
Effective Synthesis of Nucleosides Utilizing O-Acetyl-Glycosyl Chlorides as Glycosyl Donors in the Absence of Catalyst: Mechanism Revision and Application to Silyl-Hilbert-Johnson Reaction



Abstract
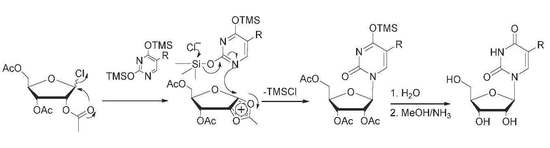
:
1. Introduction
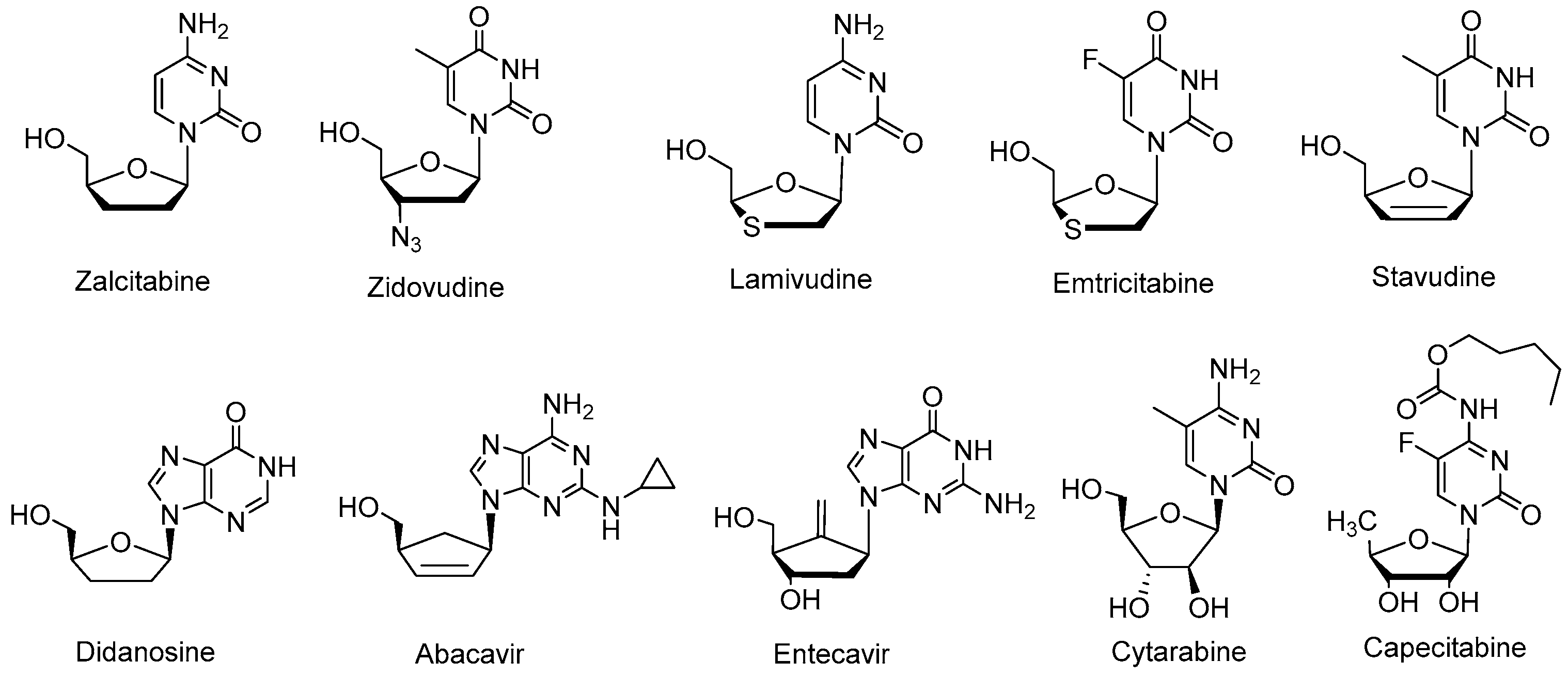
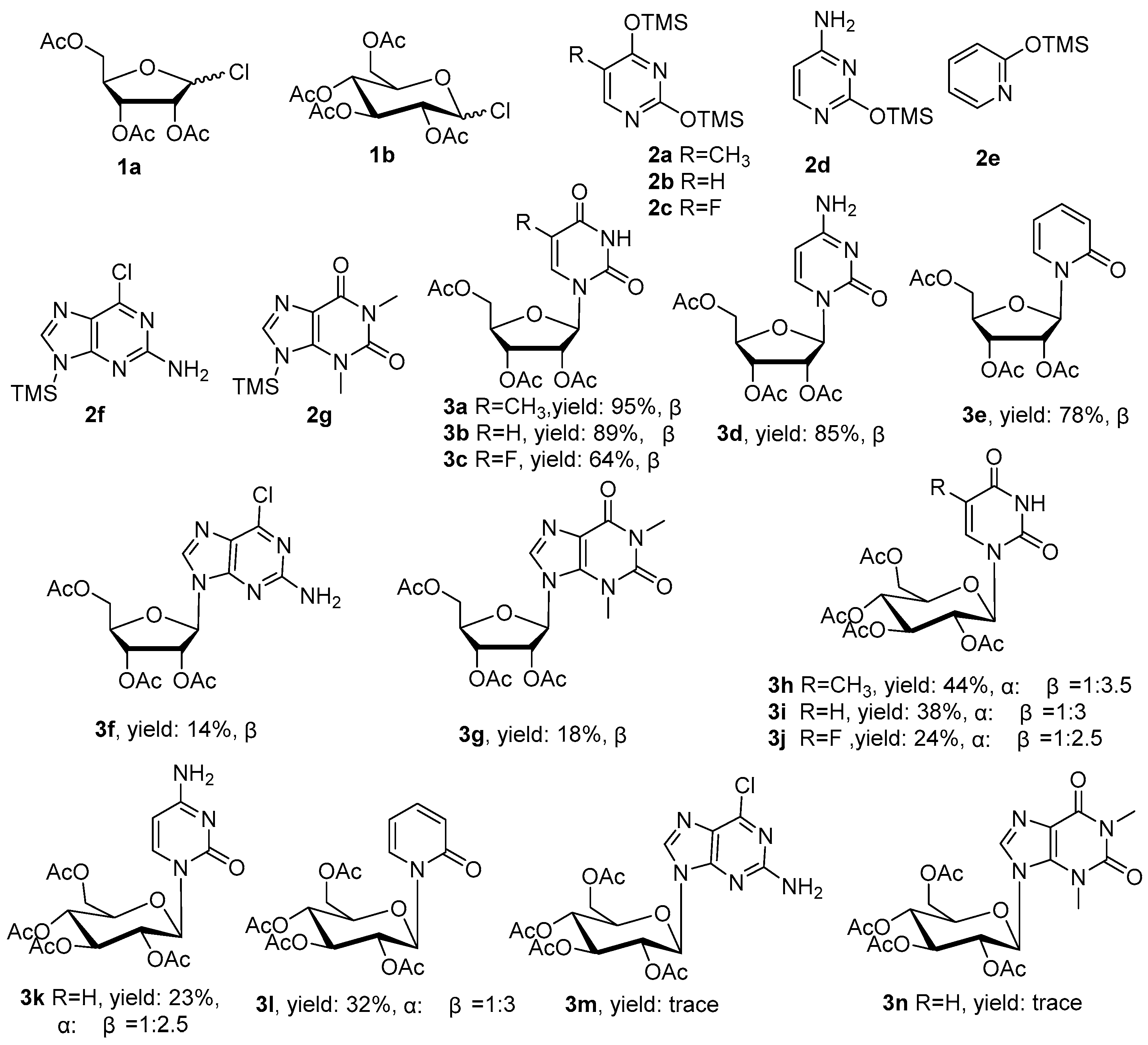
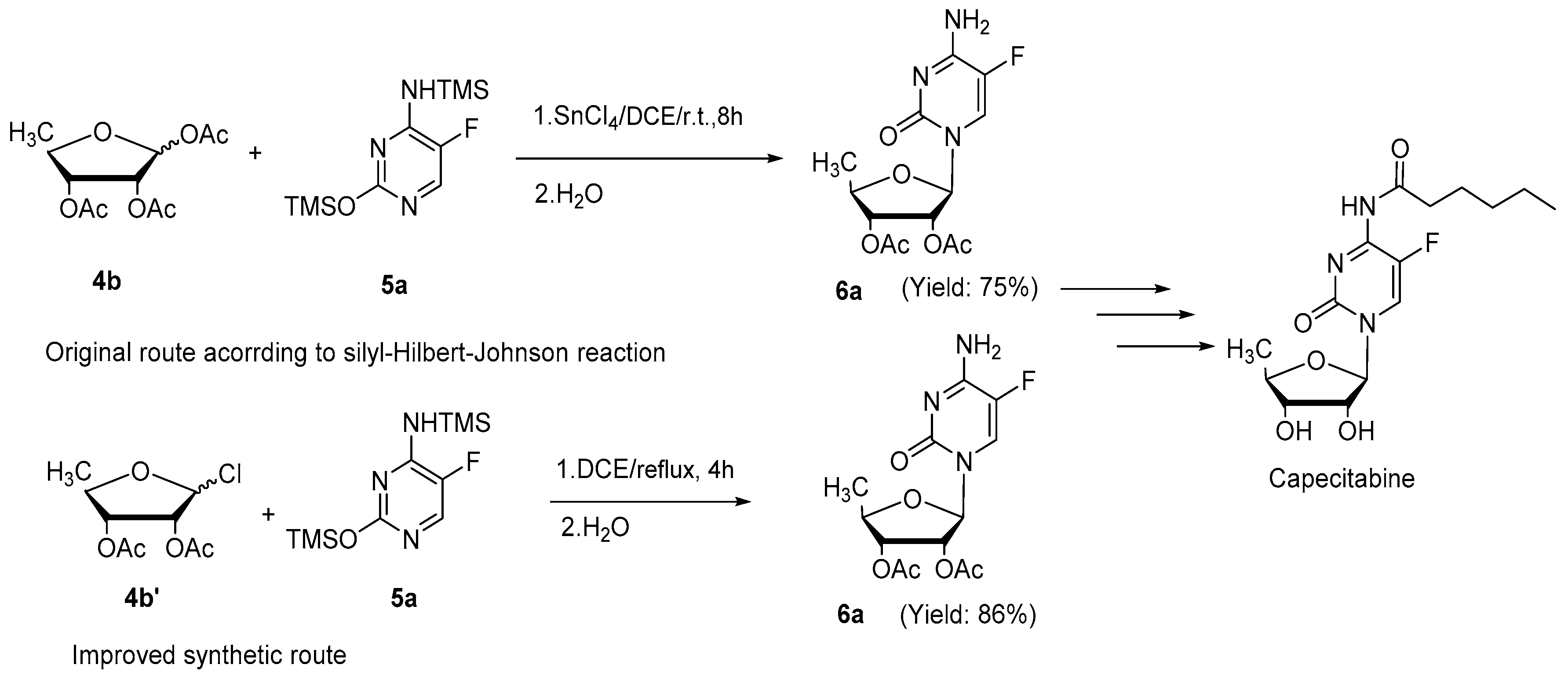
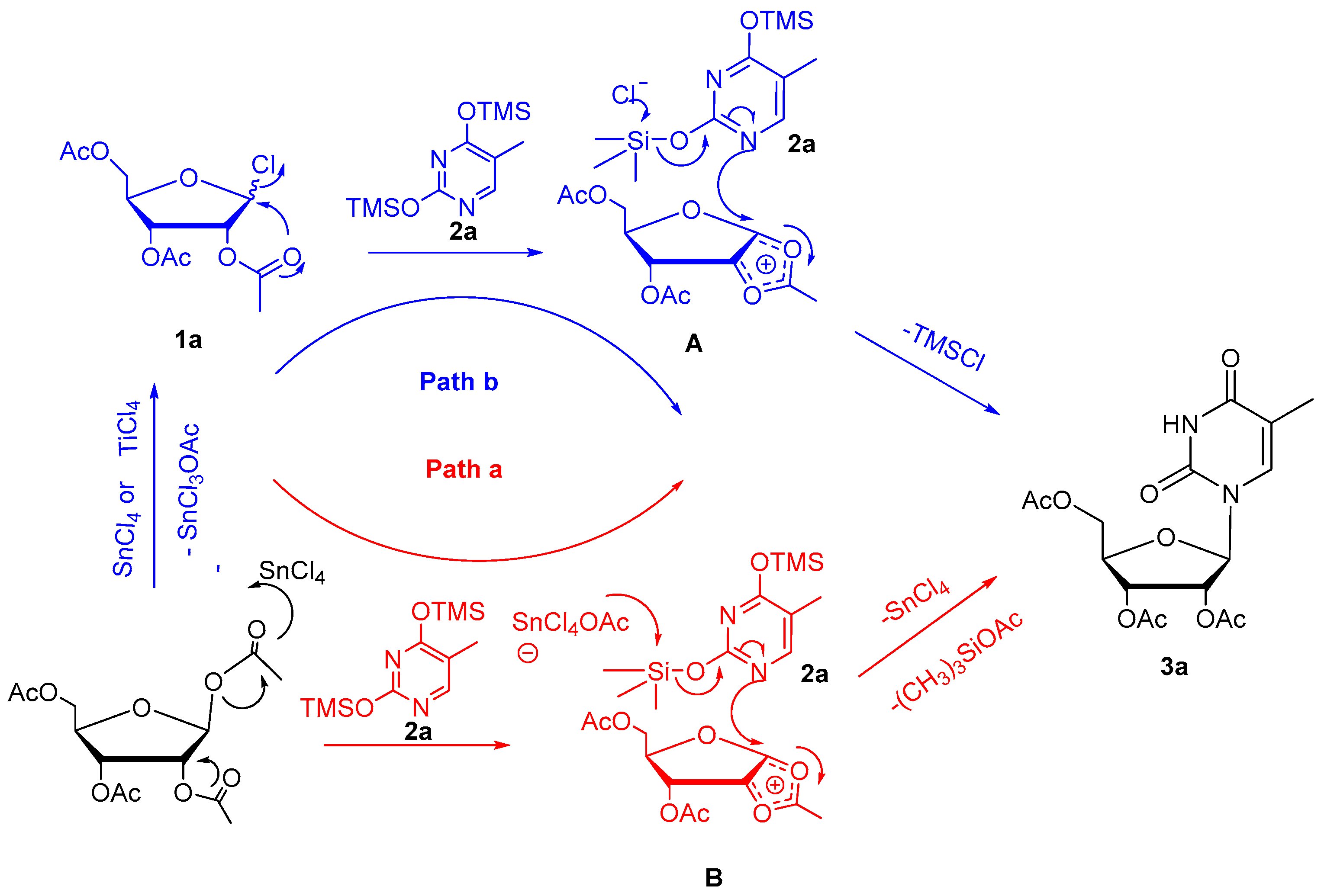
2. Results and Discussion
3. Experimental Section
General Experimental Procedure for Synthesis of Acetyl Ribosylthymine (3a)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Crespan, E.; Garbelli, A.; Amoroso, A.; Maga, G. Exploiting the Nucleotide Substrate Specificity of Repair DNA Polymerases to Develop Novel Anticancer Agents. Molecules 2011, 16, 7994–8019. [Google Scholar] [CrossRef] [PubMed]
- Robak, T.; Korycka, A.; Lech-Maranda, E.; Robak, P. Current Status of Older and New Purine Nucleoside Analogues in the Treatment of Lymphoproliferative Diseases. Molecules 2009, 14, 1183–1226. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Schwidom, D.; Exner, A.; Carlsen, P. Synthesis of Novel Homo-N-Nucleoside Analogs Composed of a Homo-1,4-Dioxane Sugar Analog and Substituted 1,3,5-Triazine Base Equivalents. Molecules 2008, 13, 3092–3106. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Carlsen, P. Enantioselective Synthesis of Homo-N-Nucleosides Containing a 1,4-Dioxane Sugar Analog. Molecules 2008, 13, 2962–2974. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-K.; Rosemeyer, H. Perspectives in Nucleoside and Nucleic Acid Chemistry. Molecules 2001, 6, 1030–1033. [Google Scholar]
- Rachakonda, S.; Cartee, L. Challenges in Antimicrobial Drug Discovery and the Potential of Nucleoside Antibiotics. Curr. Med. Chem. 2004, 11, 775–793. [Google Scholar] [CrossRef] [PubMed]
- Vitali, L.A.; Petrelli, D.; Lambertucci, C.; Prenna, M.; Volpini, R.; Cristalli, G. In vitro antibacterial activity of different adenosine analogues. J. Med. Microbiol. 2012, 61, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Xavier, N.M.; Schwarz, S.; Vaz, P.D.; Csuk, R.; Rauter, A.P. Synthesis of Purine Nucleosides from d-Glucuronic Acid Derivatives and Evaluation of Their Cholinesterase-Inhibitory Activities. Eur. J. Org. Chem. 2014, 2014, 2770–2779. [Google Scholar] [CrossRef]
- Schwarz, S.; Csuk, R.; Rauter, A.P. Microwave-assisted synthesis of novel purine nucleosides as selective cholinesterase inhibitors. Org. Biomol. Chem. 2014, 12, 2446–2456. [Google Scholar] [CrossRef] [PubMed]
- Batista, D.; Schwarz, S.; Loesche, A.; Csuk, R.; Costa, P.J.; Oliveira, M.C.; Xavier, N.M. Synthesis of glucopyranos-6′-yl purine and pyrimidine isonucleosides as potential cholinesterase inhibitors. Access to pyrimidine-linked pseudodisaccharides through Mitsunobu reaction. Pure Appl. Chem. 2016, 88, 363–379. [Google Scholar] [CrossRef]
- Lin, T.S.; Luo, M.Z.; Liu, M.C.; Pai, S.B.; Dutschman, G.E.; Cheng, Y.C. Synthesis and biological evaluation of 2′,3′-dideoxy-l-pyrimidine nucleosides as potential antiviral agents against human immunodeficiency virus (HIV) and hepatitis B virus (HBV). J. Med. Chem. 1994, 37, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Pai, S.B.; Zhu, Y.L.; Lin, J.S.; Shanmuganathan, K.; Du, J.; Wang, C.; Kim, H.; Newton, M.G.; Cheng, Y.C. Structure-activity relationships of 1-(2-deoxy-2-fluoro-β-l-arabino-furanosyl) pyrimidine nucleosides as anti-hepatitis B virus agents. J. Med. Chem. 1996, 39, 2835–2843. [Google Scholar] [CrossRef] [PubMed]
- Suzich, J.; Tamura, J.; Palmer-Hill, F.; Warrener, P.; Grakoui, A.; Rice, C.; Feinstone, S.; Collett, M. Hepatitis C virus NS3 protein polynucleotide-stimulated nucleoside triphosphatase and comparison with the related pestivirus and flavivirus enzymes. J. Virol. 1993, 67, 6152–6158. [Google Scholar] [PubMed]
- Klumpp, K.; Lévêque, V.; Le Pogam, S.; Ma, H.; Jiang, W.R.; Kang, H.; Granycome, C.; Singer, M.; Laxton, C.; Hang, J.Q. The novel nucleoside analog R1479 (4′-azidocytidine) is a potent inhibitor of NS5B-dependent RNA synthesis and hepatitis C virus replication in cell culture. J. Biol. Chem. 2006, 281, 3793–3799. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, J.; Keller, P.; Furman, P.; Miller, R.; Elion, G. Thymidine kinase from herpes simplex virus phosphorylates the new antiviral compound, 9-(2-hydroxyethoxymethyl) guanine. J. Biol. Chem. 1978, 253, 8721–8727. [Google Scholar] [PubMed]
- Smith, K.O.; Galloway, K.S.; Kennell, W.L.; Ogilvie, K.K.; Radatus, B.K. A new nucleoside analog, 9-[[2-hydroxy-1-(hydroxymethyl)ethoxyl] methyl] guanine, highly active in vitro against herpes simplex virus types 1 and 2. Antimicrob. Agents Chemother. 1982, 22, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Vince, R.; Hua, M.; Brownell, J.; Daluge, S.; Lee, F.; Shannon, W.M.; Lavelle, G.C.; Qualls, J.; Weislow, O.S.; Kiser, R. Potent and selective activity of a new carbocyclic nucleoside analog (carbovir: NSC 614846) against human immunodeficiency virus in vitro. Biochem. Biophys. Res. Commun. 1988, 156, 1046–1053. [Google Scholar] [CrossRef]
- Rabkin, C. AIDS and cancer in the era of highly active antiretroviral therapy (HAART). Eur. J. Cancer 2001, 37, 1316–1319. [Google Scholar] [CrossRef]
- Mallal, S.A.; John, M.; Moore, C.B.; James, I.R.; McKinnon, E.J. Contribution of nucleoside analogue reverse transcriptase inhibitors to subcutaneous fat wasting in patients with HIV infection. Aids 2000, 14, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.L.; Shouval, D.; Lok, A.S.; Chang, T.T.; Cheinquer, H.; Goodman, Z.; DeHertogh, D.; Wilber, R.; Zink, R.C.; Cross, A. Entecavir versus lamivudine for patients with HBeAg-negative chronic hepatitis B. N. Engl. J. Med. 2006, 354, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Miwa, M.; Ura, M.; Nishida, M.; Sawada, N.; Ishikawa, T.; Mori, K.; Shimma, N.; Umeda, I.; Ishitsuka, H. Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur. J. Cancer 1998, 34, 1274–1281. [Google Scholar] [CrossRef]
- Darrington, D.L.; Vose, J.M.; Anderson, J.R.; Bierman, P.J.; Bishop, M.R.; Chan, W.C.; Morris, M.E.; Reed, E.C.; Sanger, W.G.; Tarantolo, S.R. Incidence and characterization of secondary myelodysplastic syndrome and acute myelogenous leukemia following high-dose chemoradiotherapy and autologous stem-cell transplantation for lymphoid malignancies. J. Clin. Oncol. 1994, 12, 2527–2534. [Google Scholar] [PubMed]
- Yamaoka, N.; Aso, K.; Matsuda, K. New Syntheses of Nucleosides. 1a The Syntheses of Glycopyranosides of Purines, Pyrimidine, and Benzimidazole. J. Org. Chem. 1965, 30, 149–152. [Google Scholar] [CrossRef]
- Jungmann, O.; Pfleiderer, W. A new efficient method in nucleoside synthesis. Tetrahedron Lett. 1996, 37, 8355–8358. [Google Scholar] [CrossRef]
- Niedballa, U.; Vorbrüggen, H. Synthesis of nucleosides 17. A general synthesis of N-glycosides. 6. On the mechanism of the stannic chloride catalyzed silyl Hilbert-Johnson reaction. J. Org. Chem. 1976, 41, 2084–2086. [Google Scholar] [CrossRef] [PubMed]
- Vorbrüggen, H.; Krolikiewicz, K.; Bennua, B. Nucleoside syntheses, XXII1) Nucleoside synthesis with trimethylsilyl triflate and perchlorate as catalysts. Chem. Ber. 2007, 114, 1234–1255. [Google Scholar] [CrossRef]
- Catry, M.; Madder, A. Synthesis of Functionalised Nucleosides for Incorporation into Nucleic Acid-Based Serine Protease Mimics. Molecules 2007, 12, 114–129. [Google Scholar] [CrossRef]
- Qu, G.; Zhang, Z.; Guo, H.; Geng, M.; Xia, R. Microwave-promoted Facile and Efficient Preparation of N-(alkoxycarbonylmethyl) Nucleobases—Building Blocks for Peptide Nucleic Acids. Molecules 2007, 12, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Carlsen, P. Synthesis of Polynucleotide Analogs Containing a Polyvinyl Alcohol Backbone. Molecules 2008, 13, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Lanver, A.; Schmalz, H.-G. Microwave-Assisted Amination of a Chloropurine Derivative in the Synthesis of Acyclic Nucleoside Analogues. Molecules 2005, 10, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Shimizu, B. Studies on Synthetic Nucleosides. Agric. Biol. Chem. Tokyo 1964, 28, 224–229. [Google Scholar]
- Shimma, N.; Umeda, I.; Arasaki, M.; Murasaki, C.; Masubuchi, K.; Kohchi, Y.; Miwa, M.; Ura, M.; Sawada, N.; Tahara, H. The design and synthesis of a new tumor-selective fluoropyrimidine carbamate, capecitabine. Bioorg. Med. Chem. 2000, 8, 1697–1706. [Google Scholar] [CrossRef]
- Vorbrüggen, H.; Bennua, B. Nucleoside syntheses, XXV1) A new simplified nucleoside synthesis. Chem. Ber. 1981, 19, 1279–1286. [Google Scholar] [CrossRef]
- Vorbrueggen, H. Adventures in Silicon-Organic Chemistry. Acc. Chem. Res. 1995, 28, 509–520. [Google Scholar] [CrossRef]
- Bookser, B.C.; Raffaele, N.B. High-throughput five minute microwave accelerated glycosylation approach to the synthesis of nucleoside libraries. J. Org. Chem. 2007, 72, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Sun, J.; Zhu, Y.; Zhang, F.; Yu, B. An Efficient Approach to the Synthesis of Nucleosides: Gold (I)-Catalyzed N-Glycosylation of Pyrimidines and Purines with Glycosyl ortho-Alkynyl Benzoates. Angew. Chem. Int. Ed. 2011, 50, 4933–4936. [Google Scholar] [CrossRef] [PubMed]
- Shohda, K.; Wada, T.; Sekine, M. Detailed Studies on Trimethylselyl Triflate Mediated Glycosylation via a 3,5-O-(l,l,3,3-Tetraisopropyl-Disiloxane-l,3-diyl)-2-O-methylribofuranos-l-yl Trichloroacetimidate Intermediate. Nucleosides Nucleotides 1998, 17, 2199–2210. [Google Scholar] [CrossRef]
- Liao, J.; Sun, J.; Yu, B. Effective synthesis of nucleosides with glycosyl trifluoroacetimidates as donors. Tetrahedron Lett. 2008, 49, 5036–5038. [Google Scholar] [CrossRef]
- Liao, J.; Sun, J.; Yu, B. An improved procedure for nucleoside synthesis using glycosyl trifluoroacetimidates as donors. Carbohyd. Res. 2009, 344, 1034–1038. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.J.; Zhang, X.T.; Xing, G.W. Cheminform abstract: A general method for N-glycosylation of nucleobases promoted by (p-TOL) 2 SO/Tf 2 O with thioglycoside as donor. ChemInform 2015, 46, 12803–12806. [Google Scholar]
- Vorbrüggen, H.; Ruh-Pohlenz, C. Handbook of Nucleoside Synthesis; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2001. [Google Scholar]
- Sample Availability: Samples of all the synthesized nucleoside are available from the authors.


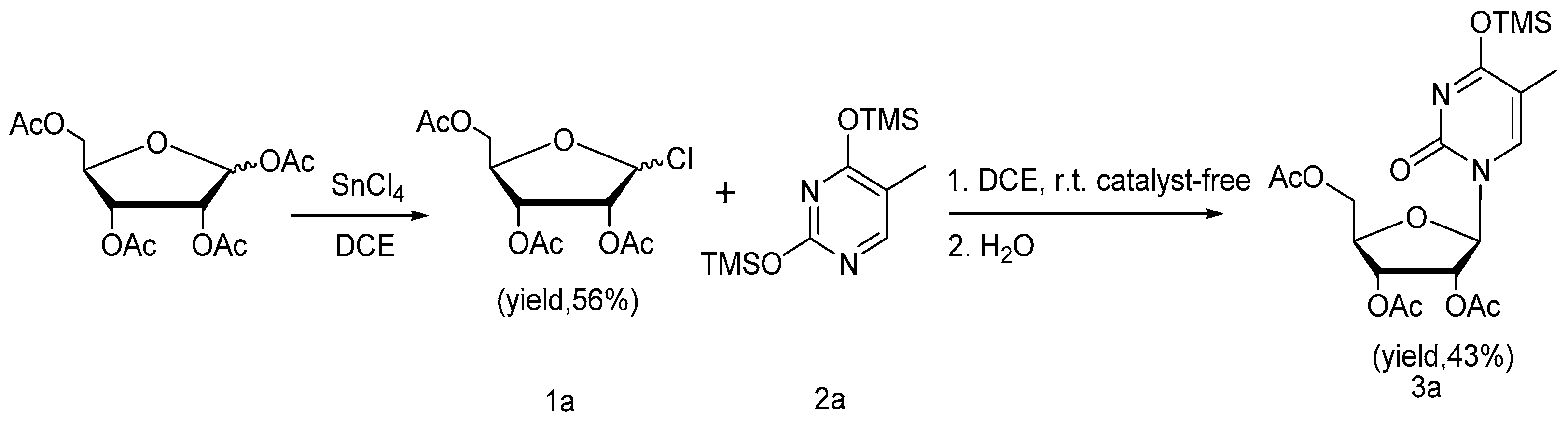

{kind=link}
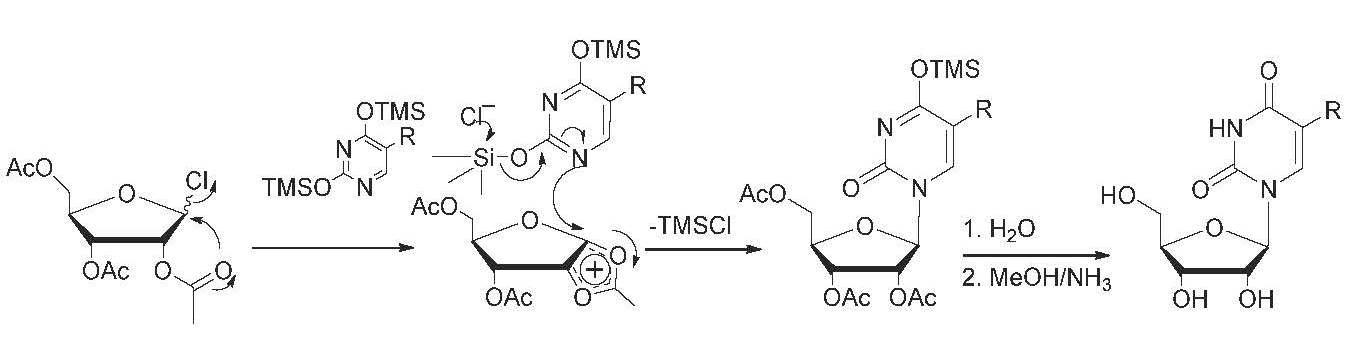
{kind=link}
{kind=link}
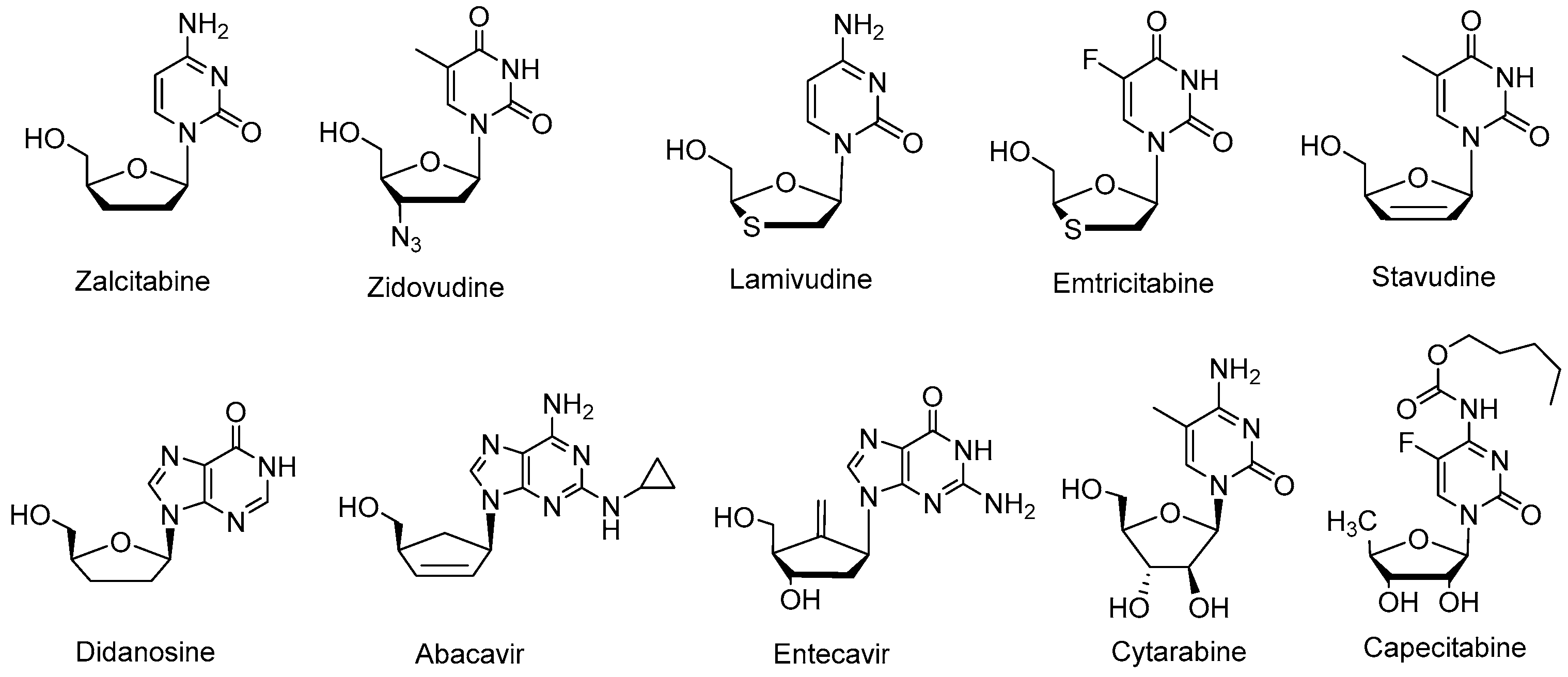
{kind=link}
{kind=link}
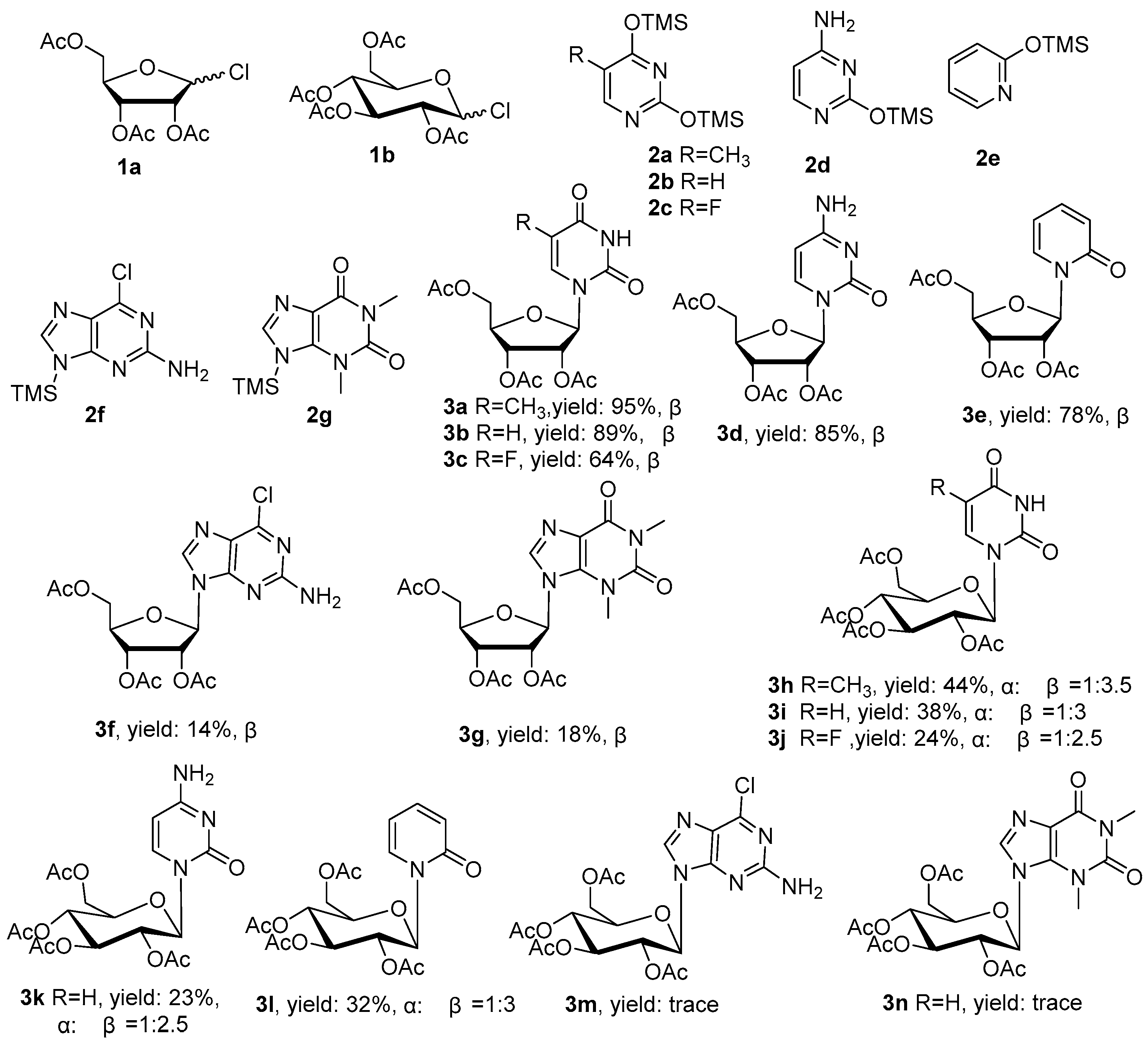
{kind=link}
{kind=link}
| Entry | Solvent | Reaction Time (h) | Temprature (°C) | Yield (%) |
|---|---|---|---|---|
| 1 | 1,2-Dichloroethane | 4 | 83 | 95 |
| 2 | Toluene | 6 | 110 | 89 |
| 3 | MeCN | 7 | 80 | 78 |
| 4 | THF | 10 | 66 | 64 |
| 5 | Ethanol | 10 | 78 | Trace |
| 6 | [Bmim]BF4 | 8 | 90 | - |
| 7 | [Bmim]PF6 | 8 | 90 | - |
| 8 | [Bmim]HSO4 | 8 | 90 | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, C.; Ju, W.; Ding, S.; Sun, H.; Mao, G. Effective Synthesis of Nucleosides Utilizing O-Acetyl-Glycosyl Chlorides as Glycosyl Donors in the Absence of Catalyst: Mechanism Revision and Application to Silyl-Hilbert-Johnson Reaction. Molecules 2017, 22, 84. https://doi.org/10.3390/molecules22010084
Liang C, Ju W, Ding S, Sun H, Mao G. Effective Synthesis of Nucleosides Utilizing O-Acetyl-Glycosyl Chlorides as Glycosyl Donors in the Absence of Catalyst: Mechanism Revision and Application to Silyl-Hilbert-Johnson Reaction. Molecules. 2017; 22(1):84. https://doi.org/10.3390/molecules22010084
Chicago/Turabian StyleLiang, Chengyuan, Weihui Ju, Shunjun Ding, Han Sun, and Gennian Mao. 2017. "Effective Synthesis of Nucleosides Utilizing O-Acetyl-Glycosyl Chlorides as Glycosyl Donors in the Absence of Catalyst: Mechanism Revision and Application to Silyl-Hilbert-Johnson Reaction" Molecules 22, no. 1: 84. https://doi.org/10.3390/molecules22010084
APA StyleLiang, C., Ju, W., Ding, S., Sun, H., & Mao, G. (2017). Effective Synthesis of Nucleosides Utilizing O-Acetyl-Glycosyl Chlorides as Glycosyl Donors in the Absence of Catalyst: Mechanism Revision and Application to Silyl-Hilbert-Johnson Reaction. Molecules, 22(1), 84. https://doi.org/10.3390/molecules22010084