Improving the Catalytic Property of the Glycoside Hydrolase LXYL-P1–2 by Directed Evolution


Abstract
:1. Introduction
2. Results
2.1. Establishment of the Methanol-Induced Plate Screening Approach for Mutants with Improved Enzyme Activities
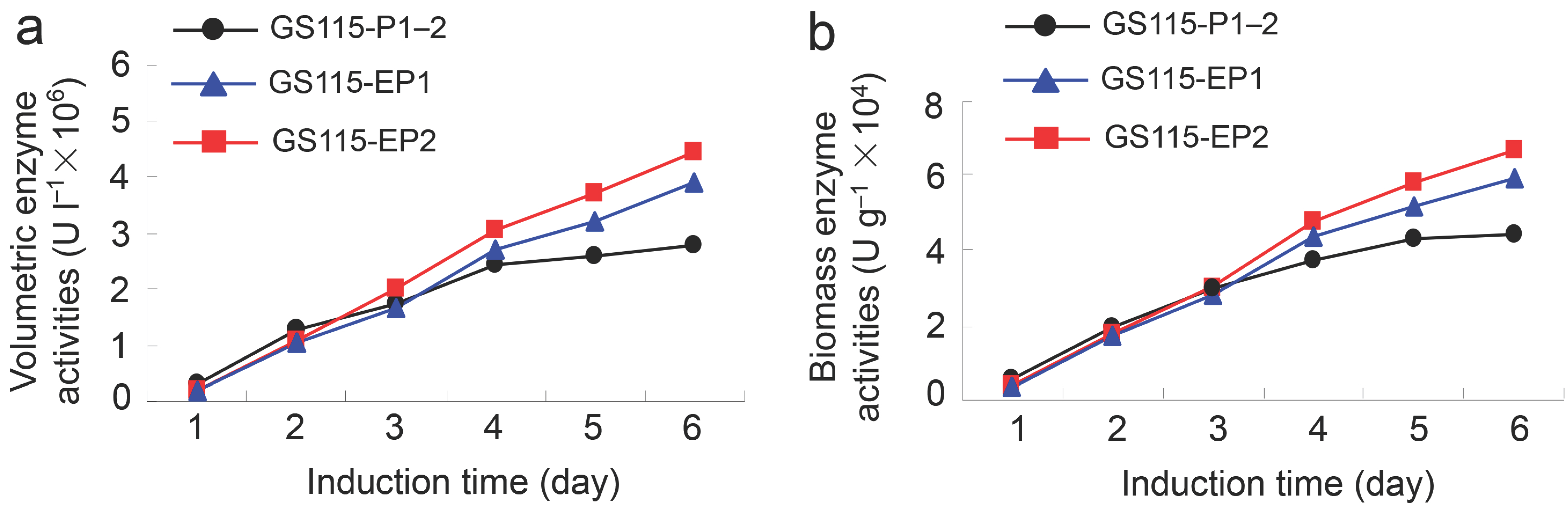
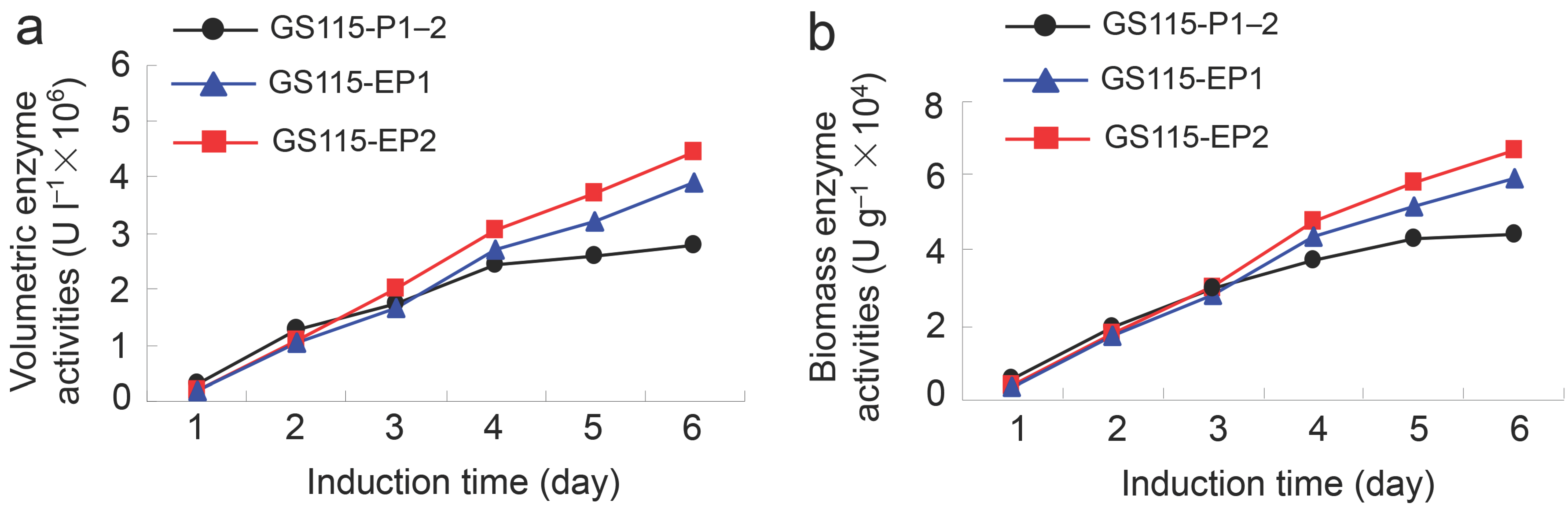
2.2. β-Xylosidase Activities of the Recombinant Yeast GS115-EP1 and GS115-EP2 against PNP-Xyl
2.3. Conversion Rates of 7-β-Xylosyltaxanes by the Recombinant Yeasts GS115-EP1 and GS115-EP2
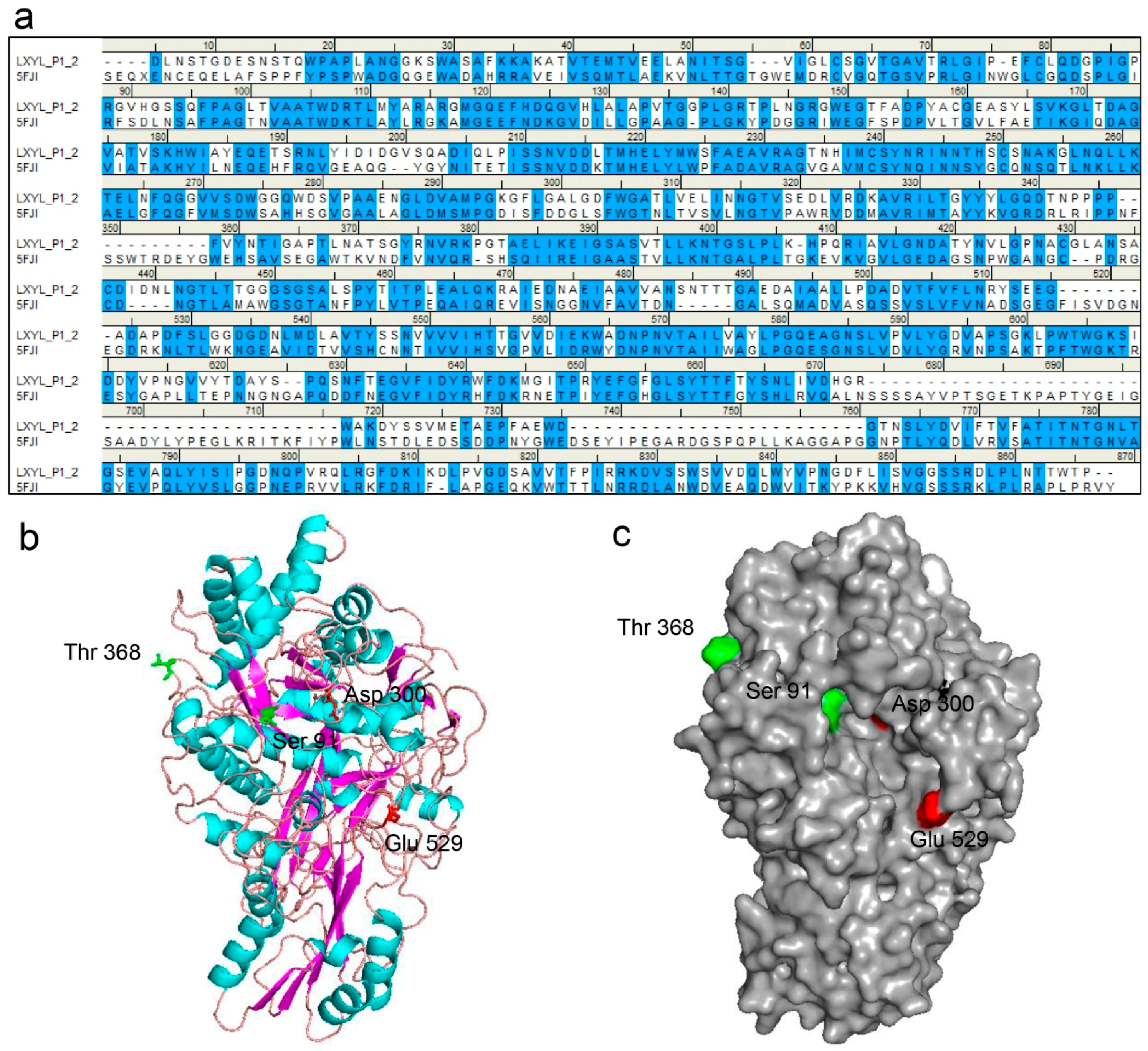
2.4. Sequencing Analysis of EP1 and EP2
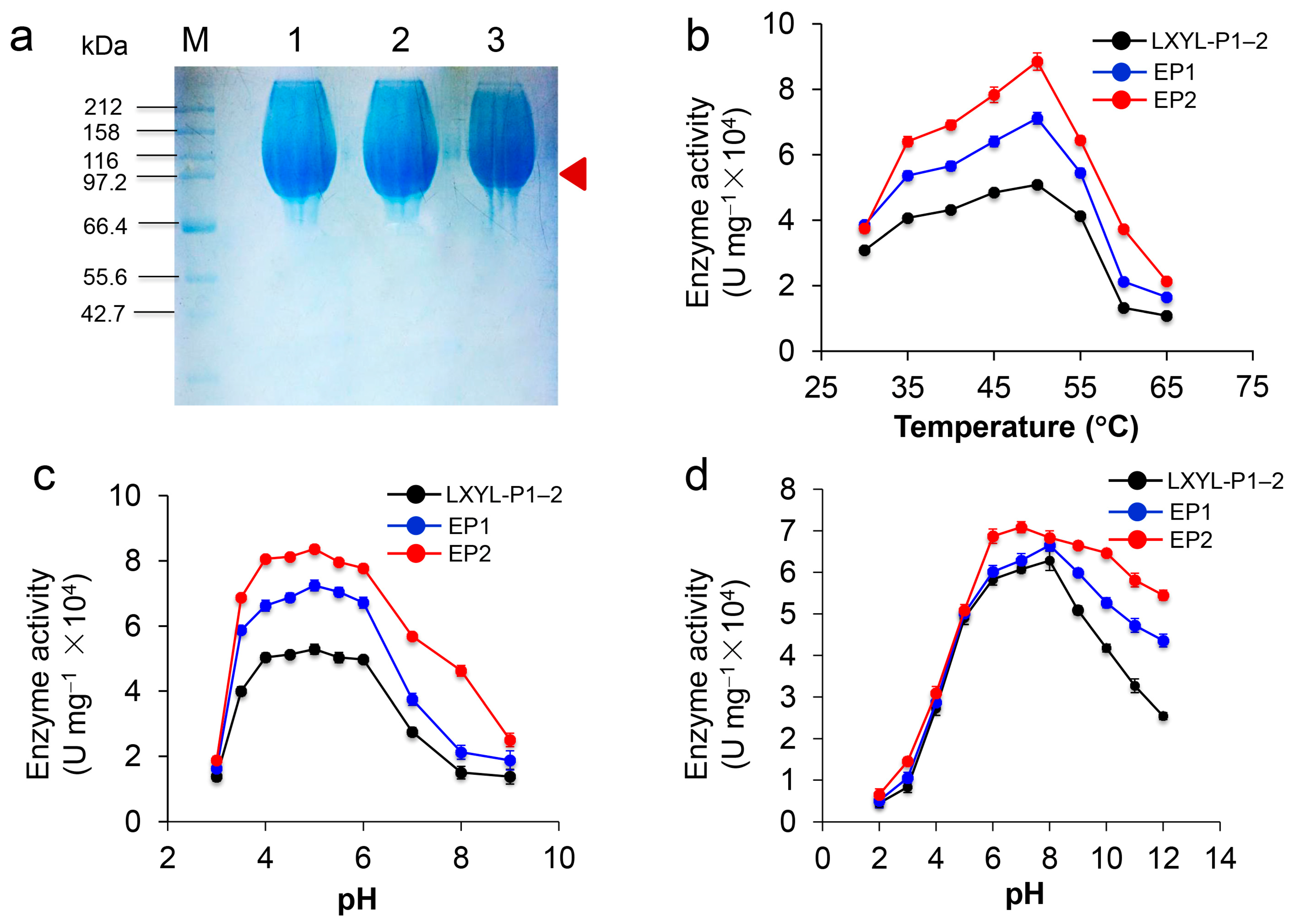
2.5. Characterization of Recombinant Proteins EP1 and EP2
2.6. Characterization and Kinetics of the Recombinant Proteins EP1 and EP2
2.7. Three-Dimensional Structure Prediction of Enzyme
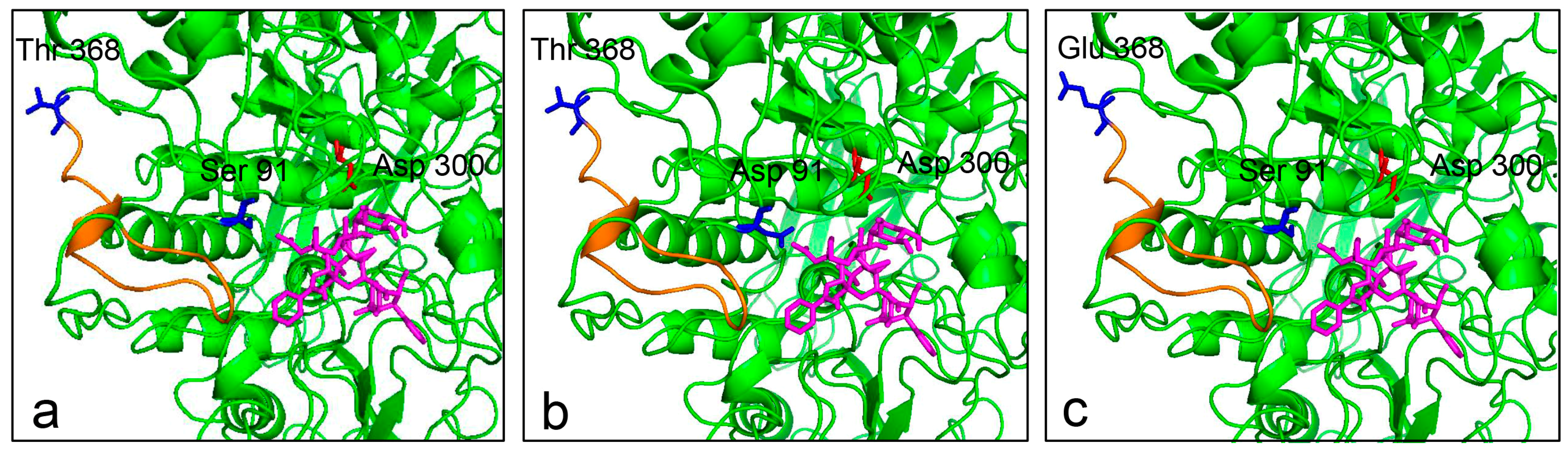
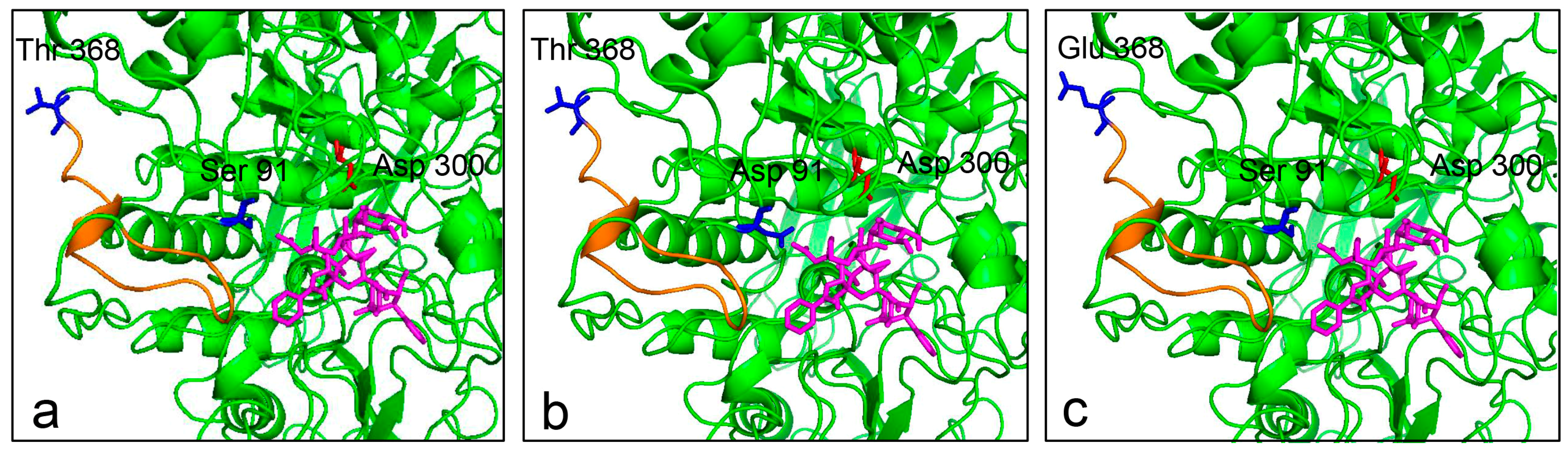
2.8. Substrate Docking Analysis of Mutants EP1 and EP2
3. Discussion
4. Materials and Methods
4.1. Plasmids and Strains
4.2. Error-Prone PCR and Construction of the Library of Variant Lxyl-p1–2 Genes
4.3. Screening of Mutants with Higher β-Xylosidase Activities
4.4. Measurement of β-Xylosidase Activity of Mutant Strains
4.5. Sequencing Analysis of Lxyl-p1–2 Variants
4.6. Enzyme Purification and SDS-PAGE Analysis
4.7. Characterization and Kinetics of the Recombinant LXYL-P1–2 Mutants
4.8. Homology Modeling and Molecular Docking
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Rowinsky, E.K.; Donehower, R.C. Paclitaxel (taxol). N. Engl. J. Med. 1995, 332, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.S.; Wu, Y.; Zhou, J.Y.; Chen, W.M.; Fang, Q.C. Qualitative and quantitative determination of taxol and related compounds in Taxus cuspidata Sieb et Zucc. Phytochem. Anal. 1993, 4, 115–119. [Google Scholar] [CrossRef]
- Liu, W.C.; Gong, T.; Zhu, P. Advances in exploring alternative Taxol sources. RSC Adv. 2016, 6, 48800–48809. [Google Scholar] [CrossRef]
- Fu, Y.J.; Sun, R.; Zu, Y.G.; Li, S.M.; Liu, W.; Efferth, T.; Gu, C.B.; Zhang, L.; Luo, H. Simultaneous determination of main taxoids in Taxus needles extracts by solid-phase extraction-high-performance liquid chromatography with pentafluorophenyl column. Biomed. Chromatogr. 2009, 23, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.L.; Zhao, R.Y.; Chen, T.J.; Yu, W.B.; Wang, F.; Cheng, K.D.; Zhu, P. Cloning and characterization of the glycoside hydrolases that remove xylosyl groups from 7-β-xylosyl-10-deacetyltaxol and its analogues. Mol. Cell. Proteom. 2013, 12, 2236–2248. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.C.; Gong, T.; Wang, Q.H.; Liang, X.; Chen, J.J.; Zhu, P. Scaling-up Fermentation of Pichia pastoris to demonstration-scale using new methanol-feeding strategy and increased air pressure instead of pure oxygen supplement. Sci. Rep. 2016, 6, 18439. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.C.; Zhu, P. Pilot studies on scale-up biocatalysis of 7-β-xylosyl-10-deacetyltaxol and its analogues by an engineered yeast. J. Ind. Microbiol. Biot. 2015, 42, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.B.; Liang, X.; Zhu, P. High-cell-density fermentation and pilot-scale biocatalytic studies of an engineered yeast expressing the heterologous glycoside hydrolase of 7-β-xylosyltaxanes. J. Ind. Microbiol. Biot. 2013, 40, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Bornscheuer, U.T.; Pohl, M. Improved biocatalysts by directed evolution and rational protein design. Curr. Opin. Chem. Biol. 2001, 5, 137–143. [Google Scholar] [CrossRef]
- Dalby, P.A. Strategy and success for the directed evolution of enzymes. Curr. Opin. Struct. Biol. 2011, 21, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Jäckel, C.; Kast, P.; Hilvert, D. Protein design by directed evolution. Annu. Rev. Biophys. 2008, 37, 153–173. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.J. Directed evolution drives the next generation of biocatalysts. Nat. Chem. Biol. 2009, 5, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.B.; Li, G.; Reetz, M.T. Enzymatic site-selectivity enabled by structure-guided directed evolution. Chem. Commun. 2017, 53, 3916–3928. [Google Scholar] [CrossRef] [PubMed]
- Biles, B.D.; Connolly, B.A. Low-fidelity Pyrococcus furiosus DNA polymerase mutants useful in error-prone PCR. Nucleic Acids Res. 2004, 32, e176. [Google Scholar] [CrossRef] [PubMed]
- Rasila, T.S.; Pajunen, M.I.; Savilahti, H. Critical evaluation of random mutagenesis by error-prone polymerase chain reaction protocols, Escherichia coli mutator strain, and hydroxylamine treatment. Anal. Biochem. 2009, 388, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.H.; Zhang, X.P.; Ebright, R.H. Random mutagenesis of gene-sized DNA molecules by use of PCR with Taq DNA polymerase. Nucleic Acids Res. 1991, 19, 6052. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Cai, G.; Hall, E.O.; Freeman, G.J. In-Fusion™ assembly: Seamless engineering of multidomain fusion proteins, modular vectors, and mutations. Biotechniques 2007, 43, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Goddard, J.P.; Reymond, J.L. Enzyme assays for high-throughput screening. Curr. Opin. Biotechnol. 2004, 15, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Reymond, J.L.; Fluxa, V.S.; Maillard, N. Enzyme assays. Chem. Commun. 2009, 1, 34–46. [Google Scholar]
- Jones, A.; Lamsa, M.; Frandsen, T.P.; Spendler, T.; Harris, P.; Sloma, A.; Xu, F.; Nielsen, J.B.; Cherry, J.R. Directed evolution of a maltogenic α-amylase from Bacillus sp. TS-25. J. Biotechnol. 2008, 134, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Sriprang, R.; Asano, K.; Gobsuk, J.; Tanapongpipat, S.; Champreda, V.; Eurwilaichitr, L. Improvement of thermostability of fungal xylanase by using site-directed mutagenesis. J. Biotechnol. 2006, 126, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Si, T.; Zhao, H.M. Biocatalyst development by directed evolution. Bioresour. Technol. 2012, 115, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Feng, S.Y.; Zhan, T.; Huang, Z.Q.; Wu, G.J.; Liu, Z.D. Improving catalytic efficiency of endo-β-1, 4-xylanase from Geobacillus stearothermophilus by directed evolution and H179 saturation mutagenesis. J. Biotechnol. 2013, 168, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.J.; Zhang, R.Z.; Liu, X.Y.; Shi, J.S.; Xu, Z.H.; Rao, Z.M. Improving the acidic stability of a β-mannanase from Bacillus subtilis by site-directed mutagenesis. Process Biochem. 2013, 48, 1166–1173. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Yi, Z.L.; Pei, X.Q.; Wu, Z.L. Improving the thermostability of Geobacillus stearothermophilus xylanase XT6 by directed evolution and site-directed mutagenesis. Bioresour. Technol. 2010, 101, 9272–9278. [Google Scholar] [CrossRef] [PubMed]
- Stephens, D.E.; Rumbold, K.; Permaul, K.; Prior, B.A.; Singh, S. Directed evolution of the thermostable xylanase from Thermomyces lanuginosus. J. Biotechnol. 2007, 127, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Veno, J.; Ahmad Kamarudin, N.H.; Mohamad Ali, M.S.; Masomian, M.; Raja Abd Rahman, R.N.Z. Directed Evolution of Recombinant C-Terminal Truncated Staphylococcus epidermidis Lipase AT2 for the Enhancement of Thermostability. Int. J. Mol. Sci. 2017, 18, 2202. [Google Scholar] [CrossRef] [PubMed]
- Li, H.X.; Zhu, P. Effect of the length of homologous sequence on the In-Fusion recombination efficiency. Chin. Med. Biotechnol. 2013, 8, 241–246. [Google Scholar]
- Li, B.J.; Wang, H.; Gong, T.; Chen, J.J.; Chen, T.J.; Yang, J.L.; Zhu, P. Improving 10-deacetylbaccatin III-10-β-O-acetyltransferase catalytic fitness for Taxol production. Nat. Commun. 2017, 8, 15544. [Google Scholar] [CrossRef] [PubMed]
- Adrian, R.R.; Marc, G.B.; Silvia, O. Computational tools for the evaluation of laboratory-engineered biocatalysts. Chem. Commun. 2017, 53, 284–297. [Google Scholar]
- Fan, Y.; Fang, W.; Xiao, Y.; Yang, X.; Zhang, Y.; Bidochka, M.J.; Pei, Y. Directed evolution for increased chitinase activity. Appl. Microbiol. Biot. 2007, 76, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Sample of the compound, 10-deacetyltaxol, is available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Conversion Rates (%) | Yields (mg mL−1) | Total Yields | ||||
|---|---|---|---|---|---|---|---|
| XDC | XDT | XDTC | DC | DT | DTC | (mg mL−1) | |
| GS115-P1–2 | 75.78 | 76.77 | 76.97 | 0.61 | 3.02 | 0.82 | 4.45 |
| GS115-EP1 | 87.41 | 83.95 | 86.01 | 0.67 | 3.27 | 0.89 | 4.83 |
| GS115-EP2 | 87.68 | 83.74 | 86.87 | 0.68 | 3.33 | 0.91 | 4.92 |
| PNP-Xyl | PNP-Glc | |||
|---|---|---|---|---|
| U mg−1 (×104) | Relative Activity (%) | U mg−1 (×104) | Relative Activity (%) | |
| LXYL-P1–2 | 5.03 (±0.14) | 100.00 | 12.05 (±0.49) | 100.00 |
| EP1 | 7.06 (±0.56) ** | 140.35 | 16.81 (±0.53) ** | 139.50 |
| EP2 | 8.12 (±0.63) ** | 161.44 | 15.82 (±0.85) ** | 131.29 |
| Vmax (μmol L−1 min−1) | Km (mmol L−1) | kcat (s−1) | kcat/Km (mmol L−1 s−1) | |
|---|---|---|---|---|
| LXYL-P1–2 | 8.16 (±0.15) | 0.51 (±0.01) | 4.89 (±0.10) | 9.67 (±0.10) |
| EP1 | 3.15 (±0.06) | 0.17 (±0.01) ** | 1.89 (±0.04) | 11.33 (±0.39) ** |
| EP2 | 3.84 (±0.05) | 0.16 (±0.01) ** | 2.30 (±0.03) | 14.17 (±0.81) ** |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.-J.; Liang, X.; Li, H.-X.; Chen, T.-J.; Zhu, P. Improving the Catalytic Property of the Glycoside Hydrolase LXYL-P1–2 by Directed Evolution. Molecules 2017, 22, 2133. https://doi.org/10.3390/molecules22122133
Chen J-J, Liang X, Li H-X, Chen T-J, Zhu P. Improving the Catalytic Property of the Glycoside Hydrolase LXYL-P1–2 by Directed Evolution. Molecules. 2017; 22(12):2133. https://doi.org/10.3390/molecules22122133
Chicago/Turabian StyleChen, Jing-Jing, Xiao Liang, Hui-Xian Li, Tian-Jiao Chen, and Ping Zhu. 2017. "Improving the Catalytic Property of the Glycoside Hydrolase LXYL-P1–2 by Directed Evolution" Molecules 22, no. 12: 2133. https://doi.org/10.3390/molecules22122133
APA StyleChen, J.-J., Liang, X., Li, H.-X., Chen, T.-J., & Zhu, P. (2017). Improving the Catalytic Property of the Glycoside Hydrolase LXYL-P1–2 by Directed Evolution. Molecules, 22(12), 2133. https://doi.org/10.3390/molecules22122133