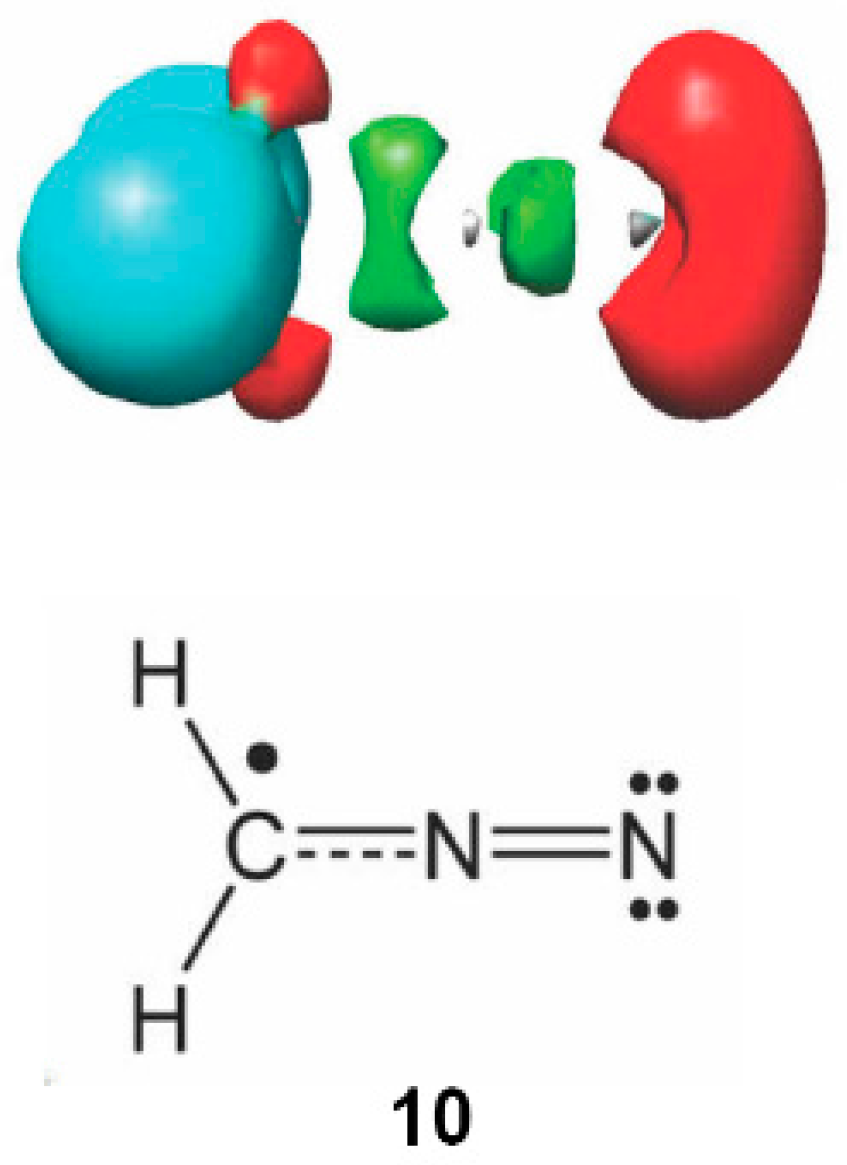
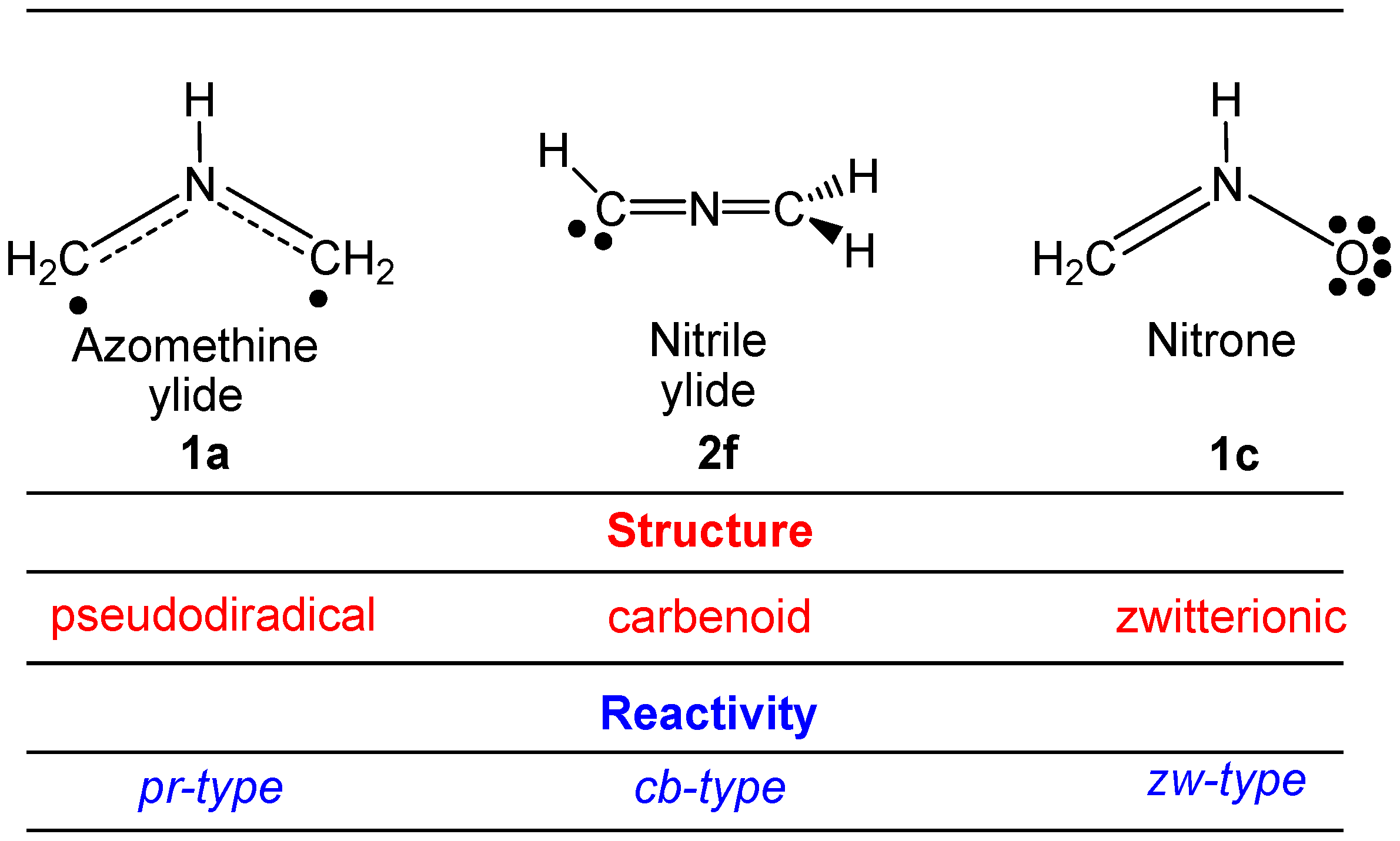
2.6. Understanding the Reactivity of AI 1b Possessing a Carbon Pseudoradical Centre
3. Conclusions
4. Computational Methods
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 32CA | [3+2] cycloaddition |
| AI | azomethine imine |
| ASD | atomic spin densities |
| A-TAC | allylic-type TAC |
| BET | Bonding Evolution Theory |
| cb-type | carbenoid-type |
| CDFT | Conceptual DFT |
| DAA | diazoalkanes |
| DCE | dicyanoethylene |
| DFT | Density Functional Theory |
| DIEM | Distortion/Interaction Energy Model |
| ED | electron-deficient |
| ELF | electron localisation function |
| FMO | Frontier Molecular Orbital |
| GEDT | global electron density transfer |
| GS | ground state |
| MC | molecular complex |
| MEDT | Molecular Electron Density Theory |
| MO | molecular orbital |
| Ni | nitrone |
| pdr-type | pseudodiradical-type |
| pmr-type | pseudoradical-type |
| P-TAC | propargylic-type TAC |
| TAC | three-atom-component |
| TCE | tetracyanoethylene |
| TS | transition state structure |
| zw-type | zwitterionic-type |
References
- Padwa, A. 1,3-Dipolar Cycloaddition Chemistry; Wiley-Interscience: New York, NY, USA, 1984. [Google Scholar]
- Padwa, A.; Pearson, W.H. Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; John Wiley & Sons, Inc.: New York, NY, USA, 2002. [Google Scholar]
- Huisgen, R. 1,3-dipolar cycloadditions. Proc. Chem. Soc. 1961, 33, 357–396. [Google Scholar]
- Huisgen, R. Kinetics and Mechanism of 1,3-Dipolar Cycloadditions. Angew. Chem. Int. Ed. Engl. 1963, 2, 633–696. [Google Scholar] [CrossRef]
- Huisgen, R. The Concerted Nature of 1,3-Dipolar Cycloadditions and the Question of Diradical Intermediates. J. Org. Chem. 1976, 41, 403–419. [Google Scholar] [CrossRef]
- Gothelf, K.V.; Jorgensen, K.A. Asymmetric 1,3-Dipolar Cycloaddition Reactions. Chem. Rev. 1998, 98, 863–909. [Google Scholar] [CrossRef] [PubMed]
- Ess, D.H.; Houk, K.N. Distortion/Interaction energy control of 1,3-dipolar cycloaddition reactivity. J. Am. Chem. Soc. 2007, 129, 10646–10647. [Google Scholar] [CrossRef] [PubMed]
- Ess, D.H.; Houk, K.N. Theory of 1,3-Dipolar Cycloadditions: Distortion/Interaction and Frontier Molecular Orbital Models. J. Am. Chem. Soc. 2008, 130, 10187–10198. [Google Scholar] [CrossRef] [PubMed]
- Hammond, G.S. A Correlation of Reaction Rates. J. Am. Chem Soc 1955, 77, 334–338. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. In homogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Duque-Noreña, M.; Chamorro, E.; Pérez, P. Understanding the carbenoid-type reactivity of nitrile ylides in [3+2] cycloaddition reactions towards electron-deficient ethylenes. A molecular electron density theory study. Theor. Chem. Acc. 2016, 135, 160. [Google Scholar] [CrossRef]
- Domingo, L.R. The Molecular Electron Density Theory: A Modern View of Molecular Reactivity in Organic Chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K. Molecular Orbitals in Chemistry, Physics, and Biology; Academic Press: New York, NY, USA, 1964. [Google Scholar]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the High Reactivity of the Azomethine Ylides in [3+2] Cycloaddition Reactions. Lett. Org. Chem. 2010, 7, 432–439. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. An MEDT study of the carbenoid-type [3+2] cycloaddition reactions of nitrile ylides with electron-deficient chiral oxazolid. Org. Biomol. Chem. 2016, 14, 10427–10436. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Emamian, S.R. Understanding the mechanisms of [3+2] cycloaddition reactions. The pseudoradical versus the zwitterionic mechanism. Tetrahedron 2014, 70, 1267–1273. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. A new model for C-C bond formation processes derived from the Molecular Electron-Density Theory in the study of the mechanism of [3+2] cycloaddition reactions of carbenoid nitrile ylides with electron-deficient ethylenes. Tetrahedron 2016, 72, 1524–1532. [Google Scholar] [CrossRef]
- Domingo, L.R.; Saez, J.A. Understanding the Electronic Reorganization along the Nonpolar [3+2] Cycloaddition Reactions of Carbonyl Ylides. J. Org. Chem. 2011, 76, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Aurell, M.J.; Pérez, P. A DFT analysis of the participation of TACs in zw-type [3+2] Cycloaddition Reactions. Tetrahedron 2014, 70, 4519–4525. [Google Scholar] [CrossRef]
- Ríos-Gutiérrez, M.; Pérez, P.; Domingo, L.R. A bonding evolution theory study of the mechanism of [3+2] cycloaddition reactions of nitrones with electron-deficient ethylenes. RSC Adv. 2015, 5, 58464–58477. [Google Scholar] [CrossRef]
- Krokidis, X.; Noury, S.; Silvi, B. Characterization of elementary chemical processes by catastrophe theory. J. Phys. Chem. A 1997, 101, 7277–7282. [Google Scholar] [CrossRef]
- Houk, K.N.; Sims, J.; Duke, R.E.; Strozier, R.W.; George, J.K. Frontier molecular orbitals of 1,3 dipoles and dipolarophiles. J. Am. Chem.Soc. 1973, 95, 7287–7301. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R. A New C-C Bond Formation Model Based on the Quantum Chemical Topology of Electron Density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Perez, P.; Contreras, R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Jaramillo, P.; Domingo, L.R.; Chamorro, E.; Perez, P. A further exploration of a nucleophilicity index based on the gas-phaseionization potentials. J. Mol. Struct. 2008, 865, 68–72. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P.; Sáez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Sáez, J.A. Unravelling the Mechanism of the Ketene-Imine Staudinger Reaction. An ELF Quantum Topological Analysis. RSC Adv. 2015, 5, 37119–37129. [Google Scholar] [CrossRef]
- Polo, V.; Andrés, J.; Berskit, S.; Domingo, L.R.; Silvi, B. Understanding reaction mechanisms in organic chemistry from catastrophe theory applied to the electron localization function topology. J. Phys. Chem. A 2008, 112, 7128–7136. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L. The nature of the chemical bond. Application of results obtained from the quantum mechanics and from a theory of paramagnetic susceptibility to the structure of molecules. J. Am. Chem. Soc. 1931, 53, 1367–1400. [Google Scholar] [CrossRef]
- Slater, J.C. Directed Valence in Polyatomic Molecules. Phys. Rev. 1931, 37, 481–489. [Google Scholar] [CrossRef]
- Domingo, L.R.; Saéz, J.A.; Zaragozá, R.J.; Arnó, M. Understanding the Participation of Quadricyclane as Nucleophile in Polar [2σ + 2σ + 2σ] Cycloadditions toward Electrophilic σ Molecules. J. Org. Chem. 2008, 73, 8791–8799. [Google Scholar] [CrossRef] [PubMed]
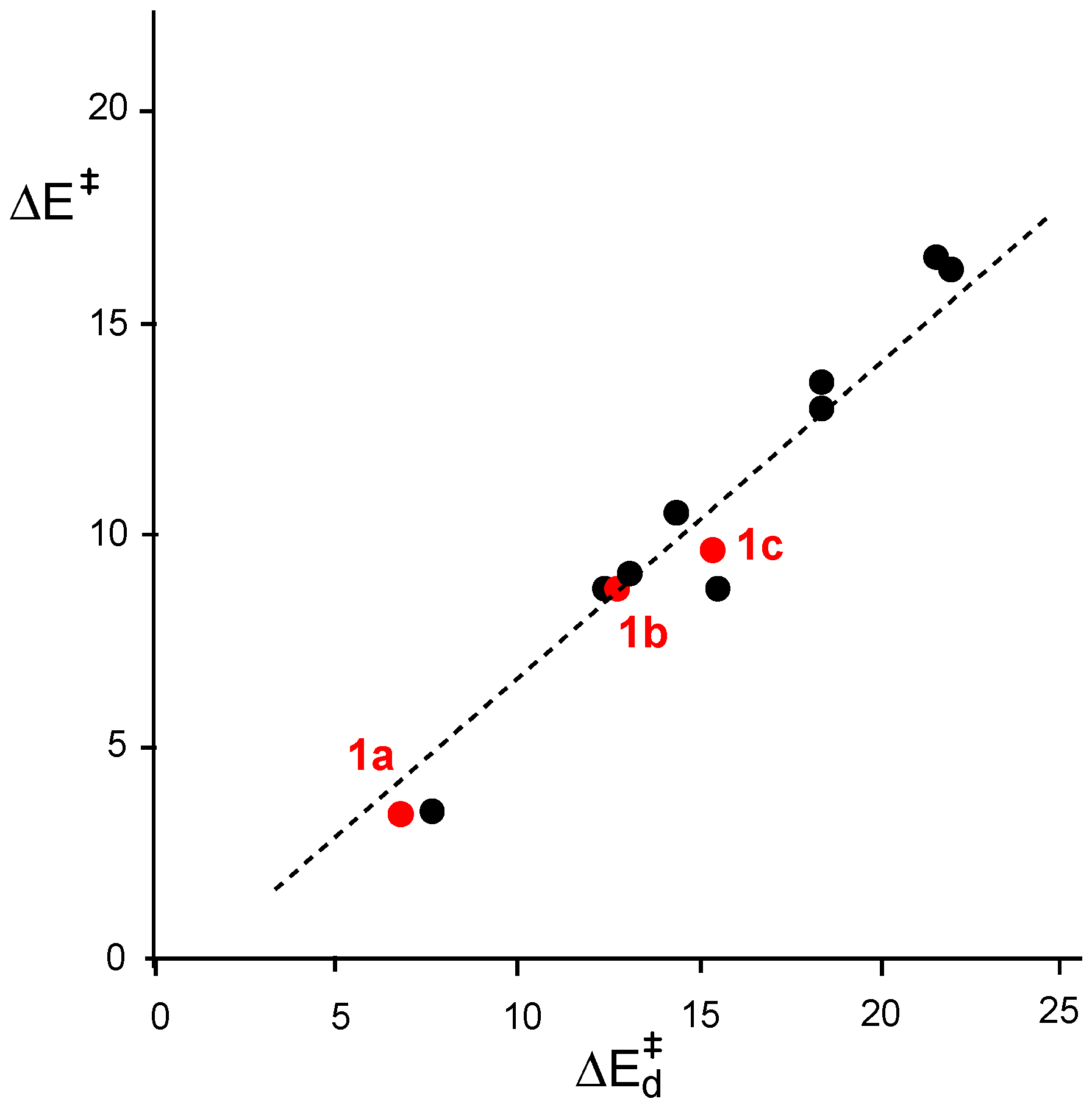
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. How does the global electron density transfer diminish activation energies in polar cycloaddition reactions? A Molecular Electron Density Theory study. Tetrahedron 2017, 73, 1718–1724. [Google Scholar] [CrossRef]
- Goldstein, E.; Beno, B.; Houk, K.N. Density functional theory prediction of the relative energies and isotope effects for the concerted and stepwise mechanisms of the Diels-Alder reaction of butadiene and ethylene. J. Am. Chem Soc 1996, 118, 6036–6043. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Emamian, S. Understanding the domino reactions between 1-diazopropan-2-one and 1,1-dinitroethylene. A molecular electron density theory study of the [3+2] cycloaddition reactions of diazoalkanes with electron-deficient ethylenes. RSC. Adv. 2017, 7, 15586–15595. [Google Scholar]
- Woodward, R.B.; Hoffmann, R. The conservation of orbital symmetry. Angew. Chem. Int. Ed. Engl. 1969, 8, 781–853. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, G.D. Hybrid Meta Density Functional Theory Methods for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions: The MPW1B95 and MPWB1K Models and Comparative Assessments for Hydrogen Bonding and van der Waals Interactions. J. Phys. Chem. A 2004, 108, 6908–6918. [Google Scholar] [CrossRef]
- Hehre, W.J.; Radom, L.; Schleyer, P.V.R.; Pople, J.A. Ab initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Schlegel, H.B. Modern Electronic Structure Theory; Yarkony, D.R., Ed.; World Scientific Publishing: Singapore, 1994. [Google Scholar]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Improved algorithms for reaction path following: Higher-order implicit algorithms. J. Chem. Phys. 1991, 95, 5853–5860. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural-population analysis. J. Chem. Phys. 1985, 83, 735–7465. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussain 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Parr, R.G.; von Szentpaly, L.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness—Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–75162. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Pérez, P. The nucleophilicity N index in organic chemistry. Org. Biomol. Chem. 2011, 9, 7168–7175. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, 1133–1138. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
Sample Availability: Not Available |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
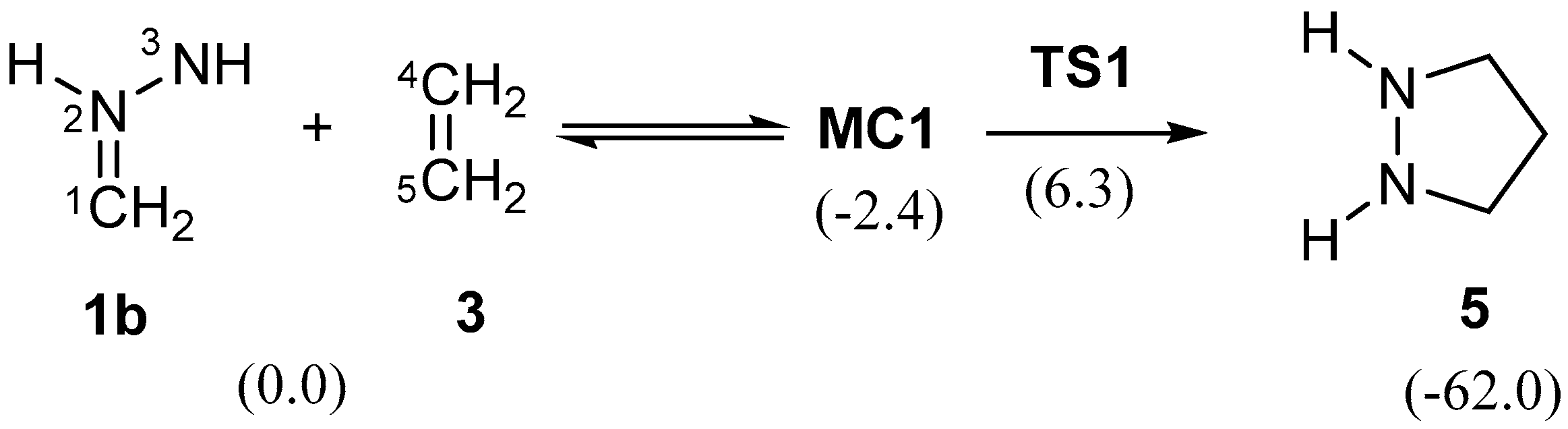{kind=link}
{kind=link}
{kind=link}
| µ | η | ω | N | pr | |
|---|---|---|---|---|---|
| AY 1a | −1.82 | 4.47 | 0.37 | 5.07 | 1.13 |
| AI 1b | −2.70 | 5.02 | 0.72 | 3.92 | 0.78 |
| Ni 1c | −3.43 | 5.55 | 1.06 | 2.92 | 0.53 |
| Ethylene 3 | −3.37 | 7.77 | 0.73 | 1.86 | |
| DCE 6 | −5.64 | 5.64 | 2.82 | 0.65 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domingo, L.R.; Ríos-Gutiérrez, M. A Molecular Electron Density Theory Study of the Reactivity of Azomethine Imine in [3+2] Cycloaddition Reactions. Molecules 2017, 22, 750. https://doi.org/10.3390/molecules22050750
Domingo LR, Ríos-Gutiérrez M. A Molecular Electron Density Theory Study of the Reactivity of Azomethine Imine in [3+2] Cycloaddition Reactions. Molecules. 2017; 22(5):750. https://doi.org/10.3390/molecules22050750
Chicago/Turabian StyleDomingo, Luis R., and Mar Ríos-Gutiérrez. 2017. "A Molecular Electron Density Theory Study of the Reactivity of Azomethine Imine in [3+2] Cycloaddition Reactions" Molecules 22, no. 5: 750. https://doi.org/10.3390/molecules22050750
APA StyleDomingo, L. R., & Ríos-Gutiérrez, M. (2017). A Molecular Electron Density Theory Study of the Reactivity of Azomethine Imine in [3+2] Cycloaddition Reactions. Molecules, 22(5), 750. https://doi.org/10.3390/molecules22050750