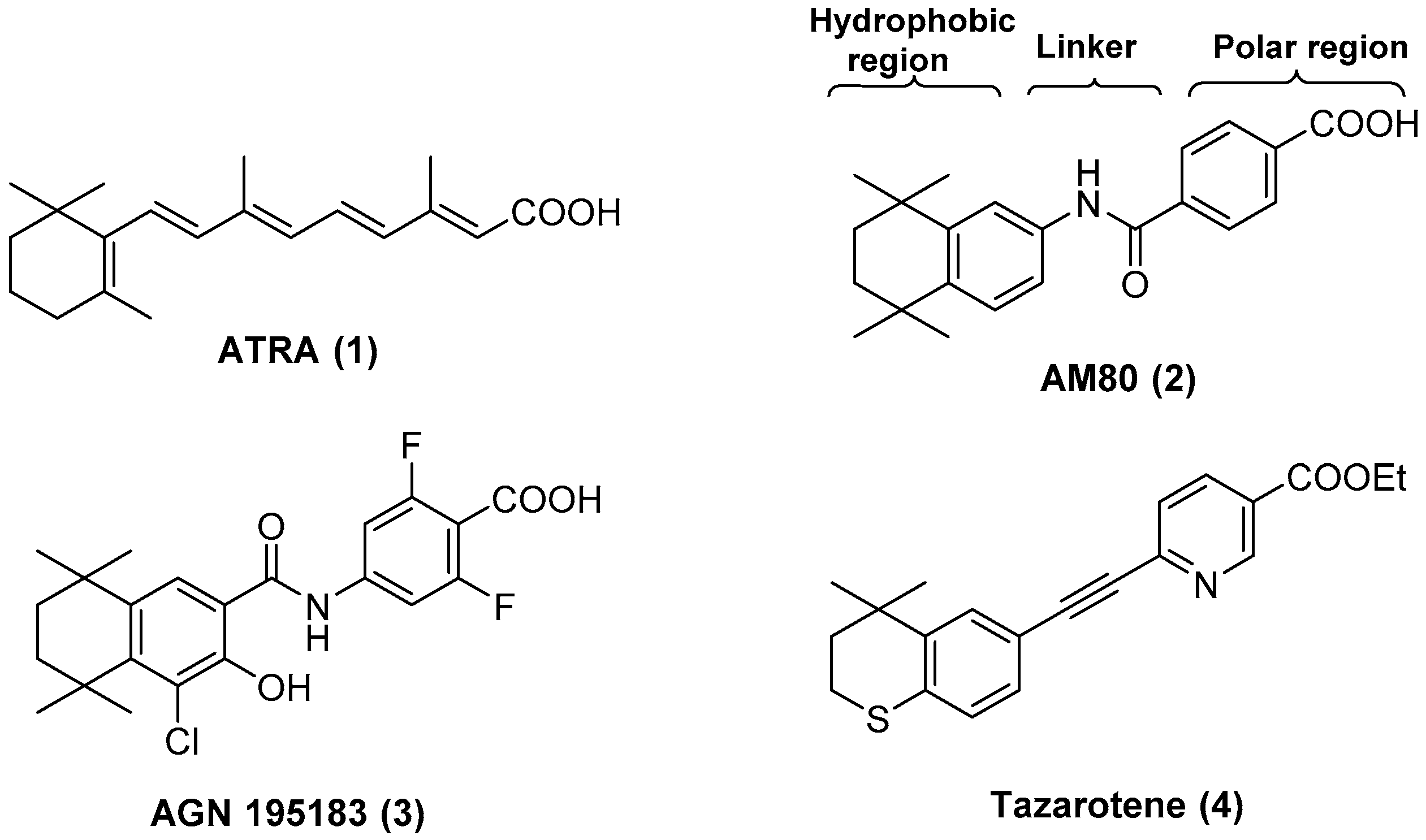
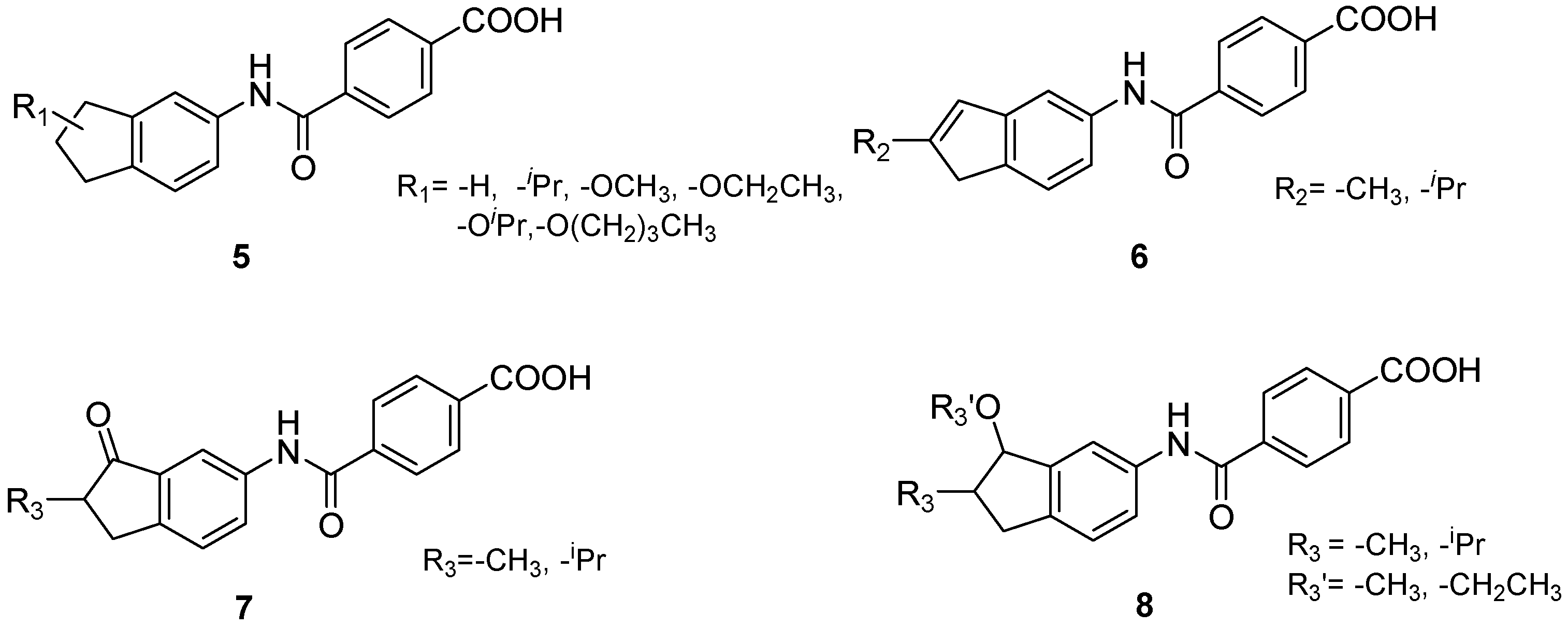
4.2. Chemistry
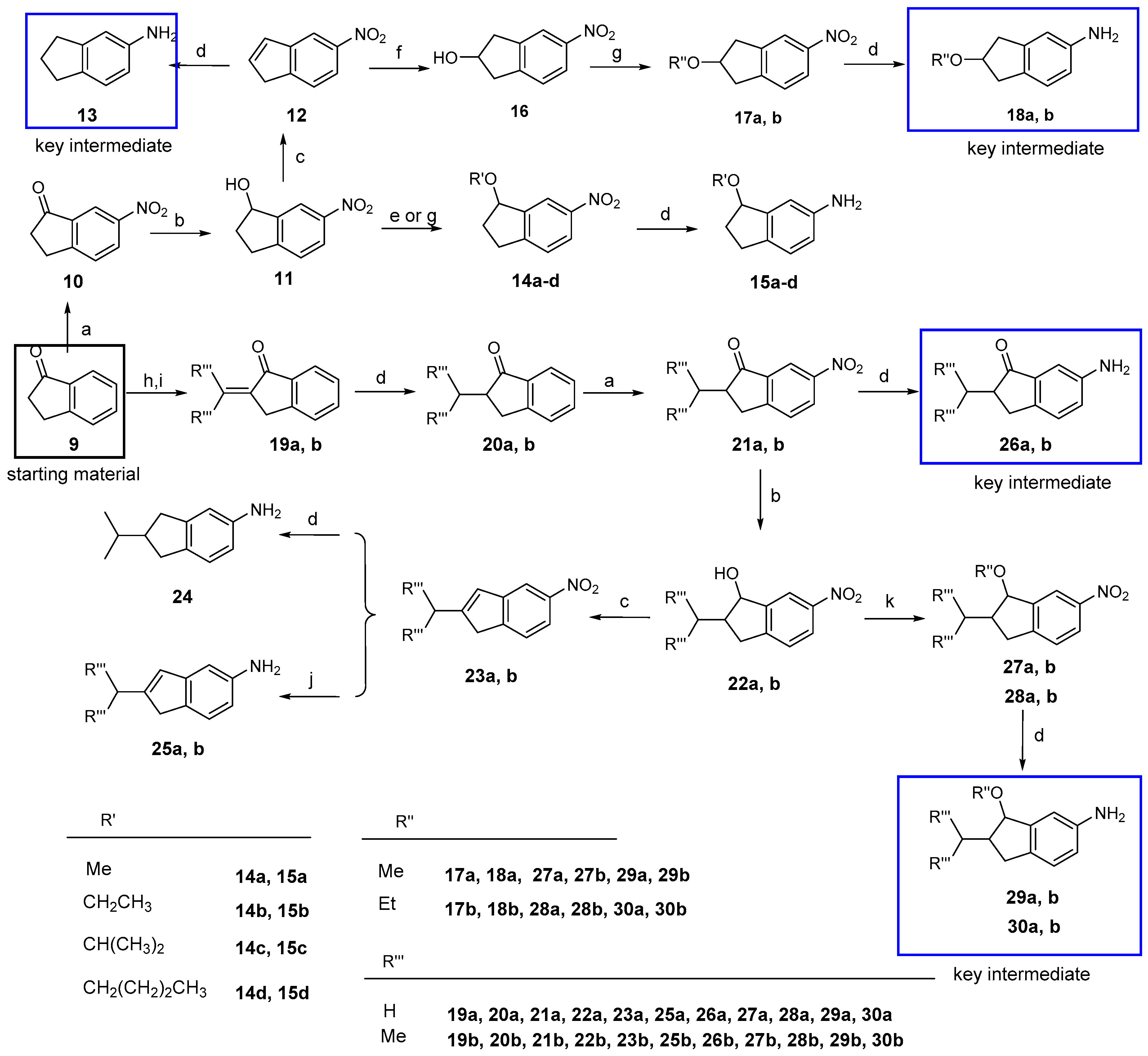
6-Nitro-2,3-dihydro-1H-inden-1-one (10). To a solution of 2,3-dihydro-1H-inden-1-one (9, 1.32 g, 10.0 mmol) in concentrated sulfuric acid (10 mL), KNO3 (1.21 g, 12.0 mmol) in concentrated sulfuric acid (10 mL) was added dropwise at −5 °C in 30 min. The mixture was stirred at −5 °C for 4 h. After adding ice water slowly, the mixture was partitioned between water and CH2Cl2. The organic layer was washed with a saturated aqueous solution of NaHCO3 and brine, dried over anhydrous Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography on silica gel (eluent: hexane/EtOAc = 7/1) to give 10 (1.10 g, 62%) as a beige solid. m.p. 71~74 °C; 1H-NMR: δ 8.59 (d, J = 1.9 Hz, 1H), 8.47 (dd, J = 8.4, 2.2 Hz, 1H), 7.68 (t, J = 8.4 Hz, 1H), 3.33–3.25 (m, 2H), 2.89–2.78 (m, 2H); ESI-MS: m/z [M + H]+ 178.
6-Nitro-2,3-dihydro-1H-inden-1-ol (11). To a solution of 10 (1.77 g, 10.0 mmol) in a mixed solution of MeOH/THF (2:1, 20 mL), NaBH4 (1.52 g, 40.0 mmol) was added in portions. The mixture was stirred at room temperature for one hour. After the addition of water (40 mL), the mixture was partitioned between water and EtOAc. The organic layer was washed with a saturated aqueous brine, dried over anhydrous Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography on silica gel (eluent: hexane/EtOAc = 2/1) to give 11 (1.64 g, 92%) as a white solid. m.p. 74~77 °C; 1H-NMR: δ 8.26 (d, J = 1.8 Hz, 1H), 8.15 (dd, J = 8.3, 2.1 Hz, 1H), 7.38 (d, J = 8.3 Hz, 1H), 5.32 (t, J = 6.2 Hz, 1H), 3.14 (m, 1H), 2.98–2.85 (m, 1H), 2.64–2.58 (m, 1H), 2.09–1.97 (m, 1H); ESI-MS: m/z [M + H]+ 180.
5-Nitro-1H-indene (12). To a solution of 11 (1.79 g, 10.0 mmol) in anhydrous toluene (20 mL), TsOH (1.72 g, 10.0 mmol) was added at room temperature and the mixture was refluxed for 2 h. The solvent was removed by distillation. After adding water (40 mL), the mixture was partitioned between water and EtOAc. The organic layer was washed with a saturated aqueous solution of NaHCO3 and brine, dried over anhydrous Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography on silica gel (eluent: hexane/EtOAc = 10/1) to give 12 (1.38 g, 86%) as a white solid, m.p. 78~81 °C; 1H-NMR: δ 8.23 (d, J = 2.1 Hz, 1H), 8.11 (dd, J = 8.2, 2.1 Hz, 1H), 7.58 (d, J = 8.2 Hz, 1H), 6.96 (d, J = 5.5 Hz, 1H), 6.76 (dt, J = 5.4, 1.9 Hz, 1H), 3.52 (s, 2H);ESI-MS: m/z [M + H]+ 162.
1-Methoxy-6-nitro-2,3-dihydro-1H-indene (14a). To a solution of 11 (90 mg, 0.5 mmol) and CH3I (0.31 mL, 5.0 mmol) in anhydrous THF (2 mL), CH3ONa (108 mg, 2.0 mmol) was added at 0 °C. The mixture was stirred at room temperature for 12 h. After adding water (20 mL), the mixture was partitioned between water and EtOAc. The organic layer was washed with saturated brine, dried over anhydrous Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography on silica gel (eluent: hexane/EtOAc = 30/1) to give 14a (39 mg, 41%) as a pale yellow liquid. 1H-NMR: δ 8.26 (d, J = 2.0 Hz, 1H), 8.17 (dd, J = 8.3, 2.2 Hz, 1H), 7.40 (d, J = 8.3 Hz, 1H), 4.87 (dd, J = 6.5, 4.5 Hz, 1H), 3.46 (s, 3H), 3.20–3.13 (m, 1H), 2.95–2.89 (m, 1H), 2.52–2.40 (m, 1H), 2.25–2.13 (m, 1H); ESI-MS: m/z [M + H]+ 194.
1-Ethoxy-6-nitro-2,3-dihydro-1H-indene (14b). The title compound was prepared (34 mg, 33%) as a pale yellow liquid from 11 and CH3CH2I in a similar method with that described for 14a. 1H-NMR: δ 8.24 (d, J = 2.1 Hz, 1H), 8.14 (dd, J = 8.3, 2.2 Hz, 1H), 7.37 (d, J = 8.3 Hz, 1H), 4.95 (q, J = 6.5 Hz, 1H), 3.70–3.60 (m, 2H), 3.18–3.12 (m, 1H), 2.93–2.84 (m, 1H), 2.50–2.43 (m, 1H), 2.19–2.12 (m, 1H), 1.27 (t, J = 7.0 Hz, 3H). ESI-MS: m/z [M + H]+ 208.
1-Isopropoxy-6-nitro-2,3-dihydro-1H-indene (14c). To a stirred solution of compound 11 (180 mg, 1.0 mmol) and isopropyl bromide (183 mg, 1.5 mmol) in anhydrous CH2Cl2 (2 mL), dry mercury oxide/tetrafluoroboric acid (190 mg, 0.5 mmol) was added. The mixture was stirred at room temperature for 2 h and then treated successively with NaHCO3 and 3 M potassium hydroxide until basic. The precipitated mercury oxide was filtered off and the filtrate was extracted with CH2Cl2. The organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography on silica gel (eluent: hexane/EtOAc = 25/1) to give 14c (42 mg, 19%) as a pale yellow liquid. 1H-NMR: δ 8.19 (d, J = 2.0 Hz, 1H), 8.12 (dd, J = 8.3, 2.2 Hz, 1H), 7.35 (d, J = 8.3 Hz, 1H), 5.03 (t, J = 6.3 Hz, 1H), 3.93–3.84 (m, 1H), 3.15–3.09 (m, 1H), 2.94–2.82 (m, 1H), 2.54–2.48 (m, 1H), 2.12–2.05 (m, 1H), 1.29 (d, J = 6.1 Hz, 3H), 1.26 (d, J = 6.1 Hz, 3H). ESI-MS: m/z [M + H]+ 222.
1-Butoxy-6-nitro-2,3-dihydro-1H-indene (14d). The title compound was prepared as a pale yellow liquid (69 mg, 29%) from 11 and 1-bromobutane in a manner similar to that described for 14c. 1H-NMR: δ 8.22 (s, 1H), 8.13 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 8.2 Hz, 1H), 4.93 (t, J = 5.5 Hz, 1H), 3.69–3.50 (m, 2H), 3.20–3.07 (m, 1H), 2.97–2.83 (m, 1H), 2.55–2.42 (m, 1H), 2.22–2.02 (m, 1H), 1.70–1.56 (m, 2H), 1.41 (m, 2H), 0.94 (t, J = 6.4 Hz, 3H);ESI-MS: m/z [M + H]+ 236.
5-Nitro-2,3-dihydro-1H-inden-2-ol (16). To a stirred solution of compound 12 (805 mg, 5.0 mmol) in anhydrous THF, diborane (10 mmol) in diethyl sulfide (5 mL) was added dropwise at 0 °C. The mixture was stirred at room temperature for 2 h. A small amount of water was added until no bubbles were generated, then 30% hydrogen peroxide (2.8 mL) was added followed by the addition of 1 N NaOH (0.6 mL). The mixture was stirred at room temperature for another 1 h. After adding water (50 mL), the mixture was partitioned between water and EtOAc. The organic layer was washed with a saturated aqueous solution of brine, dried over anhydrous Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography on silica gel (eluent: hexane/EtOAc = 3/1) to give 16 (295 mg, 33%) as a white solid. m.p. 90~92 °C; 1H-NMR: δ 8.10 (s, 1H), 8.07 (d, J = 8.2 Hz, 1H), 7.37 (d, J = 8.2 Hz, 1H), 4.85–4.77 (m, 1H), 3.34–3.24 (m, 2H), 3.01 (m, 2H); ESI-MS: m/z [M + H]+ 180.
2-Methoxy-5-nitro-2,3-dihydro-1H-indene (17a). The title compound was prepared from 16 and iodomethane in a manner similar to that described for 14a as a pale yellow liquid (41%). 1H-NMR: δ 8.08 (s, 1H), 8.04(d, J = 8.0 Hz, 1H), 7.35 (d, J = 8.2 Hz, 1H), 4.33–4.29 (m, 1H), 3.38 (s, 3H), 3.24–3.19 (m, 2H), 3.11–3.06 (m, 2H);ESI-MS: m/z [M + H]+ 194.
2-Ethoxy-5-nitro-2,3-dihydro-1H-indene (17b). The title compound was prepared from 16 and iodoethane in a manner similar to that described for 14a as a pale yellow liquid (26%). 1H-NMR: δ 7.97 (d, J = 11.5 Hz, 2H), 7.27 (s, 1H), 4.37–4.30 (m, 1H), 3.48 (q, J = 7.0 Hz, 2H), 3.15 (dd, J = 17.0, 6.3 Hz, 2H), 2.99 (dt, J = 9.1, 4.3 Hz, 2H), 1.14 (t, J = 7.0 Hz, 3H);ESI-MS: m/z [M + H]+ 208.
2-Methylene-2,3-dihydro-1H-inden-1-one (19a). To a solution of 9 (1.32 g, 10.0 mmol) and paraformaldehyde (1.50 g, 5.0 eq) in glacial acetic acid (20 mL), morpholine (0.5 mL) was added. The mixture was refluxed under nitrogen for 2 h. The glacial acetic acid was removed by distillation. After adding water (50 mL), the mixture was partitioned between water and EtOAc. The organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography on silica gel (eluent: hexane/EtOAc = 7/1) to give 19a (0.45 g, 31%) to give a yellow liquid. 1H-NMR: δ 7.89 (d, J = 7.6 Hz, 1H), 7.62 (t, J = 7.4 Hz, 1H), 7.51 (d, J = 7.6 Hz, 1H), 7.42 (t, J = 7.4 Hz, 1H), 6.39 (s, 1H), 5.65 (s, 1H), 3.78 (s, 2H); ESI-MS: m/z [M + H]+ 145.
2-(Propan-2-ylidene)-2,3-dihydro-1H-inden-1-one (19b). To a solution of 9 (1.32 g, 10.0 mmol) in anhydrous acetone (20 mL), NaOH (132 mg, 3.3 mmol) was added at room temperature. The mixture was stirred at room temperature for 4 h and then neutralized with 1 N HCl. Acetone was removed by distillation. After adding water (50 mL), the mixture was partitioned between water and EtOAc. The organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography on silica gel (eluent: hexane/EtOAc = 7/1) to give 19b (0.77 g, 45%) as a yellow solid. m.p. 98~100 °C; 1H-NMR: δ 7.81 (d, J = 7.6 Hz, 1H), 7.55 (d, J = 7.1 Hz, 1H), 7.47 (s, 1H), 7.37 (s, 1H), 3.65 (s, 2H), 2.45 (s, 3H), 2.01 (s, 3H); ESI-MS: m/z [M + H]+ 173.
2-Methyl-2,3-dihydro-1H-inden-1-one (20a). To a solution of 19a (1.44 g, 10 mmol) in EtOAc (20 mL), 10% Pd/C (20% weight of compound 19a) was added. The mixture was stirred overnight under a hydrogen atmosphere at room temperature. Insoluble materials were removed by filtration and washed with EtOAc. The filtrate was evaporated to dryness under reduced pressure to give 20a (1.43 g, 98%) as a colorless transparent liquid. 1H-NMR: δ 7.76 (d, J = 7.7 Hz, 1H), 7.59 (d, J = 7.6, 1H), 7.45 (d, J = 7.7 Hz, 1H), 7.37 (t, J = 7.4 Hz, 1H), 3.45–3.36 (m, 1H), 2.79–2.68 (m, 2H), 1.35–1.30 (m, 3H); ESI-MS: m/z [M + H]+ 147.
2-Isopropyl-2,3-dihydro-1H-inden-1-one (20b). The title compound was prepared from 19b in a manner similar to that described for 20a as a colorless transparent liquid (97%). 1H-NMR: δ 7.75 (d, J = 7.7 Hz, 1H), 7.59 (td, J = 7.6, 1.1 Hz, 1H), 7.49 (d, J = 7.7 Hz, 1H), 7.39–7.34 (m, 1H), 3.16 (dd, J = 17.4, 8.1 Hz, 1H), 2.95 (dd, J = 17.4, 4.0 Hz, 1H), 2.82–2.79 (m, 1H), 2.45–2.42 (m, 1H), 1.07 (d, J = 6.9 Hz, 3H), 0.81 (d, J = 6.8 Hz, 3H); ESI-MS: m/z [M + H]+ 175.
2-Methyl-6-nitro-2,3-dihydro-1H-inden-1-one (21a). The title compound was prepared from 20a in a manner similar to that described for 10 as a yellow solid (67%). m.p. 64~66 °C; 1H-NMR: δ 8.58 (d, J = 2.1 Hz, 1H), 8.46 (dd, J = 8.4, 2.2 Hz, 1H), 7.64 (d, J = 8.4 Hz, 1H), 3.55–3.50 (m, 1H), 2.91–2.79 (m, 2H), 1.37 (d, J = 7.3 Hz, 3H); ESI-MS: m/z [M + H]+ 192.
2-Isopropyl-6-nitro-2,3-dihydro-1H-inden-1-one (21b). The title compound was prepared from 20b in a manner similar to that described for 10 as a yellow solid (73%). m.p. 72~76 °C; 1H-NMR: δ 8.55 (d, J = 2.0 Hz, 1H), 8.44 (dd, J = 8.4, 2.2 Hz, 1H), 7.65 (d, J = 8.3 Hz, 1H), 3.28 (dd, J = 18.3, 8.2 Hz, 1H), 3.04 (dd, J = 18.3, 4.1 Hz, 1H), 2.82-2.79 (m, 1H), 2.51–2.40 (m, 1H), 1.07 (d, J = 6.9 Hz, 3H), 0.83 (d, J = 6.8 Hz, 3H); ESI-MS: m/z [M + H]+ 192.
2-Methyl-6-nitro-2,3-dihydro-1H-inden-1-ol (22a). The title compound was prepared from 21a in a manner similar to that described for 11 as a white solid (86%). m.p. 81~84 °C; 1H-NMR: δ 8.58 (d, J = 5.0 Hz, 1H), 8.06 (s, 1H), 7.45 (d, J = 8.0 Hz, 1H), 5.72 (d, J = 6.0 Hz, 1H), 4.62–4.59 (s, 1H), 3.10–3.05 (m, 1H), 2.58–2.53 (m, 1H), 2.24–2.14 (m, 1H), 1.20 (d, J = 6.7 Hz, 3H);ESI-MS: m/z [M + H]+ 194.
2-Isopropyl-6-nitro-2,3-dihydro-1H-inden-1-ol (22b). The title compound was prepared from 21b in a manner similar to that described for 11 as a white solid (89%). m.p. 90~93 °C; 1H-NMR: δ 8.15–8.04 (m, 1H), 7.70 (m, 1H), 7.41 (m, 1H), 3.22 (d, J = 3.3 Hz, 1H), 3.54 (dd, J = 17.9, 7.3 Hz, 1H), 3.15 (dd, J = 17.9, 9.8 Hz, 1H), 2.05–1.92 (m, 1H), 1.55 (m, 1H), 1.14 (t, J = 5.5 Hz, 3H), 1.09 (d, J = 6.5 Hz, 3H)); ESI-MS: m/z [M + H]+ 222.
2-Methyl-5-nitro-1H-indene (23a). The title compound was prepared from 22a in a manner similar to that described for 12 as a white solid (81%). m.p.: 68~72 °C; 1H-NMR: δ 7.76 (d, J = 7.6 Hz, 1H), 7.59 (t, J = 7.4 Hz, 1H), 7.46 (d, J = 7.7 Hz, 1H), 5.02 (d, J = 5.9 Hz, 1H), 3.41 (s, 8.7 Hz, 2H), 1.32 (d, J = 7.2 Hz, 3H); ESI-MS: m/z [M + H]+ 176.
2-Isopropyl-5-nitro-1H-indene (23b). The title compound was prepared from 22b in a manner similar to that described for 12 as a white solid (82%). m.p. 78~81 °C; 1H-NMR: δ 8.07 (d, J = 1.8 Hz, 1H), 8.00 (dd, J = 8.1, 1.8 Hz, 1H), 7.46 (d, J = 8.1 Hz, 1H), 6.56 (s, 1H), 3.44 (s, 2H), 2.90–2.73 (m, 1H), 1.25 (s, 3H), 1.24 (s, 3H); ESI-MS: m/z [M + H]+ 204.
1-Methoxy-2-methyl-6-nitro-2,3-dihydro-1H-indene (27a). To a solution of 22a (106 mg, 0.55 mmol) and trimethyl orthoformate (1 mL) in anhydrous CH2Cl2 (2 mL), bismuth trichloride (173 mg, 0.55 mmol) was added at room temperature. The mixture was stirred at room temperature for 7 h and then treated with aqueous 1 N NaHCO3 until basic. After adding water (50 mL), the mixture was partitioned between water and EtOAc. The organic layer was washed with a saturated aqueous solution of brine, dried over anhydrous Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography on silica gel (eluent: hexane/EtOAc = 30/1) to give 27a (27 mg, 24%) as a yellow oil. 1H-NMR: δ 8.22 (s, 1H), 8.15 (dd, J = 8.3, 2.1 Hz, 1H), 7.35 (d, J = 8.3 Hz, 1H), 4.42 (d, J = 4.4 Hz, 1H), 3.52 (s, 3H), 3.31–3.26 (m, 1H), 2.64–2.56 (m, 1H), 2.56–2.50 (m, 1H), 1.19 (d, J = 7.0 Hz, 3H); ESI-MS: m/z [M + H]+ 208.
1-Ethoxy-2-methyl-6-nitro-2,3-dihydro-1H-indene (27b). The title compound was prepared from 22b and triethyl orthoformate in a manner similar to that described for 27a as yellow oil (33%). 1H-NMR: δ 8.26 (s, 1H), 8.16 (d, J = 8.3 Hz, 1H), 7.38 (d, J = 8.1 Hz, 1H), 4.92 (d, J = 6.4 Hz, 1H), 3.65 (d, J = 7.0 Hz, 2H), 2.80–2.73 (m, 2H), 2.65 (q, J = 6.5, 1H), 1.33 (d, J = 6.6 Hz, 3H), 1.31–1.27 (m, 3H); ESI-MS: m/z [M + H]+ 222.
2-Isopropyl-1-methoxy-6-nitro-2,3-dihydro-1H-indene (28a). The title compound was prepared from 22a in a manner similar to that described for 27a as yellow oil (27%). 1H-NMR: δ 8.20 (d, J = 2.0 Hz, 1H), 8.18 (dd, J = 8.2, 2.2 Hz, 1H), 7.41 (d, J = 8.2 Hz, 1H), 4.51 (d, J = 3.2 Hz, 1H), 3.34 (s, 3H), 3.00–2.86 (m, 2H), 2.09–2.01 (m, 1H), 2.00–1.95 (m, 1H), 1.07 (d, J = 6.3 Hz, 3H), 1.00 (d, J = 6.6 Hz, 3H); ESI-MS: m/z [M + H]+ 236.
1-Ethoxy-2-isopropyl-6-nitro-2,3-dihydro-1H-indene (28b). The title compound was prepared from 22b and triethyl orthoformate in a manner similar to that described for 27a as yellow oil (19%). 1H-NMR: δ 8.25 (d, J = 4.4 Hz, 1H), 8.14 (dd, J = 8.3, 2.2 Hz, 1H), 7.35 (d, J = 8.3 Hz, 1H), 4.62 (d, J = 5.3 Hz, 1H), 3.20 (q, J =8.5 Hz, 2H), 2.83–2.75 (m, 2H), 2.66–2.56 (m, 1H), 2.19–2.06 (m, 1H), 1.09 (d, J = 6.5 Hz, 5H), 1.01–0.99 (m, 3H); ESI-MS: m/z [M + H]+ 250.
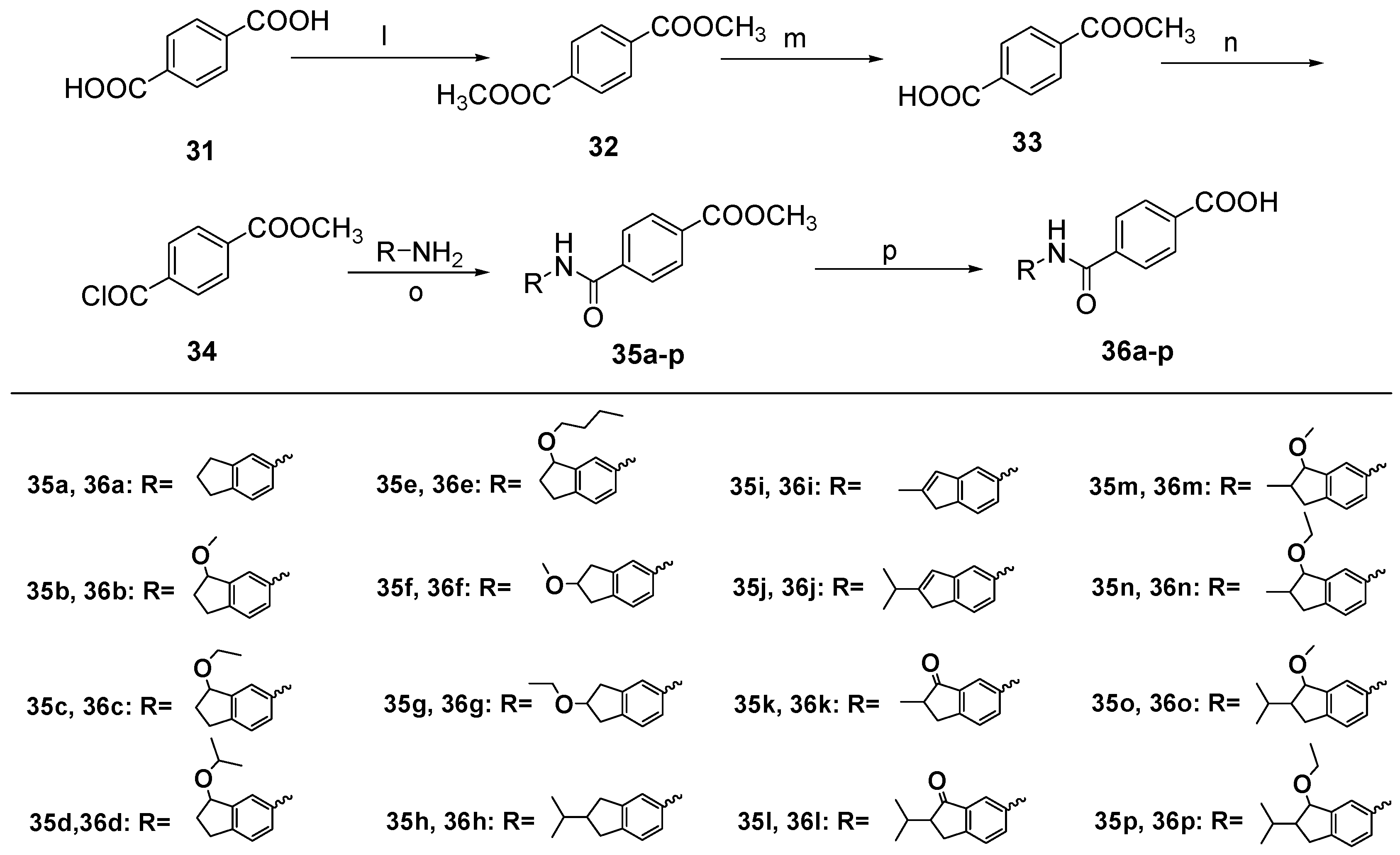
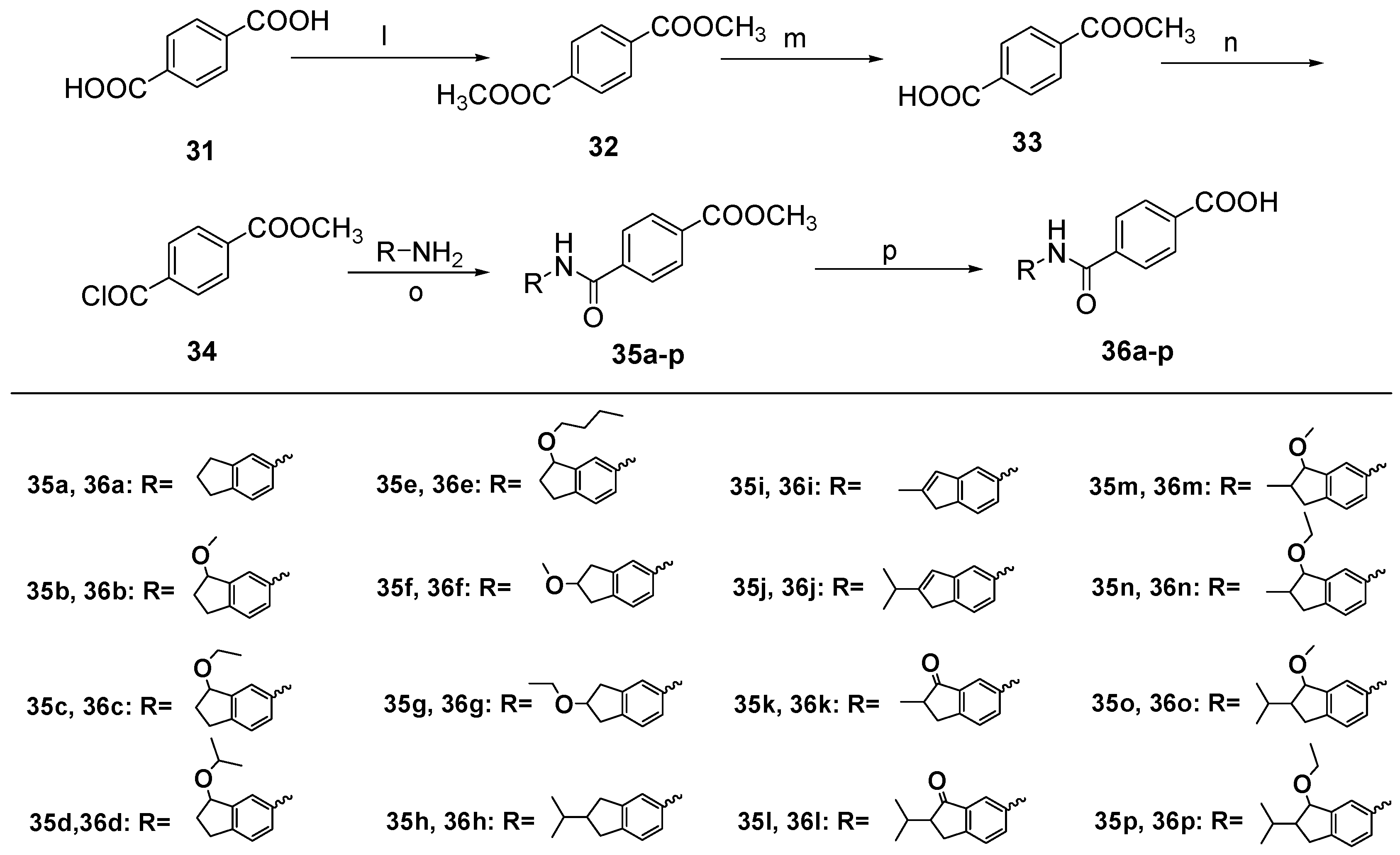
Dimethyl Terephthalate (32). To a solution of terephthalic acid (31, 6.0 g, 36.0 mmol) in methanol (150 mL), thionyl chloride (7.7 mL, 108 mmol) was added dropwise at 0 °C. The mixture was stirred at room temperature for 17 h and then saturated potassium carbonate solution was added until no bubbles were generated. The methanol was removed by distillation. After adding water (40 mL), the mixture was partitioned between water and ether. The organic layer was washed with a saturated aqueous solution of NaHCO3 and brine, dried over anhydrous Na2SO4, and concentrated under vacuum to give 32 (6.84 g, 98%) as a white solid. m.p. 141~143 °C (ether); ESI-MS: m/z [M + H]+ 195.
4-(Methoxycarbonyl)benzoic acid (33). To a solution of 32 (2.0 g, 10.0 mmol) in methanol and ether (methanol:ether = 1:1, 20 mL), a solution of KOH (0.58 g, 10.0 mmol) in methanol and water (methanol:water = 10:1, 10 mL) was added dropwise at 0 °C. The mixture was stirred at room temperature for 24 h. After adding water (50 mL), the mixture was partitioned between water and ether. Then treating the aqueous layer successively with 1 N HCl until pH = 1. The mixture was partitioned between water and EtOAc. The organic layer was washed with a saturated aqueous solution of brine, dried over anhydrous Na2SO4, and concentrated under vacuum to give 33 (1.0 g, 56%) as a white solid. m.p. 188~192 °C.
4-((2,3-Dihydro-1H-inden-5-yl)carbamoyl) benzoate (35a). To a solution of 12 (80.5 mg, 0.5 mmol) in EtOAc (20 mL), 10% Pd/C (20% net weight of compound 19a) was added. The mixture was stirred overnight under a hydrogen atmosphere at room temperature. Insoluble materials were removed by filtration and washed with EtOAc. The filtrate was evaporated to dryness under reduced pressure to give 13 as brown oil (65 mg, 98%). To a solution of 33 (150 mg, 0.8 mmol) in thionyl chloride (4 mL), a drop of pyridine was added and refluxed for 24 h. The thionyl chloride was removed by distillation and get 34 as pale yellow solid. 34 was used directly in the next reaction. To a solution of 13 (65 mg, 0.5 mmol) in anhydrous pyridine (2 mL), 34 was added in CH2Cl2 (2 mL) dropwise at 0 °C. The mixture was stirred at room temperature for 7 h and then the methanol was removed by distillation. The organic layer was treated successively with 1 N HCl until acidic. The mixture was partitioned between water and EtOAc. The organic layer was washed with a saturated aqueous solution of brine, dried over anhydrous Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography on silica gel (eluent: hexane/EtOAc = 3/1) to give 35a (118 mg, 80%) as a pale yellow solid. m.p. 138~142 °C; 1H-NMR: δ 8.15 (d, J = 6.9 Hz, 2H), 7.93 (d, J = 8.1 Hz, 2H), 7.60 (s, 1H), 7.30 (d, J = 8.1 Hz,1H), 7.22 (d, J = 7.9 Hz, 1H), 3.96 (s, 3H), 2.95–2.89 (m, 4H), 2.13–2.07 (m, 2H);ESI-MS: m/z [M + H]+ 296.
Methyl 4-((3-methoxy-2,3-dihydro-1H-inden-5-yl)carbamoyl) benzoate (35b). The title compound was prepared from 14a in a manner similar to that described for 35a as a pale yellow solid (87%). m.p. 148~151 °C; 1H-NMR: δ 8.15 (d, J = 8.3 Hz, 2H), 7.92 (d, J = 8.3 Hz, 2H), 7.86 (s, 1H), 7.72 (s, 1H), 7.50 (d, J = 7.3 Hz, 1H), 7.27 (d, J = 8.0 Hz, 1H), 4.83 (dd, J = 6.5, 4.2 Hz, 1H), 3.96 (s, 3H), 3.43 (s, 3H), 3.11–3.01 (m, 1H), 2.85–2.77 (m, 1H), 2.42–2.32 (m, 1H), 2.11–2.08 (m, 1H); ESI-MS: m/z [M + H]+ 326.
Methyl 4-((3-ethoxy-2,3-dihydro-1H-inden-5-yl)carbamoyl) benzoate (35c). The title compound was prepared from 14b in a manner similar to that described for 35a as a pale yellow solid (76%). m.p. 153~156 °C; 1H-NMR: δ 8.16 (d, J = 8.2 Hz, 2H), 7.93 (d, J = 8.1 Hz, 2H), 7.82 (s, 1H), 7.71 (s, 1H), 7.50 (d, J = 7.7 Hz, 1H), 7.24 (d, J = 8.0 Hz, 1H), 4.96–4.89 (m, 1H), 3.97 (s, 3H), 3.64 (q, J = 6.8, 2H), 3.09–3.03 (m, 1H), 2.83–2.77 (m, 1H), 2.44–2.36 (m, J = 6.7 Hz, 1H), 2.12–2.06 (m, 1H), 1.25 (t, J = 7.0 Hz, 3H); ESI-MS: m/z [M + H]+ 340.
Methyl 4-((3-isopropoxy-2,3-dihydro-1H-inden-5-yl)carbamoyl) benzoate (35d). The title compound was prepared from 14c in a manner similar to that described for 35a as a pale yellow solid (79%). m.p. 150~153 °C ; 1H-NMR: δ 8.16 (d, J = 7.7 Hz, 2H), 7.93 (d, J = 7.6 Hz, 2H), 7.80 (s, 1H), 7.50 (d, J = 7.2 Hz, 1H), 7.23 (d, J = 8.3 Hz, 1H), 5.01 (t, J = 5.9 Hz, 1H), 3.96 (s, 3H), 3.92–3.83 (m, 1H), 3.03 (s, 1H), 2.78 (dt, J = 15.7, 7.8 Hz, 1H), 2.44 (d, J = 6.2 Hz, 1H), 2.03 (dt, J = 13.1, 8.5 Hz, 1H), 1.25 (s, 6H); ESI-MS: m/z [M + H]+ 354.
Methyl 4-((3-isopropoxy-2,3-dihydro-1H-inden-5-yl)carbamoyl) benzoate (35e). The title compound was prepared from 14d in a manner similar to that described for 35a as a pale yellow solid (73%). m.p. 159~160 °C; 1H-NMR: δ 8.16 (d, J = 8.3 Hz, 2H), 7.93 (d, J = 8.2 Hz, 2H), 7.81 (s, 1H), 7.66 (s, 1H), 7.52 (d, J = 7.8 Hz, 1H), 7.24 (s, 1H), 4.93–4.85 (m, 1H), 3.96 (s, 3H), 3.57 (dd, J = 15.4, 6.7 Hz, 2H), 3.04 (ddd, J = 16.8, 11.4, 7.0 Hz, 1H), 2.79 (s, 1H), 2.40 (dq, J = 8.2, 6.2 Hz, 1H), 2.15–1.99 (m, 1H), 1.67–1.58 (m, 2H), 1.41 (dd, J = 13.2, 7.5 Hz, 2H), 0.96–0.89 (m, 3H); ESI-MS: m/z [M + H]+ 368.
Methyl 4-((3-isopropoxy-2,3-dihydro-1H-inden-5-yl)carbamoyl) benzoate (35f). The title compound was prepared from 17a in a manner similar to that described for 35a as a yellow solid (66%). m.p. 134~135 °C; 1H-NMR: δ 8.16 (d, J = 8.4 Hz, 2H), 7.92 (d, J = 8.2 Hz, 2H), 7.75 (s, 1H), 7.63 (s, 1H), 7.31 (d, J = 5.7 Hz, 1H), 4.28 (t, J = 5.3 Hz, 1H), 3.97 (s, 3H), 3.39 (s, 3H), 3.21–3.13 (m, 2H), 3.03–2.95 (m, 2H); ESI-MS: m/z [M + H]+ 326.
Methyl 4-((3-isopropoxy-2,3-dihydro-1H-inden-5-yl)carbamoyl) benzoate (35g). The title compound was prepared from 17b in a manner similar to that described for 35a as a yellow solid (71%). m.p. 137~139 °C; 1H-NMR: δ 8.16 (d, J = 8.3 Hz, 2H), 7.92 (d, J = 8.3 Hz, 2H), 7.75 (s, 1H), 7.62 (s, 1H), 7.30 (d, J = 9.3 Hz, 1H), 7.20 (d, J = 7.8 Hz, 1H), 4.37 (t, J = 5.0 Hz, 1H), 3.56 (d, J = 7.0 Hz, 2H), 3.21-3.14 (m, 2H), 3.04–2.93 (m, 2H), 1.23 (t, J = 7.0 Hz, 3H); ESI-MS: m/z [M + H]+ 340.
Methyl 4-((3-isopropoxy-2,3-dihydro-1H-inden-5-yl)carbamoyl) benzoate (35h). The title compound was prepared from 23b in a manner similar to that described for 35a as a pale yellow solid (87%). m.p. 144~149 °C; 1H-NMR: δ 8.19 (d, J = 8.3 Hz, 2H), 7.98 (s, 1H), 8.00–7.93 (m, 2H), 7.84 (s, 1H), 7.54 (d, J = 8.2 Hz, 1H), 3.98 (d, J = 4.4 Hz, 3H), 3.21–3.11 (m, 3H), 2.79–2.70 (m, 3H), 1.25 (d, J = 20.0 Hz, 6H); ESI-MS: m/z [M + H]+ 338.
Methyl 4-((3-isopropoxy-2,3-dihydro-1H-inden-5-yl)carbamoyl) benzoate (35k). The title compound was prepared from 21a in a manner similar to that described for 35a as a pale yellow solid (77%). m.p. 167~171 °C; 1H-NMR: δ 8.17 (s, 2H), 8.11 (d, J = 6.4 Hz, 1H), 7.96 (s, 2H), 7.82 (s, 1H), 7.50 (d, J = 8.2 Hz, 1H), 3.97 (s, 3H), 3.41 (dd, J = 16.4, 7.3 Hz, 1H), 2.80–2.75 (m, 1H), 2.75 (s, 1H), 1.33 (d, J = 7.3 Hz, 3H); ESI-MS: m/z [M + H]+ 324.
Methyl 4-((3-isopropoxy-2,3-dihydro-1H-inden-5-yl)carbamoyl) benzoate (35l). The title compound was prepared from 21b in a manner similar to that described for 35a as a pale yellow solid (89%). m.p. 170~174 °C; 1H-NMR: δ 8.21 (d, J = 8.2 Hz, 1H), 8.18 (d, J = 8.4 Hz, 2H), 8.00 (d, J = 8.2 Hz, 2H), 7.86 (s, 1H), 7.53 (d, J = 8.3 Hz, 1H), 3.15 (dd, J = 17.4, 8.0 Hz, 1H), 2.94 (dd, J = 17.4, 3.8 Hz, 1H), 2.72 (dt, J = 8.1, 4.1 Hz, 1H), 2.43–2.33 (m, 1H), 1.06 (d, J = 6.9 Hz, 3H), 0.79 (d, J = 6.8 Hz, 3H; ESI-MS: m/z [M + H]+ 352.
Methyl 4-((3-isopropoxy-2,3-dihydro-1H-inden-5-yl)carbamoyl) benzoate (35m). The title compound was prepared from 27a in a manner similar to that described for 35a as a pale yellow solid (91%). m.p. 147~149 °C; 1H-NMR: δ 8.16 (dd, J = 8.2, 4.4 Hz, 2H), 7.93 (d, J = 7.2 Hz, 2H), 7.73 (s, 1H), 7.48 (d, J = 7.1 Hz, 1H), 7.23 (d, J = 8.2 Hz, 1H), 4.41 (d, J = 3.6 Hz, 1H), 3.96 (s, 3H), 3.49 (s, 3H), 3.25–3.17 (m, 1H), 2.57–2.50 (m, 1H), 2.43 (dd, J = 15.8, 3.2 Hz, 1H), 1.17 (d, J = 7.0, 3H); ESI-MS: m/z [M + H]+ 340.
Methyl 4-((3-ethoxy-2-methyl-2, 3-dihydro-1H-inden-5-yl)carbamoyl) benzoate (35n). The title compound was prepared from 27b in a manner similar to that described for 35a as a pale yellow solid (86%). m.p. 140~142 °C; 1H-NMR: δ 8.16 (d, J = 8.2 Hz, 2H), 7.93 (d, J = 8.1 Hz, 2H), 7.82 (s, 1H), 7.70 (s, 1H), 7.46 (d, J = 7.4 Hz, 1H), 7.21 (d, J = 8.0 Hz, 1H), 4.49 (d, J = 4.6 Hz, 1H), 3.97 (s, 3H), 3.71 (q, J = 7.0 Hz, 2H), 3.19 (dd, J = 15.7, 7.6 Hz, 1H), 2.57–2.45 (m, 1H), 2.42 (dd, J = 15.6, 5.8 Hz, 1H), 1.27 (t, J = 6.9 Hz, 3H), 1.18 (d, J = 7.0 Hz, 3H); ESI-MS: m/z [M + H]+ 354.
4-((2-Isopropyl-3-methoxy-2,3-dihydro-1H-inden-5-yl)carbamoyl) benzoate (35o). The title compound was prepared from 28a in a manner similar to that described for 35a as a pale yellow solid (87%). m.p. 152~155 °C; 1H-NMR: δ 8.16 (d, J = 8.3 Hz, 2H), 7.94 (d, J = 8.3 Hz, 2H), 7.89 (s, 1H), 7.72 (s, 1H), 7.48 (d, J = 7.8 Hz, 1H), 7.22 (d, J = 8.1 Hz, 1H), 4.70 (d, J = 5.0 Hz, 1H), 3.97 (s, 3H), 3.48 (s, 3H), 3.08 (dd, J = 16.2, 8.4 Hz, 1H), 2.60 (dd, J = 16.2, 6.3 Hz, 1H), 2.31 (tt, J = 8.4, 6.4 Hz, 1H), 1.84 (dq, J = 13.4, 6.7 Hz, 1H), 1.00 (d, J = 6.8 Hz, 3H), 0.94 (t, J = 7.4 Hz, 3H); ESI-MS: m/z [M + H]+ 368.
Methyl 4-((3-ethoxy-2-isopropyl-2,3-dihydro-1H-inden-5-yl)carbamoyl) benzoate (35p). The title compound was prepared from 28b in a manner similar to that described for 35a as a pale yellow solid (97%). m.p. 146~149 °C; 1H-NMR: δ 8.15 (s, 2H), 7.93 (d, J = 8.2 Hz, 2H), 7.74 (s, 1H), 7.55 (s, 1H), 7.17 (d, J = 7.9 Hz, 1H), 4.56 (d, J = 5.0 Hz, 1H), 3.96 (s, 3H), 3.00 (q, J =8.0 Hz, 2H), 2.68–2.58 (m, 2H), 2.21–2.16 (m, 1H), 1.72–1.63 (m, 1H), 1.02 (d, J = 6.8 Hz, 3H), 0.98 (d, J = 6.6 Hz, 6H); ESI-MS: m/z [M + H]+ 368.
4-((2,3-Dihydro-1H-inden-5-yl)carbamoyl) benzoate (35i). To a solution of 23a (175 mg, 1.0 mmol) and Fe (392 mg, 7.0 mmol) in EtOH (20 mL), AcOH (0.8 mL) was added. The mixture was refluxing for 2 h under a nitrogen atmosphere at room temperature. Insoluble materials were removed by filtration and washed with EtOAc. The filtrate was evaporated to dryness under reduced pressure. The mixture was partitioned between water and EtOAc. The organic layer was washed with a saturated aqueous solution of brine, dried over anhydrous Na2SO4, and concentrated under vacuum. The residue 25a was used directly in the next reaction. To a solution of 33 (150 mg, 0.8 mmol) in thionyl chloride (4 mL), a drop of pyridine was added and refluxed for 24 h. The thionyl chloride was removed by distillation and get 34 as pale yellow solid. To a solution of 25a (45 mg, 0.3 mmol) in anhydrous pyridine (2 mL), 34 was added in CH2Cl2 (2 mL) dropwise at 0 °C. The mixture was stirred at room temperature for 7 h and then the methanol was removed by distillation. The organic layer was neutralized with 1 N HCl. The mixture was partitioned between water and EtOAc. The organic layer was washed with a saturated aqueous solution of brine, dried over anhydrous Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography on silica gel (eluent: hexane/EtOAc = 3/1) to give 35i (77 mg, 84%) as a pale yellow solid. m.p. 157~159 °C; 1H-NMR: δ 8.15 (d, J = 8.4 Hz, 2H), 7.94 (d, J = 8.2 Hz, 2H), 7.83 (s, 1H), 7.62 (s, 1H), 7.34 (d, J = 7.9 Hz, 1H), 6.48 (s, 1H), 3.96 (s, 3H), 3.29 (s, 2H), 2.17 (s, 3H); ESI-MS: m/z [M + H]+ 338.
Methyl 4-((2-isopropyl-1H-inden-5-yl)carbamoyl) benzoate (35j). The title compound was prepared from 23b in a manner similar to that described for 35i as a pale yellow solid (67%). m.p. 155~156 °C; 1H-NMR: δ 8.16 (d, J = 8.4 Hz, 2H), 7.94 (d, J = 8.3 Hz, 2H), 7.78 (s, 1H), 7.63 (s, 1H), 7.35 (s, 1H), 6.50 (s, 1H), 3.97 (s, 3H), 3.34 (s, 2H), 2.81–2.76 (m, 1H), 1.24 (d, J = 6.8, 3H); ESI-MS: m/z [M + H]+ 336.
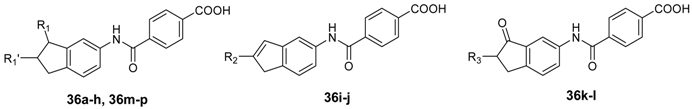
4-((2,3-Dihydro-1H-inden-5-yl)carbamoyl)benzoic acid (36a). To a solution of 35a (56 mg, 0.2 mmol) in MeOH (2 mL), 0.5 N LiOH (0.4 mL) was added dropwise at 0 °C. The mixture was stirred at room temperature for 48 h and then neutralized with 1 N HCl. The MeOH was removed by distillation. After adding water (10 mL), the mixture was partitioned between water and EtOAc. The organic layer was washed with a saturated aqueous solution of brine, dried over anhydrous Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography on silica gel (eluent: hexane/EtOAc/AcOH = 30/10/1) to give 36a (52 mg, 92%) as a white solid, purity: 97%. m.p. >250 °C; 1H-NMR: δ 13.26 (s, 1H), 10.30 (s, 1H), 8.14–7.92 (m, 4H), 7.68 (s, 1H), 7.47 (d, J = 8.0 Hz, 1H), 7.19 (d, J = 8.1 Hz, 1H), 2.93–2.76 (m, 4H), 2.07–1.97 (m, 2H); ESI-MS: m/z [M − H]− 280.
4-((3-Methoxy-2,3-dihydro-1H-inden-5-yl)carbamoyl)benzoic acid (36b). The title compound was prepared from 35b in a manner similar to that described for 36a as a white solid (91%), purity: 96%. m.p. >250 °C; 1H-NMR: δ 10.38 (s, 1H), 8.15–8.01 (m, 4H), 7.86 (s, 1H), 7.63 (d, J = 8.1 Hz, 1H), 7.25 (d, J = 8.2 Hz, 1H), 4.86–4.72 (m, 1H), 3.33(s, 3H),3.00–2.87 (m, 1H), 2.81–2.69 (m, 1H), 2.36–2.32 (m, 1H), 1.99–1.93 (m, 1H); ESI-MS: m/z [M − H]− 310. HRMS (ESI) calcd [M + H]+ for C18H18NO4 312.1230, found 312.1234.
4-((3-Methoxy-2,3-dihydro-1H-inden-5-yl)carbamoyl)benzoic acid (36c). The title compound was prepared from 35c in a manner similar to that described for 36a as a white solid (99%), purity: 95%. m.p. >250 °C; 1H-NMR: δ 10.37 (s, 1H), 8.07–8.03 (m, 4H), 7.81 (s, 1H), 7.63 (dd, J = 8.0, 2.0 Hz, 1H), 7.23 (d, J = 8.2 Hz, 1H), 4.89–4.83 (m, 1H), 3.60–3.50 (m, 2H), 2.94–2.88 (m, 1H), 2.76–2.69 (m, 1H), 2.36–2.30 (m, 1H), 1.95–1.89 (m, 1H), 1.15 (t, J = 7.0 Hz, 3H); ESI-MS: m/z [M − H]− 324. HRMS (ESI) calcd [M + H]+ for C19H20NO4 326.1387, found 326.1392.
4-((3-Isopropoxy-2,3-dihydro-1H-inden-5-yl)carbamoyl)benzoic acid (36d). The title compound was prepared from 35d in a manner similar to that described for 36a as a white solid (97%), purity: 97%. m.p. >250 °C; 1H-NMR: δ 13.24 (s, 1H), 10.36 (s, 1H), 8.05 (s, 4H), 7.74 (s, 1H), 7.64 (dd, J = 8.1, 1.9 Hz, 1H), 7.21 (d, J = 8.2 Hz, 1H), 4.96 (t, J = 6.1 Hz, 1H), 3.93–3.74 (m, 2H), 3.85–3.79 (m, 1H), 2.94–2.84 (m, 1H), 2.73–2.67 (m, 1H), 2.40–2.34 (m, 1H), 1.87–1.81 (m, 1H), 1.17 (d, J = 6.0, 3H) , 1.16 (d, J = 6.5, 3H); ESI-MS: m/z [M − H]− 338. HRMS (ESI) calcd [M + H]+ for C20H22NO4 340.1543, found 340.1550.
4-((3-Butoxy-2,3-dihydro-1H-inden-5-yl)carbamoyl)benzoic acid (36e). The title compound was prepared from 35e in a manner similar to that described for 36a as a white solid (90%). purity: 96%. m.p. >250 °C; 1H-NMR: δ 13.26 (s, 1H), 10.37 (s, 1H), 8.05 (d, J = 2.1 Hz, 4H), 7.79 (s, 1H), 7.63 (dd, J = 8.0 Hz, 1H), 7.21 (d, J = 8.0 Hz, 1H), 4.89–4.80 (m, 1H), 3.51 (td, J = 6.5, 2.5 Hz, 2H), 2.98–2.85 (m, 1H), 2.78–2.66 (m, 1H), 2.38–2.30 (m, 1H), 1.94–1.86 (m, 1H), 1.56–1.42 (m, 2H), 1.39–1.31 (m, 2H), 0.88 (t, J = 7.4 Hz, 3H); ESI-MS: m/z [M − H]− 352. HRMS (ESI) calcd [M + H]+ for C21H24NO4 354.1700, found 354.1703.
4-((2-Methoxy-2,3-dihydro-1H-inden-5-yl)carbamoyl)benzoic acid (36f). The title compound was prepared from 35f in a manner similar to that described for 36a as a white solid (86%), purity: 98%. m.p. >250 °C; 1H-NMR: δ 8.15 (d, J = 8.3 Hz, 2H), 7.92 (d, J = 8.2 Hz, 2H), 7.80 (s, 1H), 7.62 (s, 1H), 7.31 (d, J = 7.8 Hz, 1H), 7.21 (d, J = 8.1 Hz, 1H), 4.31–4.23 (m, 1H), 3.96 (s, 3H), 3.38 (s, 3H), 3.20–3.12 (m, 2H), 3.03-2.94 (m, 2H); ESI-MS: m/z [M − H]− 310. HRMS (ESI) calcd [M + H]+ for C18H18NO4 312.1230, found 312.1240.
4-((2-Ethoxy-2,3-dihydro-1H-inden-5-yl)carbamoyl)benzoic acid (36g). The title compound was prepared from 35g in a manner similar to that described for 36a as a white solid (97%), purity: 97%. m.p. >250 °C; 1H-NMR: δ 10.37 (s, 1H), 8.12–8.01 (m, 4H), 7.82 (s, 1H), 7.65 (dd, J = 8.1, 1.7 Hz, 1H), 7.24 (d, J = 8.2 Hz, 1H), 4.94–4.80 (m, 1H), 3.62–3.54 (m, 2H), 2.97–2.88 (m, 1H), 2.78–2.69 (m, 1H), 2.37–2.31 (m, 1H), 1.98–1.90 (m, 1H), 1.16 (t, J = 7.0 Hz, 3H); ESI-MS: m/z [M − H]− 324. HRMS (ESI) calcd [M + H]+ for C19H20NO4 326.1387, found 326.1396.
4-((2-Isopropyl-2,3-dihydro-1H-inden-5-yl)carbamoyl)benzoic acid (36h). The title compound was prepared from 35h in a manner similar to that described for 36a as a white solid (94%), purity: 96%. m.p. >250 °C; 1H-NMR: δ 8.07–8.02 (m, 4H), 7.63 (s, 1H), 7.47 (d, J = 7.9 Hz, 1H), 7.15 (d, J = 8.1 Hz, 1H), 2.98–2.90 (m, 2H), 2.66–2.52 (m, 2H), 2.15–2.10 (m, 1H), 1.68–1.10 (m, 1H), 0.95 (d, J = 6.5 Hz, 3H), 0.94 (d, J = 6.5 Hz, 3H); ESI-MS: m/z [M − H]− 322. HRMS (ESI) calcd [M + H]+ for C20H22NO3 324.1594, found 324.1600.
4-((2-Methyl-1H-inden-5-yl)carbamoyl)benzoic acid (36i). The title compound was prepared from 35i in a manner similar to that described for 36a as a white solid (98%), purity: 95%. m.p. >250 °C; 1H-NMR: 10.31 (s, 1H), 8.07–8.02 (m, 4H), 7.78 (s, 1H), 7.48 (d, J = 6.4 Hz, 1H), 7.26 (d, J = 8.1 Hz, 1H), 6.51 (s, 1H), 3.29 (s, 2H), 2.12 (s, 3H); ESI-MS: m/z [M − H]− 292. HRMS (ESI) calcd [M + H]+ for C18H16NO3 294.1125, found 294.1129.
4-((2-Isopropyl-1H-inden-5-yl)carbamoyl)benzoic acid (36j). The title compound was prepared from 35j in a manner similar to that described for 36a as a white solid (83%), purity: 95%. m.p. >250 °C; 1H-NMR: δ 13.25 (s, 1H), 10.34 (s, 1H), 8.09–8.06 (m, 4H), 7.72 (s, 1H), 7.47 (dd, J = 8.0, 1.5 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 6.53 (s, 1H), 3.37 (s, 2H), 2.79–2.72 (m, 1H), 1.20 (d, J = 6.5, 3H), 1.19 (d, J = 6.5, 3H); ESI-MS: m/z [M − H]− 320. HRMS (ESI) calcd [M + H]+ for C20H20NO3 322.1438, found 322.1440.
4-((2-Methyl-3-oxo-2,3-dihydro-1H-inden-5-yl)carbamoyl)benzoic acid (36k). The title compound was prepared from 35k in a manner similar to that described for 36a as a white solid (92%), purity: 95%. m.p. >250 °C (AcOH–EtOAc–hexane); 1H-NMR: δ 10.60 (s, 1H), 8.09–8.04 (m, 4H), 8.07 (d, J = 2.8 Hz, 1H), 8.00 (dd, J = 8.3, 2.1 Hz, 1H), 7.56 (d, J = 8.4 Hz, 1H), 3.38–3.36 (m, J = 7.4 Hz, 1H), 2.78–2.72 (m, 1H), 2.70–2.65 (m, 1H), 1.20 (d, J = 7.4 Hz, 3H); ESI-MS: m/z [M − H]− 308. HRMS (ESI) calcd. [M + H]+ for C18H16NO4 310.1074, found 310.1069.
4-((2-Isopropyl-3-oxo-2,3-dihydro-1H-inden-5-yl)carbamoyl)benzoic acid (36l). The title compound was prepared from 35l in a manner similar to that described for 36a as a white solid (97%), purity: 98%. m.p. >250 °C; 1H-NMR: δ 13.30 (s, 1H), 10.61 (s, 1H), 8.14 (d, J = 1.8 Hz, 1H), 8.08 (d, J = 3.9 Hz, 4H), 8.00 (dd, J = 8.3, 2.0 Hz, 1H), 7.60 (d, J = 8.3 Hz, 1H), 3.13 (dd, J = 17.4, 8.0 Hz, 1H), 2.88 (dd, J = 17.5, 3.8 Hz, 1H), 2.73 (dt, J = 8.0, 4.1 Hz, 1H), 2.30–2.24 (m, 1H), 1.01 (d, J = 6.9 Hz, 3H), 0.74 (d, J = 6.8 Hz, 3H); ESI-MS: m/z [M − H]− 336. HRMS (ESI) calcd [M + H]+ for C20H20NO4 338.1387, found 338.1384.
4-((3-Methoxy-2-methyl-2,3-dihydro-1H-inden-5-yl)carbamoyl)benzoic acid (36m). The title compound was prepared from 35m in a manner similar to that described for 36a as a white solid (89%), purity: 99%. m.p. >250 °C (AcOH–EtOAc–hexane); 1H-NMR: δ 13.26 (s, 1H), 10.36 (s, 1H), 8.06–8.04 (m, 4H), 7.84 (s, 1H), 7.63 (dd, J = 8.1, 1.8 Hz, 1H), 7.23 (d, J = 8.0, 1H), 4.37 (d, J = 4.3 Hz, 1H), 3.40 (s, 3H), 3.12–3.07 (m, 1H), 2.45–2.34 (m, 2H), 1.12 (d, J = 6.8 Hz, 3H); ESI-MS: m/z [M − H]− 324. HRMS (ESI) calcd [M + H]+ for C19H20NO4 326.1387, found 326.1392.
4-((3-Ethoxy-2-methyl-2,3-dihydro-1H-inden-5-yl)carbamoyl)benzoic acid (36n). The title compound was prepared from 35n in a manner similar to that described for 36a as a white solid (97%), purity: 98%. m.p. >250 °C; 1H-NMR: δ 13.25 (s, 1H), 10.36 (s, 1H), 8.13–8.00 (m, 4H), 7.80 (s, 1H), 7.64 (d, J = 8.1 Hz, 1H), 7.21 (d, J = 8.1 Hz, 1H), 4.44 (d, J = 4.5 Hz, 1H), 3.68 (q, J = 7.0 Hz, 2H), 3.10–3.05 (m, 1H), 2.40–3.34 (m, 2H), 1.18 (t, J = 7.0 Hz, 3H), 1.13 (d, J = 6.6 Hz, 3H); ESI-MS: m/z [M − H]− 338. HRMS (ESI) calcd [M + H]+ for C20H22NO4 340.1543, found 340.1549.
4-((2-Isopropyl-3-methoxy-2,3-dihydro-1H-inden-5-yl)carbamoyl)benzoic acid (36o). The title compound was prepared from 35o in a manner similar to that described for 36a as a white solid (98%), purity: 97%. m.p. >250 °C; 1H-NMR: δ 13.21 (s, 1H), 10.34 (s, 1H), 8.10–8.02 (m, 4H), 7.84 (s, 1H), 7.64 (dd, J = 8.5, 1.5 Hz , 1H), 7.20 (d, J = 8.2 Hz, 1H), 4.65 (d, J = 3.3 Hz, 1H), 3.39 (s, 3H), 3.01–2.96 (m, 1H), 2.55–2.53 (m, 1H), 2.23–2.15 (m, 1H), 1.83–1.76 (m, 1H), 0.95 (d, J = 6.7 Hz, 3H), 0.90 (d, J = 6.7 Hz, 3H); ESI-MS: m/z [M − H]− 352. HRMS (ESI) calcd [M + H]+ for C21H24NO4 354.1700, found 354.1704.
4-((3-Ethoxy-2-isopropyl-2,3-dihydro-1H-inden-5-yl)carbamoyl)benzoic acid (36p). The title compound was prepared from 35p in a manner similar to that described for 36a as a white solid (98%), purity: 98%. m.p. >250 °C; 1H-NMR: δ 13.28 (s, 1H), 10.36 (s, 1H), 8.11–7.99 (m, 4H), 7.80 (s, 1H), 7.66–7.58 (dd, J = 8.0, 2.0 Hz, 1H), 7.18 (d, J = 8.2 Hz, 1H), 4.69 (d, J = 5.6 Hz, 1H), 3.72–3.57 (m, 2H), 3.33–3.31 (m, 1H), 2.94 (q, J = 8.4 Hz, 1H), 2.18–2.12 (m, 1H), 1.81–1.76 (m, 1H), 1.16 (t, J = 7.0 Hz, 3H), 0.95 (d, J = 6.7 Hz, 3H), 0.89 (d, J = 6.7 Hz, 3H); ESI-MS: m/z [M − H]− 366. HRMS (ESI) calcd [M + H]+ for C22H26NO4 368.1856, found 368.1863.



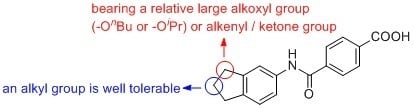
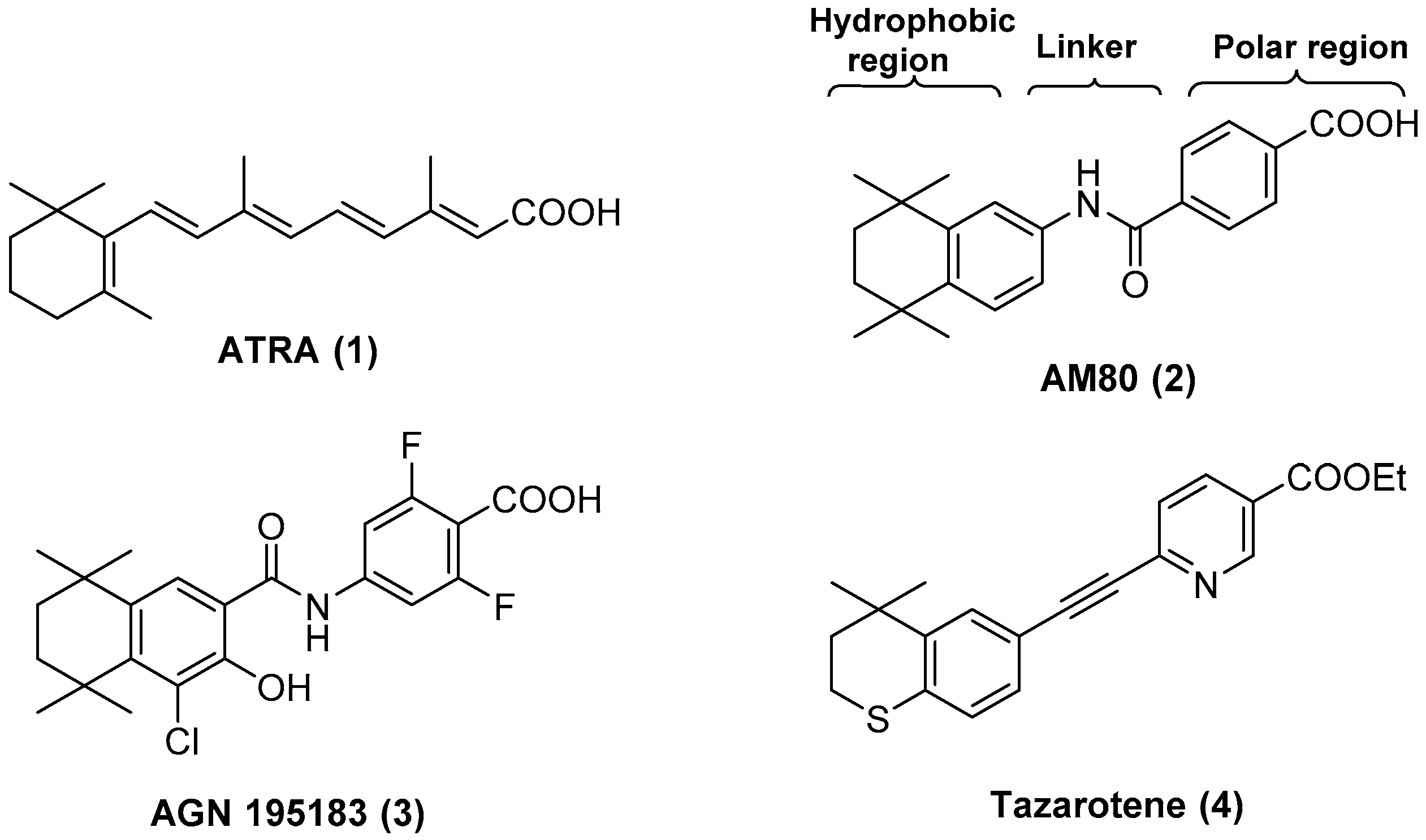
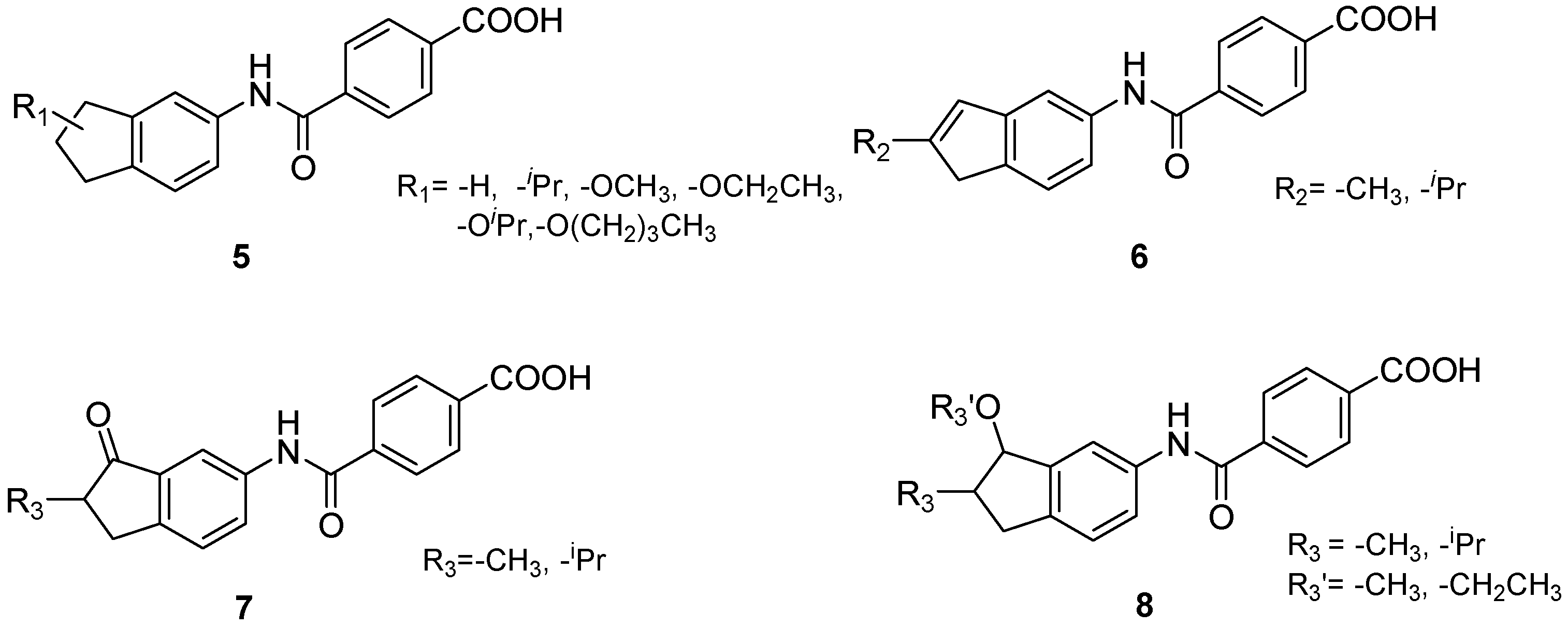
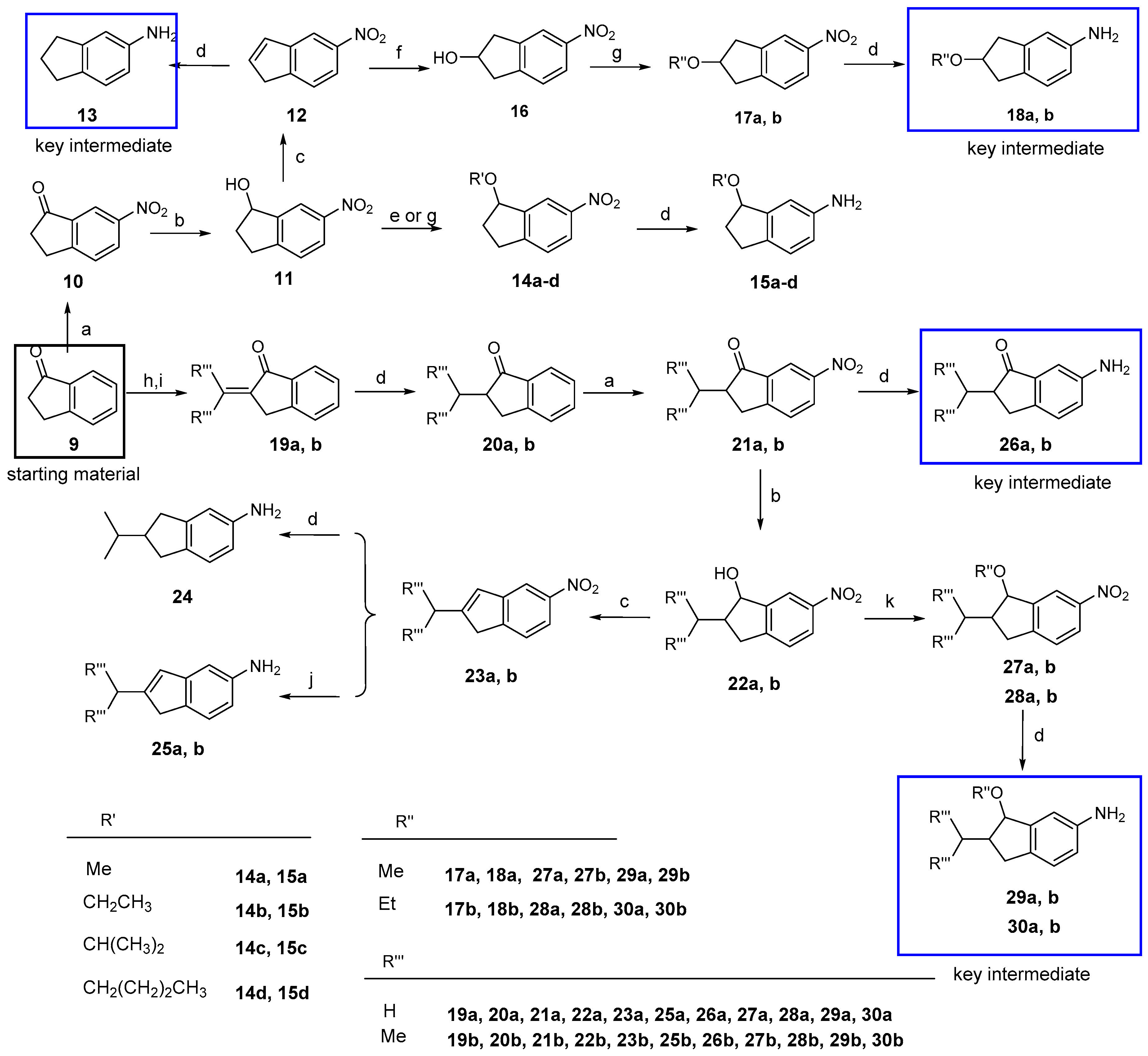

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}