Vanadium Compounds as PTP Inhibitors

Abstract
1. Introduction
2. Protein Tyrosine Phosphatases (PTPs)
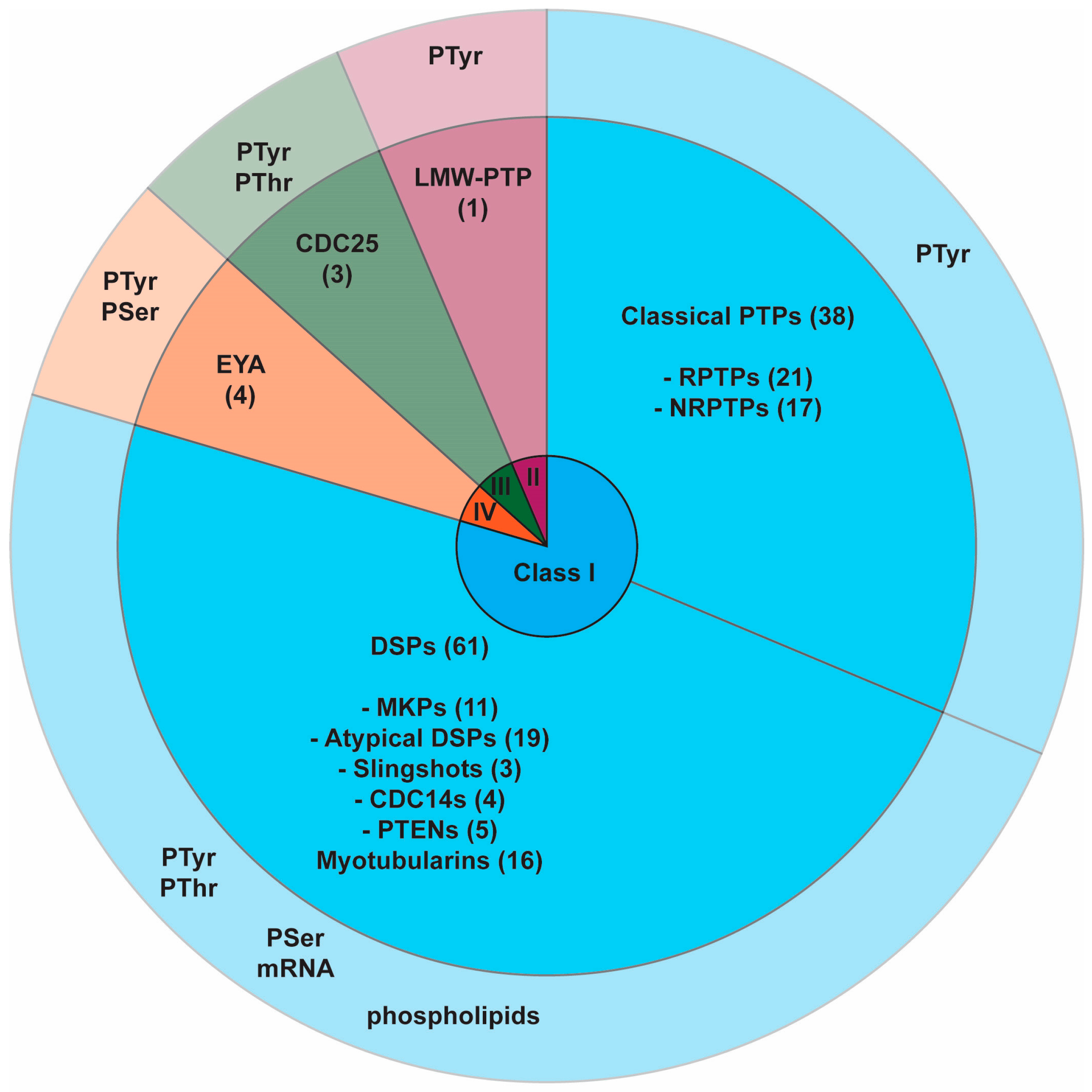
2.1. PTP Superfamily
2.2. PTP Inhibition
3. Vanadium
3.1. Mechanisms of PTP Inhibition
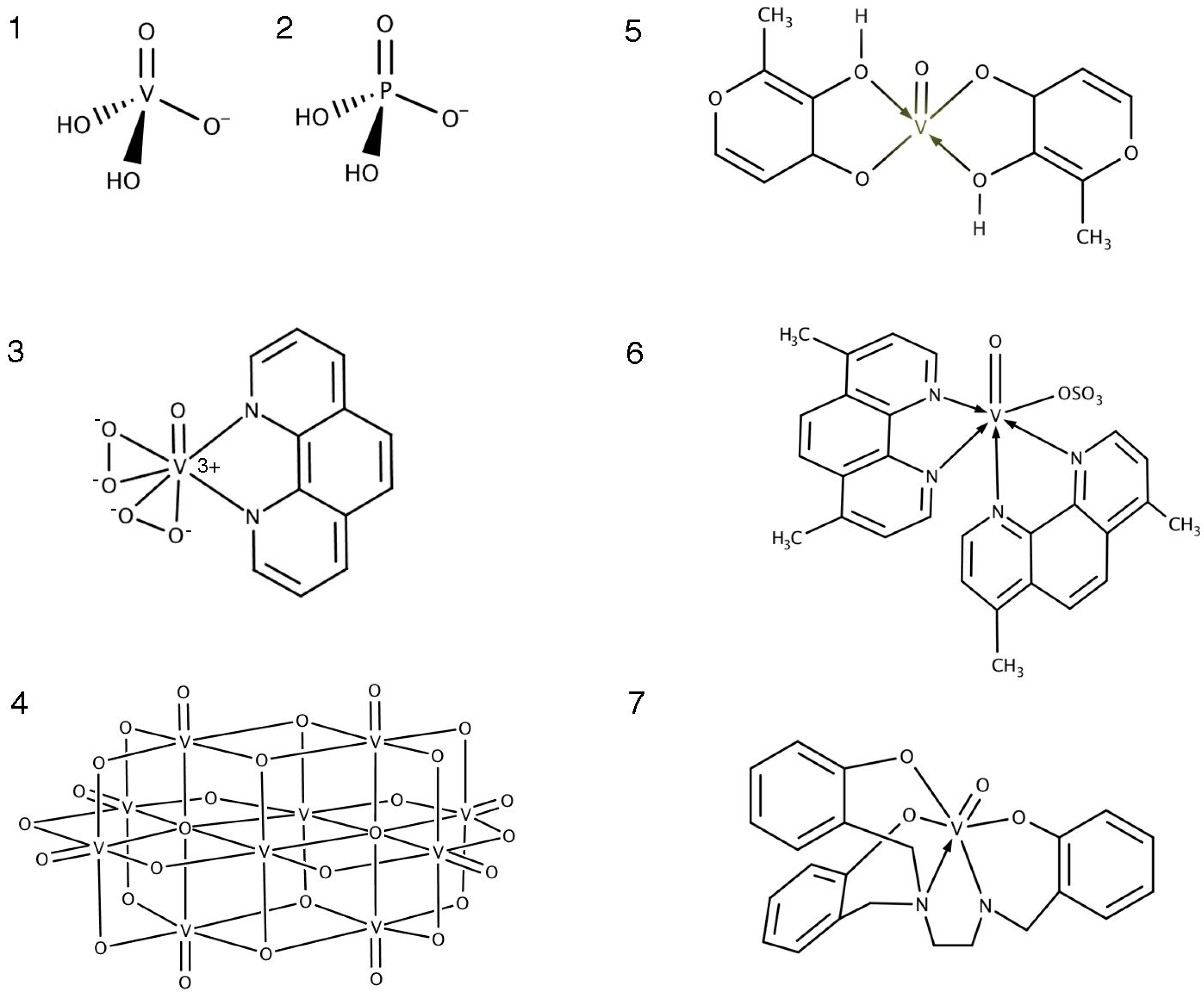
3.2. Oxidovanadium Compounds
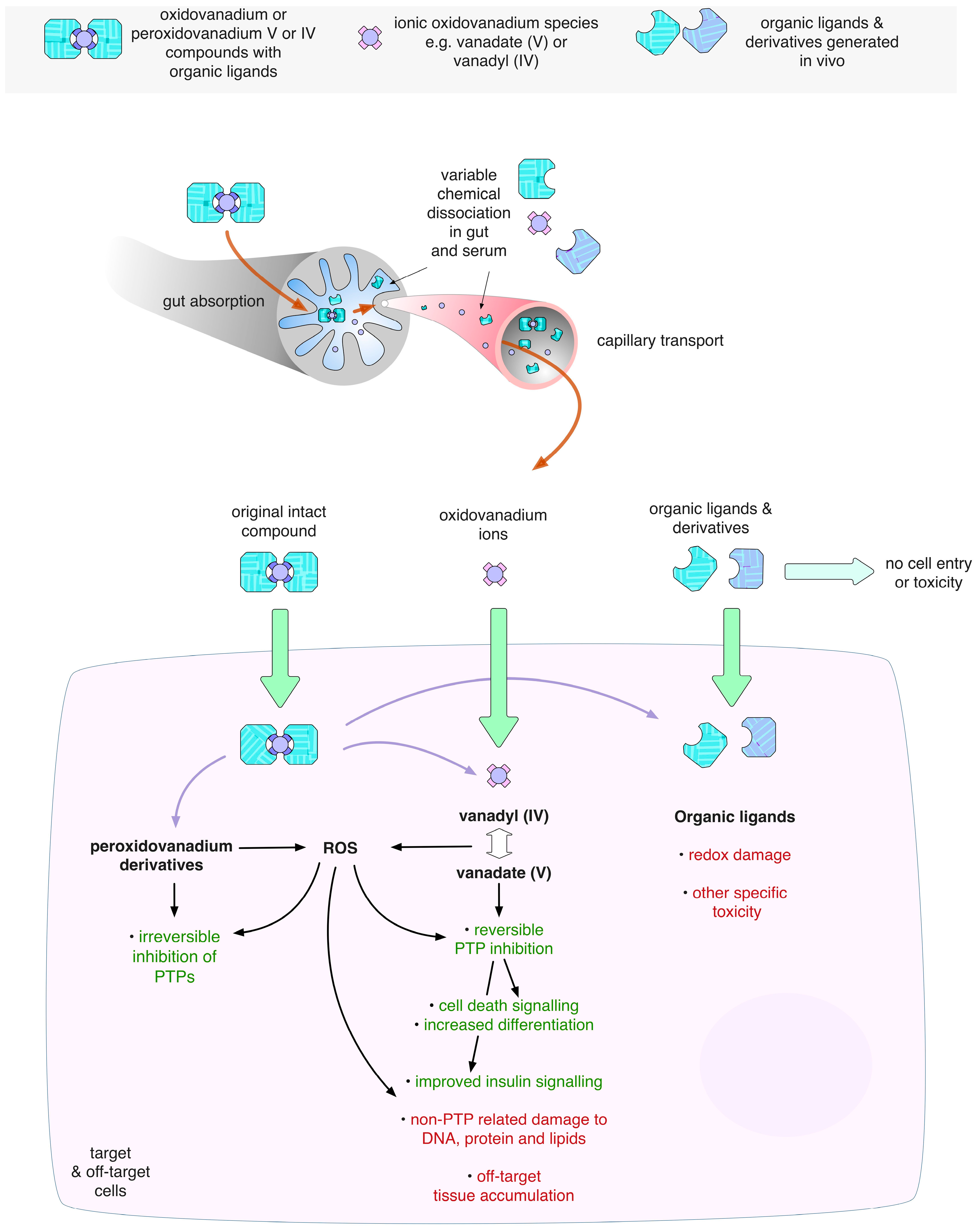
3.3. Ligand Toxicity
4. Vanadium in Diabetes
4.1. Insulin-Like Effects of Vanadium
4.2. Clinical Trials
5. Vanadium in Cancer
5.1. Phosphotyrosine Signalling in Cancer
5.2. Anti-Cancer Activity of Vanadium
5.3. Non-PTP Inhibition Mechanisms of Vanadium
5.4. Systemic Toxicities Associated with Vanadium
6. Future of Vanadium Research
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pessoa, J.C. Thirty years through vanadium chemistry. J. Inorg. Biochem. 2015, 147, 4–24. [Google Scholar] [CrossRef] [PubMed]
- Rehder, D. Perspectives for vanadium in health issues. Future Med. Chem. 2016, 8, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Levina, A.; Lay, P.A. Stabilities and biological activities of vanadium drugs: What is the nature of the active species? Chem. Asian J. 2017, 12, 1692–1699. [Google Scholar] [CrossRef] [PubMed]
- Aureliano, M. Decavanadate toxicology and pharmacological activities: V10 or V1, both or none? Oxid. Med. Cell. Longev. 2016, 2016, 6103457. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Guan, K.L.; Dixon, J.E. Evidence for protein-tyrosine-phosphatase catalysis proceeding via a cysteine-phosphate intermediate. J. Biol. Chem. 1991, 266, 17026–17030. [Google Scholar] [PubMed]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. Small-molecule kinase inhibitors: An analysis of fda-approved drugs. Drug Discov. Today 2016, 21, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Charbonneau, H.; Tonks, N.K.; Kumar, S.; Diltz, C.D.; Harrylock, M.; Cool, D.E.; Krebs, E.G.; Fischer, E.H.; Walsh, K.A. Human placenta protein-tyrosine-phosphatase: Amino acid sequence and relationship to a family of receptor-like proteins. Proc. Natl. Acad. Sci. USA 1989, 86, 5252–5256. [Google Scholar] [CrossRef] [PubMed]
- Guan, K.L.; Haun, R.S.; Watson, S.J.; Geahlen, R.L.; Dixon, J.E. Cloning and expression of a protein-tyrosine-phosphatase. Proc. Natl. Acad. Sci. USA 1990, 87, 1501–1505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y. Drugging the undruggable: Therapeutic potential of targeting protein tyrosine phosphatases. Acc. Chem. Res. 2017, 50, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J.; Jensen, M.R.; Gauss, C.M.; Page, R.; Blackledge, M.; et al. Targeting the disordered c terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.; Morrice, N.; Grant, L.; Le Sommer, S.; Lees, E.K.; Mody, N.; Wilson, H.M.; Delibegovic, M. Pharmacological inhibition of protein tyrosine phosphatase 1B protects against atherosclerotic plaque formation in the LDLR-/- mouse model of atherosclerosis. Clin. Sci. (Lond.) 2017, 131, 2489–2501. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.A. Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods Enzymol. 1991, 201, 477–482. [Google Scholar] [PubMed]
- Peters, K.G.; Davis, M.G.; Howard, B.W.; Pokross, M.; Rastogi, V.; Diven, C.; Greis, K.D.; Eby-Wilkens, E.; Maier, M.; Evdokimov, A.; et al. Mechanism of insulin sensitization by BMOV (bis maltolato oxo vanadium); unliganded vanadium (VO4) as the active component. J. Inorg. Biochem. 2003, 96, 321–330. [Google Scholar] [CrossRef]
- Rehder, D. The potentiality of vanadium in medicinal applications. Future Med Chem 2012, 4, 1823–1837. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, H.A.; Balassa, J.J.; Tipton, I.H. Abnormal trace metals in man—Vanadium. J. Chronic Dis. 1963, 16, 1047–1071. [Google Scholar] [CrossRef]
- Crans, D.C.; Smee, J.J.; Gaidamauskas, E.; Yang, L. The chemistry and biochemistry of vanadium and the biological activities exerted by vanadium compounds. Chem. Rev. 2004, 104, 849–902. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, A.M. Vanadium in cancer treatment. Crit. Rev. Oncol. Hematol. 2002, 42, 249–265. [Google Scholar] [CrossRef]
- Scrivens, P.J.; Alaoui-Jamali, M.A.; Giannini, G.; Wang, T.; Loignon, M.; Batist, G.; Sandor, V.A. Cdc25a-inhibitory properties and antineoplastic activity of bisperoxovanadium analogues. Mol. Cancer Ther. 2003, 2, 1053–1059. [Google Scholar] [PubMed]
- Nechay, B.R. Mechanisms of action of vanadium. Annu. Rev. Pharmacol. Toxicol. 1984, 24, 501–524. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Shimomura, S.; Fukuzawa, K.; Ishizu, K. Detection of oxovanadium (IV) and characterization of its ligand environment in subcellular fractions of the liver of rats treated with pentavalent vanadium(V). Biochem. Biophys. Res. Commun. 1980, 96, 293–298. [Google Scholar] [CrossRef]
- Ostman, A.; Frijhoff, J.; Sandin, A.; Böhmer, F.D. Regulation of protein tyrosine phosphatases by reversible oxidation. J. Biochem. 2011, 150, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Barford, D. The role of cysteine residues as redox-sensitive regulatory switches. Curr. Opin. Struct. Biol. 2004, 14, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Reytman, L.; Braitbard, O.; Hochman, J.; Tshuva, E.Y. Highly effective and hydrolytically stable vanadium(V) amino phenolato antitumor agents. Inorg. Chem. 2016, 55, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Tiago, T.; Martel, P.; Gutierrez-Merino, C.; Aureliano, M. Binding modes of decavanadate to myosin and inhibition of the actomyosin atpase activity. Biochim. Biophys. Acta 2007, 1774, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Fraqueza, G.; Batista de Carvalho, L.A.; Marques, M.P.; Maia, L.; Ohlin, C.A.; Casey, W.H.; Aureliano, M. Decavanadate, decaniobate, tungstate and molybdate interactions with sarcoplasmic reticulum Ca2+-atpase: Quercetin prevents cysteine oxidation by vanadate but does not reverse atpase inhibition. Dalton Trans. 2012, 41, 12749–12758. [Google Scholar] [CrossRef] [PubMed]
- Soares, S.S.; Gutierrez-Merino, C.; Aureliano, M. Decavanadate induces mitochondrial membrane depolarization and inhibits oxygen consumption. J. Inorg. Biochem. 2007, 101, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Soares, S.S.; Gutierrez-Merino, C.; Aureliano, M. Mitochondria as a target for decavanadate toxicity in sparus aurata heart. Aquat. Toxicol. 2007, 83, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Aureliano, M.; Gandara, R.M. Decavanadate effects in biological systems. J. Inorg. Biochem. 2005, 99, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Aureliano, M.; Crans, D.C. Decavanadate (V10O286−) and oxovanadates: Oxometalates with many biological activities. J. Inorg. Biochem. 2009, 103, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.H.; Orvig, C. Vanadium in diabetes: 100 years from phase 0 to phase I. J. Inorg. Biochem. 2006, 100, 1925–1935. [Google Scholar] [CrossRef] [PubMed]
- McNeill, J.H.; Yuen, V.G.; Hoveyda, H.R.; Orvig, C. Bis(maltolato)oxovanadium(IV) is a potent insulin mimic. J. Med. Chem. 1992, 35, 1489–1491. [Google Scholar] [CrossRef] [PubMed]
- Setyawati, I.A.; Thompson, K.H.; Yuen, V.G.; Sun, Y.; Battell, M.; Lyster, D.M.; Vo, C.; Ruth, T.J.; Zeisler, S.; McNeill, J.H.; et al. Kinetic analysis and comparison of uptake, distribution, and excretion of 48V-labeled compounds in rats. J. Appl. Physiol. (1985) 1998, 84, 569–575. [Google Scholar]
- D’Cruz, O.J.; Uckun, F.M. Metvan: A novel oxovanadium(IV) complex with broad spectrum anticancer activity. Expert Opin. Investig. Drugs 2002, 11, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Narla, R.K.; Chen, C.L.; Dong, Y.; Uckun, F.M. In vivo antitumor activity of bis(4,7-dimethyl-1,10-phenanthroline) sulfatooxovanadium(Iv) (METVAN [VO(SO4)(Me2-Phen)2]). Clin. Cancer Res. 2001, 7, 2124–2133. [Google Scholar] [PubMed]
- Dong, Y.; Narla, R.K.; Sudbeck, E.; Uckun, F.M. Synthesis, X-ray structure, and anti-leukemic activity of oxovanadium(IV) complexes. J. Inorg. Biochem. 2000, 78, 321–330. [Google Scholar] [CrossRef]
- Le, M.; Rathje, O.; Levina, A.; Lay, P.A. High cytotoxicity of vanadium(IV) complexes with 1,10-phenanthroline and related ligands is due to decomposition in cell culture medium. J. Biol. Inorg. Chem. 2017, 22, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Sanna, D.; Ugone, V.; Micera, G.; Buglyó, P.; Bíró, L.; Garribba, E. Speciation in human blood of metvan, a vanadium based potential anti-tumor drug. Dalton Trans. 2017, 46, 8950–8967. [Google Scholar] [CrossRef] [PubMed]
- Coyle, B.; Kinsella, P.; McCann, M.; Devereux, M.; O’Connor, R.; Clynes, M.; Kavanagh, K. Induction of apoptosis in yeast and mammalian cells by exposure to 1,10-phenanthroline metal complexes. Toxicol. In Vitro 2004, 18, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Crooks, P.A.; Wei, X.; de Leon, J. Toxicity of dipyridyl compounds and related compounds. Crit. Rev. Toxicol. 2004, 34, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Rozzo, C.; Sanna, D.; Garribba, E.; Serra, M.; Cantara, A.; Palmieri, G.; Pisano, M. Antitumoral effect of vanadium compounds in malignant melanoma cell lines. J. Inorg. Biochem. 2017, 174, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Banting, F.G.; Best, C.H.; Collip, J.B.; Campbell, W.R.; Fletcher, A.A. Pancreatic extracts in the treatment of diabetes mellitus. Can. Med. Assoc. J. 1922, 12, 141–146. [Google Scholar] [PubMed]
- Somerville, J.; Davies, B. Effect of vanadium on serum cholesterol. Am. Heart J. 1962, 64, 54–56. [Google Scholar] [CrossRef]
- Heyliger, C.E.; Tahiliani, A.G.; McNeill, J.H. Effect of vanadate on elevated blood glucose and depressed cardiac performance of diabetic rats. Science 1985, 227, 1474–1477. [Google Scholar] [CrossRef] [PubMed]
- Poucheret, P.; Verma, S.; Grynpas, M.D.; McNeill, J.H. Vanadium and diabetes. Mol. Cell. Biochem. 1998, 188, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Mehdi, M.Z. Insulino-mimetic and anti-diabetic effects of vanadium compounds. Diabet. Med. 2005, 22, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.M.; Pickering, R.M.; Lewith, G.T. A systematic review of vanadium oral supplements for glycaemic control in type 2 diabetes mellitus. QJM 2008, 101, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.H.; Lichter, J.; LeBel, C.; Scaife, M.C.; McNeill, J.H.; Orvig, C. Vanadium treatment of type 2 diabetes: A view to the future. J. Inorg. Biochem. 2009, 103, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Mehdi, M.Z.; Pandey, S.K.; Théberge, J.F.; Srivastava, A.K. Insulin signal mimicry as a mechanism for the insulin-like effects of vanadium. Cell Biochem. Biophys. 2006, 44, 73–81. [Google Scholar] [CrossRef]
- Goldfine, A.B.; Simonson, D.C.; Folli, F.; Patti, M.E.; Kahn, C.R. Metabolic effects of sodium metavanadate in humans with insulin-dependent and noninsulin-dependent diabetes mellitus in vivo and in vitro studies. J. Clin. Endocrinol. Metab. 1995, 80, 3311–3320. [Google Scholar] [PubMed]
- Goldfine, A.B.; Patti, M.E.; Zuberi, L.; Goldstein, B.J.; LeBlanc, R.; Landaker, E.J.; Jiang, Z.Y.; Willsky, G.R.; Kahn, C.R. Metabolic effects of vanadyl sulfate in humans with non-insulin-dependent diabetes mellitus: In vivo and in vitro studies. Metabolism 2000, 49, 400–410. [Google Scholar] [CrossRef]
- Cohen, N.; Halberstam, M.; Shlimovich, P.; Chang, C.J.; Shamoon, H.; Rossetti, L. Oral vanadyl sulfate improves hepatic and peripheral insulin sensitivity in patients with non-insulin-dependent diabetes mellitus. J. Clin. Investig. 1995, 95, 2501–2509. [Google Scholar] [CrossRef] [PubMed]
- Halberstam, M.; Cohen, N.; Shlimovich, P.; Rossetti, L.; Shamoon, H. Oral vanadyl sulfate improves insulin sensitivity in niddm but not in obese nondiabetic subjects. Diabetes 1996, 45, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Chen, X.; Ruiz, J.; van Rossum, G.D.; Turco, S. Effects of vanadyl sulfate on carbohydrate and lipid metabolism in patients with non-insulin-dependent diabetes mellitus. Metabolism 1996, 45, 1130–1135. [Google Scholar] [CrossRef]
- Cusi, K.; Cukier, S.; DeFronzo, R.A.; Torres, M.; Puchulu, F.M.; Redondo, J.C. Vanadyl sulfate improves hepatic and muscle insulin sensitivity in type 2 diabetes. J. Clin. Endocrinol. Metab. 2001, 86, 1410–1417. [Google Scholar] [CrossRef] [PubMed]
- Domingo, J.L. Vanadium and diabetes. What about vanadium toxicity? Mol. Cell. Biochem. 2000, 203, 185–187. [Google Scholar] [CrossRef] [PubMed]
- He, R.J.; Yu, Z.H.; Zhang, R.Y.; Zhang, Z.Y. Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol. Sin. 2014, 35, 1227–1246. [Google Scholar] [CrossRef] [PubMed]
- Hollander, M.C.; Blumenthal, G.M.; Dennis, P.A. Pten loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 2011, 11, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the pten tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- MacKeigan, J.P.; Murphy, L.O.; Blenis, J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat. Cell Biol. 2005, 7, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Coad, J.; Ducatman, B.; Agazie, Y.M. Shp2 is up-regulated in breast cancer cells and in infiltrating ductal carcinoma of the breast, implying its involvement in breast oncogenesis. Histopathology 2008, 53, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Yu, Y.; Zheng, S.; Zhao, X.; Dong, Q.; He, Z.; Liang, Y.; Lu, Q.; Fang, Y.; Gan, X.; et al. Overexpression of Shp2 tyrosine phosphatase is implicated in leukemogenesis in adult human leukemia. Blood 2005, 106, 3142–3149. [Google Scholar] [CrossRef] [PubMed]
- Bentires-Alj, M.; Paez, J.G.; David, F.S.; Keilhack, H.; Halmos, B.; Naoki, K.; Maris, J.M.; Richardson, A.; Bardelli, A.; Sugarbaker, D.J.; et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004, 64, 8816–8820. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.D.; Agazie, Y.M. Inhibition of SHP2 leads to mesenchymal to epithelial transition in breast cancer cells. Cell Death Differ. 2008, 15, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.G.; Dubé, N.; Read, M.; Penney, J.; Paquet, M.; Han, Y.; Kennedy, B.P.; Muller, W.J.; Tremblay, M.L. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat. Genet. 2007, 39, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.G.; Dubé, N.; Hardy, S.; Tremblay, M.L. Inside the human cancer tyrosine phosphatome. Nat. Rev. Cancer 2011, 11, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Elson, A. Stepping out of the shadows: Oncogenic and tumor-promoting protein tyrosine phosphatases. Int. J. Biochem. Cell Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kieler, J.; Gromek, A.; Nissen, N.I. Studies on the antineoplastic effect of vanadium salts. Acta Chir. Scand. Suppl. 1965, 343, 154–164. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.S.; Kuriakose, M.; Somasundaram, V.; Shenoi, V.; Kurup, M.R.; Srinivas, P. The molecular response of vanadium complexes of nicotinoyl hydrazone in cervical cancers—A possible interference with hpv oncogenic markers. Life Sci. 2014, 116, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ma, Y.; Xu, Z.; Wang, D.; Zhao, B.; Pan, H.; Wang, J.; Xu, D.; Zhao, X.; Pan, S.; et al. Sodium orthovanadate inhibits growth of human hepatocellular carcinoma cells in vitro and in an orthotopic model in vivo. Cancer Lett. 2014, 351, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Leon, I.E.; Di Virgilio, A.L.; Porro, V.; Muglia, C.I.; Naso, L.G.; Williams, P.A.; Bollati-Fogolin, M.; Etcheverry, S.B. Antitumor properties of a vanadyl(IV) complex with the flavonoid chrysin [VO(chrysin)2EtOH]2 in a human osteosarcoma model: The role of oxidative stress and apoptosis. Dalton Trans. 2013, 42, 11868–11880. [Google Scholar] [CrossRef] [PubMed]
- Leon, I.E.; Porro, V.; Di Virgilio, A.L.; Naso, L.G.; Williams, P.A.; Bollati-Fogolín, M.; Etcheverry, S.B. Antiproliferative and apoptosis-inducing activity of an oxidovanadium(IV) complex with the flavonoid silibinin against osteosarcoma cells. J. Biol. Inorg. Chem. 2014, 19, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Molinuevo, M.S.; Cortizo, A.M.; Etcheverry, S.B. Vanadium(IV) complexes inhibit adhesion, migration and colony formation of UMR106 osteosarcoma cells. Cancer Chemother. Pharmacol. 2008, 61, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Strianese, M.; Basile, A.; Mazzone, A.; Morello, S.; Turco, M.C.; Pellecchia, C. Therapeutic potential of a pyridoxal-based vanadium(IV) complex showing selective cytotoxicity for cancer versus healthy cells. J. Cell. Physiol. 2013, 228, 2202–2209. [Google Scholar] [CrossRef] [PubMed]
- Petanidis, S.; Kioseoglou, E.; Domvri, K.; Zarogoulidis, P.; Carthy, J.M.; Anestakis, D.; Moustakas, A.; Salifoglou, A. In vitro and ex vivo vanadium antitumor activity in (TGF-β)-induced emt. Synergistic activity with carboplatin and correlation with tumor metastasis in cancer patients. Int. J. Biochem. Cell Biol. 2016, 74, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Petanidis, S.; Kioseoglou, E.; Hadzopoulou-Cladaras, M.; Salifoglou, A. Novel ternary vanadium-betaine-peroxido species suppresses H-ras and matrix metalloproteinase-2 expression by increasing reactive oxygen species-mediated apoptosis in cancer cells. Cancer Lett. 2013, 335, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.S.; Ghosh, B.; Rana, A.; Chatterjee, M. Suppression of cell proliferation, induction of apoptosis and cell cycle arrest: Chemopreventive activity of vanadium in vivo and in vitro. Int. J. Cancer 2007, 120, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Navara, C.S.; Benyumov, A.; Vassilev, A.; Narla, R.K.; Ghosh, P.; Uckun, F.M. Vanadocenes as potent anti-proliferative agents disrupting mitotic spindle formation in cancer cells. Anticancer Drugs 2001, 12, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Ajeawung, N.F.; Faure, R.; Jones, C.; Kamnasaran, D. Preclinical evaluation of dipotassium bisperoxo (picolinato) oxovanadate v for the treatment of pediatric low-grade gliomas. Future Oncol. 2013, 9, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Dąbroś, W.; Adamczyk, A.; Ciurkot, K.; Kordowiak, A.M. Vanadium compounds affect growth and morphology of human rhabdomyosarcoma cell line. Pol. J. Pathol. 2011, 62, 262–268. [Google Scholar] [PubMed]
- Meshkini, A.; Yazdanparast, R. Chemosensitization of human leukemia K562 cells to taxol by a vanadium-salen complex. Exp. Mol. Pathol. 2010, 89, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Clark, O.; Daga, S.; Stoker, A.W. Tyrosine phosphatase inhibitors combined with retinoic acid can enhance differentiation of neuroblastoma cells and trigger ERK- and AKT-dependent, p53-independent senescence. Cancer Lett. 2013, 328, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Clark, O.; Park, I.; Di Florio, A.; Cichon, A.C.; Rustin, S.; Jugov, R.; Maeshima, R.; Stoker, A.W. Oxovanadium-based inhibitors can drive redox-sensitive cytotoxicity in neuroblastoma cells and synergise strongly with buthionine sulfoximine. Cancer Lett. 2015, 357, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; D’Cruz, O.J.; Narla, R.K.; Uckun, F.M. Apoptosis-inducing vanadocene compounds against human testicular cancer. Clin. Cancer Res. 2000, 6, 1536–1545. [Google Scholar] [PubMed]
- Liu, T.T.; Liu, Y.J.; Wang, Q.; Yang, X.G.; Wang, K. Reactive-oxygen-species-mediated Cdc25C degradation results in differential antiproliferative activities of vanadate, tungstate, and molybdate in the PC-3 human prostate cancer cell line. J. Biol. Inorg. Chem. 2012, 17, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Scalese, G.; Mosquillo, M.F.; Rostán, S.; Castiglioni, J.; Alho, I.; Pérez, L.; Correia, I.; Marques, F.; Costa Pessoa, J.; Gambino, D. Heteroleptic oxidovanadium(IV) complexes of 2-hydroxynaphtylaldimine and polypyridyl ligands against trypanosoma cruzi and prostate cancer cells. J. Inorg. Biochem. 2017, 175, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, S.; Hać, S.; Wyrzykowski, D.; Zauszkiewicz-Pawlak, A.; Inkielewicz-Stępniak, I. Selective cytotoxicity of vanadium complexes on human pancreatic ductal adenocarcinoma cell line by inducing necroptosis, apoptosis and mitotic catastrophe process. Oncotarget 2017, 8, 60324–60341. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.X.; Hong, Y.H.; Yang, X.G. Bis(acetylacetonato)-oxidovanadium(IV) and sodium metavanadate inhibit cell proliferation via ROS-induced sustained MAPK/ERK activation but with elevated AKT activity in human pancreatic cancer AsPC-1 cells. J. Biol. Inorg. Chem. 2016, 21, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Banerjee, K.; Banerjee, A.; Sarkar, A.; Ahir, M.; Adhikary, A.; Chatterjee, M.; Choudhuri, S.K. Induction of apoptosis in human colorectal cancer cell line, HCT-116 by a vanadium- schiff base complex. Biomed. Pharmacother. 2017, 92, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, T.; Swamy, A.H.; Chatterjee, A.; Rana, B.; Shyamsundar, A.; Chatterjee, M. Molecular basis of vanadium-mediated inhibition of hepatocellular preneoplasia during experimental hepatocarcinogenesis in rats. J. Cell. Biochem. 2007, 101, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, T.; Chatterjee, A.; Dhachinamoorthi, D.; Srivastawa, S.; Panayappan, L.; Chatterjee, M. Vanadium limits the expression of proliferating cell nuclear antigen and inhibits early DNA damage during diethylnitrosamine-induced hepatocellular preneoplasia in rats. Environ. Mol. Mutagen. 2006, 47, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, T.; Chatterjee, A.; Rana, A.; Dhachinamoorthi, D.; Kumar, P.A.; Chatterjee, M. Carcinogen-induced early molecular events and its implication in the initiation of chemical hepatocarcinogenesis in rats: Chemopreventive role of vanadium on this process. Biochim. Biophys. Acta 2007, 1772, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, T.; Chatterjee, A.; Saralaya, M.G.; Chatterjee, M. Chemopreventive effect of vanadium in a rodent model of chemical hepatocarcinogenesis: Reflections in oxidative DNA damage, energy-dispersive X-ray fluorescence profile and metallothionein expression. J. Biol. Inorg. Chem. 2006, 11, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, T.; Pandey, N.; Chatterjee, A.; Ghosh, B.; Rana, B.; Chatterjee, M. Molecular basis of anticlastogenic potential of vanadium in vivo during the early stages of diethylnitrosamine-induced hepatocarcinogenesis in rats. Mutat. Res. 2006, 609, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A.; Roy, S.; Chatterjee, M. Characterization of selective induction and alteration of xenobiotic biotransforming enzymes by vanadium during diethylnitrosamine-induced chemical rat liver carcinogenesis. Oncol. Res. 1999, 11, 41–53. [Google Scholar] [PubMed]
- Chakraborty, T.; Chatterjee, A.; Rana, A.; Rana, B.; Palanisamy, A.; Madhappan, R.; Chatterjee, M. Suppression of early stages of neoplastic transformation in a two-stage chemical hepatocarcinogenesis model: Supplementation of vanadium, a dietary micronutrient, limits cell proliferation and inhibits the formations of 8-hydroxy-2′-deoxyguanosines and DNA strand-breaks in the liver of sprague-dawley rats. Nutr. Cancer 2007, 59, 228–247. [Google Scholar] [PubMed]
- Chakraborty, T.; Ghosh, S.; Datta, S.; Chakraborty, P.; Chatterjee, M. Vanadium suppresses sister-chromatid exchange and DNA-protein crosslink formation and restores antioxidant status and hepatocellular architecture during 2-acetylaminofluorene-induced experimental rat hepatocarcinogenesis. J. Exp. Ther. Oncol. 2003, 3, 346–362. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, T.; Samanta, S.; Ghosh, B.; Thirumoorthy, N.; Chatterjee, M. Vanadium induces apoptosis and modulates the expressions of metallothionein, Ki-67 nuclear antigen, and p53 during 2-acetylaminofluorene-induced rat liver preneoplasia. J. Cell. Biochem. 2005, 94, 744–762. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Chatterjee, M.; Ghosh, B.; Rajkumar, M.; Rana, A. Vanadium and 1, 25 (OH)2 vitamin D 3 combination in inhibitions of 1,2, dimethylhydrazine-induced rat colon carcinogenesis. Biochim. Biophys. Acta 2008, 1780, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Swamy, V.; Suresh, D.; Rajkumar, M.; Rana, B.; Rana, A.; Chatterjee, M. Protective effects of vanadium against dmh-induced genotoxicity and carcinogenesis in rat colon: Removal of O6-methylguanine DNA adducts, p53 expression, inducible nitric oxide synthase downregulation and apoptotic induction. Mutat. Res. 2008, 650, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Kanna, P.S.; Mahendrakumar, C.B.; Indira, B.N.; Srivastawa, S.; Kalaiselvi, K.; Elayaraja, T.; Chatterjee, M. Chemopreventive effects of vanadium toward 1,2-dimethylhydrazine-induced genotoxicity and preneoplastic lesions in rat colon. Environ. Mol. Mutagen. 2004, 44, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.J.; Chasteen, N.D.; Meeker, L.D. Dietary vanadyl(IV) sulfate inhibits chemically-induced mammary carcinogenesis. Carcinogenesis 1984, 5, 849–851. [Google Scholar] [CrossRef] [PubMed]
- Sankar Ray, R.; Roy, S.; Ghosh, S.; Kumar, M.; Chatterjee, M. Suppression of cell proliferation, DNA protein cross-links, and induction of apoptosis by vanadium in chemical rat mammary carcinogenesis. Biochim. Biophys. Acta 2004, 1675, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Sankar Ray, R.; Roy, S.; Samanta, S.; Maitra, D.; Chatterjee, M. Protective role of vanadium on the early process of rat mammary carcinogenesis by influencing expression of metallothionein, ggt-positive foci and DNA fragmentation. Cell Biochem. Funct. 2005, 23, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.S.; Basu, M.; Ghosh, B.; Samanta, K.; Chatterjee, M. Vanadium, a versatile biochemical effector in chemical rat mammary carcinogenesis. Nutr. Cancer 2005, 51, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Manna, S.; Das, S.; Chatterjee, M.; Janarthan, M. Combined supplementation of vanadium and fish oil suppresses tumor growth, cell proliferation and induces apoptosis in dmba-induced rat mammary carcinogenesis. J. Cell. Biochem. 2011, 112, 2327–2339. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A.; Oinam, S.; Basu, M.; Chatterjee, M. Vanadium chemoprevention of 7,12-dimethylbenz(a)anthracene-induced rat mammary carcinogenesis: Probable involvement of representative hepatic phase i and ii xenobiotic metabolizing enzymes. Breast Cancer Res. Treat. 2000, 63, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Köpf-Maier, P.; Wagner, W.; Hesse, B.; Köpf, H. Tumor inhibition by metallocenes: Activity against leukemias and detection of the systemic effect. Eur. J. Cancer 1981, 17, 665–669. [Google Scholar] [CrossRef]
- Bishayee, A.; Waghray, A.; Patel, M.A.; Chatterjee, M. Vanadium in the detection, prevention and treatment of cancer: The in vivo evidence. Cancer Lett. 2010, 294, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhai, F.; Wang, X.; Li, D.; Zhang, H.; Li, R.; Song, L. Synthesis and biological evaluation of decavanadate na4co(h2o)6v10o28.18h2o. Biomed. Pharmacother. 2009, 63, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Galani, A.; Tsitsias, V.; Stellas, D.; Psycharis, V.; Raptopoulou, C.P.; Karaliota, A. Two novel compounds of vanadium and molybdenum with carnitine exhibiting potential pharmacological use. J. Inorg. Biochem. 2015, 142, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C.; Josephson, L.; Warner, R.; Yanagisawa, M.; Lechene, C.; Guidotti, G. Vanadate is a potent (Na,K)-ATPase inhibitor found in ATP derived from muscle. J. Biol. Chem. 1977, 252, 7421–7423. [Google Scholar] [PubMed]
- Collauto, A.; Mishra, S.; Litvinov, A.; Mchaourab, H.S.; Goldfarb, D. Direct spectroscopic detection of atp turnover reveals mechanistic divergence of abc exporters. Structure 2017, 25, 1264–1274.e1263. [Google Scholar] [CrossRef] [PubMed]
- Willsky, G.R.; White, D.A.; McCabe, B.C. Metabolism of added orthovanadate to vanadyl and high-molecular-weight vanadates by saccharomyces cerevisiae. J. Biol. Chem. 1984, 259, 13273–13281. [Google Scholar] [PubMed]
- Harding, M.M.; Mokdsi, G. Antitumour metallocenes: Structure-activity studies and interactions with biomolecules. Curr. Med. Chem. 2000, 7, 1289–1303. [Google Scholar] [CrossRef] [PubMed]
- Köpf-Maier, P.; Wagner, W.; Liss, E. Induction of cell arrest at G1/S and in G2 after treatment of ehrlich ascites tumor cells with metallocene dichlorides and cis-platinum in vitro. J. Cancer Res. Clin. Oncol. 1983, 106, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Rehder, D. Vanadium. Its role for humans. Met. Ions Life Sci 2013, 13, 139–169. [Google Scholar] [PubMed]
- Korbecki, J.; Baranowska-Bosiacka, I.; Gutowska, I.; Chlubek, D. Vanadium compounds as pro-inflammatory agents: Effects on cyclooxygenases. Int. J. Mol. Sci. 2015, 16, 12648–12668. [Google Scholar] [CrossRef] [PubMed]
- Ress, N.B.; Chou, B.J.; Renne, R.A.; Dill, J.A.; Miller, R.A.; Roycroft, J.H.; Hailey, J.R.; Haseman, J.K.; Bucher, J.R. Carcinogenicity of inhaled vanadium pentoxide in F344/N rats and B6C3F1 mice. Toxicol. Sci. 2003, 74, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Boulassel, B.; Sadeg, N.; Roussel, O.; Perrin, M.; Belhadj-Tahar, H. Fatal poisoning by vanadium. Forensic Sci. Int. 2011, 206, e79–e81. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jie, X.; Guo, Y.; Zhang, X.; Wang, J.; Xue, C. Green synthesis of oxovanadium(IV)/chitosan nanocomposites and its ameliorative effect on hyperglycemia, insulin resistance, and oxidative stress. Biol. Trace Elem. Res. 2016, 169, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Lichawska, M.E.; Bodek, K.H.; Jezierska, J.; Kufelnicki, A. Coordinative interaction of microcrystalline chitosan with oxovanadium (IV) ions in aqueous solution. Chem. Cent. J. 2014, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Kremer, L.E.; McLeod, A.I.; Aitken, J.B.; Levina, A.; Lay, P.A. Vanadium(V) and -(IV) complexes of anionic polysaccharides: Controlled release pharmaceutical formulations and models of vanadium biotransformation products. J. Inorg. Biochem. 2015, 147, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cheng, L.; Dong, Z.; Chao, Y.; Lei, H.; Zhao, H.; Wang, J.; Liu, Z. Degradable vanadium disulfide nanostructures with unique optical and magnetic functions for cancer theranostics. Angew. Chem. Int. Ed. Engl. 2017, 56, 12991–12996. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Torchilin, V.P. Targeted pharmaceutical nanocarriers for cancer therapy and imaging. AAPS J. 2007, 9, E128–E147. [Google Scholar] [CrossRef] [PubMed]
- Ivankovic, S.; Music, S.; Gotic, M.; Ljubesic, N. Cytotoxicity of nanosize V2O5 particles to selected fibroblast and tumor cells. Toxicol. In Vitro 2006, 20, 286–294. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Tumour Cell Type | Compound | Effect | Suggested Mechanism |
|---|---|---|---|
| Cervical [71] | Nicotinoyl hydrazine vanadium complexes (50–100 μM) | Increased apoptosis | p53 induction |
| Hepatocellular carcinoma (HCC) [72] | Sodium orthovanadate (15–30 μM) | Decreased Proliferation G2/M arrest Increased Apoptosis | |
| Osteosarcoma [73,74] | Oxidovanadium flavonoid complexes (10–100 μM) | Increased apoptosis DNA damage Cell cycle arrest | ROS production DNA strand breaks |
| Osteosarcoma [75] | Vanadium (IV) complexes (2.5–5 μM) | Reduced cell adhesion and migration Reduced colony formation | Reduced actin polymerisation via suppressed PKA activity |
| Malignant melanoma [42] | Pyridinone ligated oxidovanadium complexes (1–100 μM) | Reduced proliferation Increased apoptosis Cell cycle arrest | |
| Lung and Melanoma [76] | Pyridoxylideneiminato vanadium (50 μM) | Increased apoptosis | ROS production |
| Lung and breast [77] | Vanadium (V)-peroxido-betaine (25–50 μM) | Reduced migration Increased cell death | Reduced TGFβ mediated EMT |
| Lung and Breast [78] | Vanadium-peroxido-betaine (100–400 μM) | Increased apoptosis DNA damage | ROS production Reduced HRAS and MMP2 expression |
| Breast [79] | Ammonium monovanadate (100–250 μM) | Apoptosis and cell cycle arrest | |
| Breast [80] | Vanadocene dichloride (10–20 μM) | Reduced proliferation G2/M arrest | |
| Glioma [81] | Picolinato-bis(peroxido)oxidovanadate (V) (Bpv(pic)) (5–20 μM) | Reduced proliferation S phase and G2/M accumulation Increased apoptosis Reduced migration and invasion | Inhibition of PTP expression and activity |
| Rhabdomyosarcoma [82] | BMOV and vanadium salts (10–40 μM) | Growth inhibition | |
| CML [83] | VO-salen (6–32 μM) | Reduced proliferation G2/M arrest Chemosensitisation to taxol | |
| Neuroblastoma [84,85] | BMOV (10 μM) | Cytotoxicity Differentiation | PTP inhibition |
| Testicular [86] | Vanadocene dichloride (100 μM) | Apoptosis | |
| Prostate [87] | Vanadate (25–100 μM) | G2/M arrest Growth inhibition | ROS mediated CDC25C degradation |
| Ovarian and Prostate [88] | Heteroleptic Schiff base vanadium complexes (1–150 μM) | Cytotoxicity | Disrupted mitotic spindle formation |
| Pancreas [89] | Phenanthroline/quinolone ligated vanadium (1–100 μM) | Increased apoptosis and necroptosis G2/M arrest | ROS production |
| Pancreas [90] | Bis(acetylacetonato)-oxidovanadium (IV) (1–400 μM) | Reduced proliferation G2/M arrest | ROS production ERK pathway activation |
| Colorectal [91] | Schiff base vanadium complex (20 μg/mL) | Increased apoptosis G2/M arrest | GSH depletion ROS production DNA damage |
| Model | Compound | Effect | Suggested Mechanism |
|---|---|---|---|
| DEN rat liver model [92,93,94,95,96,97] | Ammonium metavanadate (0.5 ppm/4.27 μM in drinking water) | Chemopreventative—reduced proliferation and premalignant nodule incidence | Reduced DNA damage Increased expression of drug metabolising enzymes |
| 2-AAF rat liver model [98,99,100] | Ammonium monovanadate/ammonium metavanadate (0.5 ppm/4.27 μM in drinking water) | Chemopreventative—reduced tumour incidence, reduced proliferation and increased apoptosis | Reduced DNA damage Increased expression of drug metabolising enzymes Induction of p53 |
| Orthotopic Hepatocellular carcinoma mouse model [72] | Sodium orthovanadate (10–20 mg/kg) | Reduced cell proliferation and tumour volume | |
| DMH rat colon model [101,102] | Vanadium (0.5 ppm/4.27 μM in drinking water) | Chemopreventative—reduced proliferation, increased apoptosis | Reduced DNA damage Induction of p53 |
| DMH rat colon model [103] | Ammonium monovanadate (0.5 ppm/4.27 μM in drinking water) | Chemopreventative—reduced tumour incidence | Reduced DNA damage |
| MNU rat mammary model [104] | Vanadyl sulphate (25 ppm in feed) | Chemopreventative—reduced tumour incidence and increase survival | |
| DMBA rat mammary model [79,105,106,107] | Ammonium monovanadate/ammonium metavanadate (0.5 ppm/4.27 μM in drinking water) | Chemopreventative—reduced tumour incidence and size. Reduced proliferation and increased apoptosis | Reduced DNA damage Induction of p53 |
| DMBA rat mammary model [108] | Ammonium monovanadate (0.5 ppm/4.27 μM in drinking water) | Chemopreventative—reduced tumour incidence, reduced proliferation and increased apoptosis | Reduced DNA damage Induction of p53 |
| DMBA rat mammary model [109] | Ammonium monovanadate (0.5 ppm/4.27 μM in drinking water) | Chemopreventative—reduced tumour incidence | Increased expression of drug metabolising enzymes |
| MDA-MB-231 mouse breast cancer xenograft model [35,36] | Metvan (10 mg/kg intraperitoneal) | Reduced tumour progression and increased apoptosis | Induction of oxidative damage |
| DA3 mouse breast cancer xenograft model [20] | Bisperoxidovanadium compounds (20 mg/kg intraperitoneal) | Reduced tumour growth | CDC25A inhibition leading to cell cycle arrest and apoptosis |
| U87 mouse glioblastoma xenograft model [35,36] | Metvan (10 mg/kg intraperitoneal) | Reduced tumour progression and increased apoptosis | Induction of oxidative damage |
| L1210 injected mice (leukemia) [110] | Vanadocene dichloride (10–130 mg/kg) | Increased life span |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irving, E.; Stoker, A.W. Vanadium Compounds as PTP Inhibitors. Molecules 2017, 22, 2269. https://doi.org/10.3390/molecules22122269
Irving E, Stoker AW. Vanadium Compounds as PTP Inhibitors. Molecules. 2017; 22(12):2269. https://doi.org/10.3390/molecules22122269
Chicago/Turabian StyleIrving, Elsa, and Andrew W. Stoker. 2017. "Vanadium Compounds as PTP Inhibitors" Molecules 22, no. 12: 2269. https://doi.org/10.3390/molecules22122269
APA StyleIrving, E., & Stoker, A. W. (2017). Vanadium Compounds as PTP Inhibitors. Molecules, 22(12), 2269. https://doi.org/10.3390/molecules22122269