
Exploring the Antitumor Mechanism of High-Dose Cytarabine through the Metabolic Perturbations of Ribonucleotide and Deoxyribonucleotide in Human Promyelocytic Leukemia HL-60 Cells

 ,
,
Abstract
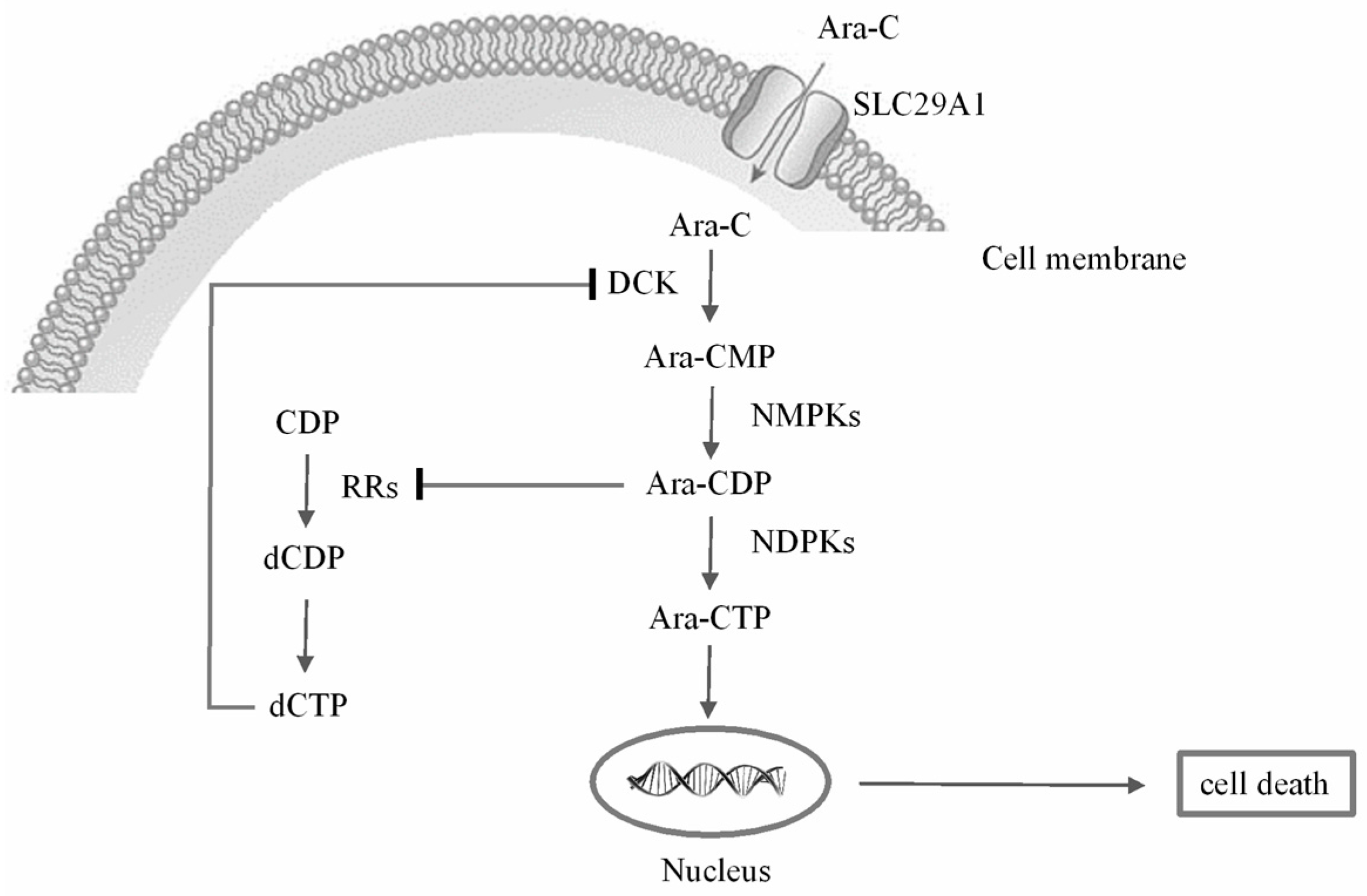
:1. Introduction
2. Results and Discussion
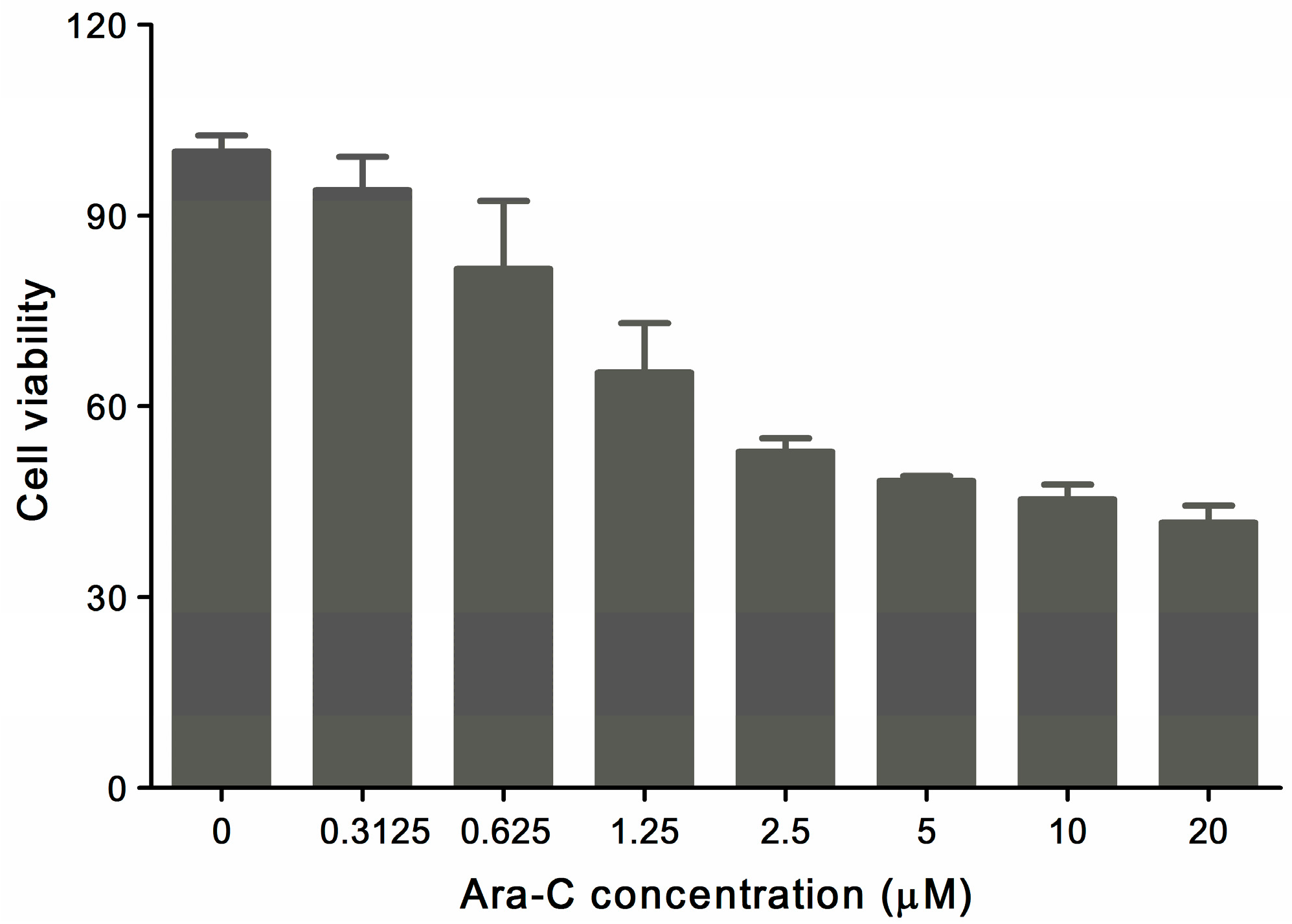
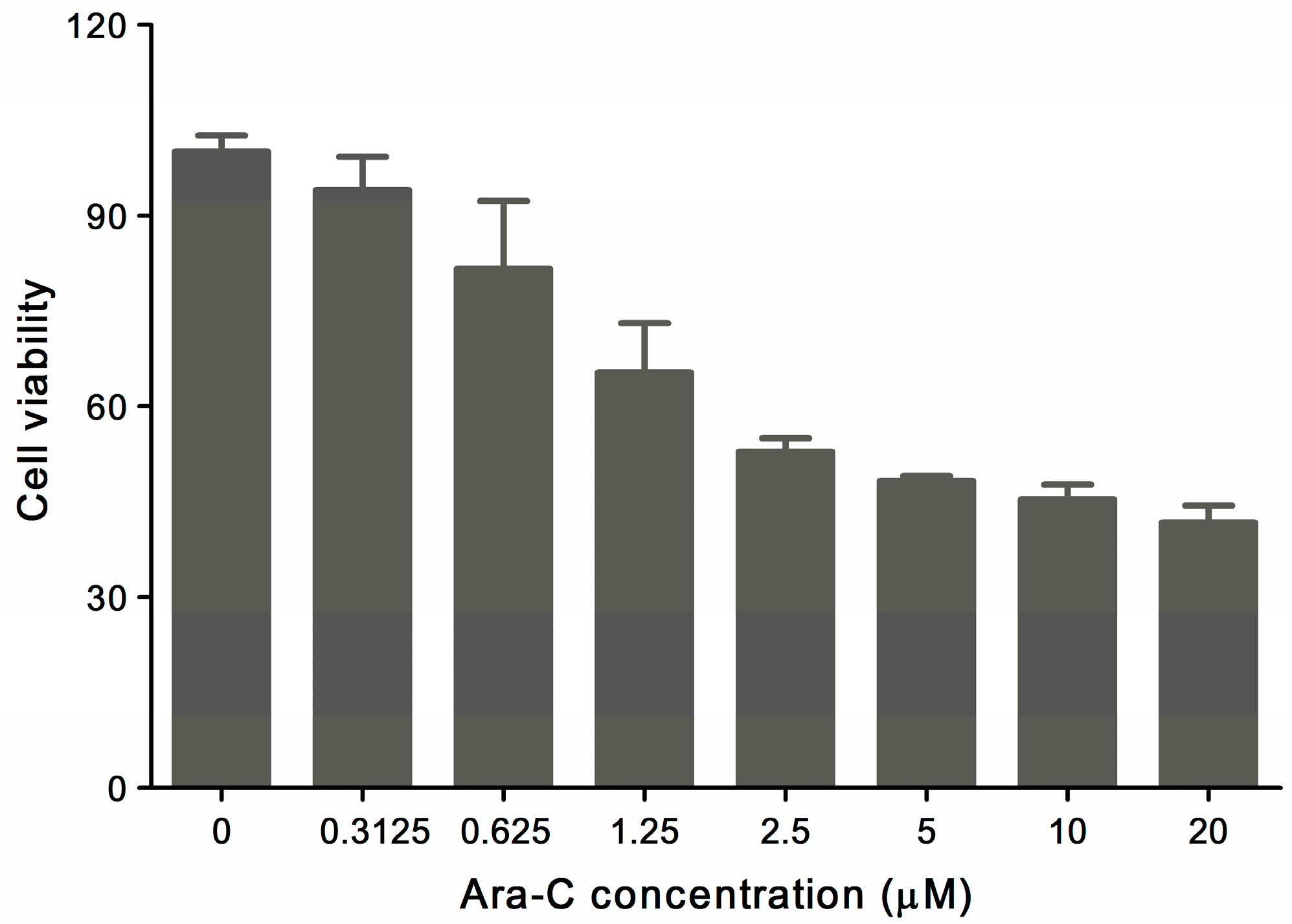
2.1. MTT Assay
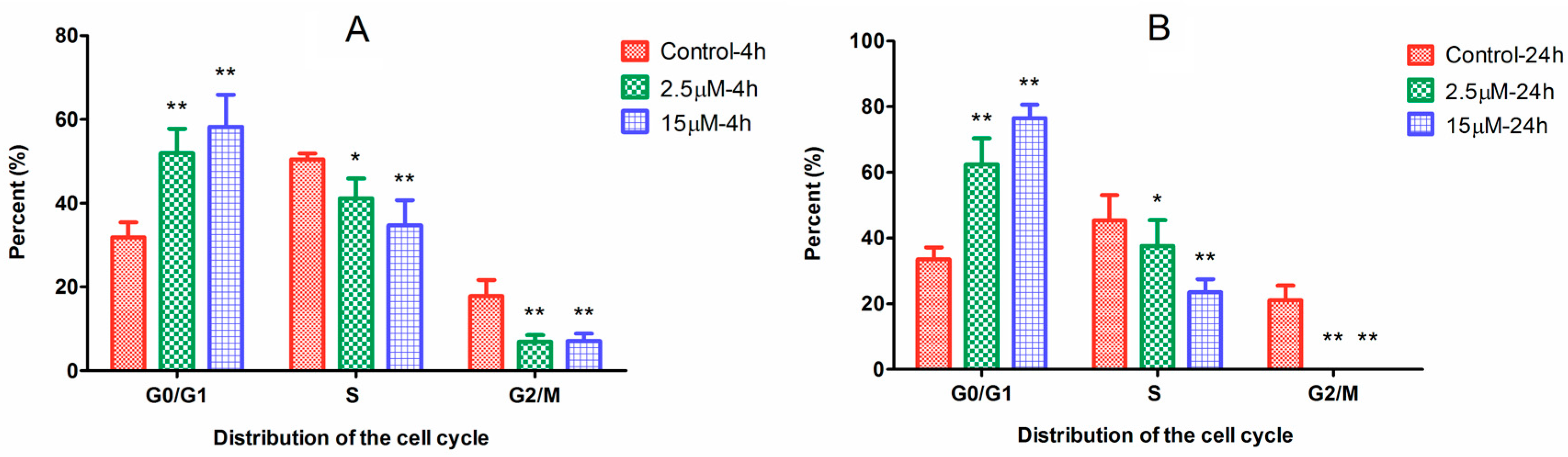
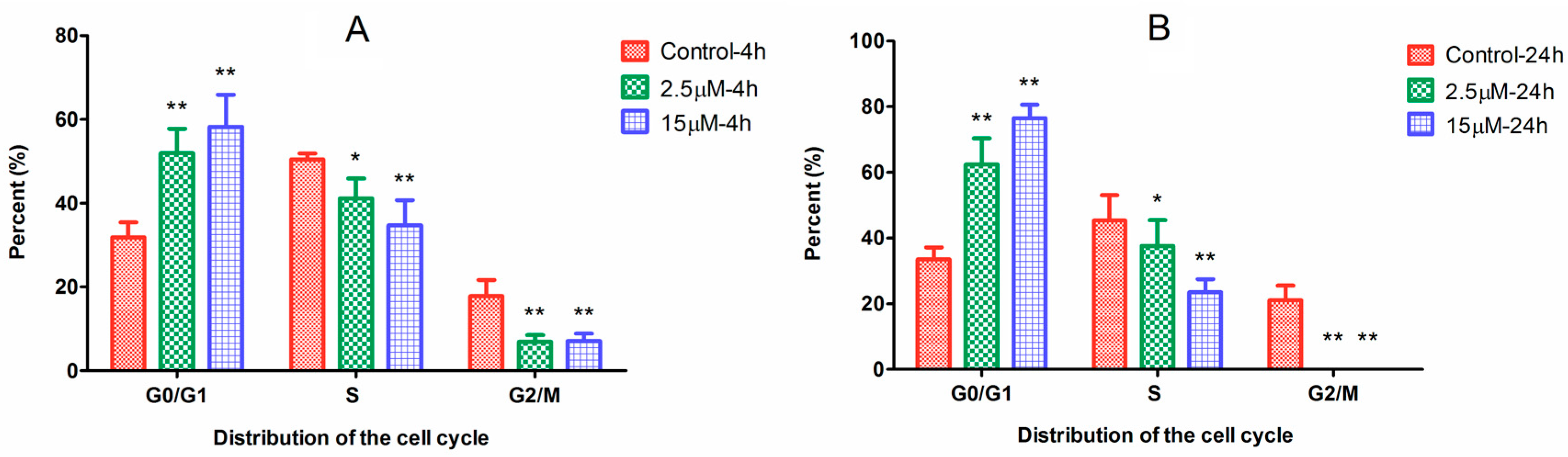
2.2. Cell Cycle Arrest Induced by Ara-C
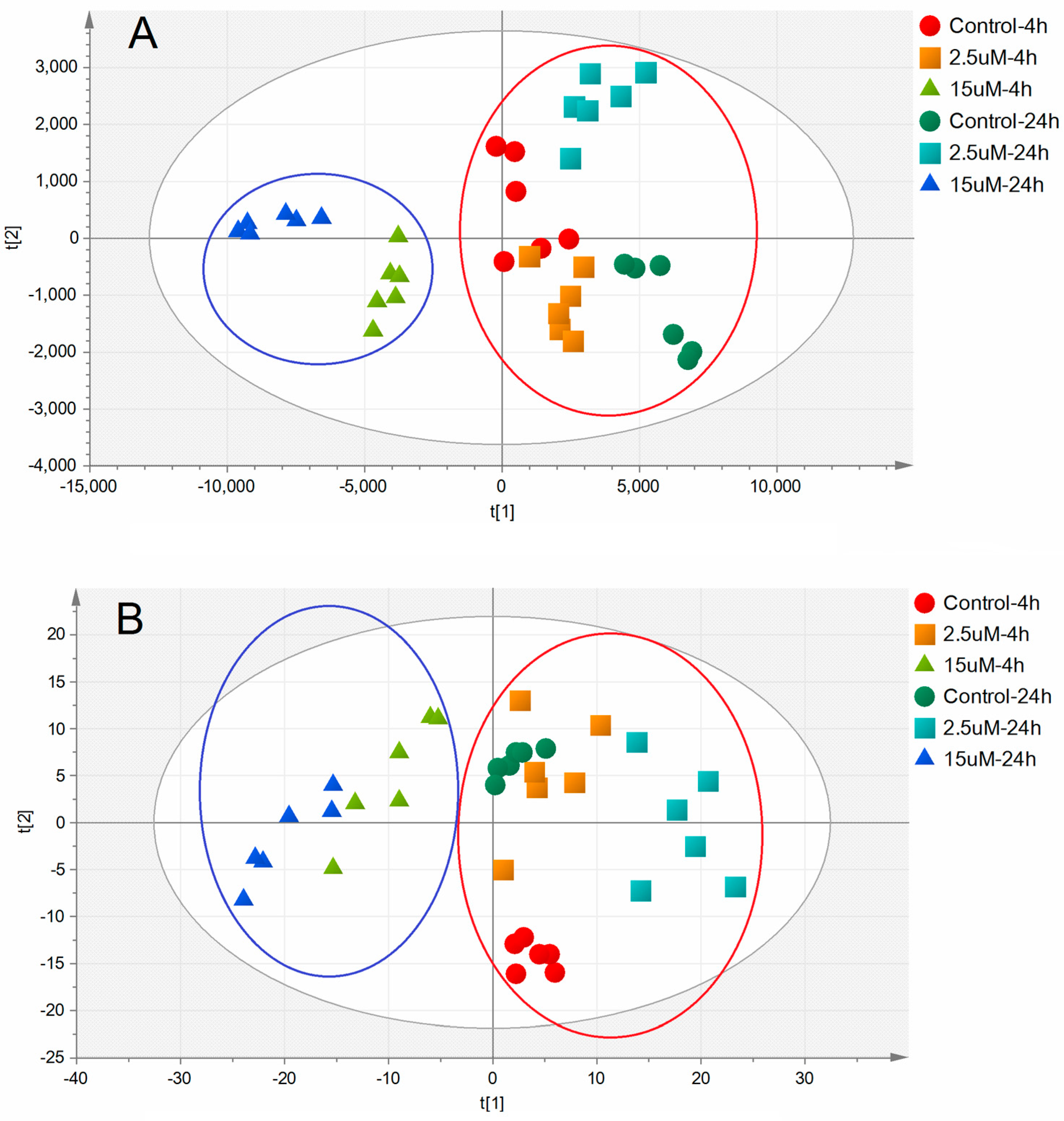
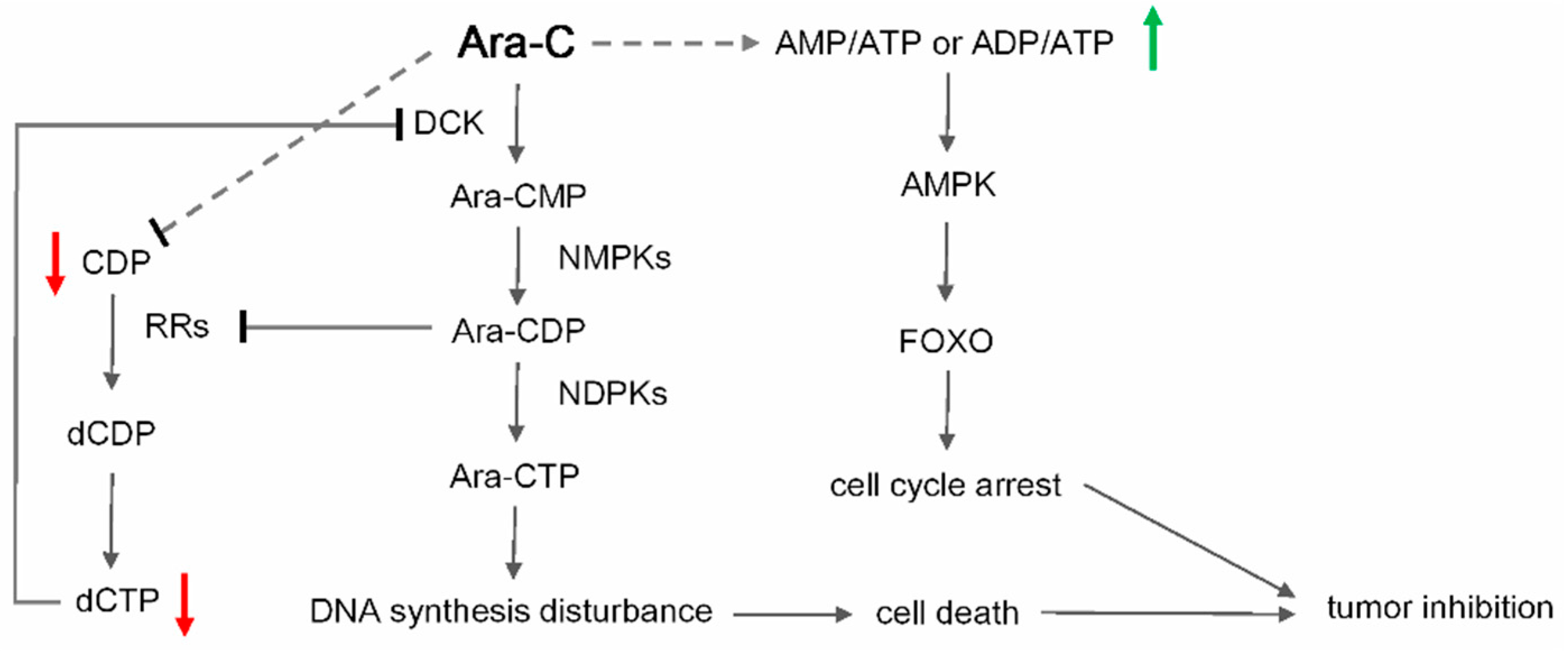
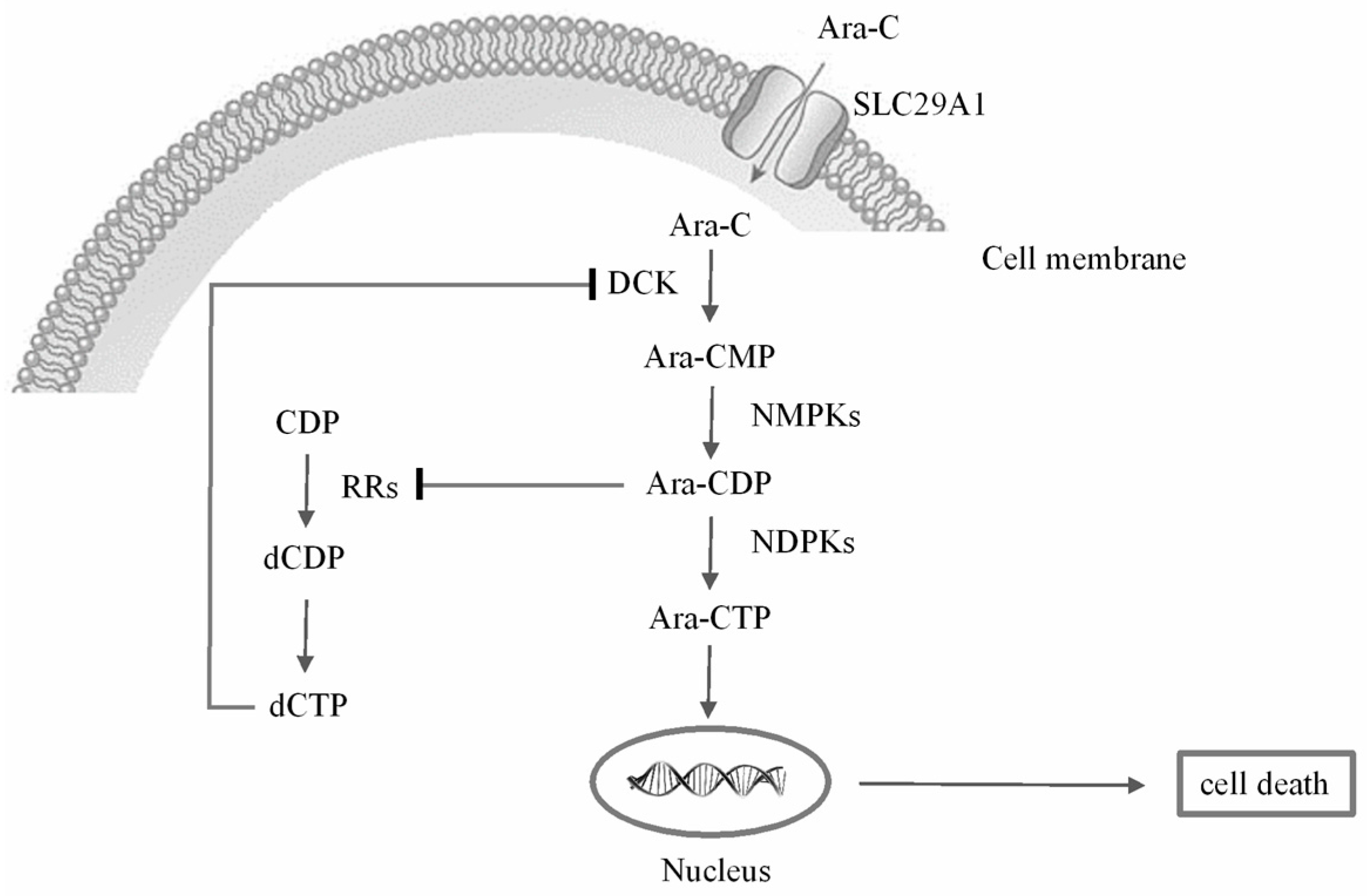
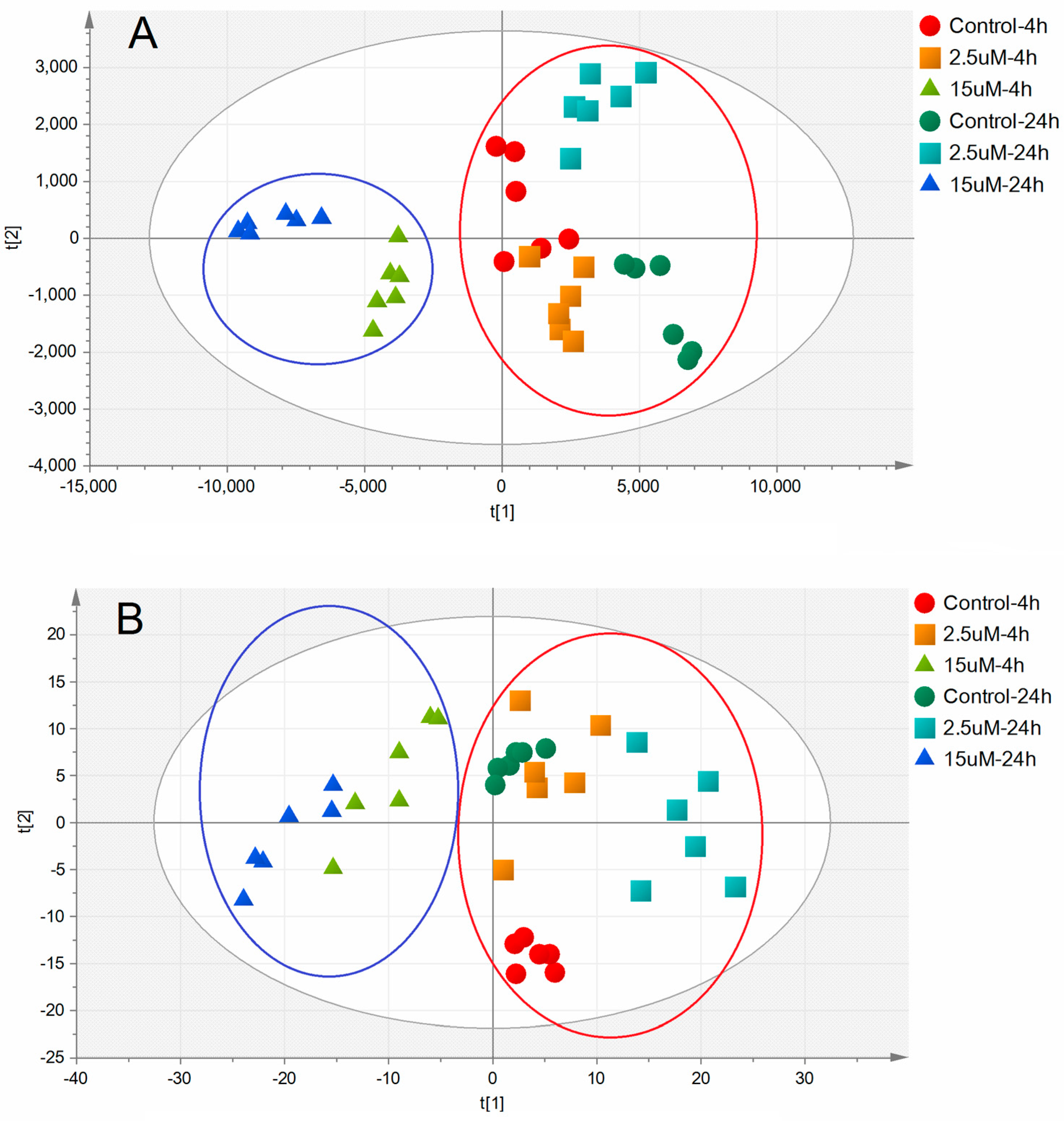
2.3. Metabolic Alterations Following Treatment with Ara-C
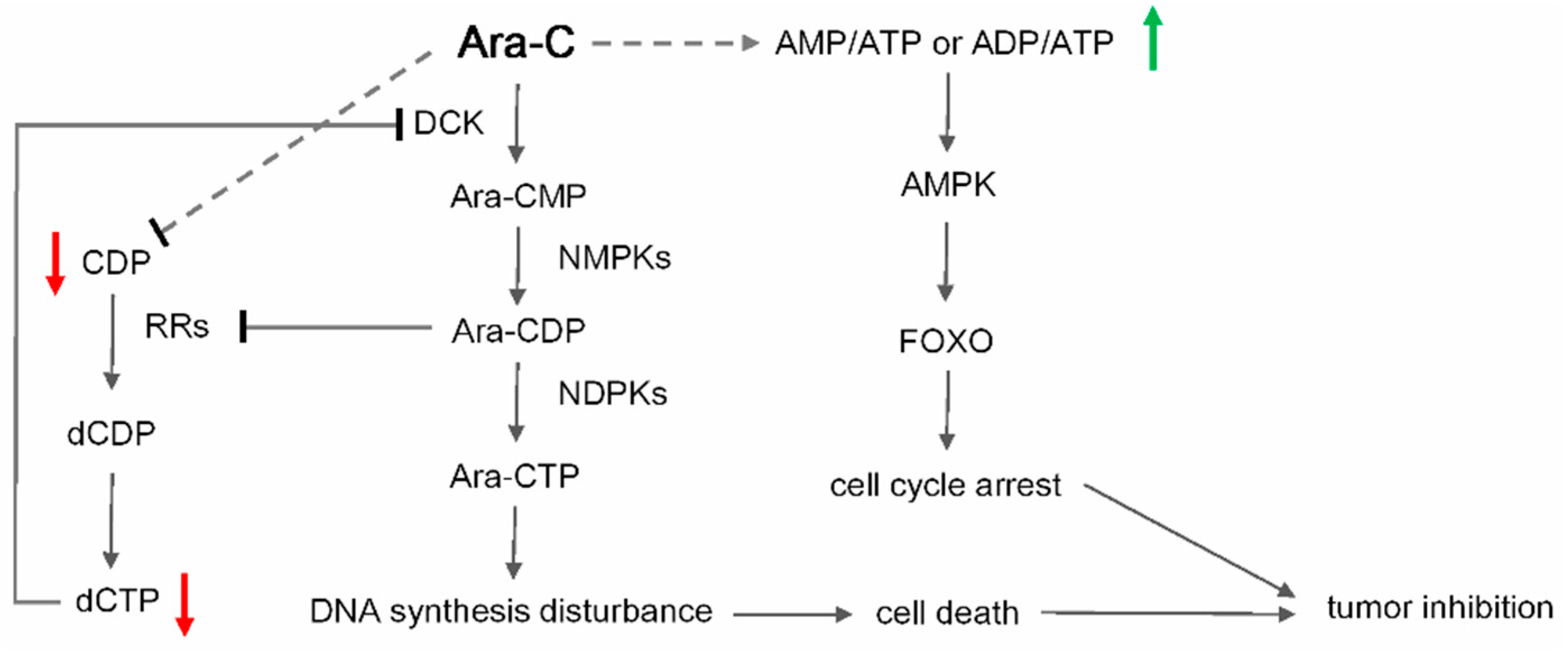
2.4. Metabolic Biomarkers and Pathway Analysis
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Cell Culture
3.3. MTT Assay
3.4. Cell Cycle Analysis
3.5. Sample Preparation
3.6. LC-MS/MS Analysis
3.7. Statistics Analysis and Potential Metabolic Biomarkers Determination
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kumar, C.C. Genetic abnormalities and challenges in the treatment of acute myeloid leukemia. Genes Cancer 2011, 2, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, S.; Ohtake, S.; Fujisawa, S.; Kiyoi, H.; Shinagawa, K.; Usui, N.; Sakura, T.; Miyamura, K.; Nakaseko, C.; Miyazaki, Y.; et al. A randomized comparison of 4 courses of standard-dose multiagent chemotherapy versus 3 courses of high-dose cytarabine alone in postremission therapy for acute myeloid leukemia in adults: The JALSG AML201 Study. Blood 2011, 117, 2366–2372. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, S.A.; Cadman, E.C.; Capizzi, R.L.; Skeel, R.T.; Bertino, J.R.; McIntosh, S. High dose cytosine arabinoside (HDARAC) in refractory acute leukemia. Cancer 1979, 44, 1189–1193. [Google Scholar] [CrossRef]
- Kern, W.; Aul, C.; Maschmeyer, G.; Schönrock-Nabulsi, R.; Ludwig, W.D.; Bartholomäus, A.; Bettelheim, P.; Wörmann, B.; Büchner, T.; Hiddemann, W. Superiority of high-dose over intermediate-dose cytosine arabinoside in the treatment of patients with high-risk acute myeloid leukemia: Results of an age-adjusted prospective randomized comparison. Leukemia 1998, 12, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Braess, J.; Wegendt, C.; Jahns-Streubel, G.; Kern, W.; Keye, S.; Unterhalt, M.; Schleyer, E.; Hiddemann, W. Successful modulation of high-dose cytosine arabinoside metabolism in acute myeloid leukaemia by haematopoietic growth factors: No effect of ribonucleotide reductase inhibitors fludarabine and gemcitabine. Br. J. Haematol. 2000, 109, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Reese, N.D.; Schiller, G.J. High-dose cytarabine (HD araC) in the treatment of leukemias: A review. Curr. Hematol. Malig. Rep. 2013, 8, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Hartford, C.M.; Duan, S.; Delaney, S.M.; Mi, S.; Kistner, E.O.; Lamba, J.K.; Huang, R.S.; Dolan, M.E. Population-specific genetic variants important in susceptibility to cytarabine arabinoside cytotoxicity. Blood 2009, 113, 2145–2153. [Google Scholar] [CrossRef] [PubMed]
- Lamba, J.K.; Crews, K.; Pounds, S.; Schuetz, E.G.; Gresham, J.; Gandhi, V.; Plunkett, W.; Rubnitz, J.; Ribeiro, R. Pharmacogenetics of deoxycytidine kinase: Identification and characterization of novel genetic variants. J. Pharmacol. Exp. Ther. 2007, 323, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Cros, E.; Jordheim, L.; Dumontet, C.; Galmarini, C.M. Problems related to resistance to cytarabine in acute myeloid leukemia. Leuk. Lymphoma 2004, 45, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Emadi, A.; Karp, J.E. The clinically relevant pharmacogenomic changes in acute myelogenous leukemia. Pharmacogenomics 2012, 13, 1257–1269. [Google Scholar] [CrossRef] [PubMed]
- Kufe, D.W.; Munroe, D.; Herrick, D.; Egan, E.; Spriggs, D. Effects of 1-beta-d-arabinofuranosylcytosine incorporation on eukaryotic DNA template function. Mol. Pharmacol. 1984, 26, 128–134. [Google Scholar] [PubMed]
- Lamba, J.K. Genetic factors influencing cytarabine therapy. Pharmacogenomics 2009, 10, 1657–1674. [Google Scholar] [CrossRef] [PubMed]
- Hubeek, I.; Stam, R.W.; Peters, G.J.; Broekhuizen, R.; Meijerink, J.P.; van Wering, E.R.; Gibson, B.E.; Creutzig, U.; Zwaan, C.M.; Cloos, J.; et al. The human equilibrative nucleoside transporter 1 mediates invitro cytarabine sensitivity in childhood acutemyeloid leukaemia. Br. J. Cancer 2005, 93, 1388–1394. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Zhou, B.; Chu, B.; Yen, Y. Ribonucleotide reductase inhibitors and future drug design. Curr. Cancer Drug Targets 2006, 6, 409–431. [Google Scholar] [CrossRef] [PubMed]
- Mayer, R.J.; Davis, R.B.; Schiffer, C.A.; Berg, D.T.; Powell, B.L.; Schulman, P.; Omura, G.A.; Moore, J.O.; McIntyre, O.R.; Frei, E. Intensive postremission chemotherapy in adults with acute myeloid Leukemia. N. Engl. J. Med. 1994, 331, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Plunkett, W.; Liliemark, J.O.; Adams, T.M.; Nowak, B.; Estey, E.; Kantarjian, H.; Keating, M.J. Saturation of 1-beta-d-arabinofuranosylcytosine 5′-triphosphate accumulation in leukemia cells during high-dose 1-beta-d-arabinofuranosylcytosine therapy. Cancer Res. 1987, 47, 3005–3011. [Google Scholar] [PubMed]
- Robak, T.; Korycka, A.; Kasznicki, M.; Wrzesien-Kus, A.; Smolewski, P. Purine nucleoside analogues for the treatment of hematological malignancies: Pharmacology and clinical applications. Curr Cancer Drug Targets 2005, 5, 421–444. [Google Scholar] [CrossRef] [PubMed]
- Carnrot, C.; Wehelie, R.; Eriksson, S.; Bolske, G.; Wang, L. Molecular characterization of thymidine kinase from Ureaplasma urealyticum: Nucleoside analogues as potent inhibitors of mycoplasma growth. Mol. Microbiol. 2003, 50, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Reichard, P. Ribonucleotide reductase and deoxyribonucleotide pools. Basic Life Sci. 1985, 31, 33–45. [Google Scholar] [PubMed]
- Fay, J.R.; Steele, V.; Crowell, J.A. Energy Homeostasis and Cancer Prevention: The AMP-Activated Protein Kinase. Cancer Prev. Res. 2009, 2, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; de Mattos, S.F.; van der Horst, A.; Klompmaker, R.; Kops, G.J.; Lam, E.W.; Burgering, B.M.; Medema, R.H. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol. Cell. Biol. 2002, 22, 7842–7852. [Google Scholar] [CrossRef] [PubMed]
- Paik, J.H.; Kollipara, R.; Chu, G.; Ji, H.; Xiao, Y.; Ding, Z.; Miao, L.; Tothova, Z.; Horner, J.W.; Carrasco, D.R.; et al. Foxos are lineage-restricted redundant tumor suppressors and critical regulators of endothelial cell homeostasis. Cell 2007, 128, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef] [PubMed]
- Essafi, A.; Fernandez deMattos, S.; Hassen, Y.A.; Soeiro, I.; Mufti, G.J.; Thomas, N.S.; Medema, R.H.; Lam, E.W. Direct transcriptional regulation of BIM by FoxO3a mediates STI571-induced apoptosis in BCR-ABL-expressing cells. Oncogene 2005, 24, 2317–2329. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, S.; Jagani, Z.; Kitidis, C.; Lodish, H.F.; Khosravi-Far, R. Cytokines and BCR-ABL mediate suppression of TRAIL-induced apoptosis through inhibition of forkhead FOXO3a transcription factor. Proc. Natl. Acad. Sci. USA 2003, 100, 6523–6528. [Google Scholar] [CrossRef] [PubMed]
- De Fernandez Mattos, S.; Essafi, A.; Soeiro, I.; Pietersen, A.M.; Birkenkamp, K.U.; Edwards, C.S.; Martino, A.; Nelson, B.H.; Francis, J.M.; Jones, M.C.; et al. FoxO3a and BCR-ABL regulate cyclin D2 transcription through a STAT5/BCL6-dependent mechanism. Mol. Cell. Biol. 2004, 24, 10058–10071. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tan, S.; Paintsil, E.; Dutschman, G.E.; Gullen, E.A.; Chu, E.; Cheng, Y.C. Analysis of deoxyribonucleotide pools in human cancer cell lines using a liquid chromatography coupled with tandem mass spectrometry technique. Biochem. Pharmacol. 2011, 82, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not Available.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control-4 h | 2.5 μM-4 h | 15 μM-4 h | Control-24 h | 2.5 μΜ-24 h | 15 μΜ-24 h | |
|---|---|---|---|---|---|---|
| ATP | 14,558.32 ± 685.79 | 15,117.47 ± 498.53 | 9428.09 ± 508.33 ** | 17,822.85 ± 441.51 | 17,085.81 ± 1050.99 | 5934.37 ± 507.32 ** |
| ADP | 1002.55 ± 217.5 | 768.84 ± 99.63 | 489.81 ± 72.97 ** | 2551.64 ± 288.24 | 3239.08 ± 804.21 | 738.05 ± 153.52 ** |
| AMP | 52.35 ± 21.93 | 54.4 ± 11.51 | 46.33 ± 8.71 | 190.5 ± 36.57 | 349.7 ± 84.19 * | 195.34 ± 62.2 |
| CTP | 1686.13 ± 369.04 | 2675.99 ± 507.97 ** | 2126.18 ± 310.6 | 2001.85 ± 349.47 | 2308.94 ± 620.87 | 1187.43 ± 156.51 ** |
| CDP | 136.78 ± 46.11 | 133.47 ± 24.9 | 102.6 ± 29.83 * | 362.11 ± 83.62 | 481.79 ± 210.33 | 115.73 ± 35.45 ** |
| CMP | 46.67 ± 12.03 | 54.39 ± 18.45 | 49.57 ± 15.37 | 66.44 ± 10.55 | 181.33 ± 105.04 * | 111.8 ± 51.61 |
| GTP | 2169.34 ± 589.95 | 2923.08 ± 951.26 | 1911.77 ± 262.31 | 3453.18 ± 213.16 | 2297.68 ± 689.24 * | 1301.35 ± 532.45 ** |
| GDP | 252 ± 35.18 | 243 ± 27.63 | 192.55 ± 20.4 | 607.68 ± 76.64 | 842.26 ± 126.22 ** | 331.8 ± 41.29 ** |
| GMP | 5.54 ± 1.74 | 3.7 ± 0.83 | 3.17 ± 0.98 | 18.39 ± 3.16 | 42.2 ± 18.64 * | 17.79 ± 8.25 |
| UTP | 4907.34 ± 1309.09 | 6819.2 ± 935.39 * | 4269.28 ± 446.38 | 9065.78 ± 1205.95 | 4986.46 ± 685.95 ** | 1817.11 ± 259.88 ** |
| UDP | 257.54 ± 100.54 | 180.96 ± 46.09 | 135.55 ± 24.21 * | 674.1 ± 103.75 | 671.86 ± 266.56 | 179.18 ± 64.47 ** |
| UMP | 6.83 ± 3.21 | 5.22 ± 1.55 | 4.19 ± 1.32 | 20.43 ± 4.2 | 27.28 ± 9.79 | 9.61 ± 4.56 ** |
| AMP/ATP | 0.003 ± 0.0 | 0.003 ± 0 | 0.005 ± 0 * | 0.011 ± 0.002 | 0.02 ± 0.006 | 0.034 ± 0.005 ** |
| Control-4 h | 2.5 μM-4 h | 15 μM-4 h | Control-24 h | 2.5 μΜ-24 h | 15 μΜ-24 h | |
|---|---|---|---|---|---|---|
| dATP | 6.77 ± 1.25 | 12.31 ± 3.14 ** | 9.22 ± 1.08 * | 9.95 ± 1.15 | 14.81 ± 3.17 * | 7.1 ± 1.37 ** |
| dADP | 0 | 0 | 0 | 0.015 ± 0.006 | 0.048 ± 0.042 | 0.003 ± 0.001 ** |
| dAMP | 0 | 0 | 0 | 0 | 0.01 ± 0 | 0 |
| dCTP | 22.39 ± 8.85 | 20.75 ± 3.42 | 10.63 ± 1.73 ** | 16.55 ± 2.23 | 34.55 ± 5.77 ** | 7.83 ± 1.08 ** |
| dCDP | 0.009 ± 0 | 0.004 ± 0.003 * | 0 ** | 0.014 ± 0.006 | 0.063 ± 0.038 * | 0 ** |
| dCMP | 0 | 0 | 0 | 0.016 ± 0.004 | 0.006 ± 0.005 * | 0 ** |
| dGTP | 1.88 ± 0.8 | 3.68 ± 2.76 | 2.14 ± 1.24 | 5.33 ± 2.39 | 2.94 ± 2.11 | 0.66 ± 0.56 ** |
| dGDP | 0.12 ± 0.07 | 0.07 ± 0.04 * | 0.05 ± 0.01 * | 0.66 ± 0.06 | 0.66 ± 0.2 | 0.04 ± 0.03 ** |
| dGMP | 0.022 ± 0.021 | 0.103 ± 0.061 * | 0.029 ± 0.011 | 0.168 ± 0.048 | 0.234 ± 0.111 | 0.047 ± 0.042 ** |
| dTTP | 25.54 ± 3.59 | 41.22 ± 6.19 ** | 32.76 ± 6.67 | 41.3 ± 3 | 50.92 ± 6.57 | 20.83 ± 5.18 ** |
| dTDP | 0.91 ± 0.29 | 0.99 ± 0.12 | 0.66 ± 0.15 * | 1.18 ± 0.24 | 3.57 ± 0.61 ** | 1.15 ± 0.57 |
| dTMP | 0 | 0.011 ± 0.007 * | 0.01 ± 0.006 * | 0.012 ± 0.002 | 0.038 ± 0.02 ** | 0.008 ± 0.008 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Guo, J.-R.; Chen, Q.-Q.; Wang, C.-Y.; Zhang, W.-J.; Yao, M.-C.; Zhang, W. Exploring the Antitumor Mechanism of High-Dose Cytarabine through the Metabolic Perturbations of Ribonucleotide and Deoxyribonucleotide in Human Promyelocytic Leukemia HL-60 Cells. Molecules 2017, 22, 499. https://doi.org/10.3390/molecules22030499
Li Z, Guo J-R, Chen Q-Q, Wang C-Y, Zhang W-J, Yao M-C, Zhang W. Exploring the Antitumor Mechanism of High-Dose Cytarabine through the Metabolic Perturbations of Ribonucleotide and Deoxyribonucleotide in Human Promyelocytic Leukemia HL-60 Cells. Molecules. 2017; 22(3):499. https://doi.org/10.3390/molecules22030499
Chicago/Turabian StyleLi, Zheng, Jian-Ru Guo, Qian-Qian Chen, Cai-Yun Wang, Wei-Jia Zhang, Mei-Cun Yao, and Wei Zhang. 2017. "Exploring the Antitumor Mechanism of High-Dose Cytarabine through the Metabolic Perturbations of Ribonucleotide and Deoxyribonucleotide in Human Promyelocytic Leukemia HL-60 Cells" Molecules 22, no. 3: 499. https://doi.org/10.3390/molecules22030499
APA StyleLi, Z., Guo, J.-R., Chen, Q.-Q., Wang, C.-Y., Zhang, W.-J., Yao, M.-C., & Zhang, W. (2017). Exploring the Antitumor Mechanism of High-Dose Cytarabine through the Metabolic Perturbations of Ribonucleotide and Deoxyribonucleotide in Human Promyelocytic Leukemia HL-60 Cells. Molecules, 22(3), 499. https://doi.org/10.3390/molecules22030499