Rational Engineering of a Flavoprotein Oxidase for Improved Direct Oxidation of Alcohols to Carboxylic Acids

 ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Bacterial Cultivation and Enzyme Production
3.2. Site-Directed Mutagenesis
3.3. Biotransformation
3.4. Aldehyde Hydration
3.5. Kinetics
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pickl, M.; Fuchs, M.; Glueck, S.M.; Faber, K. The substrate tolerance of alcohol oxidases. Appl. Microbiol. Biotechnol. 2015, 99, 6617–6642. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, F.; Arends, I.W.C.E.; Buehler, K.; Schallmey, A.; Bühler, B. Enzyme-mediated oxidations for the chemist. Green Chem. 2011, 13, 226–265. [Google Scholar] [CrossRef]
- Holec, C.; Neufeld, K.; Pietruszka, J. P450 BM3 Monooxygenase as an efficient NAD(P)H-oxidase for regeneration of nicotinamide cofactors in ADH-Catalysed PreparativeScale Biotransformations. Adv. Synth. Catal. 2016, 358, 1810–1819. [Google Scholar] [CrossRef]
- Dijkman, W.P.; Fraaije, M.W. Discovery and characterization of a 5-hydroxymethylfurfural oxidase from Methylovorus sp. strain MP688. Appl. Environ. Microbiol. 2014, 80, 1082–1890. [Google Scholar] [CrossRef] [PubMed]
- Dijkman, W.P.; Groothuis, D.E.; Fraaije, M.W. Enzyme-catalyzed oxidation of 5-hydroxymethylfurfural to furan-2,5-dicarboxylic acid. Angew. Chem. Int. Ed. 2014, 53, 6515–6518. [Google Scholar] [CrossRef] [PubMed]
- Werpy, T.; Petersen, G. Top Value Added Chemicals from Biomass; Results of Screening for Potential Candidates from Sugars and Synthesis Gas; U.S. Department of Energy: Washington, DC, USA, 2004; Volume 1.
- Ewing, T.A.; Dijkman, W.P.; Vervoort, J.M.; Fraaije, M.W.; Van Berkel, W.J.H. The oxidation of thiols by favoprotein oxidases: A biocatalytic route to reactive thiocarbonyls. Angew. Chem. Int. Ed. 2014, 53, 13206–13209. [Google Scholar] [CrossRef] [PubMed]
- Dijkman, W.P.; Binda, C.; Fraaije, M.W.; Mattevi, A. Structure-based enzyme tailoring of 5-hydroxymethylfurfural oxidase. ACS Catal. 2015, 5, 1833–1839. [Google Scholar] [CrossRef]
- Hernández-Ortega, A.; Ferreira, P.; Martínez, A.T. Fungal aryl-alcohol oxidase: A peroxide-producing flavoenzyme involved in lignin degradation. Appl. Microbiol. Biotechnol. 2012, 93, 1395–1410. [Google Scholar] [CrossRef] [PubMed]
- Guillén, F.; Martinez, A.T.; Martinez, M.J. Substrate specificity and properties of the aryl-alcohol oxidase from the ligninolytic fungus Pleurotus eryngii. Eur. J. Biochem. 1992, 209, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.; Hernández-Ortega, A.; Herguedas, B.; Rencoret, J.; Gutiérrez, A.; Martínez, M.J.; Jiménez-Barbero, J.; Medina, M.; Martínez, A.T. Kinetic and chemical characterization of aldehyde oxidation by fungal aryl-alcohol oxidase. Biochem. J. 2010, 425, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Bulter, T.; Alcalde, M.; Petrounia, I.P.; Arnold, F.H. Modification of galactose oxidase to introduce glucose 6-oxidase activity. ChemBioChem 2002, 3, 781–783. [Google Scholar] [CrossRef]
- Siebum, A.; van Wijk, A.; Schoevaart, R.; Kieboom, T. Galactose oxidase and alcohol oxidase: Scope and limitations for the enzymatic synthesis of aldehydes. J. Mol. Catal. B Enzym. 2006, 41, 141–145. [Google Scholar] [CrossRef]
- Whittaker, J.W. Free radical catalysis by galactose oxidase. Chem. Rev. 2003, 103, 2347–2363. [Google Scholar] [CrossRef]
- Bechi, B.; Herter, S.; McKenna, S.; Riley, C.; Leimkühler, S.; Turner, N.J.; Carnell, A.J. Catalytic bio-chemo and bio-bio tandem oxidation reactions for amide and carboxylic acid synthesis. Green Chem. 2014, 16, 4524–4529. [Google Scholar] [CrossRef]
- Van Hellemond, E.W.; Vermote, L.; Koolen, W.; Sonke, T.; Zandvoort, E.; Heuts, D.P.H.M.; Janssen, D.B.; Fraaije, M.W. Exploring the biocatalytic scope of alditol oxidase from Streptomyces coelicolor. Adv. Synth. Catal. 2009, 351, 1523–1530. [Google Scholar] [CrossRef]
- Fan, F.; Gadda, G. On the catalytic mechanism of choline oxidase. J. Am. Chem. Soc. 2005, 127, 2067–2074. [Google Scholar] [CrossRef] [PubMed]
- Fersht, A.R.; Shi, J.-P.; Knill-Jones, J.; Lowe, D.M.; Wilkinson, A.J.; Blow, D.M.; Brick, P.; Carter, P.; Waye, M.M.Y.; Winter, G. Hydrogen bonding and biological specificity analysed by protein engineering. Nature 1985, 314, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, A.R.G.; Sivaraman, J.; Li, Y.; Larocque, R.; Matte, A.; Schrag, J.D.; Cygler, M. Mechanism of action and NAD+-binding mode revealed by the crystal structure of l-histidinol dehydrogenase. Proc. Natl. Acad. Sci. USA 2002, 99, 1859–1864. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Ortega, A.; Ferreira, P.; Merino, P.; Medina, M.; Guallar, V.; Martínez, A.T. Stereoselective hydride transfer by aryl-alcohol oxidase, a member of the GMC superfamily. ChemBioChem 2012, 13, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Kunjapur, A.M.; Tarasova, Y.; Prather, K.L.J. Synthesis and accumulation of aromatic aldehydes in an engineered strain of Escherichia coli. J. Am. Chem. Soc. 2014, 136, 11644–11654. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, G.M.; Atsumi, S. Toward aldehyde and alkane production by removing aldehyde reductase activity in Escherichia coli. Metab. Eng. 2014, 25, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Mittelstädt, G.; Iobbi-Nivol, C.; Saggu, M.; Lendzian, F.; Hildebrandt, P.; Leimkühler, S. A periplasmic aldehyde oxidoreductase represents the first molybdopterin cytosine dinucleotide cofactor containing molybdo-flavoenzyme from Escherichia coli. FEBS J. 2009, 276, 2762–2774. [Google Scholar] [CrossRef] [PubMed]
- McClelland, R.A.; Coe, M. Structure-reactivity effects in the hydration of benzaldehydes. J. Am. Chem. Soc. 1983, 105, 2718–2725. [Google Scholar] [CrossRef]
- Velonia, K.; Smonou, I. Dismutation of aldehydes catalyzed by alcohol dehydrogenases. J. Chem. Soc. Perkin Trans. 2000, 1, 2283–2287. [Google Scholar] [CrossRef]
- Rungsrisuriyachai, K.; Gadda, G. On the role of histidine 351 in the Reaction of alcohol oxidation catalyzed by choline oxidase. Biochemistry 2008, 47, 6762–6769. [Google Scholar] [CrossRef] [PubMed]
- Heuts, D.P.H.M.; van Hellemond, E.W.; Janssen, D.B.; Fraaije, M.W. Discovery, characterization, and kinetic analysis of an alditol oxidase from Streptomyces coelicolor. J. Biol. Chem. 2007, 282, 20283–20291. [Google Scholar] [CrossRef] [PubMed]
- Bayer, T.; Milker, S.; Wiesinger, T.; Winkler, M.; Mihovilovic, M.D.; Rudroff, F. In vivo synthesis of polyhydroxylated compounds from a ‘hidden reservoir’ of toxic aldehyde species. ChemCatChem 2017, 9, 1–6. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1a–5a and 1c–5c are available from the authors. |
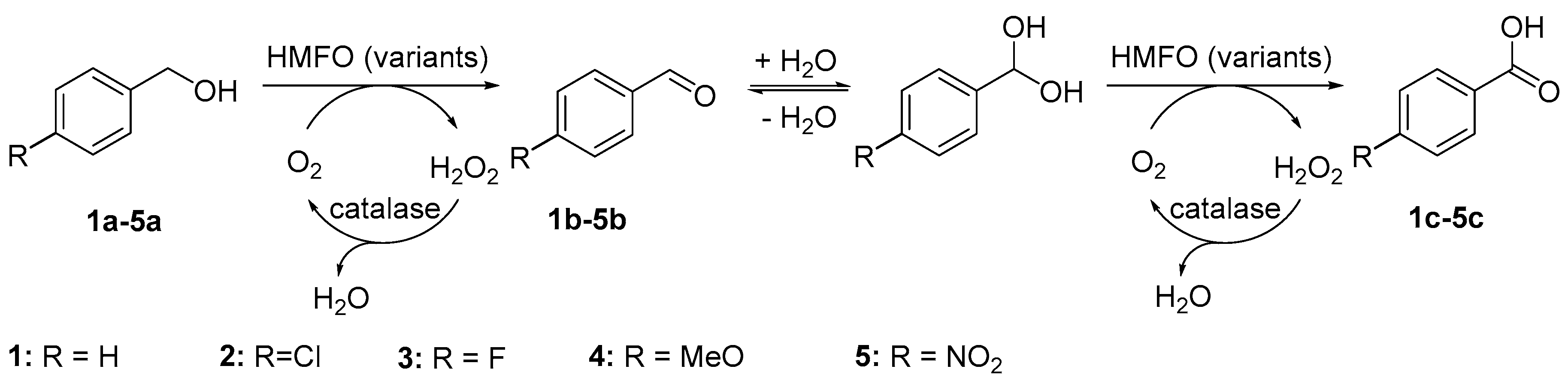
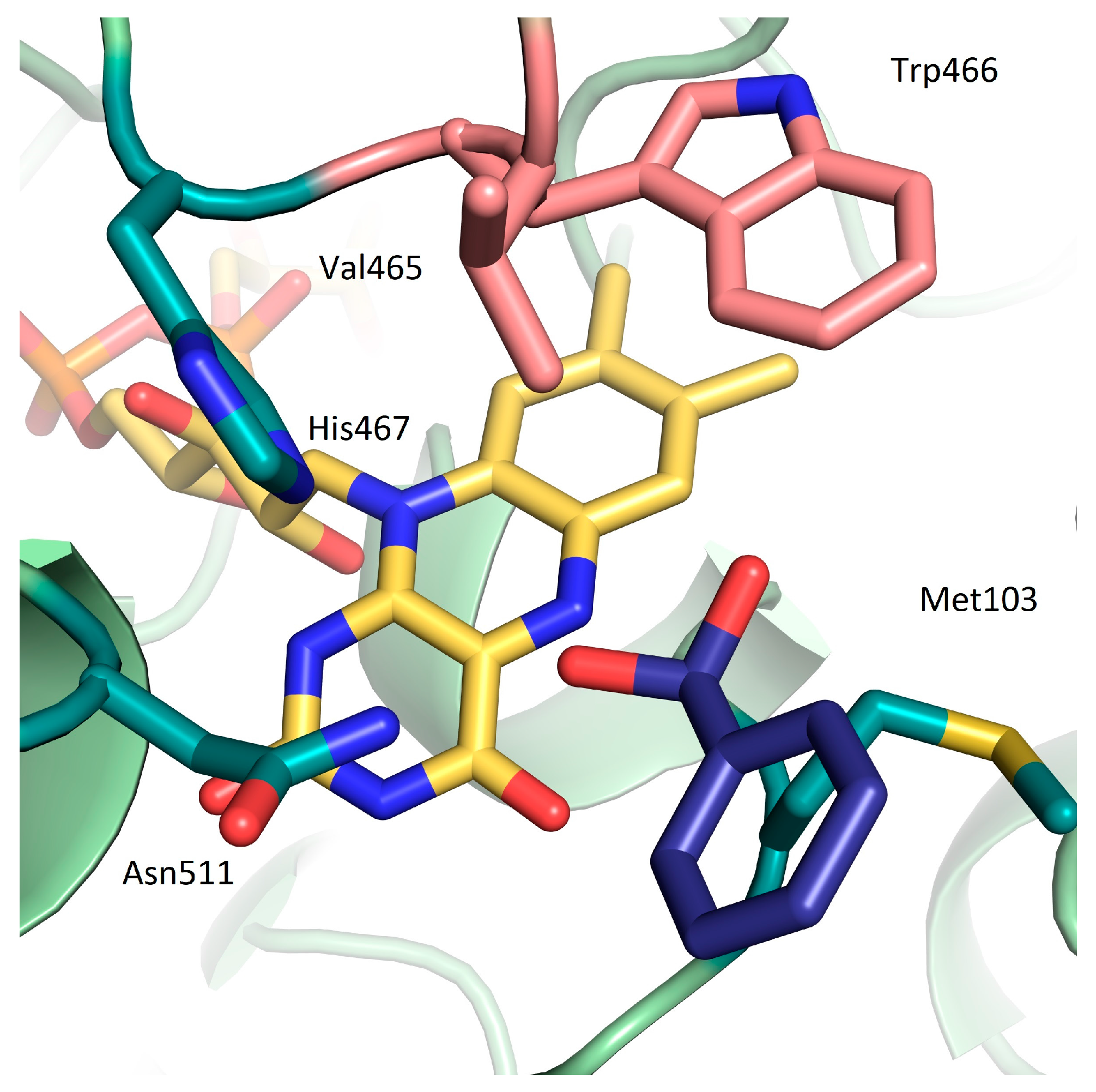
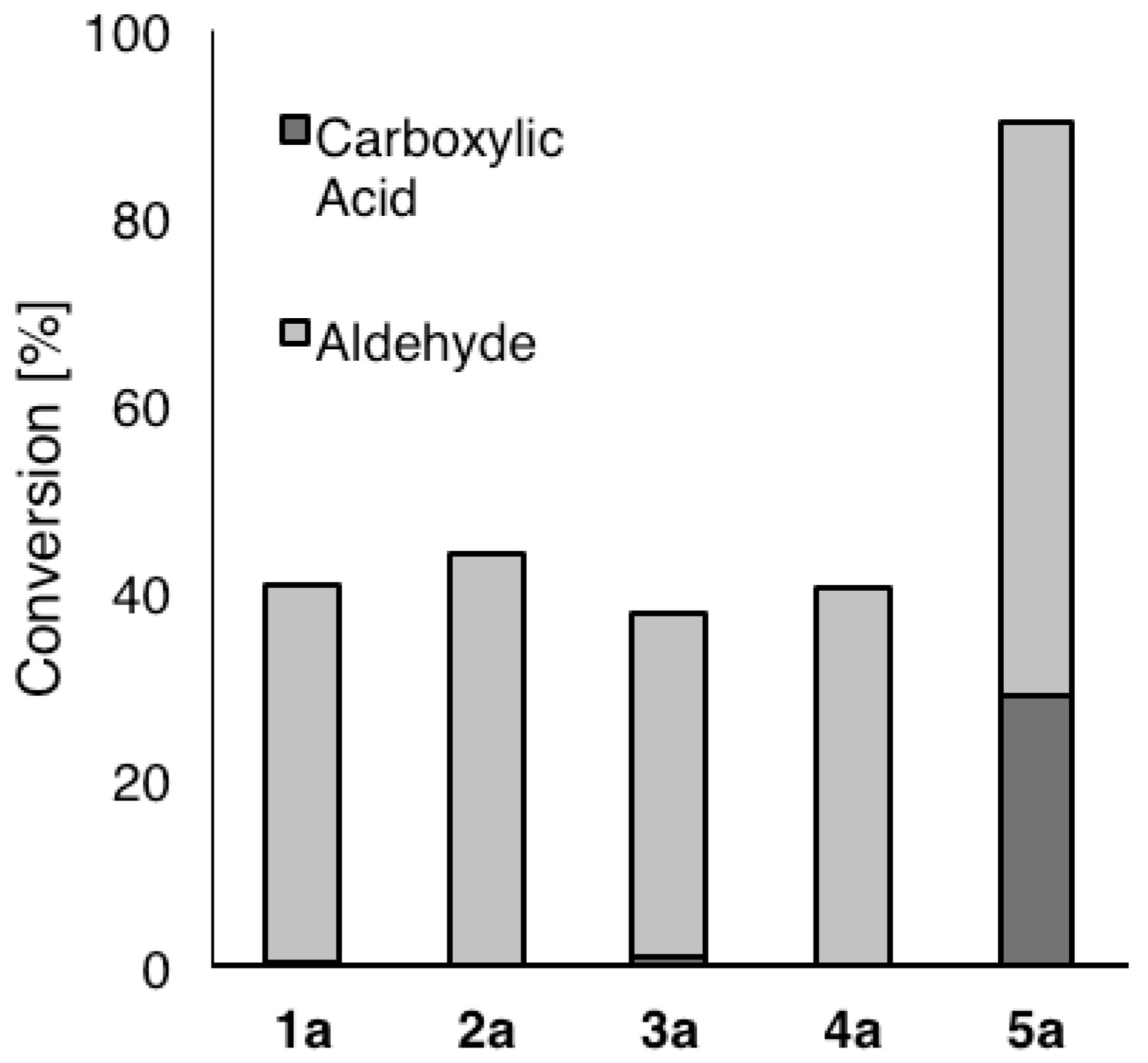
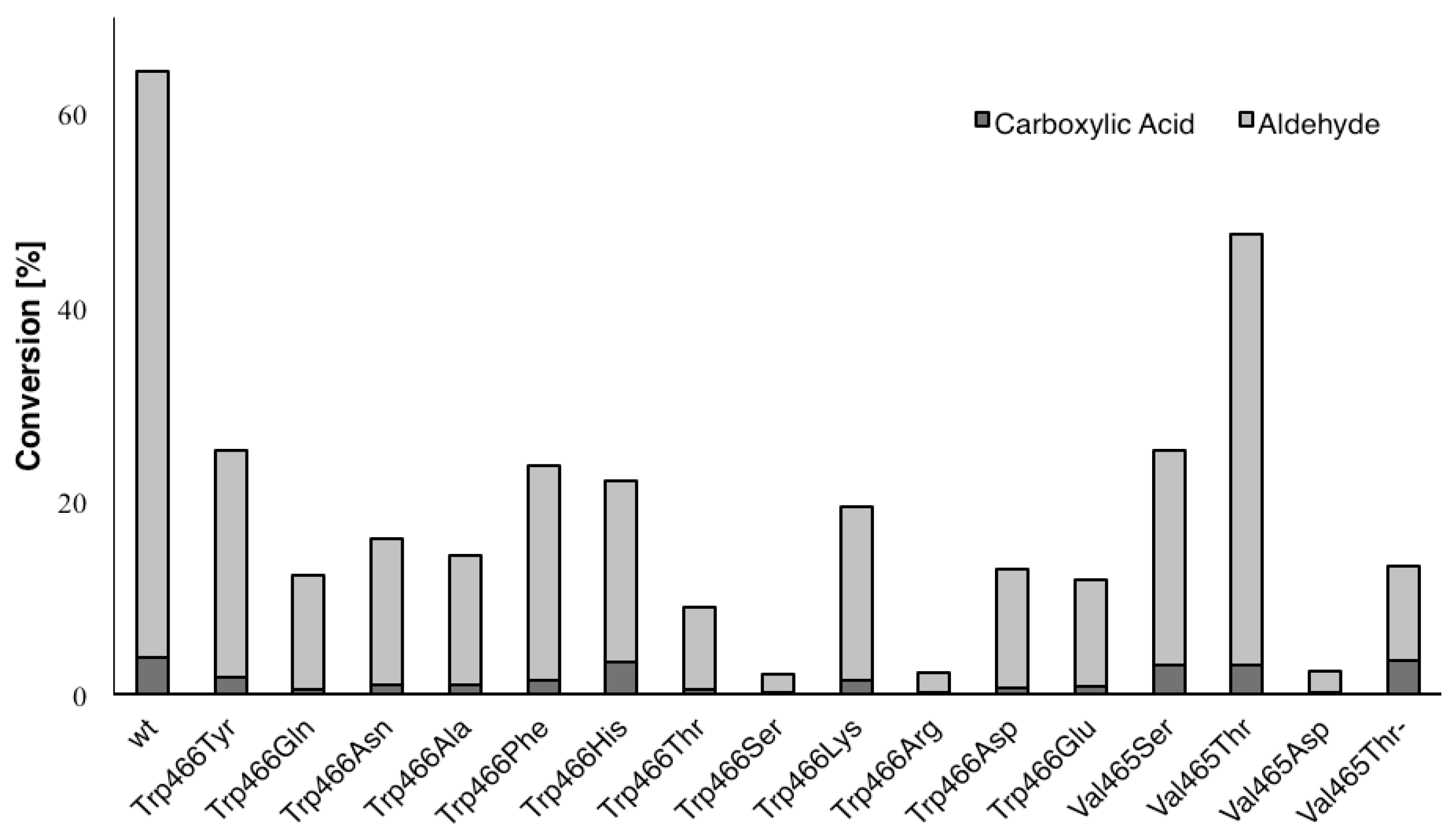

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Ratio (%) a |
|---|---|
| Wild type | 6 |
| Trp466Gln | 5 |
| Trp466His | 18 |
| Trp466Ser | 18 |
| Trp466Arg | 17 |
| Val465Ser | 14 |
| Val465Thr | 7 |
| Val465Asp | 16 |
| Val465Thr/Trp466His | 37 |
| Substrate | HMFO Variant | kcat (app) (s−1) | KM (app) (mM) | kcat (app)/KM (app) (s−1·M−1) | |
|---|---|---|---|---|---|
| 5a | 4-nitrobenzyl alcohol | Wild type | 7.4 | 0.26 | 28,000 |
| Trp466His | 0.02 | 0.40 | 50 | ||
| Val465Thr | 1.7 | 0.03 | 57,000 | ||
| 5b | 4-nitrobenzaldehyde | Wild type | 0.3 | 0.31 | 970 |
| Trp466His | 0.003 | 0.16 | 20 | ||
| Val465Thr | <0.0001 | n.d. | - | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pickl, M.; Winkler, C.K.; Glueck, S.M.; Fraaije, M.W.; Faber, K. Rational Engineering of a Flavoprotein Oxidase for Improved Direct Oxidation of Alcohols to Carboxylic Acids. Molecules 2017, 22, 2205. https://doi.org/10.3390/molecules22122205
Pickl M, Winkler CK, Glueck SM, Fraaije MW, Faber K. Rational Engineering of a Flavoprotein Oxidase for Improved Direct Oxidation of Alcohols to Carboxylic Acids. Molecules. 2017; 22(12):2205. https://doi.org/10.3390/molecules22122205
Chicago/Turabian StylePickl, Mathias, Christoph K. Winkler, Silvia M. Glueck, Marco W. Fraaije, and Kurt Faber. 2017. "Rational Engineering of a Flavoprotein Oxidase for Improved Direct Oxidation of Alcohols to Carboxylic Acids" Molecules 22, no. 12: 2205. https://doi.org/10.3390/molecules22122205
APA StylePickl, M., Winkler, C. K., Glueck, S. M., Fraaije, M. W., & Faber, K. (2017). Rational Engineering of a Flavoprotein Oxidase for Improved Direct Oxidation of Alcohols to Carboxylic Acids. Molecules, 22(12), 2205. https://doi.org/10.3390/molecules22122205