Participation of the Halogens in Photochemical Reactions in Natural and Treated Waters


Abstract
1. Introduction
2. Sources and Speciation of RHS Produced from Halide Ions
2.1. Sensitized Photolysis
2.2. Oxidation of Halide Ions by Secondary Photo-Products
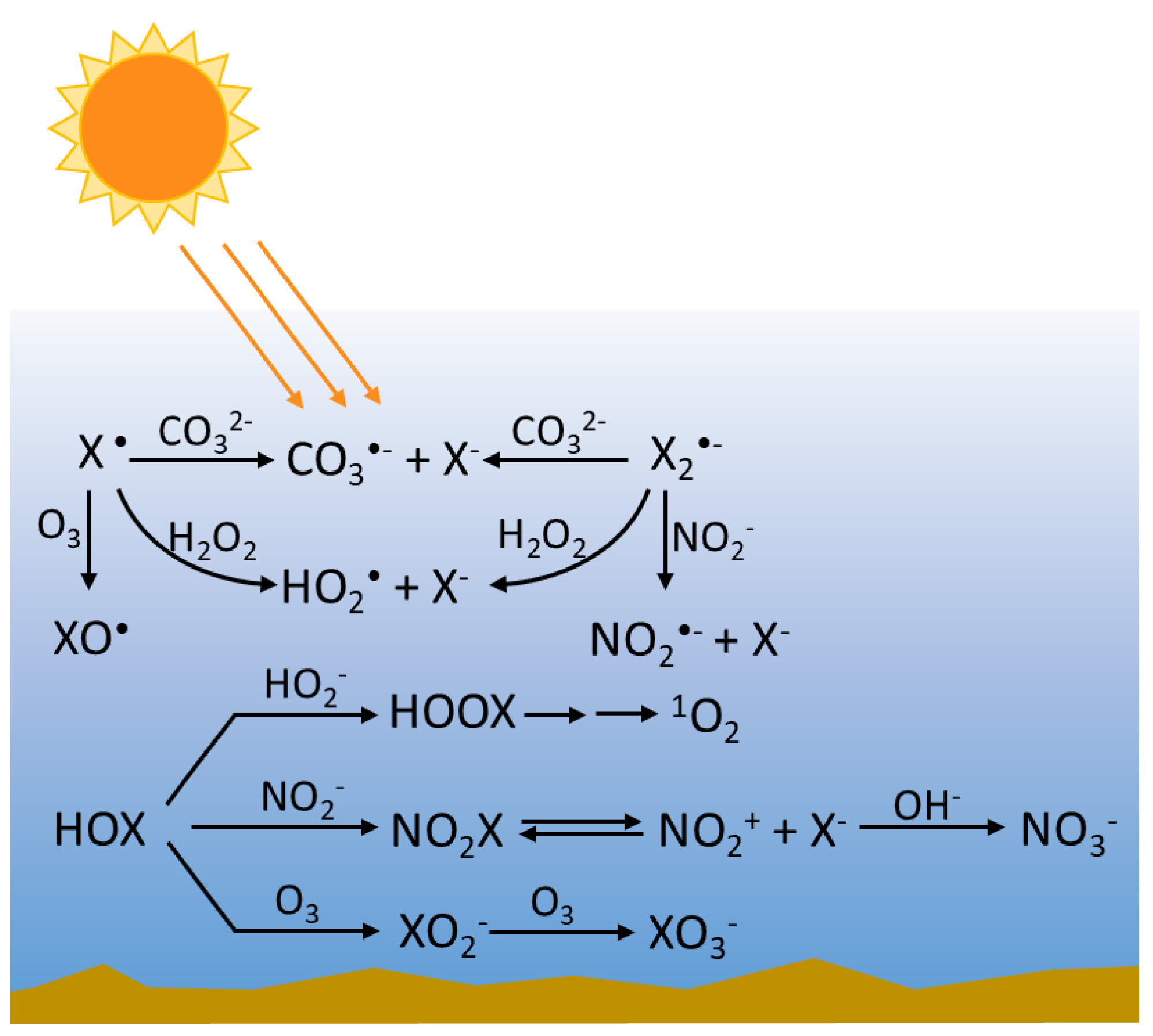
2.3. Heterogeneous Reactions Leading to RHS
- (i)
- The pH is often more acidic in the bulk liquid phase of aerosols than in terrestrial water bodies. By contrast, the air-liquid interface can be significantly more basic than the bulk aerosol phase; for example, it is known that the pH is 7 at the surface of bulk water at pH 3 [32].
- (ii)
- The heavier halide ions (Br−, I−) concentrate at the air-liquid interface. Evidence exists for unique chemical reactions close to the air-liquid interface [33].
- (iii)
- Particles may become depleted in bromide and iodide with respect to chloride, so that the chemistry can change over time.
- (iv)
- Reactions may be sensitive to humidity which governs film thickness.
2.4. Speciation and Interconversion of RHS in Waters
3. Reactions of RHS
3.1. Photolysis of nrRHS (X2, X3−, HOX)
3.2. Reactions of RHS with Inorganic Species
3.2.1. Radical RHS
3.2.2. Non-Radical RHS
4. Involvement of Halogen Species in Organic Matter Processing and Transformations of Organic Compounds
4.1. Impact of Halide Ions on Photoexcitation and Photobleaching of DOM
4.2. Reactions of RHS with Organic Compounds
4.2.1. Radical RHS
4.2.2. Non-Radical RHS
4.3. Photo-Initiated Incorporation of Halogen into Organic Compounds under Natural Conditions
4.3.1. Incorporation of Halogen into Simple, Defined Molecules
4.3.2. Incorporation of Halogen into Bulk DOM
5. Impacts of Halides on Water Treatment Processes
5.1. Hydroxyl Radical-Based AOPs
5.2. The UV/Chlorine AOP
5.3. Sulfate Radical-Based AOPs
6. Concluding Remarks
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Grebel, J.E.; Pignatello, J.J.; Mitch, W.A. Effect of halide ions and carbonates on organic contaminant degradation by hydroxyl radical-based advanced oxidation processes in saline waters. Environ. Sci. Technol. 2010, 44, 6822–6828. [Google Scholar] [CrossRef] [PubMed]
- Luther, G.W., III; Swartz, C.B.; Ullman, W.J. Direct determination of iodide in seawater by cathodic stripping square wave voltammetry. Anal. Chem. 1988, 60, 1721–1724. [Google Scholar] [CrossRef]
- Plewa, M.J.; Muellner, M.G.; Richardson, S.D.; Fasano, F.; Buettner, K.M.; Woo, Y.-T.; McKague, A.B.; Wagner, E.D. Occurrence, synthesis, and mammalian cell cytotoxicity and genotoxicity of haloacetamides: An emerging class of nitrogenous drinking water disinfection byproducts. Environ. Sci. Technol. 2007, 42, 955–961. [Google Scholar] [CrossRef]
- Richardson, S.D.; Plewa, M.J.; Wagner, E.D.; Schoeny, R.; DeMarini, D.M. Occurrence, genotoxicity, and carcinogenicity of regulated and emerging disinfection by-products in drinking water: A review and roadmap for research. Mutat. Res. Rev. Mutat. Res. 2007, 636, 178–242. [Google Scholar] [CrossRef] [PubMed]
- Simpson, W.R.; von Glasow, R.; Riedel, K.; Anderson, P.; Ariya, P.; Bottenheim, J.; Burrows, J.; Carpenter, L.J.; Frieß, U.; Goodsite, M.E.; et al. Halogens and their role in polar boundary-layer ozone depletion. Atmos. Chem. Phys. 2007, 7, 4375–4418. [Google Scholar] [CrossRef]
- Pratt, K.A.; Custard, K.D.; Shepson, P.B.; Douglas, T.A.; Pohler, D.; General, S.; Zielcke, J.; Simpson, W.R.; Platt, U.; Tanner, D.J.; et al. Photochemical production of molecular bromine in Arctic surface snowpacks. Nat. Geosci. 2013, 6, 351–356. [Google Scholar] [CrossRef]
- Jammoul, A.; Dumas, S.; D’Anna, B.; George, C. Photoinduced oxidation of sea salt halides by aromatic ketones: A source of halogenated radicals. Atmos. Chem. Phys. 2009, 9, 4229–4237. [Google Scholar] [CrossRef]
- Parker, K.M.; Mitch, W.A. Halogen radicals contribute to photooxidation in coastal and estuarine waters. Proc. Natl. Acad. Sci. USA 2016, 113, 5868–5873. [Google Scholar] [CrossRef] [PubMed]
- Zepp, R.G.; Schlotzhauer, P.F.; Sink, R.M. Photosensitized transformations involving electronic energy transfer in natural waters: Role of humic substances. Environ. Sci. Technol. 1985, 19, 74–81. [Google Scholar] [CrossRef]
- McNeill, K.; Canonica, S. Triplet state dissolved organic matter in aquatic photochemistry: Reaction mechanisms, substrate scope, and photophysical properties. Environ. Sci. Process Impacts 2016, 18, 1381–1399. [Google Scholar] [CrossRef] [PubMed]
- Canonica, S. Oxidation of aquatic organic contaminants induced by excited triplet states. Chim. Int. J. Chem. 2007, 61, 641–644. [Google Scholar] [CrossRef]
- Loeff, I.; Rabani, J.; Treinin, A.; Linschitz, H. Charge transfer and reactivity of nπ* and ππ* organic triplets, including anthraquinonesulfonates, in interactions with inorganic anions: A comparative study based on classical Marcus theory. J. Am. Chem. Soc. 1993, 115, 8933–8942. [Google Scholar] [CrossRef]
- Isse, A.A.; Lin, C.Y.; Coote, M.L.; Gennaro, A. Estimation of standard reduction potentials of halogen atoms and alkyl halides. J. Phys. Chem. B 2011, 115, 678–684. [Google Scholar] [CrossRef] [PubMed]
- De Laurentiis, E.; Minella, M.; Maurino, V.; Minero, C.; Mailhot, G.; Sarakha, M.; Brigante, M.; Vione, D. Assessing the occurrence of the dibromide radical (Br2−) in natural waters: Measures of triplet-sensitised formation, reactivity, and modelling. Sci. Total Environ. 2012, 439, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.K.; Linschitz, H.; Treinin, A. Interaction of halide and pseudohalide ions with triplet benzophenone-4-carboxylate: Kinetics and radical yields. J. Phys. Chem. 1988, 92, 5151–5159. [Google Scholar] [CrossRef]
- Wardman, P. Reduction potentials of one-electron couples involving free radicals in aqueous solution. J. Phys. Chem. Ref. Data 1989, 18, 1637–1755. [Google Scholar] [CrossRef]
- Mopper, K.; Zhou, X. Hydroxyl radical photoproduction in the sea and its potential impact on marine processes. Science 1990, 250, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, P.P.; Blough, N.V. Photochemical formation of hydroxyl radical by constituents of natural waters. Environ. Sci. Technol. 1998, 32, 2947–2953. [Google Scholar] [CrossRef]
- Sun, L.; Qian, J.; Blough, N.V.; Mopper, K. Insights into the photoproduction sites of hydroxyl radicals by dissolved organic matter in natural waters. Environ. Sci. Technol. Lett. 2015, 2, 352–356. [Google Scholar] [CrossRef]
- Jayson, G.G.; Parsons, B.J.; Swallow, A.J. Some Simple, Highly reactive, inorganic chlorine derivatives in aqueous solution. J. Chem. Soc., Faraday Trans. I 1973, 69, 1597–1607. [Google Scholar] [CrossRef]
- Pignatello, J. Dark and photoassisted Fe3+-catalyzed degradation of chlorophenoxy herbicides by hydrogen peroxide. Environ. Sci. Technol. 1992, 26, 944–951. [Google Scholar] [CrossRef]
- Ryerson, T.; Trainer, M.; Angevine, W.; Brock, C.; Dissly, R.; Fehsenfeld, F.; Frost, G.; Goldan, P.; Holloway, J.; Hübler, G. Effect of petrochemical industrial emissions of reactive alkenes and NOx on tropospheric ozone formation in Houston, Texas. J. Geophys. Res. Atmos. 2003, 108, 4249. [Google Scholar] [CrossRef]
- Zhang, R.; Lei, W.; Tie, X.; Hess, P. Industrial emissions cause extreme urban ozone diurnal variability. Proc. Natl. Acad. Sci. USA 2004, 101, 6346–6350. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Shao, M.; Che, W.; Zhang, L.; Zhong, L.; Zhang, Y.; Streets, D. Speciated VOC emission inventory and spatial patterns of ozone formation potential in the Pearl River Delta, China. Environ. Sci. Technol. 2009, 43, 8580–8586. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Schurter, L.M.; Muller, C.E.; Aloisio, S.; Francisco, J.S.; Margerum, D.W. Kinetics and mechanisms of aqueous ozone reactions with bromide, sulfite, hydrogen sulfite, iodide, and nitrite ions. Inorg. Chem. 2001, 40, 4436–4442. [Google Scholar] [CrossRef] [PubMed]
- Haag, W.R.; Hoigne, J. Ozonation of bromide-containing waters: Kinetics of formation of hypobromous acid and bromate. Environ. Sci. Technol. 1983, 17, 261–267. [Google Scholar] [CrossRef]
- Kumar, A.; Borgen, M.; Aluwihare, L.I.; Fenical, W. Ozone-activated halogenation of mono- and dimethylbipyrrole in seawater. Environ. Sci. Technol. 2017, 51, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Enami, S.; Vecitis, C.D.; Cheng, J.; Hoffmann, M.R.; Colussi, A.J. Global inorganic source of atmospheric bromine. J. Phys. Chem. A 2007, 111, 8749–8752. [Google Scholar] [CrossRef] [PubMed]
- Haag, W.R.; Gassman, E. Singlet oxygen in surface waters-Part I: Furfuryl alcohol as a trapping agent. Chemosphere 1984, 13, 631–640. [Google Scholar] [CrossRef]
- Finlayson-Pitts, B.J. The tropospheric chemistry of sea salt: A molecular-level view of the chemistry of NaCl and NaBr. Chem. Rev. 2003, 103, 4801–4822. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.J. Heterogeneous reactions on salts. Chem. Rev. 2003, 103, 4823–4882. [Google Scholar] [CrossRef] [PubMed]
- Mishra, H.; Enami, S.; Nielsen, R.J.; Stewart, L.A.; Hoffmann, M.R.; Goddard, W.A.; Colussi, A.J. Brønsted basicity of the air–water interface. Proc. Natl. Acad. Sci. USA 2012, 109, 18679–18683. [Google Scholar] [CrossRef] [PubMed]
- Enami, S.; Hoffmann, M.R.; Colussi, A.J. Halogen radical chemistry at aqueous interfaces. J. Phys. Chem. A 2016, 120, 6242–6248. [Google Scholar] [CrossRef] [PubMed]
- Wayne, R.P.; Barnes, I.; Biggs, P.; Burrows, J.P.; Canosa-Mas, C.E.; Hjorth, J.; Le Bras, G.; Moortgat, G.K.; Perner, D.; Poulet, G.; et al. The nitrate radical: Physics, chemistry, and the atmosphere. Atmos. Environ. Part A 1991, 25, 1–203. [Google Scholar] [CrossRef]
- Poskrebyshev, G.A.; Huie, R.E.; Neta, P. The rate and equilibrium constants for the reaction NO3• + Cl- ⇄ NO3− + Cl• in aqueous solutions. J. Phys. Chem. A 2003, 107, 1964–1970. [Google Scholar] [CrossRef]
- Neta, P.; Huie, R.E. Rate constants for reactions of NO3 radicals in aqueous solutions. J. Phys. Chem. 1986, 90, 4644–4648. [Google Scholar] [CrossRef]
- Schweitzer, F.; Mirabel, P.; George, C. Multiphase chemistry of N2O5, ClNO2, and BrNO2. J. Phys. Chem. A 1998, 102, 3942–3952. [Google Scholar] [CrossRef]
- Roberts, J.M.; Osthoff, H.D.; Brown, S.S.; Ravishankara, A.R. N2O5 Oxidizes chloride to Cl2 in acidic atmospheric aerosol. Science 2008, 321, 1059. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Shi, Q.; Davidovits, P.; Worsnop, D.R.; Zahniser, M.S.; Kolb, C.E. Reactive Uptake of Cl2(g) and Br2(g) by aqueous surfaces as a function of Br− and I− ion concentration: The effect of chemical reaction at the interface. J. Phys. Chem. 1995, 99, 8768–8776. [Google Scholar] [CrossRef]
- Wang, T.X.; Kelley, M.D.; Cooper, J.N.; Beckwith, R.C.; Margerum, D.W. Equilibrium, kinetic, and UV-spectral characteristics of aqueous bromine chloride, bromine, and chlorine species. Inorg. Chem. 1994, 33, 5872–5878. [Google Scholar] [CrossRef]
- Ianni, J.C. Kintecus V6.01. Available online: www.kintecus.com (accessed on 30 July 2016).
- Bichsel, Y.; von Gunten, U. Formation of iodo-trihalomethanes during disinfection and oxidation of iodide containing waters. Environ. Sci. Technol. 2000, 34, 2784–2791. [Google Scholar] [CrossRef]
- Troy, R.C.; Margerum, D.W. Non-metal redox kinetics: Hypobromite and hypobromous acid reactions with iodide and with sulfite and the hydrolysis of bromosulfate. Inorg. Chem. 1991, 30, 3538–3543. [Google Scholar] [CrossRef]
- Bichsel, Y.; von Gunten, U. Hypoiodous acid: Kinetics of the buffer-catalyzed disproportionation. Water Res. 2000, 34, 3197–3203. [Google Scholar] [CrossRef]
- Barkley, R.A.; Thompson, T.G. The total Iodine and Iodate-iodine content of sea-water. Deep Sea Res. 1960, 7, 24–34. [Google Scholar] [CrossRef]
- Chen, Z.; Megharaj, M.; Naidu, R. Speciation of iodate and iodide in seawater by non-suppressed ion chromatography with inductively coupled plasma mass spectrometry. Talanta 2007, 72, 1842–1846. [Google Scholar] [CrossRef] [PubMed]
- Kurylo, M.J.; Ouellette, P.A.; Laufer, A.H. Measurements of the pressure dependence of the hydroperoxy (HO2) radical self-disproportionation reaction at 298 K. J. Phys. Chem. 1986, 90, 437–440. [Google Scholar] [CrossRef]
- Barnes, R.J.; Lock, M.; Coleman, J.; Sinha, A. Observation of a new absorption band of HOBr and its atmospheric implications. J. Phys. Chem. 1996, 100, 453–457. [Google Scholar] [CrossRef]
- Haugen, H.K.; Weitz, E.; Leone, S.R. Accurate quantum yields by laser gain vs absorption spectroscopy: Investigation of Br/Br* channels in photofragmentation of Br2 and IBr. J. Phys. Chem. 1985, 83, 3402–3412. [Google Scholar] [CrossRef]
- Gershgoren, E.; Banin, U.; Ruhman, S. Caging and geminate recombination following photolysis of triiodide in solution. J. Phys. Chem. A 1998, 102, 9–16. [Google Scholar] [CrossRef]
- Callow, A.; Griffith, R.; McKeown, A. The photo-reaction between bromine and hydrogen peroxide in aqueous solution. Trans. Faraday Soc. 1939, 35, 412–420. [Google Scholar] [CrossRef]
- Treinin, A.; Hayon, E. Charge transfer spectra of halogen atoms in water. Correlation of the electronic transition energies of iodine, bromine, chlorine, hydroxyl, and hydrogen radicals with their electron affinities. J. Am. Chem. Soc. 1975, 97, 1716–1721. [Google Scholar] [CrossRef]
- Forsyth, J.E.; Zhou, P.; Mao, Q.; Asato, S.S.; Meschke, J.S.; Dodd, M.C. Enhanced inactivation of bacillus subtilis spores during solar photolysis of free available chlorine. Environ. Sci. Technol. 2013, 47, 12976–12984. [Google Scholar] [CrossRef] [PubMed]
- Jenkin, M.; Cox, R.; Hayman, G. Kinetics of the reaction of IO radicals with HO2 radicals at 298 K. Chem. Phys. Lett. 1991, 177, 272–278. [Google Scholar] [CrossRef]
- Francisco, J.S.; Hand, M.R.; Williams, I.H. Ab initio study of the electronic spectrum of HOBr. J. Phys. Chem. 1996, 100, 9250–9253. [Google Scholar] [CrossRef]
- Minaev, B.F. The singlet-triplet absorption and photodissociation of the HOCl, HOBr, and HOI molecules calculated by the MCSCF quadratic response method. J. Phys. Chem. A 1999, 103, 7294–7309. [Google Scholar] [CrossRef]
- Biedenkapp, D.; Hartshorn, L.G.; Bair, E.J. The O (1D)+ H2O reaction. Chem. Phys. Lett. 1970, 5, 379–381. [Google Scholar] [CrossRef]
- Poskrebyshev, G.A.; Neta, P.; Huie, R.E. Temperature dependence of the acid dissociation constant of the hydroxyl radical. J. Phys. Chem. A 2002, 106, 11488–11491. [Google Scholar] [CrossRef]
- Buxton, G.; Subhani, M. Radiation chemistry and photochemistry of oxychlorine ions. Part 2.—Photodecomposition of aqueous solutions of hypochlorite ions. J. Chem. Soc. Faraday Trans. 1 1972, 68, 958–969. [Google Scholar] [CrossRef]
- Orlando, J.J.; Burkholder, J.B. Gas-phase UV/Visible absorption spectra of HOBr and Br2O. J. Phys. Chem. 1995, 99, 1143–1150. [Google Scholar] [CrossRef]
- Rowley, D.M.; Mössinger, J.C.; Cox, R.A.; Jones, R.L. The UV-visible absorption cross-sections and atmospheric photolysis rate of HOI. JAtC 1999, 34, 137–151. [Google Scholar]
- Palmer, D.A.; Van Eldik, R. Spectral characterization and kinetics of formation of hypoiodous acid in aqueous solution. Inorg. Chem. 1986, 25, 928–931. [Google Scholar] [CrossRef]
- Deborde, M.; von Gunten, U. Reactions of chlorine with inorganic and organic compounds during water treatment—Kinetics and mechanisms: A critical review. Water Res. 2008, 42, 13–51. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Pignatello, J.J.; Ma, J.; Mitch, W.A. Comparison of halide impacts on the efficiency of contaminant degradation by sulfate and hydroxyl radical-based advanced oxidation processes (AOPs). Environ. Sci. Technol. 2014, 48, 2344–2351. [Google Scholar] [CrossRef] [PubMed]
- Minakata, D.; Kamath, D.; Maetzold, S. Mechanistic insight into the reactivity of chlorine-derived radicals in the aqueous-phase UV–chlorine advanced oxidation process: Quantum mechanical calculations. Environ. Sci. Technol. 2017, 51, 6918–6926. [Google Scholar] [CrossRef] [PubMed]
- Von Gunten, U.; Oliveras, Y. Kinetics of the reaction between hydrogen peroxide and hypobromous acid: Implication on water treatment and natural systems. Water Res. 1997, 31, 900–906. [Google Scholar] [CrossRef]
- Cerkovnik, J.; Plesničar, B. Recent Advances in the chemistry of hydrogen trioxide (HOOOH). Chem. Rev. 2013, 113, 7930–7951. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.W.; Margerum, D.W. Non-metal redox kinetics: A reexamination of the mechanism of the reaction between hypochlorite and nitrite ions. Inorg. Chem. 1991, 30, 4845–4851. [Google Scholar] [CrossRef]
- Lahoutifard, N.; Lagrange, P.; Lagrange, J.; Scott, S.L. Kinetics and mechanism of nitrite oxidation by HOBr/BrO− in atmospheric water and comparison with oxidation by HOCl/ClO−. J. Phys. Chem. A 2002, 106, 11891–11896. [Google Scholar] [CrossRef]
- Von Gunten, U. Ozonation of drinking water: Part II. Disinfection and by-product formation in presence of bromide, iodide or chlorine. Water Res. 2003, 37, 1469–1487. [Google Scholar] [CrossRef]
- Environmental Protection Agency, USA. National drinking water regulations: Disinfectants and disinfection byproducts. Fed. Regist. 1998, 63, 69390–69476. [Google Scholar]
- Grebel, J.E.; Pignatello, J.J.; Mitch, W.A. Impact of halide ions on natural organic matter-sensitized photolysis of 17-β-estradiol in saline waters. Environ. Sci. Technol. 2012, 46, 7128–7134. [Google Scholar] [CrossRef] [PubMed]
- Grebel, J.E.; Pignatello, J.J.; Mitch, W.A. Sorbic acid as a quantitative probe for the formation, scavenging and steady-state concentrations of the triplet-excited state of organic compounds. Water Res. 2011, 45, 6535–6544. [Google Scholar] [CrossRef] [PubMed]
- Parker, K.M.; Pignatello, J.J.; Mitch, W.A. Influence of salinity on triplet-state natural organic matter loss by energy transfer and electron transfer pathways. Environ. Sci. Technol. 2013, 47, 10987–10994. [Google Scholar] [CrossRef] [PubMed]
- Glover, C.M.; Rosario-Ortiz, F.L. Impact of halides on the photoproduction of reactive intermediates from organic matter. Environ. Sci. Technol. 2013, 47, 13949–13956. [Google Scholar] [CrossRef] [PubMed]
- Grebel, J.E.; Pignatello, J.J.; Song, W.; Cooper, W.J.; Mitch, W.A. Impact of halides on the photobleaching of dissolved organic matter. Mar. Chem. 2009, 115, 134–144. [Google Scholar] [CrossRef]
- Song, G.; Li, Y.; Hu, S.; Li, G.; Zhao, R.; Sun, X.; Xie, H. Photobleaching of chromophoric dissolved organic matter (CDOM) in the Yangtze River estuary: Kinetics and effects of temperature, pH, and salinity. Environ. Sci. Process Impacts 2017, 19, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.A.; Weber, E.J. Reaction Mechanisms in Environmental Organic Chemistry; Lewis Publishers: Boca Ratan, FL, USA, 1994. [Google Scholar]
- Wicktor, F.; Donati, A.; Herrmann, H.; Zellner, R. Laser based spectroscopic and kinetic investigations of reactions of the Cl atom with oxygenated hydrocarbons in aqueous solution. Phys. Chem. Chem. Phys. 2003, 5, 2562–2572. [Google Scholar] [CrossRef]
- Hasegawa, K.; Neta, P. Rate constants and mechanisms of reactions of Cl2− radicals. J. Phys. Chem. 1978, 82, 854–857. [Google Scholar] [CrossRef]
- Ershov, B.G.; Kelm, M.; Gordeev, A.V.; Janata, E. A pulse radiolysis study of the oxidation of Br- by dichloro radical anion in aqueous solution: Formation and properties of chlorobromo radical anion. Phys. Chem. Chem. Phys. 2002, 4, 1872–1875. [Google Scholar] [CrossRef]
- Lee, Y.; Yoon, J.; von Gunten, U. Kinetics of the oxidation of phenols and phenolic endocrine disruptors during water treatment with ferrate (Fe(VI)). Environ. Sci. Technol. 2005, 39, 8978–8984. [Google Scholar] [CrossRef] [PubMed]
- Criquet, J.; Rodriguez, E.M.; Allard, S.; Wellauer, S.; Salhi, E.; Joll, C.A.; von Gunten, U. Reaction of bromine and chlorine with phenolic compounds and natural organic matter extracts—Electrophilic aromatic substitution and oxidation. Water Res. 2015, 85, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Gribble, G.W. Naturally Occurring Organohalogen Compopunds--A Comprehensive Update; Springer: Wien, Austria; New York, NY, USA, 2010. [Google Scholar]
- Jeffers, P.M.; Wolfe, N.L. On the degradation of methyl bromide in sea water. Geophys. Res. Lett. 1996, 23, 1773–1776. [Google Scholar] [CrossRef]
- Moore, R.M. A photochemical source of methyl chloride in saline waters. Environ. Sci. Technol. 2008, 42, 1933–1937. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.M.; Zafiriou, O.C. Photochemical production of methyl iodide in seawater. J. Geophys. Res. Atmos. 1994, 99, 16415–16420. [Google Scholar] [CrossRef]
- Martino, M.; Mills, G.P.; Woeltjen, J.; Liss, P.S. A new source of volatile organoiodine compounds in surface seawater. Geophys. Res. Lett. 2009, 36. [Google Scholar] [CrossRef]
- Jones, C.E.; Carpenter, L.J. Solar Photolysis of CH2I2, CH2ICl, and CH2IBr in water, saltwater, and seawater. Environ. Sci. Technol. 2006, 40, 1372. [Google Scholar] [CrossRef]
- Anastasio, C.; Matthew, B.M. A chemical probe technique for the determination of reactive halogen species in aqueous solution: Part 2—Chloride solutions and mixed bromide/chloride solutions. Atmos. Chem. Phys. 2006, 6, 2439–2451. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, H.; Quan, X.; Zhang, Y.; Chen, S. Formation of chlorinated intermediate from bisphenol a in surface saline water under simulated solar light irradiation. Environ. Sci. Technol. 2009, 43, 7712–7717. [Google Scholar] [CrossRef] [PubMed]
- Tamtam, F.; Chiron, S. New insight into photo-bromination processes in saline surface waters: The case of salicylic acid. Sci. Total Environ. 2012, 435, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Narukawa, M.; Kawamura, K.; Hatsushika, H.; Yamazaki, K.; Li, S.-M.; Bottenheim, J.W.; Anlauf, K.G. Measurement of halogenated dicarboxylic acids in the arctic aerosols at polar sunrise. J. Atmos. Chem. 2003, 44, 323–335. [Google Scholar] [CrossRef]
- Méndez-Díaz, J.D.; Shimabuku, K.K.; Ma, J.; Enumah, Z.O.; Pignatello, J.J.; Mitch, W.A.; Dodd, M.C. Sunlight-driven photochemical halogenation of dissolved organic matter in seawater: A natural abiotic source of organobromine and organoiodine. Environ. Sci. Technol. 2014, 48, 7418–7427. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Yin, Y.; Cao, D.; Liu, J. Probing and comparing the photobromination and photoiodination of dissolved organic matter by using ultra-high-resolution mass spectrometry. Environ. Sci. Technol. 2017, 51, 5464–5472. [Google Scholar] [CrossRef] [PubMed]
- Jortner, J.; Ottolenghi, M.; Stein, G. On the photochemistry of aqueous solutions of chloride, bromide, and iodide ions. J. Phys. Chem. 1964, 68, 247–255. [Google Scholar] [CrossRef]
- Kalmar, J.; Doka, E.; Lente, G.; Fabian, I. Aqueous photochemical reactions of chloride, bromide, and iodide ions in a diode-array spectrophotometer. Autoinhibition in the photolysis of iodide ions. Dalton Trans. 2014, 43, 4862–4870. [Google Scholar] [CrossRef] [PubMed]
- Kiwi, J.; Lopez, A.; Nadtochenko, V. Mechanism and kinetics of the OH-radical intervention during Fenton oxidation in the presence of a significant amount of radical scavenger (Cl−). Environ. Sci. Technol. 2000, 34, 2162–2168. [Google Scholar] [CrossRef]
- Pignatello, J.J.; Oliveros, E.; MacKay, A. Advanced oxidation processes for organic contaminant destruction based on the Fenton reaction and related chemistry. Crit. Rev. Env. Sci. Technol. 2006, 36, 1–84. [Google Scholar] [CrossRef]
- Machulek, A.; Moraes, J.E.F.; Vautier-Giongo, C.; Silverio, C.A.; Friedrich, L.C.; Nascimento, C.A.O.; Gonzalez, M.C.; Quina, F.H. Abatement of the inhibitory effect of chloride anions on the photo-fenton process. Environ. Sci. Technol. 2007, 41, 8459–8463. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.; Ramjaun, S.N.; Wang, Z.; Liu, J. Photocatalytic degradation and chlorination of azo dye in saline wastewater: Kinetics and AOX formation. Chem. Eng. J. 2012, 192, 171–178. [Google Scholar] [CrossRef]
- Yamazaki, S.; Tanimura, T.; Yoshida, A.; Hori, K. Reaction mechanism of photocatalytic degradation of chlorinated ethylenes on porous TiO2 pellets: Cl radical-initiated mechanism. J. Phys. Chem. A 2004, 108, 5183–5188. [Google Scholar] [CrossRef]
- Kormann, C.; Bahnemann, D.; Hoffmann, M.R. Photolysis of chloroform and other organic molecules in aqueous titanium dioxide suspensions. Environ. Sci. Technol. 1991, 25, 494–500. [Google Scholar] [CrossRef]
- Wu, Z.; Fang, J.; Xiang, Y.; Shang, C.; Li, X.; Meng, F.; Yang, X. Roles of reactive chlorine species in trimethoprim degradation in the UV/chlorine process: Kinetics and transformation pathways. Water Res. 2016, 104, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Heeb, M.B.; Criquet, J.; Zimmermann-Steffens, S.G.; von Gunten, U. Oxidative treatment of bromide-containing waters: Formation of bromine and its reactions with inorganic and organic compounds—A critical review. Water Res. 2014, 48, 15–42. [Google Scholar] [CrossRef] [PubMed]
- Benter, T.; Feldmann, C.R.; Kirchner, U.; Schmidt, M.; Schmidt, S.; Schindler, R. UV/VIS-absorption Spectra of HOBr and CH3OBr; Br(2P3/2) atom yields in the photolysis of HOBr. Ber. Bunsenges. Phys. Chem. 1995, 99, 1144–1147. [Google Scholar] [CrossRef]
- Criquet, J.; Allard, S.; Salhi, E.; Joll, C.A.; Heitz, A.; von Gunten, U. Iodate and iodo-trihalomethane formation during chlorination of iodide-containing waters: Role of bromide. Environ. Sci. Technol. 2012, 46, 7350–7357. [Google Scholar] [CrossRef] [PubMed]
- Allard, S.; Tan, J.; Joll, C.A.; von Gunten, U. Mechanistic study on the formation of Cl−/Br−/I− trihalomethanes during chlorination/chloramination combined with a theoretical cytotoxicity evaluation. Environ. Sci. Technol. 2015, 49, 11105–11114. [Google Scholar] [CrossRef] [PubMed]
- Mark, G.; Schuchmann, M.N.; Schuchmann, H.-P.; von Sonntag, C. The photolysis of potassium peroxodisulphate in aqueous solution in the presence of tert-butanol: A simple actinometer for 254 nm radiation. J. Photochem. Photobiol. A Chem. 1990, 55, 157–168. [Google Scholar] [CrossRef]
- Baxendale, J.; Wilson, J. The photolysis of hydrogen peroxide at high light intensities. Trans. Faraday Soc. 1957, 53, 344–356. [Google Scholar] [CrossRef]
- Guan, Y.-H.; Ma, J.; Li, X.-C.; Fang, J.-Y.; Chen, L.-W. Influence of pH on the formation of sulfate and hydroxyl radicals in the UV/peroxymonosulfate system. Environ. Sci. Technol. 2011, 45, 9308–9314. [Google Scholar] [CrossRef] [PubMed]
- Beitz, T.; Bechmann, W.; Mitzner, R. Investigations of reactions of selected azaarenes with radicals in water. 2. Chlorine and bromine radicals. J. Phys. Chem. A 1998, 102, 6766–6771. [Google Scholar] [CrossRef]
- Canonica, S.; Kohn, T.; Mac, M.; Real, F.J.; Wirz, J.; von Gunten, U. Photosensitizer method to determine rate constants for the reaction of carbonate radical with organic compounds. Environ. Sci. Technol. 2005, 39, 9182–9188. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Pignatello, J.J.; Ma, J.; Mitch, W.A. Effect of matrix components on UV/H2O2 and UV/S2O82− advanced oxidation processes for trace organic degradation in reverse osmosis brines from municipal wastewater reuse facilities. Water Res. 2016, 89, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.Y.; Shang, C. Bromate formation from bromide oxidation by the UV/Persulfate process. Environ. Sci. Technol. 2012, 46, 8976–8983. [Google Scholar] [CrossRef] [PubMed]
- Lutze, H.V.; Bakkour, R.; Kerlin, N.; von Sonntag, C.; Schmidt, T.C. Formation of bromate in sulfate radical based oxidation: Mechanistic aspects and suppression by dissolved organic matter. Water Res. 2014, 53, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Fawell, J.; Walker, M. Approaches to determining regulatory values for carcinogens with particular reference to bromate. Toxicology 2006, 221, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Kläning, U.K.; Wolff, T. Laser flash photolysis of HCIO, CIO−, HBrO, and BrO− in aqueous solution. reactions of Cl-and Br-atoms. Ber. Bunsenges. Phys. Chem. 1985, 89, 243–245. [Google Scholar] [CrossRef]
- Fortnum, D.H.; Battaglia, C.J.; Cohen, S.R.; Edwards, J.O. The kinetics of the oxidation of halide ions by monosubstituted peroxides. J. Am. Chem. Soc. 1960, 82, 778–782. [Google Scholar] [CrossRef]
- Lente, G.; Kalmár, J.; Baranyai, Z.; Kun, A.; Kék, I.; Bajusz, D.; Takács, M.; Veres, L.; Fábián, I. One-versus two-electron oxidation with peroxomonosulfate ion: Reactions with iron(II), vanadium(IV), halide ions, and photoreaction with cerium(III). Inorg. Chem. 2009, 48, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jiang, J.; Zhou, Y.; Pang, S.-Y.; Gao, Y.; Jiang, C.; Ma, J.; Jin, Y.; Yang, Y.; Liu, G.; et al. Kinetics of oxidation of iodide (I−) and hypoiodous acid (HOI) by peroxymonosulfate (PMS) and formation of iodinated products in the PMS/I−/NOM system. Environ. Sci. Technol. Lett. 2017, 4, 76–82. [Google Scholar] [CrossRef]
- Pan, Y.; Cheng, S.; Yang, X.; Ren, J.; Fang, J.; Shang, C.; Song, W.; Lian, L.; Zhang, X. UV/chlorine treatment of carbamazepine: Transformation products and their formation kinetics. Water Res. 2017, 116, 254–265. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |







{kind=link}
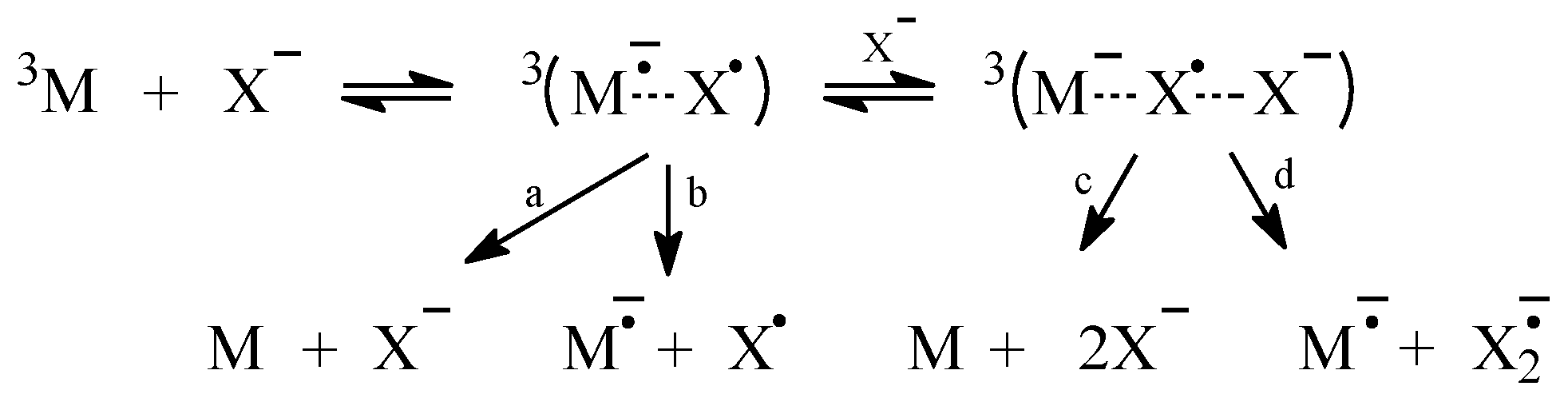{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RHS/Cl2 | Br2 | BrCl | Cl3− | BrCl2− | Br2Cl− | Br3− | HOBr/OBr− | HOCl/OCl− |
|---|---|---|---|---|---|---|---|---|
| Wastewater | 4.01 × 103 | 2.27 | 0.0257 | 33.5 | 417 | 3.5 | 0.95 × 109 (1.74 × 1010) * | 2.57 × 105 (5.92 × 105) * |
| Seawater | 1.04 × 104 | 24.7 | 0.0982 | 533 | 3800 | 145 | 6.42 × 109 (7.08 × 109) * | 1.79 × 105 (6.2 × 105) * |
| Compound | Proposed Origin | References |
|---|---|---|
| CH3Cl | (a) nucleophilic displacement by chloride on CH3I and/or CH3Br in seawater; (b) is produced on irradiation of lignin-like DOM model compounds (4-methoxy-1-naphthol; syringic acid; 2-methoxyphenol; 3,4,5-trimethoxy benzoic acid; and2-methoxyhydroquinone) in chloride solution | (a) [85] (b) [86] |
| CH3I | formed after simulated solar irradiation of filtered seawater; production was enhanced when samples were degassed or iodide was added; proposed origin is recombination of CH3• and I• radicals. | [87] |
| CH2I2, CHI3, and CHI2Cl | formed by reactions of DOM with HOI generated via oxidation of I− by O3 | [88] |
| CH2ICl | photolysis product of CH2I2 in seawater | [89] |
| Cl-CH2CH(OH)CH2OH and Br-CH2CH(OH)CH2OH | CH2=CHCH2OH reaction with reactive halogen species | [90] |
| 3-Cl and 3,3-diCl bisphenol A | solar irradiation of bisphenol A in coastal seawater and saline solution containing 0.13–0.66 mM Fe(III) and fulvic acid; Cl2−• was detected by its absorption spectrum, and OH• as its DMPO adduct by EPR spectroscopy; proposed source of halogen radicals: FeIIICl− → FeII + Cl• or FeIIIOH− → FeII + OH•, followed by OH• + Cl− → Cl•. | [91] |
| 5-bromo-and 3,5-dibromosalicylic acids | solar irradiation of salicylic acid in artificial seawater and brackish lagoon water | [92] |
| mixed poly-brominated/chlorinated bipyrroles | irradiation of 1,1-dimethyl-2,2′-bipyrrole and 1’-methyl-1,2’bipyrrole in ozonated seawater; proposed oxidation of Br− and I− by O3 to form HOX/X2. | [27] |
| halogenated dicarboxylic acids | isolated from arctic aerosols; unclear whether transformations occurred in the liquid phase | [93] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Pignatello, J.J. Participation of the Halogens in Photochemical Reactions in Natural and Treated Waters. Molecules 2017, 22, 1684. https://doi.org/10.3390/molecules22101684
Yang Y, Pignatello JJ. Participation of the Halogens in Photochemical Reactions in Natural and Treated Waters. Molecules. 2017; 22(10):1684. https://doi.org/10.3390/molecules22101684
Chicago/Turabian StyleYang, Yi, and Joseph J. Pignatello. 2017. "Participation of the Halogens in Photochemical Reactions in Natural and Treated Waters" Molecules 22, no. 10: 1684. https://doi.org/10.3390/molecules22101684
APA StyleYang, Y., & Pignatello, J. J. (2017). Participation of the Halogens in Photochemical Reactions in Natural and Treated Waters. Molecules, 22(10), 1684. https://doi.org/10.3390/molecules22101684