Design and Synthesis of New Benzothiazole Compounds as Selective hMAO-B Inhibitors

 and
and
Abstract
:1. Introduction
2. Results and Discussion
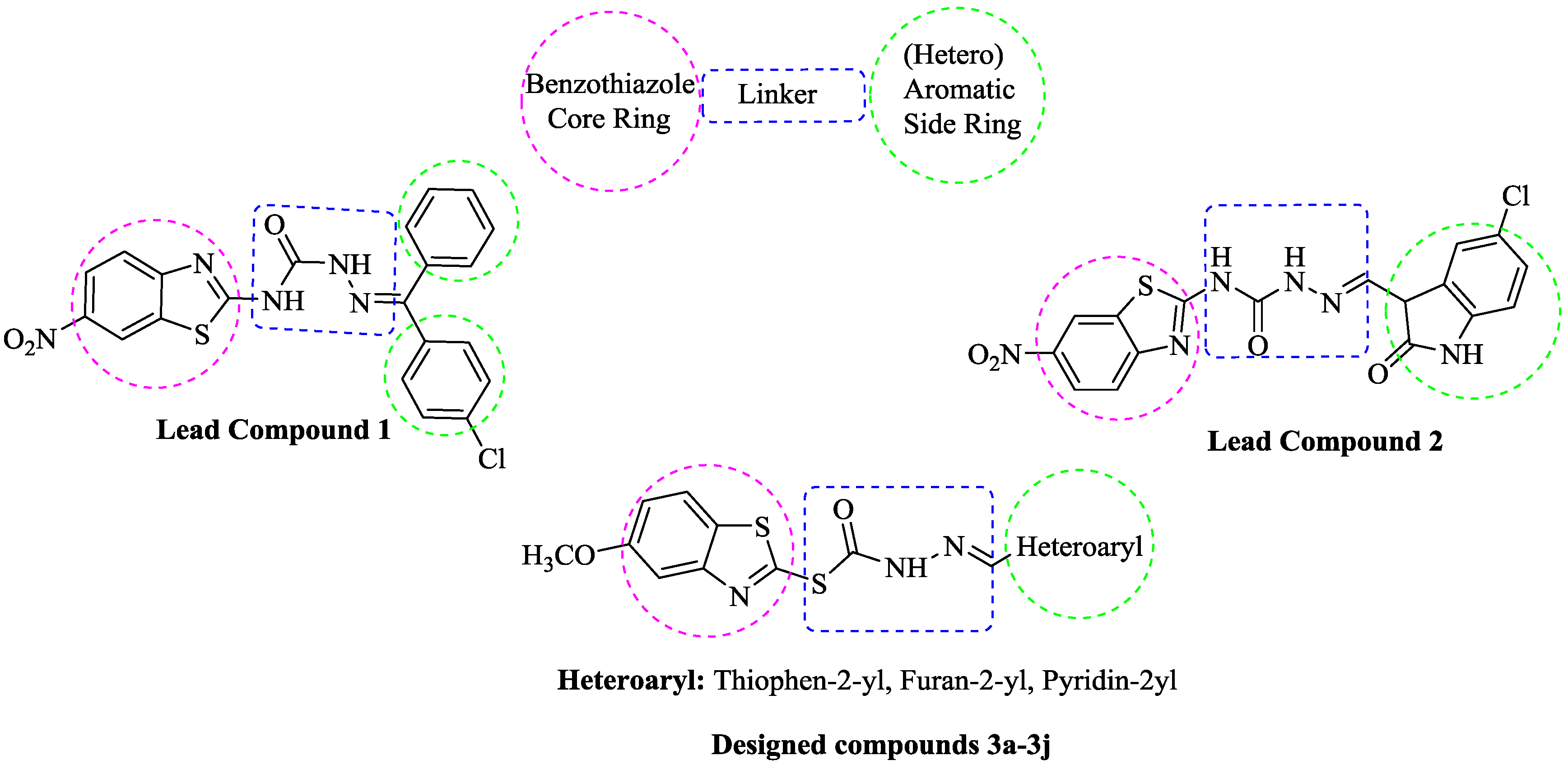
2.1. Chemistry
2.2. Enzymatic Studies
2.2.1. MAO-A and MAO-B Inhibition Assay
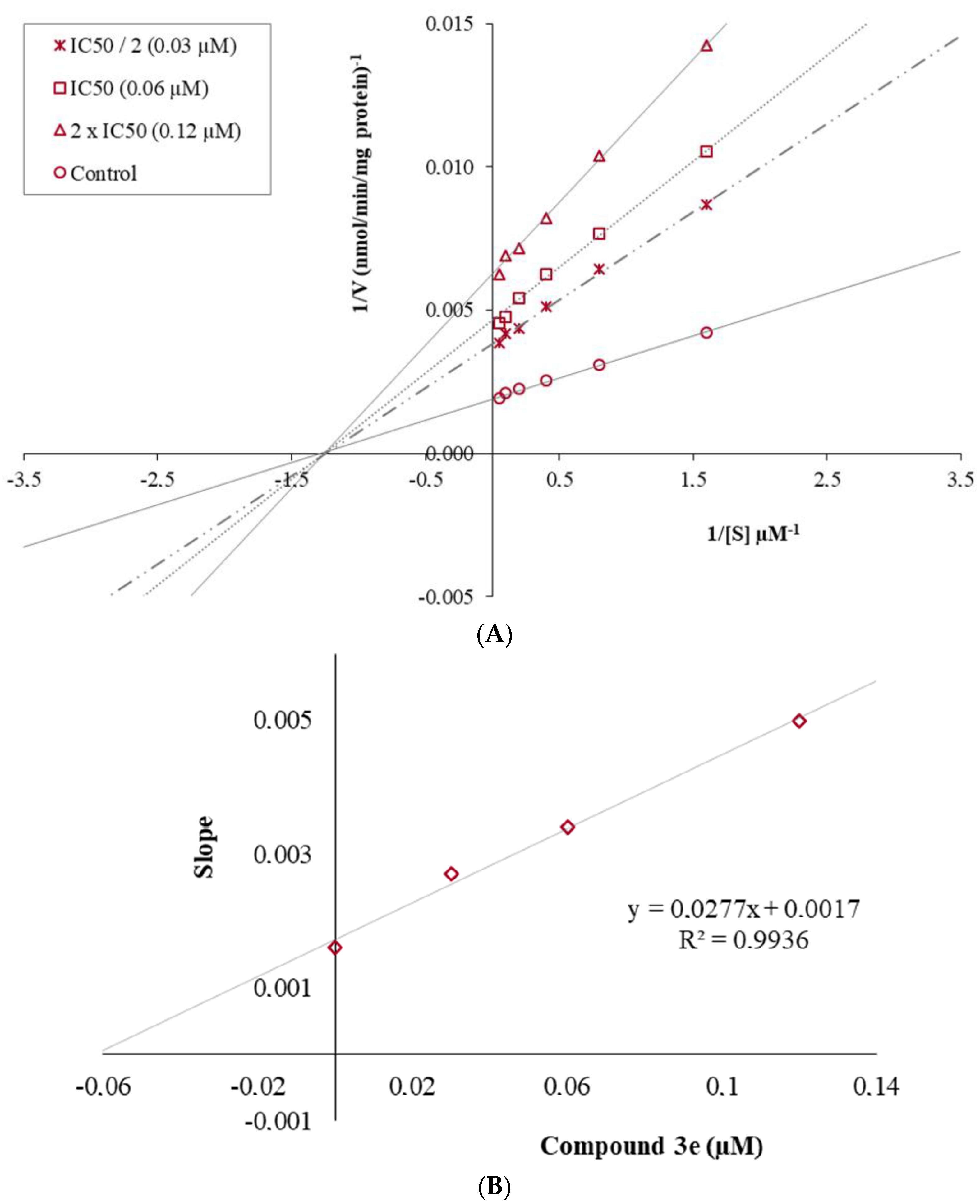
2.2.2. Enzyme Kinetic Studies
2.3. Cytotoxicity Test
2.4. Prediction of ADME Parameters and BBB Permeability
2.5. Molecular Docking Studies
3. Materials and Methods
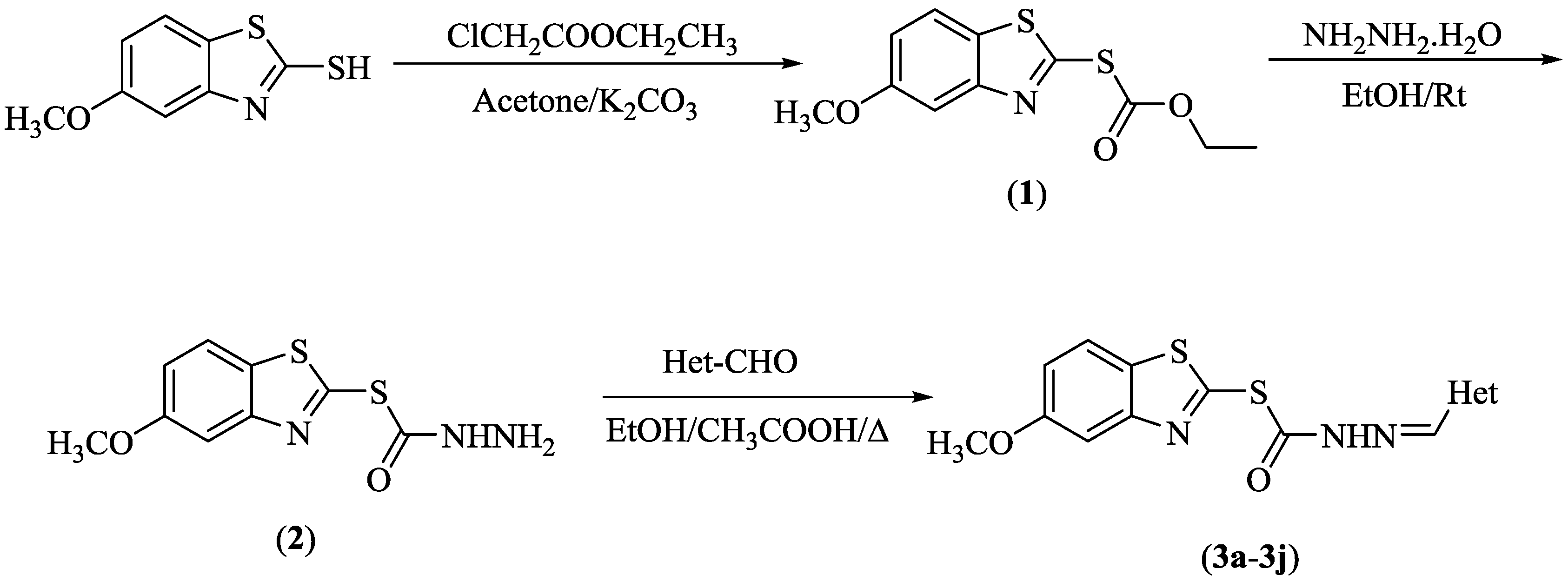
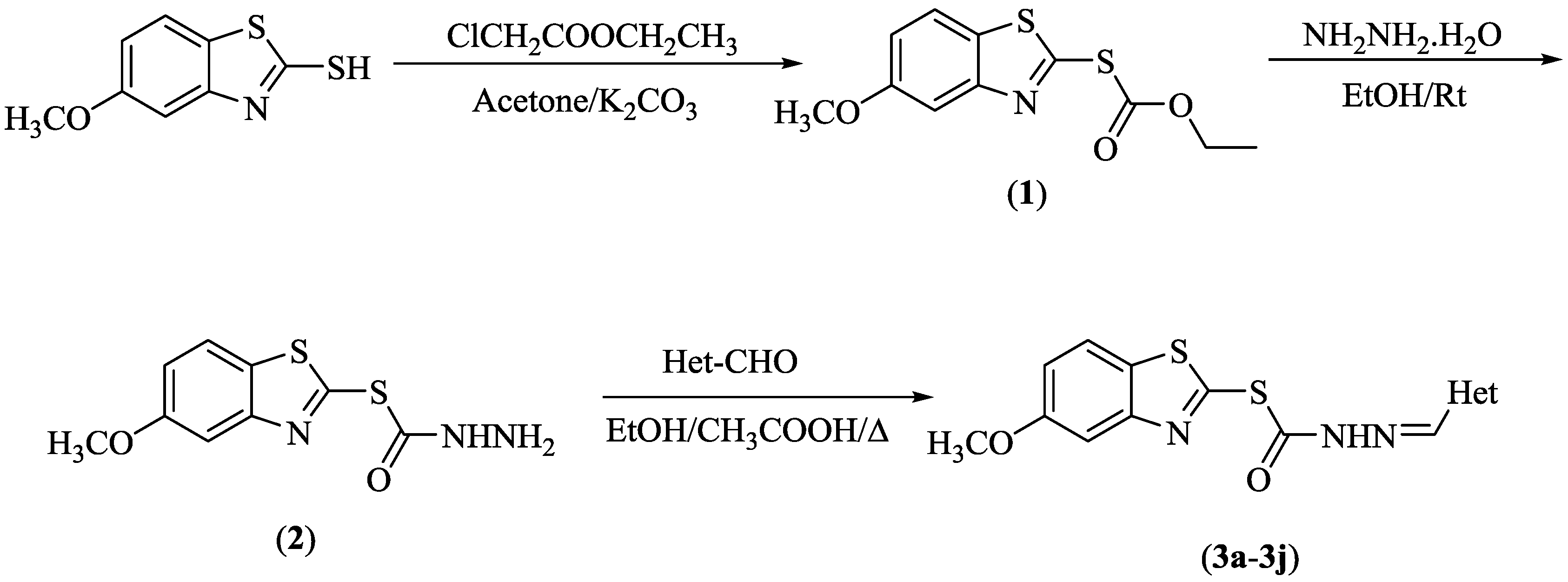
3.1. Chemistry
3.1.1. Synthesis of O-Ethyl S-(5-Methoxybenzothiazol-2-yl) Carbonothioate (1)
3.1.2. Synthesis of S-(5-Methoxybenzothiazol-2-yl) Hydrazinecarbothioate (2)
3.1.3. General Procedure for the Synthesis of the Target Compounds (3a–3j)
3.2. Activity Studies
3.2.1. MAO-A and MAO-B Inhibition Assay
3.2.2. Enzyme Kinetic Studies
3.3. Cytotoxicity Test
3.4. Prediction of ADME Parameters and BBB Permeability
3.5. Molecular Docking Studies
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.; Mattevi, A. Structures of Human Monoamine Oxidase B Complexes with Selective Noncovalent Inhibitors: Safinamide and Coumarin Analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Jang, B.K.; Cho, N.C.; Park, J.H.; Yeon, S.K.; Ju, E.J.; Lee, Y.S.; Han, G.; Pae, A.N.; Kim, D.J.; et al. Synthesis of a series of unsaturated ketone derivatives as selective and reversible monoamine oxidase inhibitors. Bioorg. Med. Chem. 2015, 23, 6486–6496. [Google Scholar] [CrossRef] [PubMed]
- Finberg, J.P.M.; Rabey, J.M. Inhibitors of MAO-A and MAO-B in Psychiatry and Neurology. Front. Pharmacol. 2016, 7, 340. [Google Scholar] [CrossRef] [PubMed]
- Livingston, M.G.; Livingston, H.M. Monoamine oxidase inhibitors. An update on drug interactions. Drug Saf. 1996, 14, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Fiedorowicz, J.G.; Swartz, K.L. The Role of Monoamine Oxidase Inhibitors in Current Psychiatric Practice. J. Psychiatr. Pract. 2004, 10, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Laux, G. MAO-inhibitors in Parkinson’s Disease. Exp. Neurobiol. 2011, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Alexander, G.E. Biology of Parkinson’s disease: Pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin. Neurosci. 2004, 6, 259–280. [Google Scholar] [PubMed]
- Dickson, D.W. Parkinson’s Disease and Parkinsonism: Neuropathology. Cold Spring Harb. Perspect. Med. 2012, 2, a009258. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, M.M. Medical Management of Parkinson’s Disease. Pharm. Ther. 2008, 33, 590–594. [Google Scholar]
- Dezsi, L.; Vecsei, L. Monoamine Oxidase B Inhibitors in Parkinson’s Disease. CNS Neurol. Disord. Drug Targets 2017, 16, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Teo, K.C.; Ho, S.L. Monoamine oxidase-B (MAO-B) inhibitors: Implications for disease-modification in Parkinson’s disease. Transl. Neurodegener. 2013, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Robakis, D.; Fahn, S. Defining the Role of the Monoamine Oxidase-B Inhibitors for Parkinson’s Disease. CNS Drugs 2015, 29, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, J.; Yang, X.; Cai, P.; Liu, Q.; Wang, K.D.G.; Kong, L.; Wang, X. Neuroprotective effects of benzyloxy substituted small molecule monoamine oxidase B inhibitors in Parkinson’s disease. Bioorg. Med. Chem. 2016, 24, 5929–5940. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Newton-Vinson, P.; Hubalek, F.; Edmondson, D.E.; Mattevi, A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat. Struct. Biol. 2002, 9, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Newton-Vinson, P.; Hubalek, F.; Edmondson, D.E.; Mattevi, A. Crystal structure of human monoamine oxidase B, a drug target enzyme monotopically inserted into the mitochondrial outer membrane. FEBS Lett. 2004, 564, 225–228. [Google Scholar] [CrossRef]
- Tripathi, R.K.; Goshain, O.; Ayyannan, S.R. Design, synthesis, in vitro MAO-B inhibitory evaluation, and computational studies of some 6-nitrobenzothiazole-derived semicarbazones. ChemMedChem 2013, 8, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Kaya, B.; Sağlık, B.N.; Levent, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis of some novel 2-substituted benzothiazole derivatives containing benzylamine moiety as monoamine oxidase inhibitory agents. J. Enzyme Inhib. Med. Chem. 2016, 31, 1654–1661. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.K.; Ayyannan, S.R. Design, Synthesis, and Evaluation of 2-Amino-6-nitrobenzothiazole-Derived Hydrazones as MAO Inhibitors: Role of the Methylene Spacer Group. ChemMedChem 2016, 11, 1551–1567. [Google Scholar] [CrossRef] [PubMed]
- Lammana, C.; Sinicropi, M.S.; Pietrangeli, P.; Corbo, F.; Franchini, C.; Mondovi, B.; Perrone, M.G.; Scilimati, A. Synthesis and biological evaluation of 3-alkyloxazolidine-2-ones as reversible MAO inhibitors. Arkivoc 2004, 5, 118–130. [Google Scholar]
- Chimenti, P.; Petzer, A.; Carradori, S.; D’Ascenzio, M.; Silvestri, R.; Alcaro, S.; Ortuso, F.; Petzer, J.P.; Secci, D. Exploring 4-substituted-2-thiazolylhydrazones from 2-, 3-, and 4-acetylpyridine as selective and reversible hMAO-B inhibitors. Eur. J. Med. Chem. 2013, 66, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Can, Ö.D.; Osmaniye, D.; Demir Özkay, Ü.; Sağlık, B.N.; Levent, S.; Ilgın, S.; Baysal, M.; Özkay, Y.; Kaplancıklı, Z.A. MAO enzymes inhibitory activity of new benzimidazole derivatives including hydrazone and propargyl side chains. Eur. J. Med. Chem. 2017, 131, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Can, N.Ö.; Osmaniye, D.; Levent, S.; Sağlık, B.N.; İnci, B.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis of New Hydrazone Derivatives for MAO Enzymes Inhibitory Activity. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- International Organization for Standardization. International Organization for Standardization, Biological Evaluation of Medical Devices-Part 5: Tests for in Vitro Cytotoxicity ISO-10993-5, 3rd ed.; International Organization for Standardization: Geneva, Switzerland, 2009. [Google Scholar]
- De Waterbeemd, H.V.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2013, 2, 192–204. [Google Scholar]
- Molinspiration Cheminformatics, Bratislava, Slovak Republic. Available online: http://www.molinspiration.com/services/properties.html (accessed on 20 July 2017).
- Carpenter, T.S.; Kirshner, D.A.; Lau, E.Y.; Wong, S.E.; Nilmeier, J.P.; Lightstone, F.C. A Method to Predict Blood-Brain Barrier Permeability of Drug-Like Compounds Using Molecular Dynamics Simulations. Biophys. J. 2014, 107, 630–641. [Google Scholar] [CrossRef] [PubMed]
- About the Blood-brain Barrier (BBB) Prediction Server. Available online: http://www.cbligand.org/BBB/index.php (accessed on 20 July 2017).
- Protein Data Bank Server. Available online: http://www.pdb.org (accessed on 14 July 2017).
- Karaca Gençer, H.; Acar Çevik, U.; Kaya Çavuşoğlu, B.; Sağlık, B.N.; Levent, S.; Atlı, Ö.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Design, synthesis, and evaluation of novel 2-phenylpropionic acid derivatives as dual COX inhibitory-antibacterial agents. J. Enzyme Inhib. Med. Chem. 2017, 32, 732–745. [Google Scholar] [CrossRef] [PubMed]
- Demir Özkay, Ü.; Can, Ö.D.; Sağlık, B.N.; Acar Çevik, U.; Levent, S.; Özkay, Y.; Ilgın, S.; Atlı, Ö. Design, synthesis, and AChE inhibitory activity of new benzothiazole-piperazines. Bioorg. Med. Chem. Lett. 2016, 26, 5387–5394. [Google Scholar] [CrossRef] [PubMed]
- Karaca Gençer, H.; Levent, S.; Acar Çevik, U.; Özkay, Y.; Ilgın, S. New 1,4-dihydro[1,8]naphthyridine derivatives as DNA gyrase inhibitors. Bioorg. Med. Chem. Lett. 2016, 27, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Maestro, Version 10.6, Schrödinger, LLC: New York, NY, USA, 2016.
- Schrödinger, Version 2016-2, LLC: New York, NY, USA, 2016.
- LigPrep, Version 3.8, Schrödinger, LLC: New York, NY, USA, 2016.
- Glide, Version 7.1, Schrödinger, LLC: New York, NY, USA, 2016.
- Toprakçı, M.; Yelekçi, K. Docking studies on monoamine oxidase-B inhibitors: Estimation of inhibition constants (Ki) of a series of experimentally tested compounds. Bioorg. Med. Chem. Lett. 2005, 15, 4438–4446. [Google Scholar] [CrossRef] [PubMed]
- Gökhan-Kelekçi, N.; Özgün Şimşek, Ö.; Ercan, A.; Yelekçi, K.; Sibel Şahin, Z.; Işık, Ş.; Uçar, G.; Bilgin, A.A. Synthesis and molecular modeling of some novel hexahydroindazole derivatives as potent monoamine oxidase inhibitors. Bioorg. Med. Chem. 2009, 17, 6761–6772. [Google Scholar] [CrossRef] [PubMed]
- Evranos-Aksöz, B.; Yabanoğlu-Çiftçi, S.; Uçar, G.; Yelekçi, K.; Ertan, R. Monoamine oxidase inhibitory activities and docking studies. Bioorg. Med. Chem. Lett. 2014, 24, 3278–3284. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1, 2, and 3a–3j are available from the authors. |
| Compounds | Het |
|---|---|
| 3a | Thiophen-2yl |
| 3b | 3-Methylthiophen-2yl |
| 3c | 5-Methylthiophen-2yl |
| 3d | 5-Bromothiophen-2yl |
| 3e | 5-Nitrothiophen-2yl |
| 3f | Furan-2yl |
| 3g | 5-Methylfuran-2yl |
| 3h | 5-Nitrofuran-2yl |
| 3i | Pyridine-3yl |
| 3j | Pyridine-4yl |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | MAO A Inhibition % | MAO B Inhibition % | ||
|---|---|---|---|---|
| 10−3 M | 10−4 M | 10−3 M | 10−4 M | |
| 3a | 25.00 | 19.02 | 60.37 | 52.15 |
| 3b | 30.08 | 20.45 | 30.46 | 26.88 |
| 3c | 14.10 | 10.75 | 25.75 | 18.20 |
| 3d | 18.88 | 12.71 | 23.97 | 17.75 |
| 3e | 22.12 | 18.55 | 87.28 | 83.50 |
| 3f | 30.77 | 18.30 | 75.66 | 68.30 |
| 3g | 16.69 | 11.07 | 18.20 | 11.23 |
| 3h | 25.89 | 21.30 | 82.58 | 79.10 |
| 3i | 19.88 | 10.55 | 32.02 | 28.75 |
| 3j | 22.15 | 17.28 | 34.60 | 30.50 |
| Moclobemide | 91.42 ± 4.60 | 77.86 ± 3.71 | - | - |
| Selegiline | - | - | 97.69 ± 4.16 | 94.42 ± 3.89 |
| Comp. | MAO B IC50 (µM) |
|---|---|
| 3a | 15.450 ± 0.398 |
| 3e | 0.060 ± 0.002 |
| 3f | 0.963 ± 0.033 |
| 3h | 0.075 ± 0.003 |
| Selegiline | 0.044 ± 0.002 |
| Comp. | IC50 (µM) |
|---|---|
| 3e | 19.002 ± 1.029 |
| Comp. | MW (g/mol) | logP | TPSA (ANG2) | HBA | HBD | Vol (ANG3) | Vio | BBB |
|---|---|---|---|---|---|---|---|---|
| 3a | 363.49 | 4.10 | 63.59 | 6 | 1 | 291.83 | 0 | + |
| 3b | 377.52 | 4.47 | 63.59 | 6 | 1 | 308.39 | 0 | + |
| 3c | 377.52 | 4.32 | 63.59 | 6 | 1 | 308.39 | 0 | + |
| 3d | 442.38 | 5.03 | 63.59 | 6 | 1 | 309.72 | 1 | + |
| 3e | 408.49 | 4.18 | 109.41 | 7 | 1 | 315.17 | 0 | + |
| 3f | 347.42 | 3.46 | 76.73 | 6 | 1 | 282.69 | 0 | + |
| 3g | 361.45 | 3.68 | 76.73 | 6 | 1 | 299.25 | 0 | + |
| 3h | 392.42 | 3.54 | 122.55 | 7 | 1 | 306.02 | 0 | + |
| 3i | 358.45 | 2.96 | 76.48 | 6 | 1 | 296.96 | 0 | + |
| 3j | 358.45 | 2.91 | 76.48 | 6 | 1 | 296.96 | 0 | + |
| Selegiline | 187.29 | 2.64 | 3.24 | 1 | 0 | 202.64 | 0 | + |
| Clorgiline | 272.18 | 3.74 | 12.47 | 2 | 0 | 238.91 | 0 | + |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilgın, S.; Osmaniye, D.; Levent, S.; Sağlık, B.N.; Acar Çevik, U.; Çavuşoğlu, B.K.; Özkay, Y.; Kaplancıklı, Z.A. Design and Synthesis of New Benzothiazole Compounds as Selective hMAO-B Inhibitors. Molecules 2017, 22, 2187. https://doi.org/10.3390/molecules22122187
Ilgın S, Osmaniye D, Levent S, Sağlık BN, Acar Çevik U, Çavuşoğlu BK, Özkay Y, Kaplancıklı ZA. Design and Synthesis of New Benzothiazole Compounds as Selective hMAO-B Inhibitors. Molecules. 2017; 22(12):2187. https://doi.org/10.3390/molecules22122187
Chicago/Turabian StyleIlgın, Sinem, Derya Osmaniye, Serkan Levent, Begüm Nurpelin Sağlık, Ulviye Acar Çevik, Betül Kaya Çavuşoğlu, Yusuf Özkay, and Zafer Asım Kaplancıklı. 2017. "Design and Synthesis of New Benzothiazole Compounds as Selective hMAO-B Inhibitors" Molecules 22, no. 12: 2187. https://doi.org/10.3390/molecules22122187
APA StyleIlgın, S., Osmaniye, D., Levent, S., Sağlık, B. N., Acar Çevik, U., Çavuşoğlu, B. K., Özkay, Y., & Kaplancıklı, Z. A. (2017). Design and Synthesis of New Benzothiazole Compounds as Selective hMAO-B Inhibitors. Molecules, 22(12), 2187. https://doi.org/10.3390/molecules22122187