
Transition Metal Complexes Coordinated by Water Soluble Phosphane Ligands: How Cyclodextrins Can Alter the Coordination Sphere?


Abstract
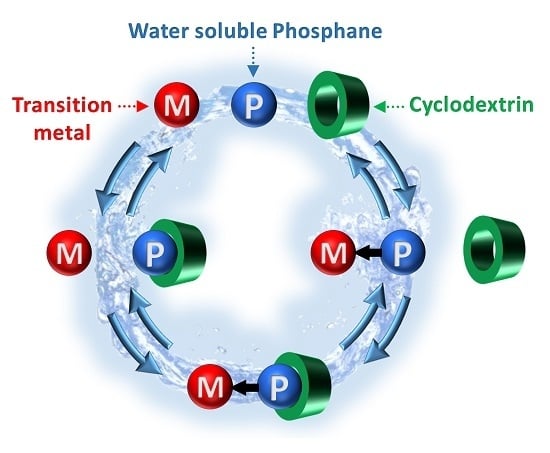
:
1. Introduction
2. Results and Discussion
2.1. Platinum Complexes
2.1.1. Complex Synthesis
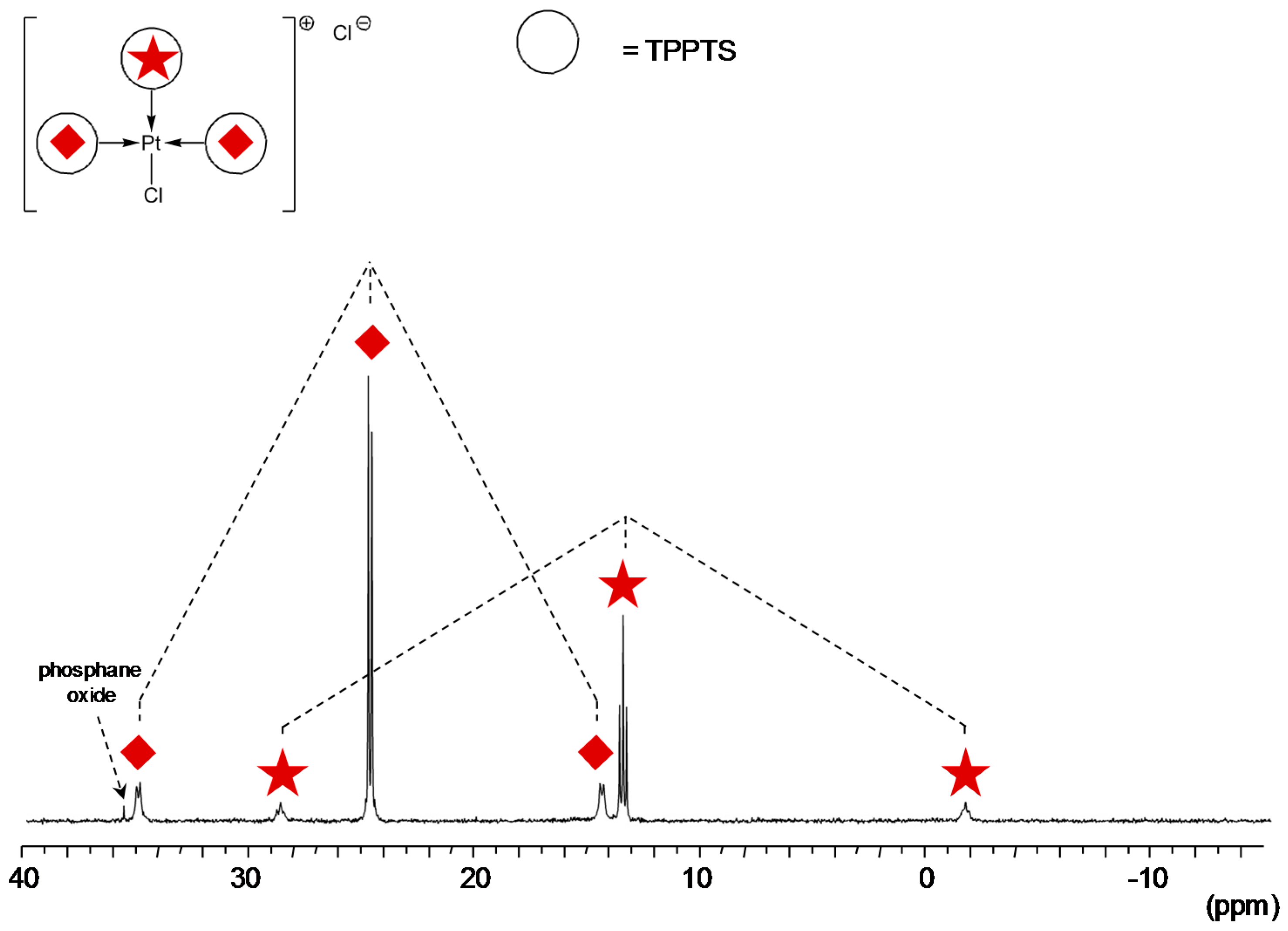
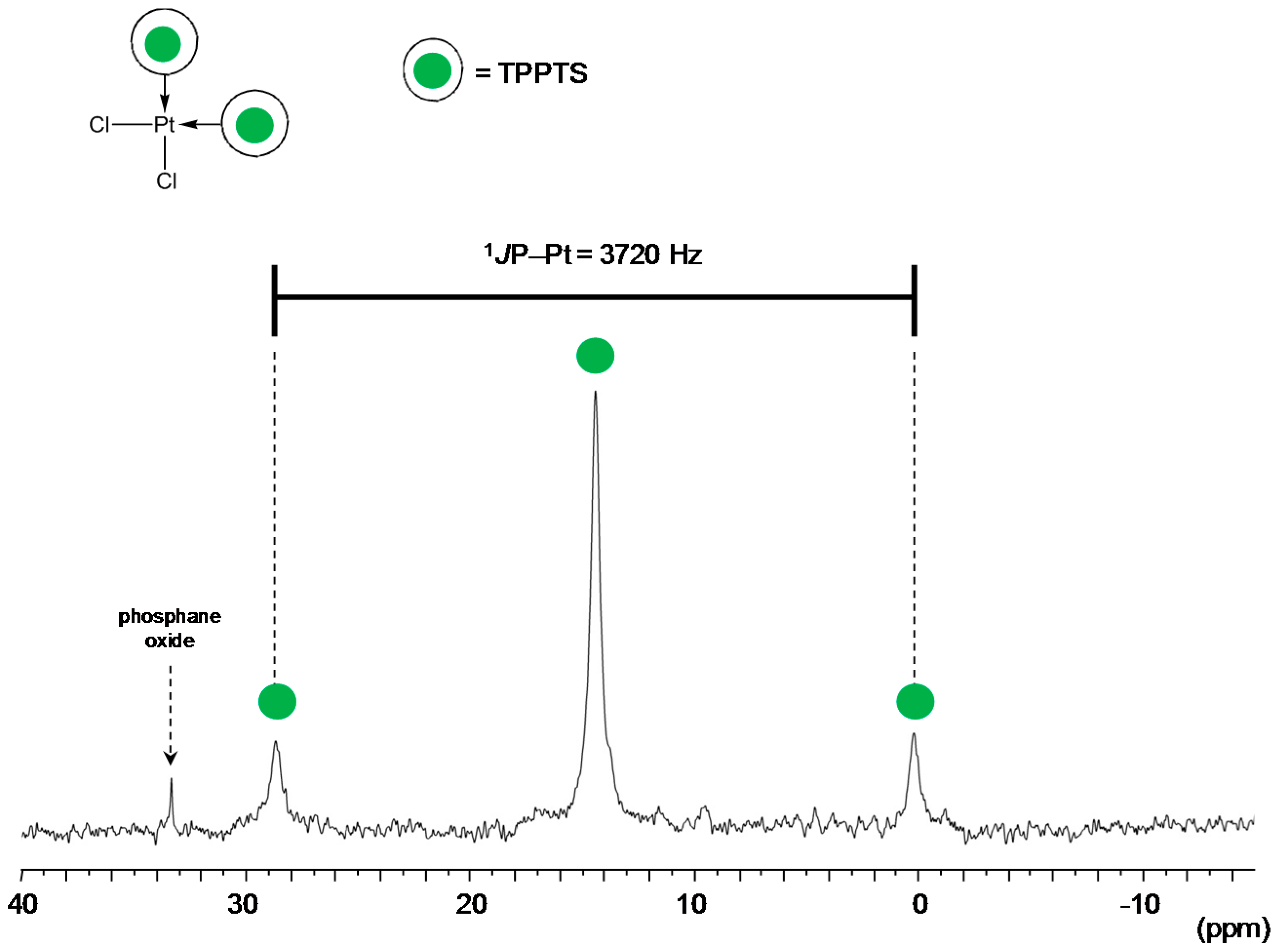
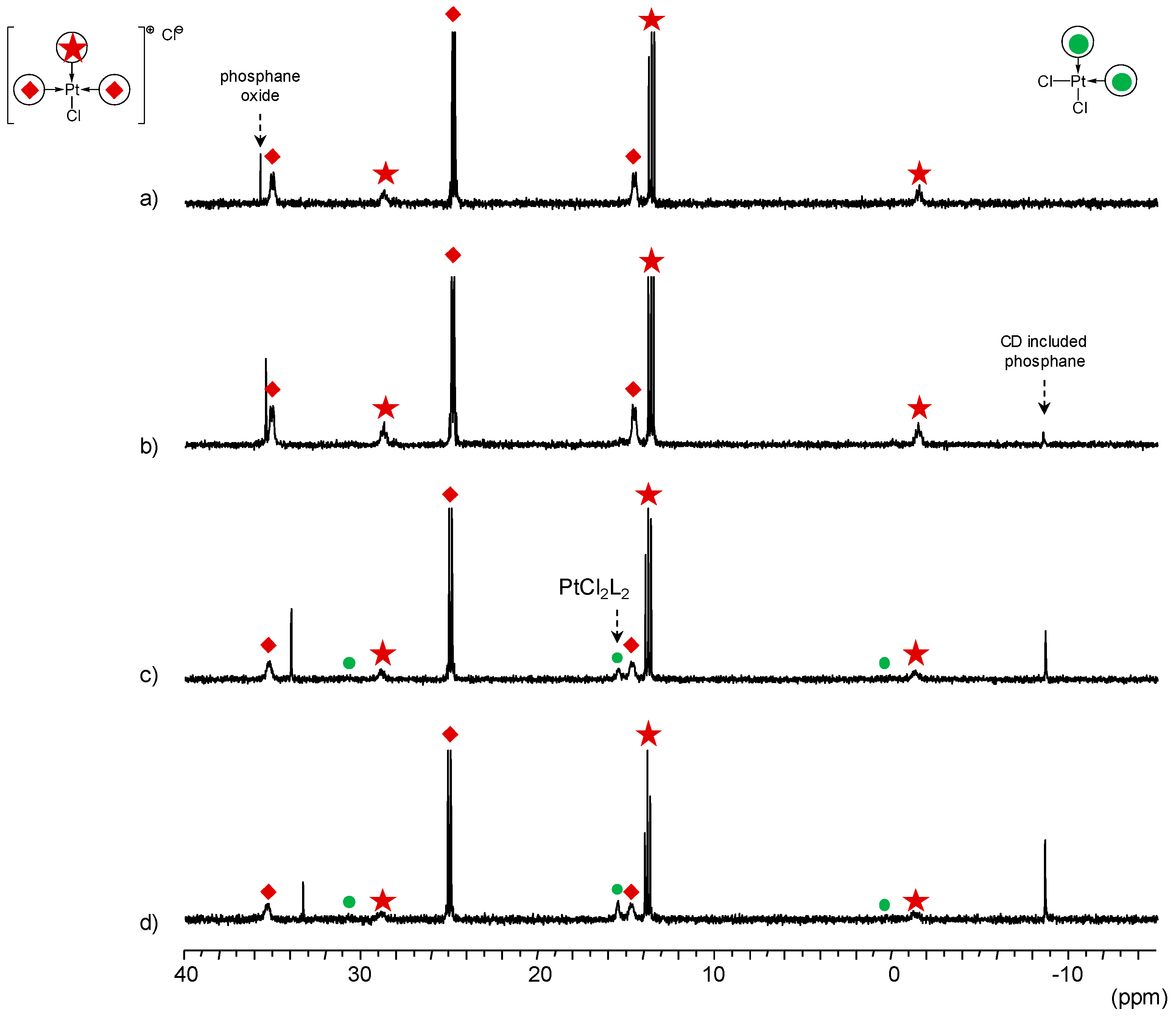
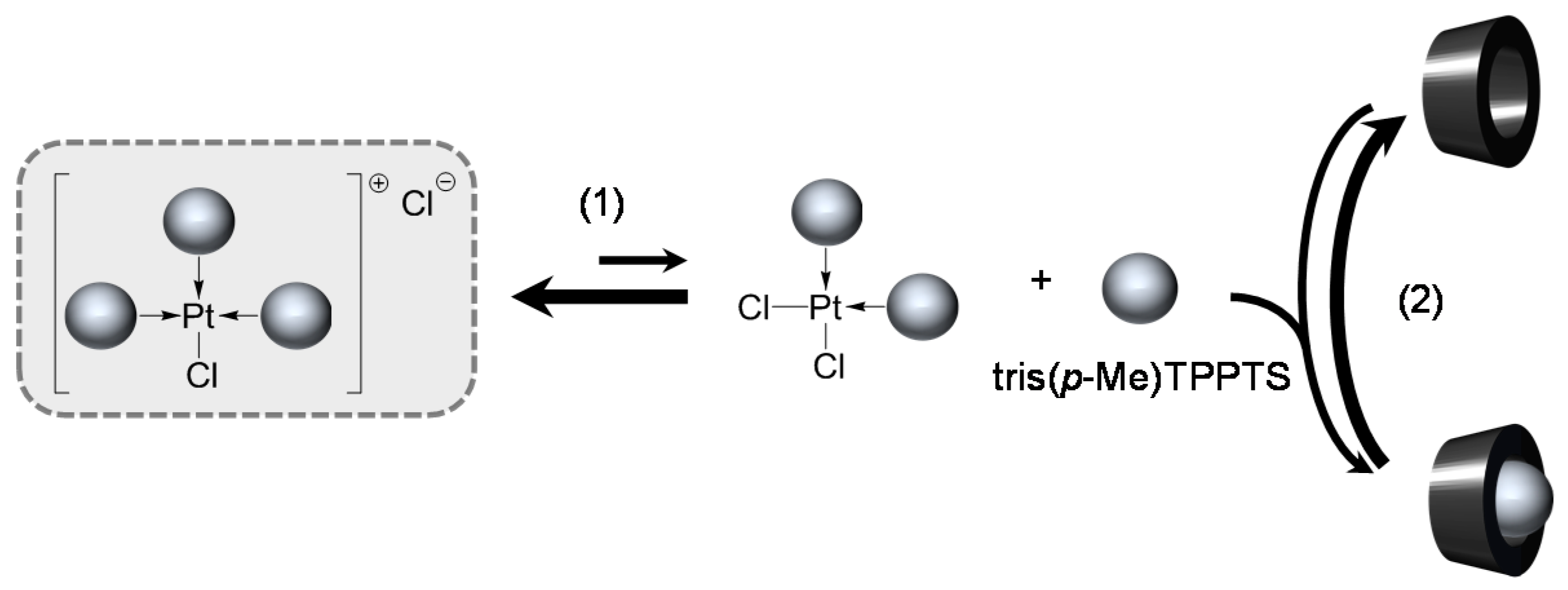
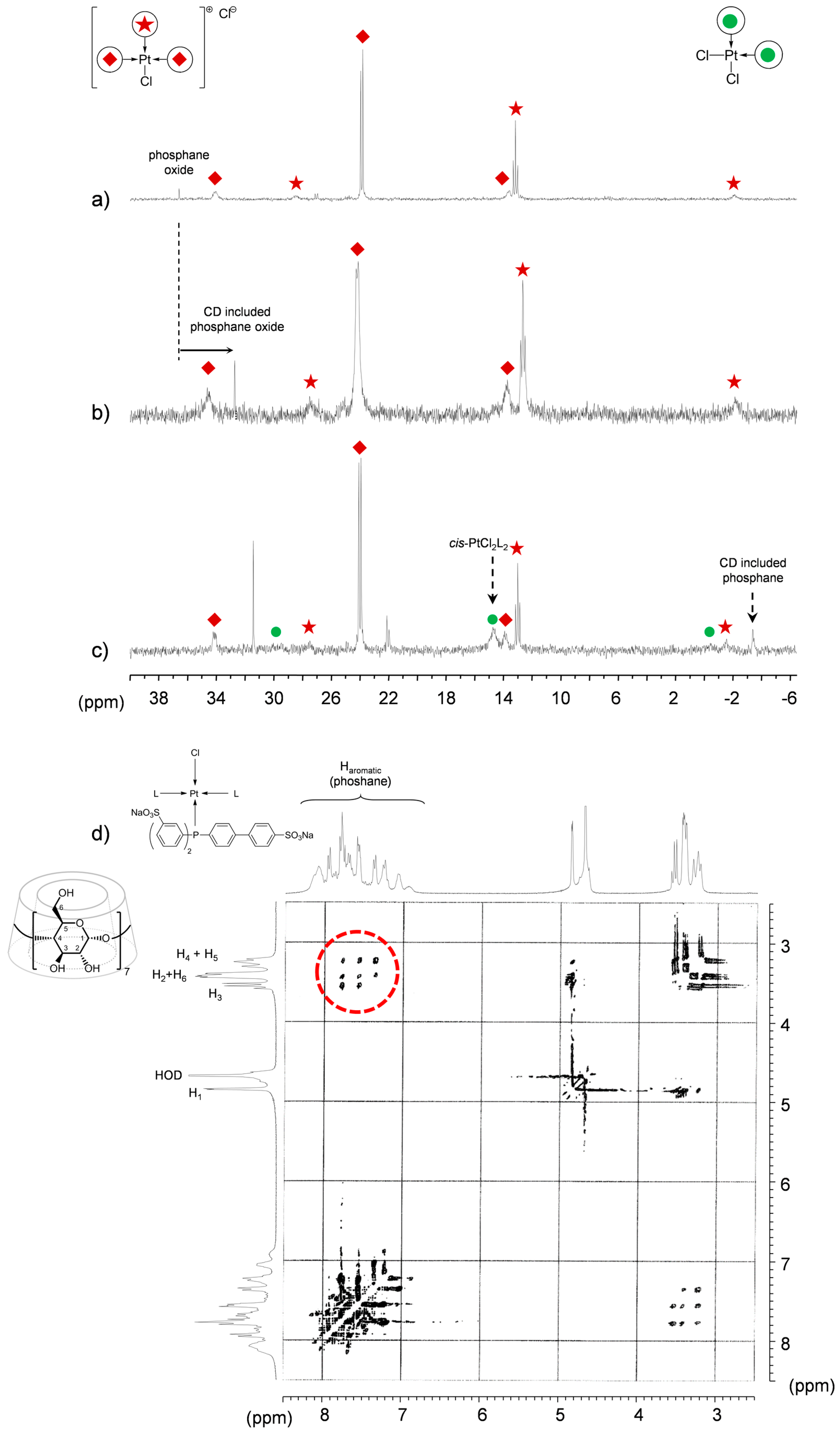
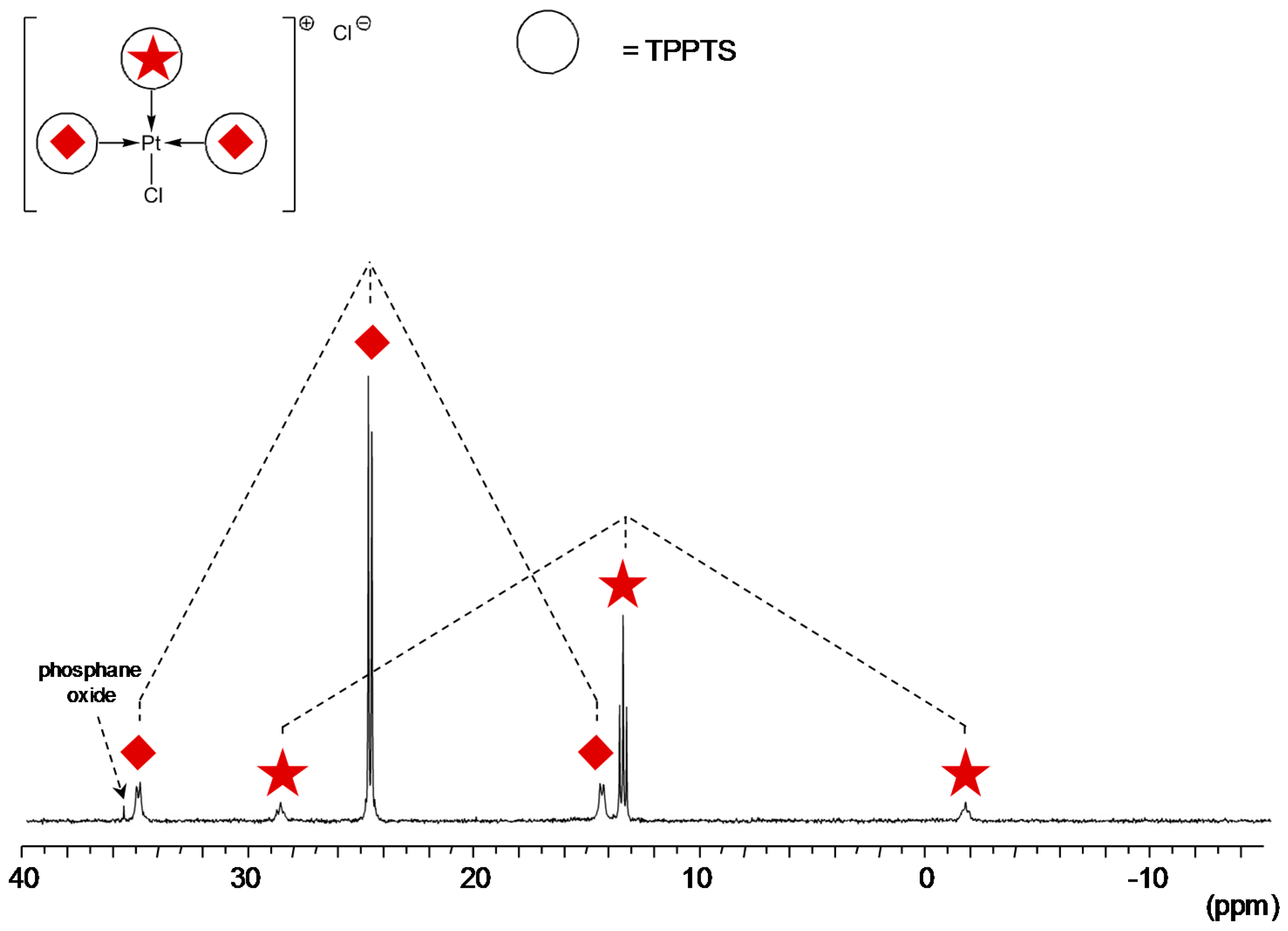
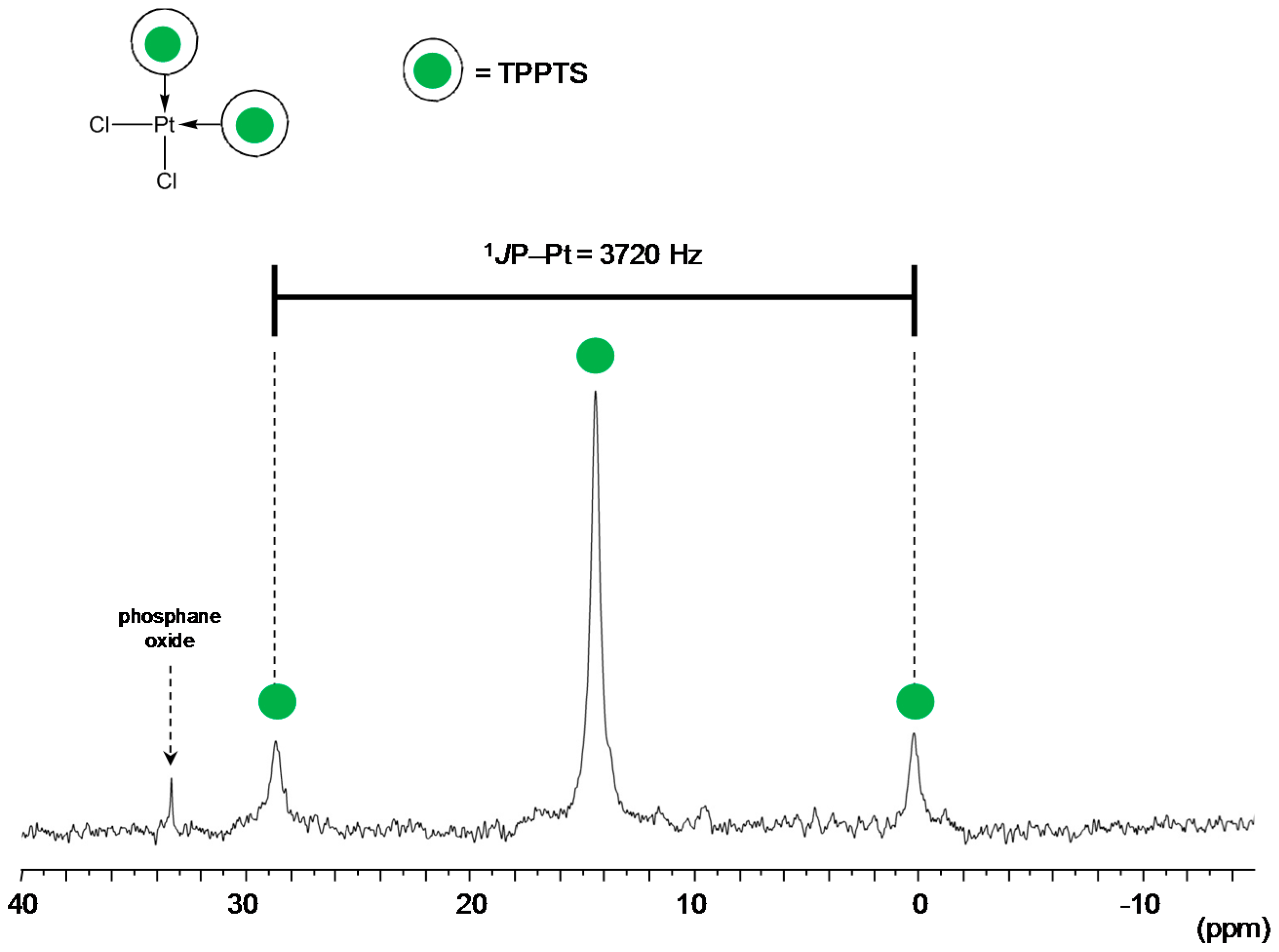
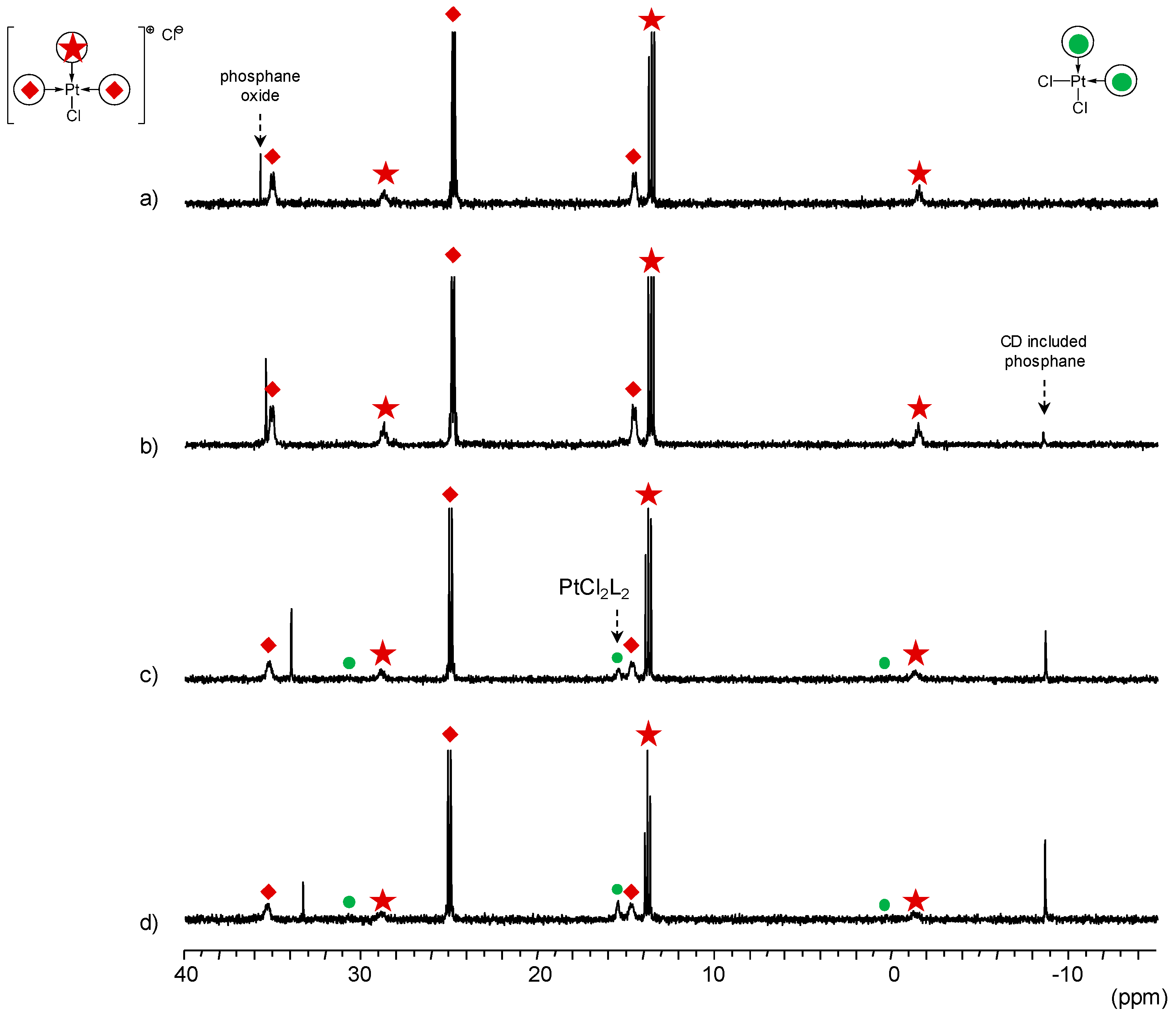
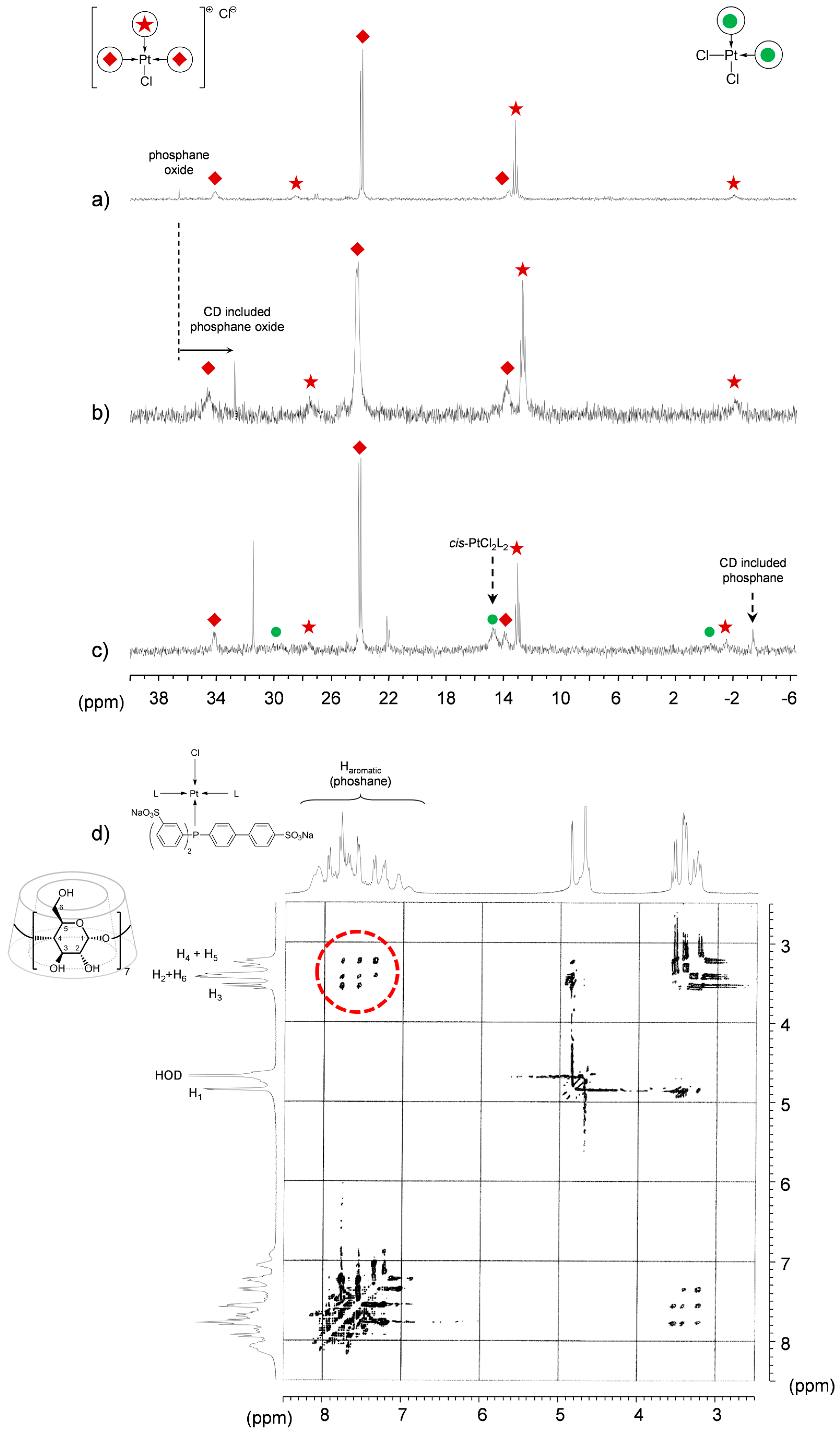
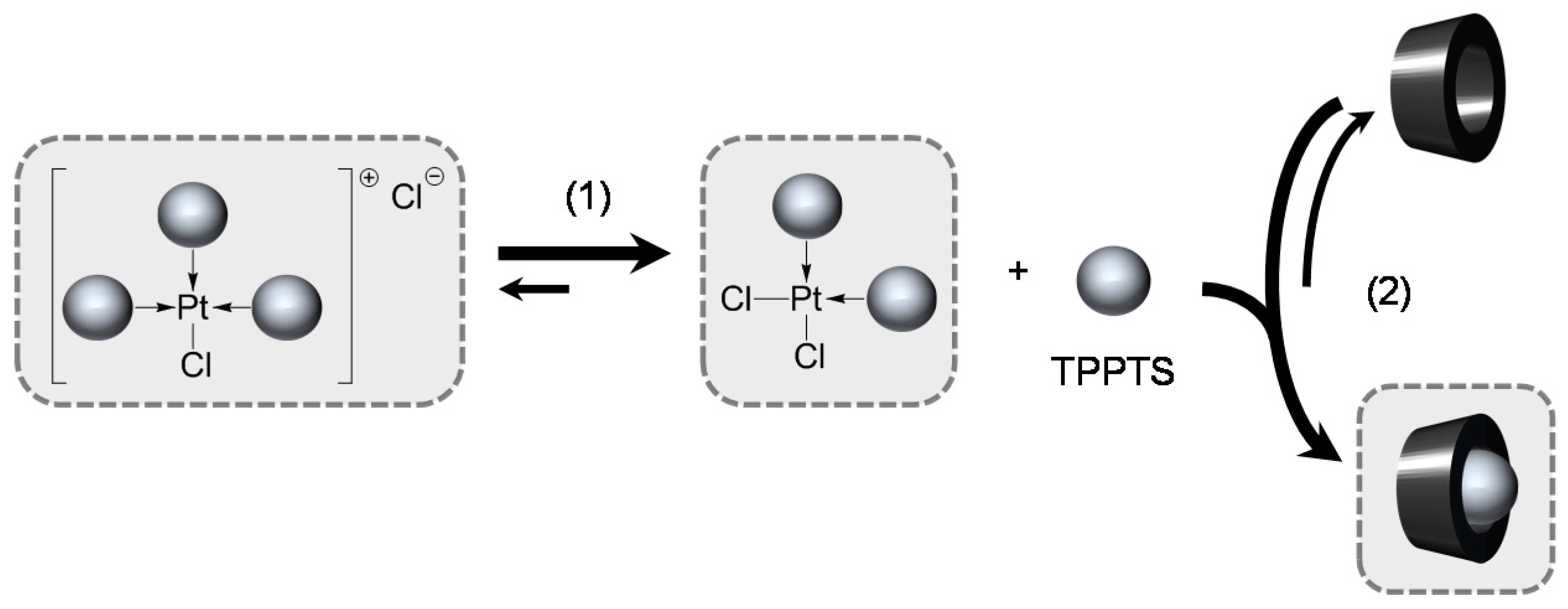
TPPTS Ligand
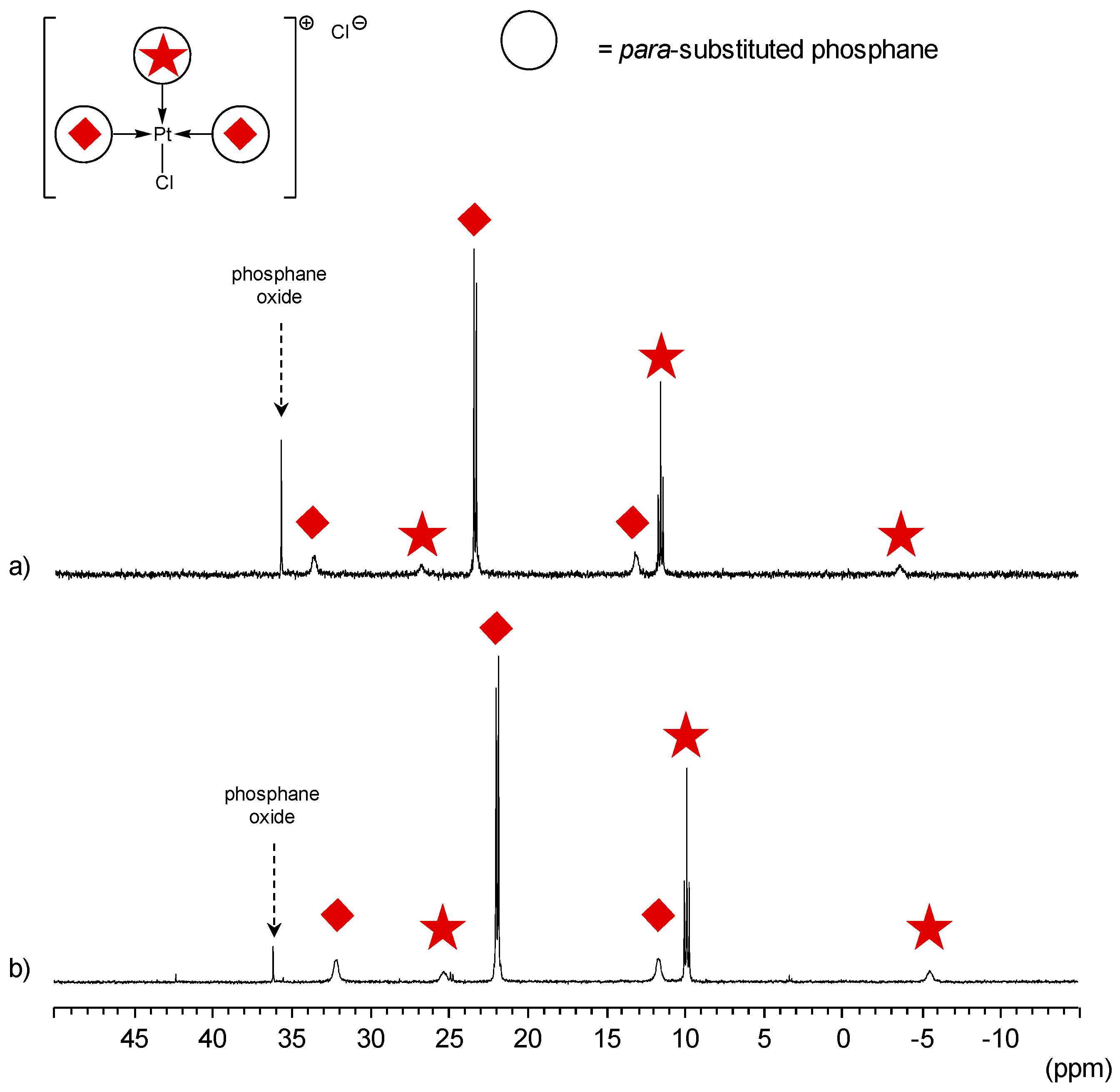
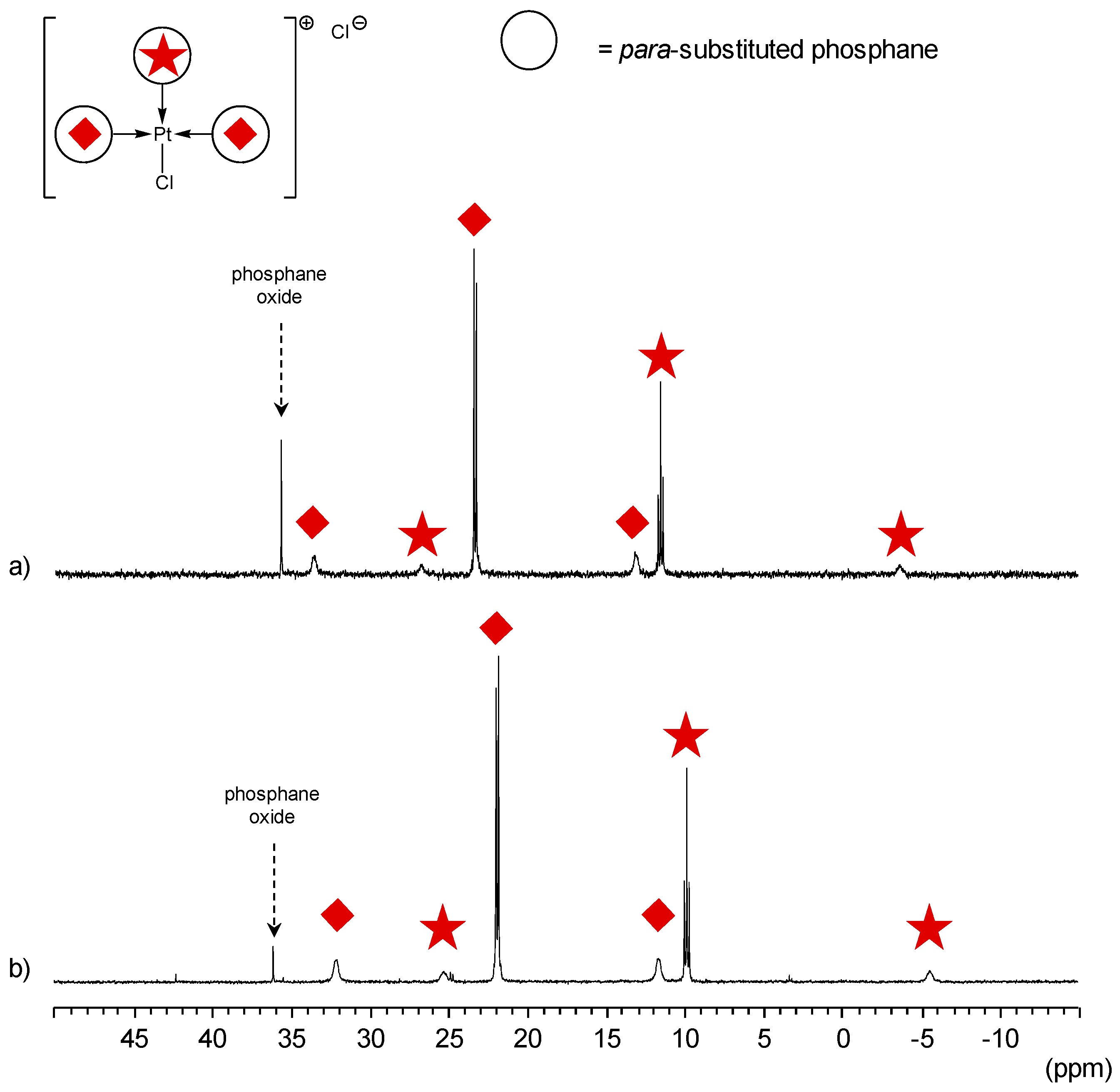
para-Substituted TPPTS Derivatives
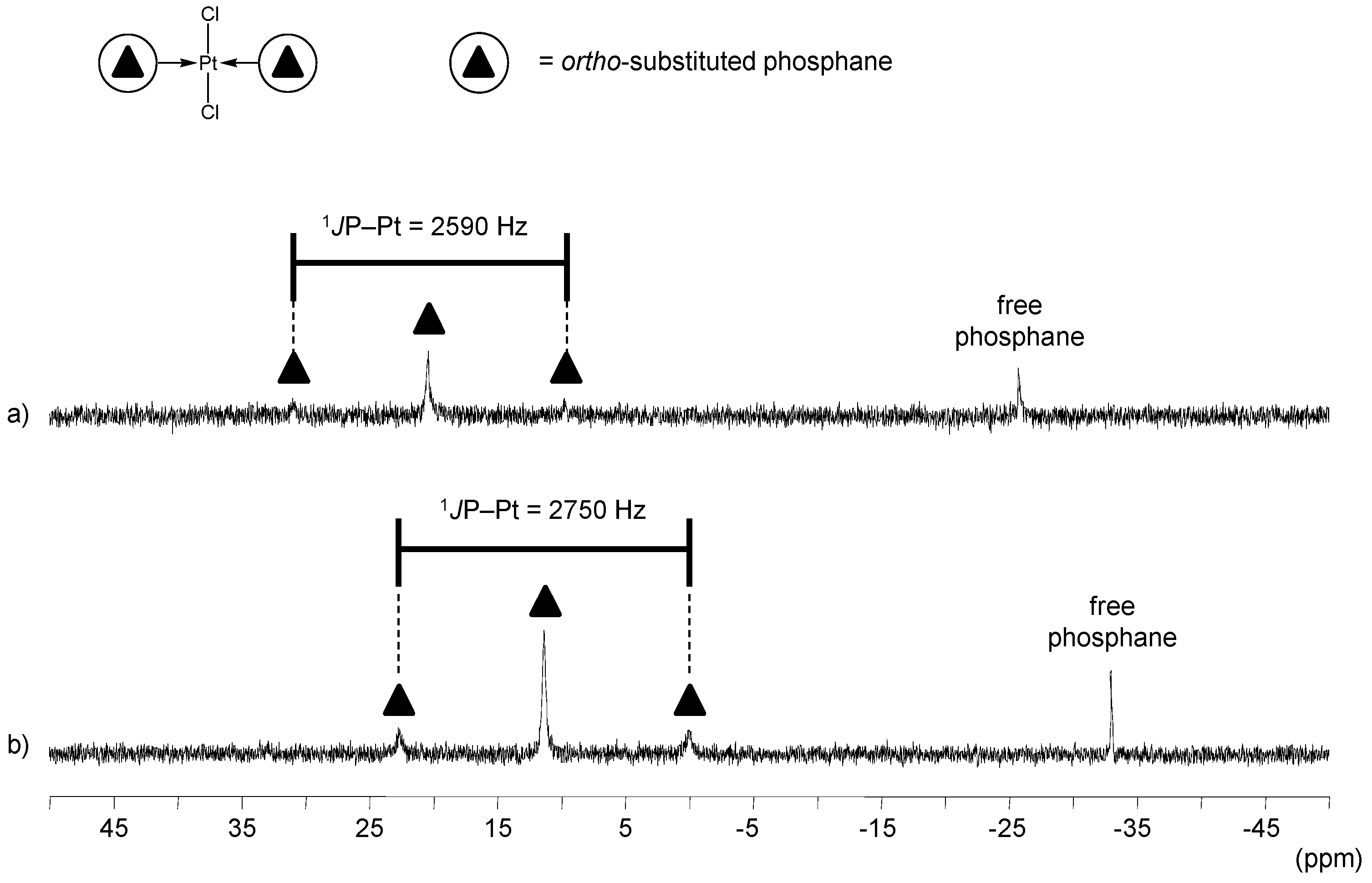
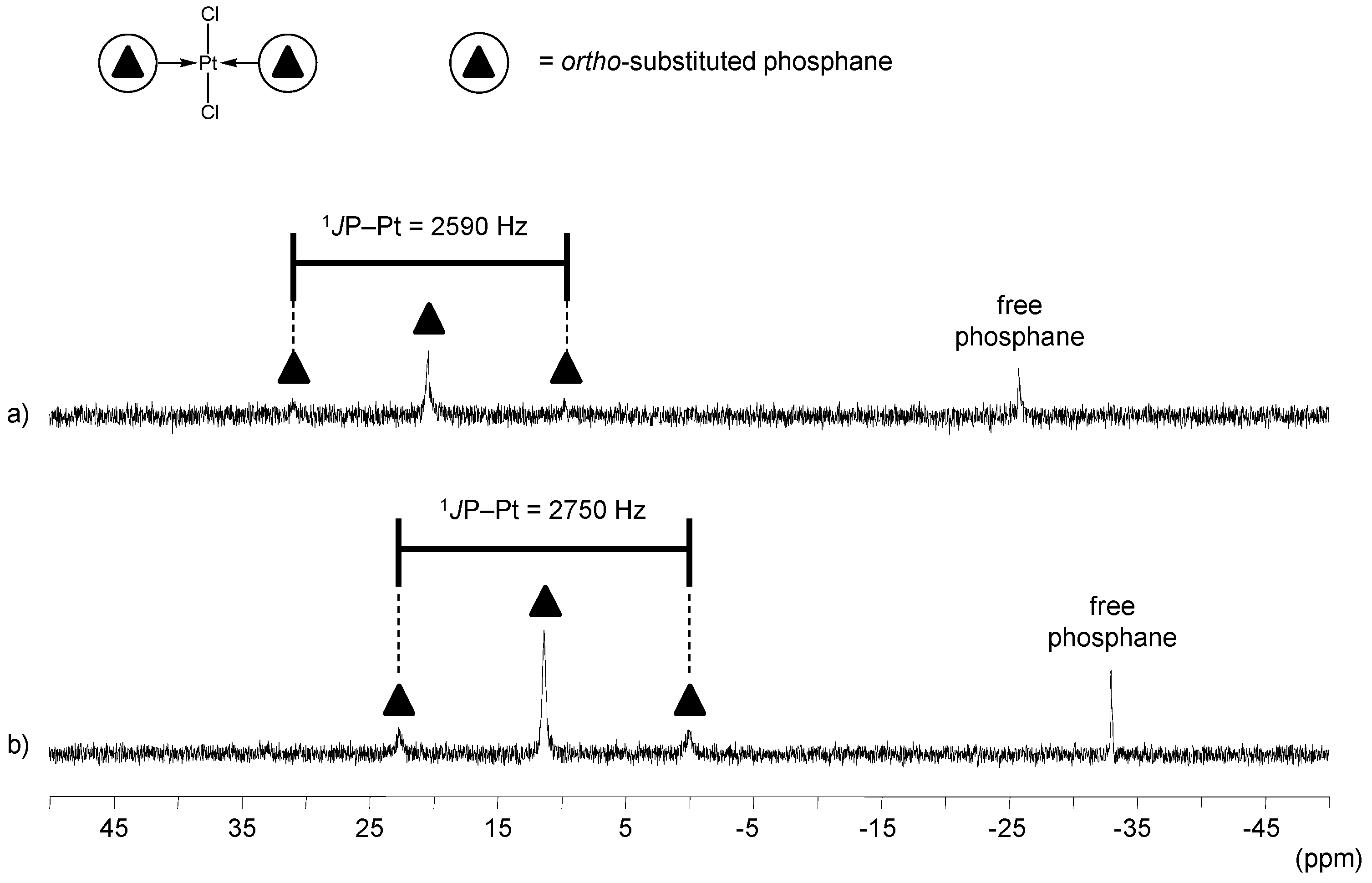
ortho-Substituted TPPTS Derivatives
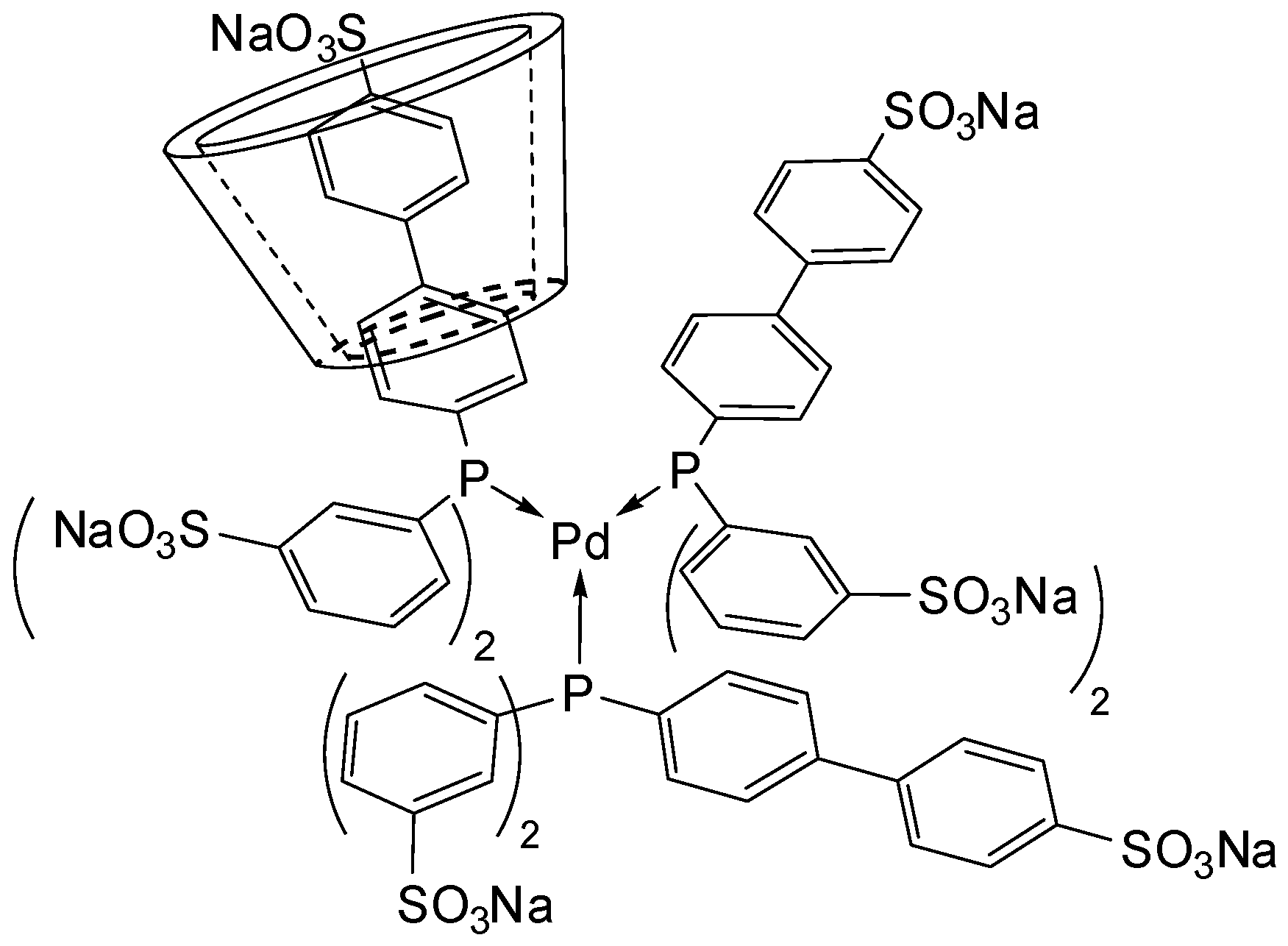
Trisulfonated Biphenylphosphane Ligands
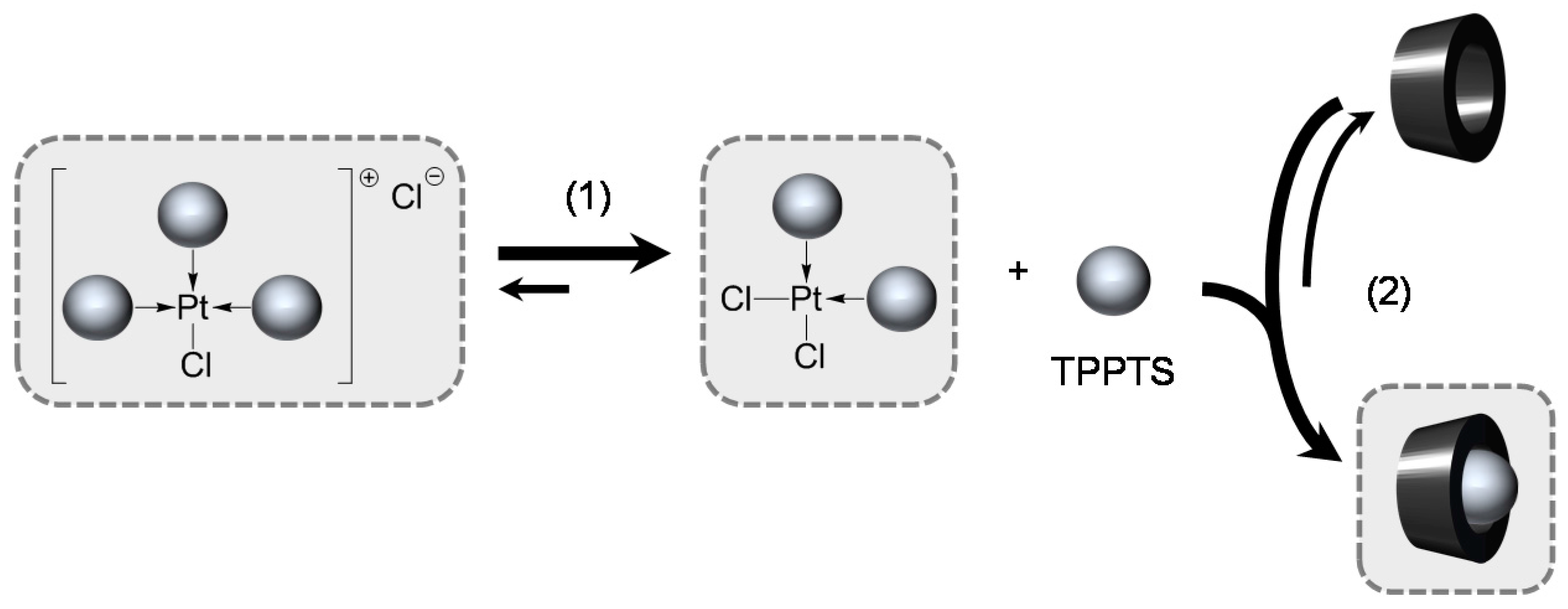
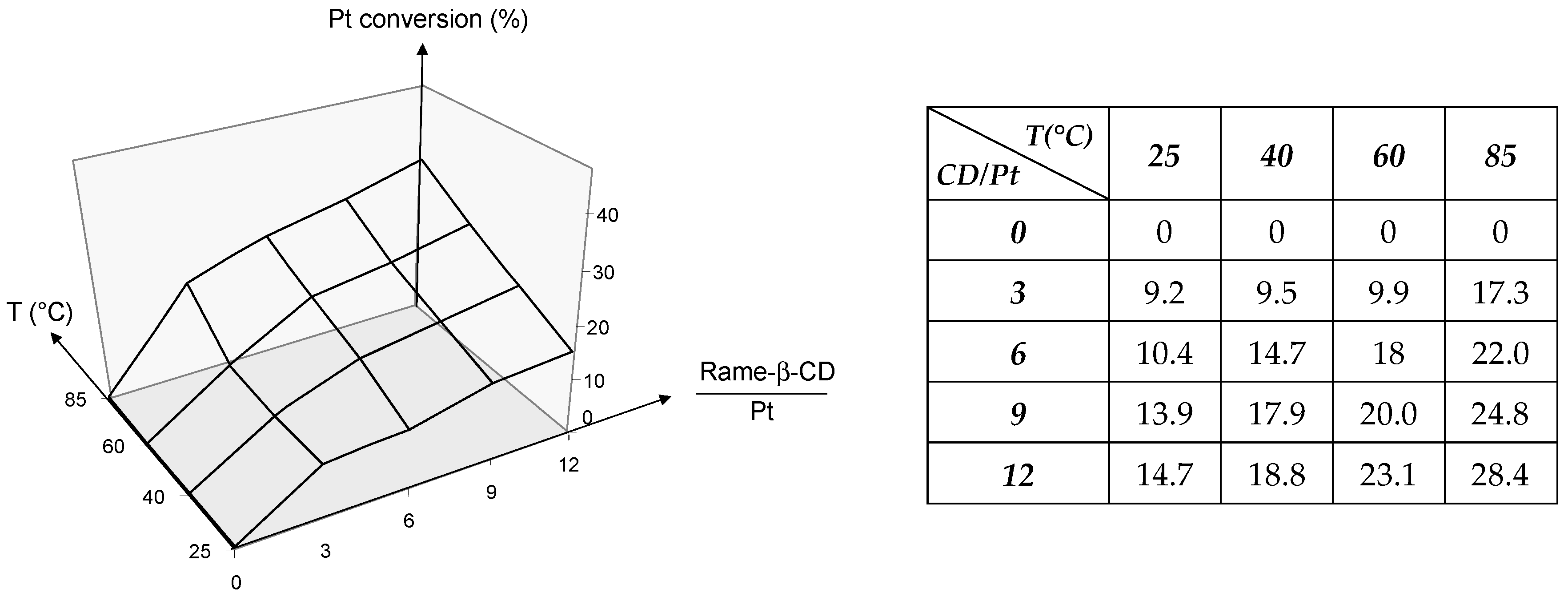
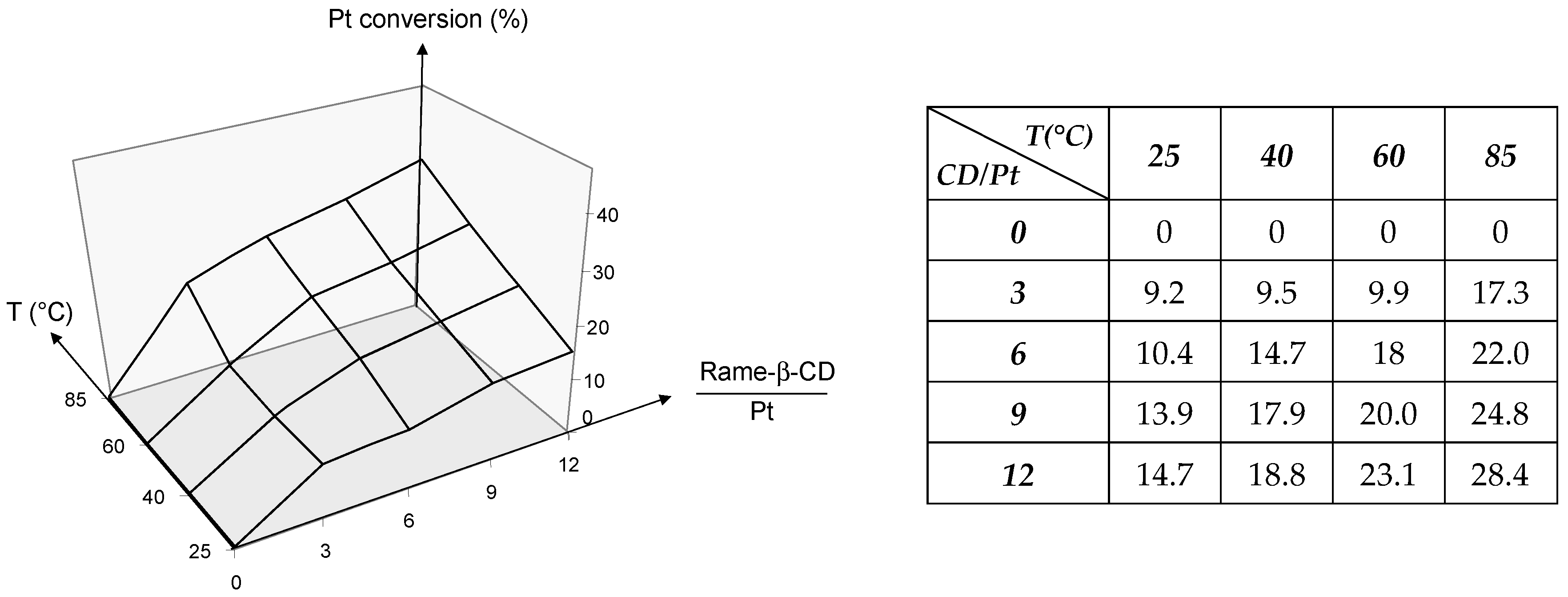
2.1.2. RAME-β-CD Effects on the Coordination Behaviour of the Ligands
TPPTS Ligand
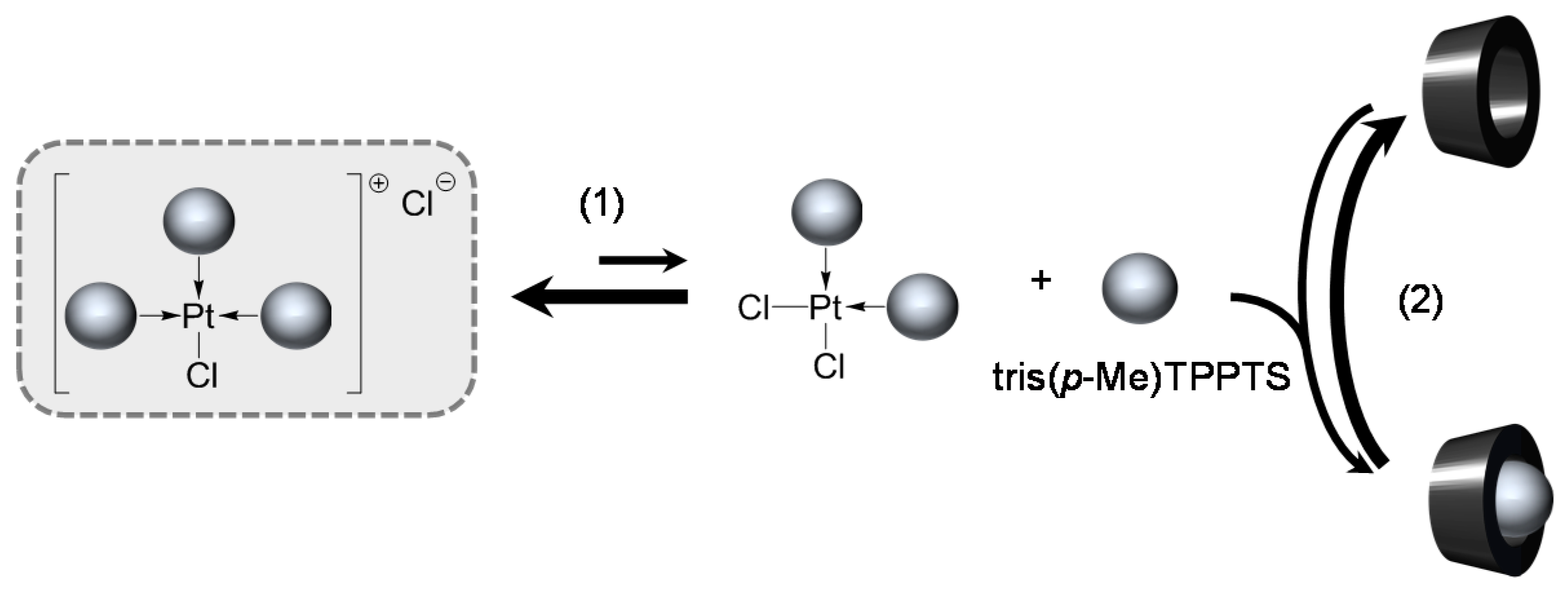
para-Substituted TPPTS Derivatives
Ortho-Substituted TPPTS Derivatives
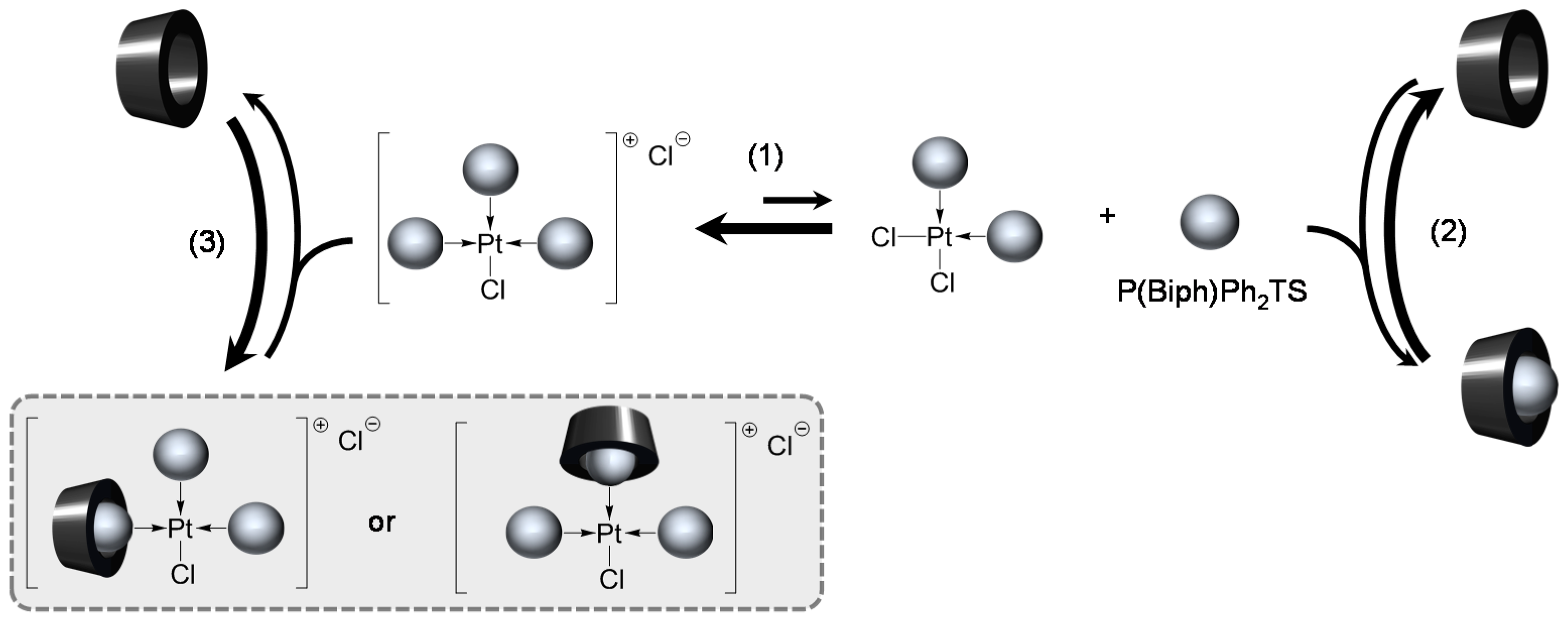
Trisulfonated Biphenylphosphane Ligands
2.2. Palladium Complexes
2.2.1. Para-Substituted TPPTS Derivatives
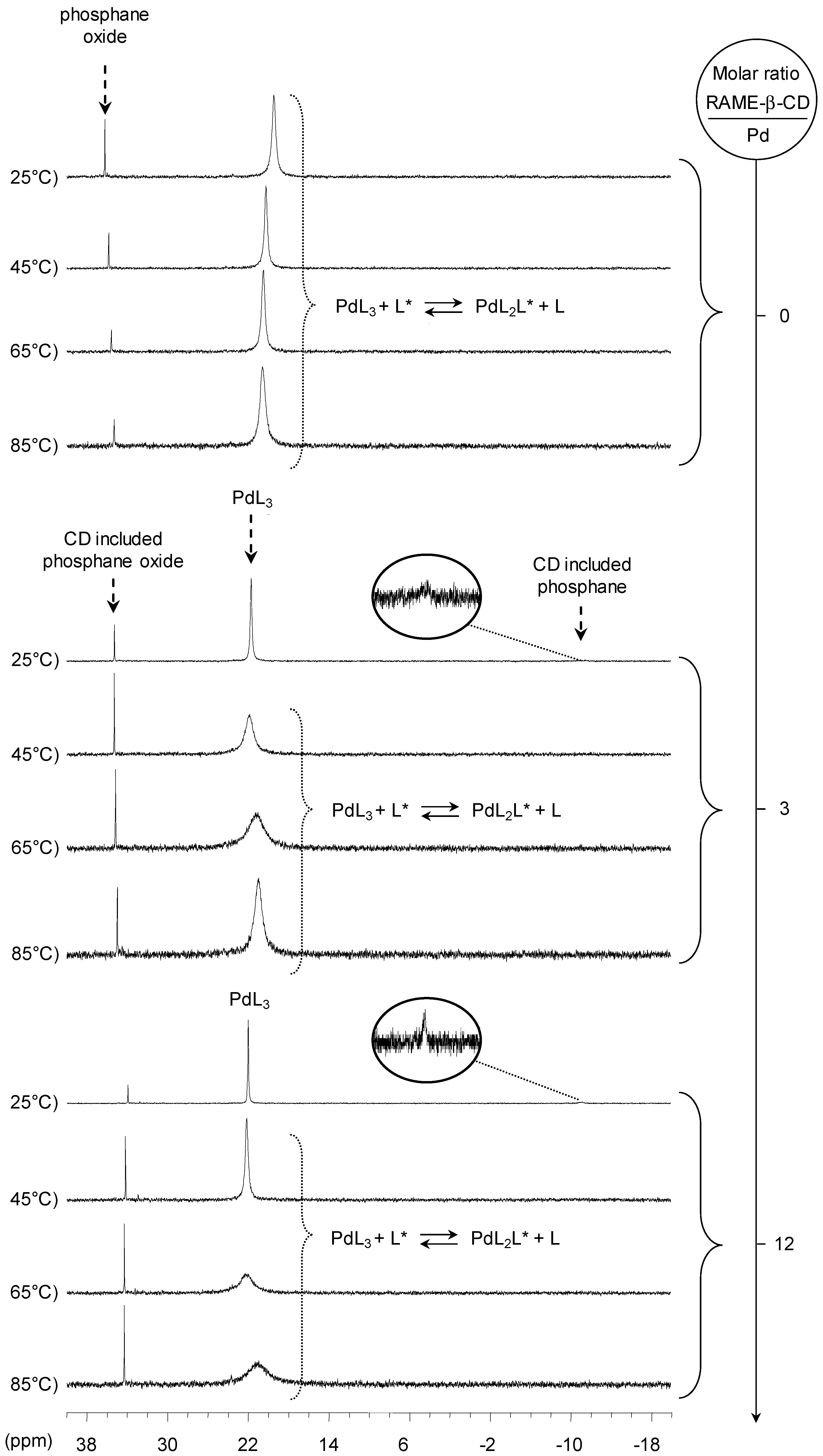
[Pd(0)/tris(p-Me)TPPTS] Complex
[Pd(0)/tris(p-OMe)TPPTS] Complex
2.2.2. ortho-Substituted Ligands
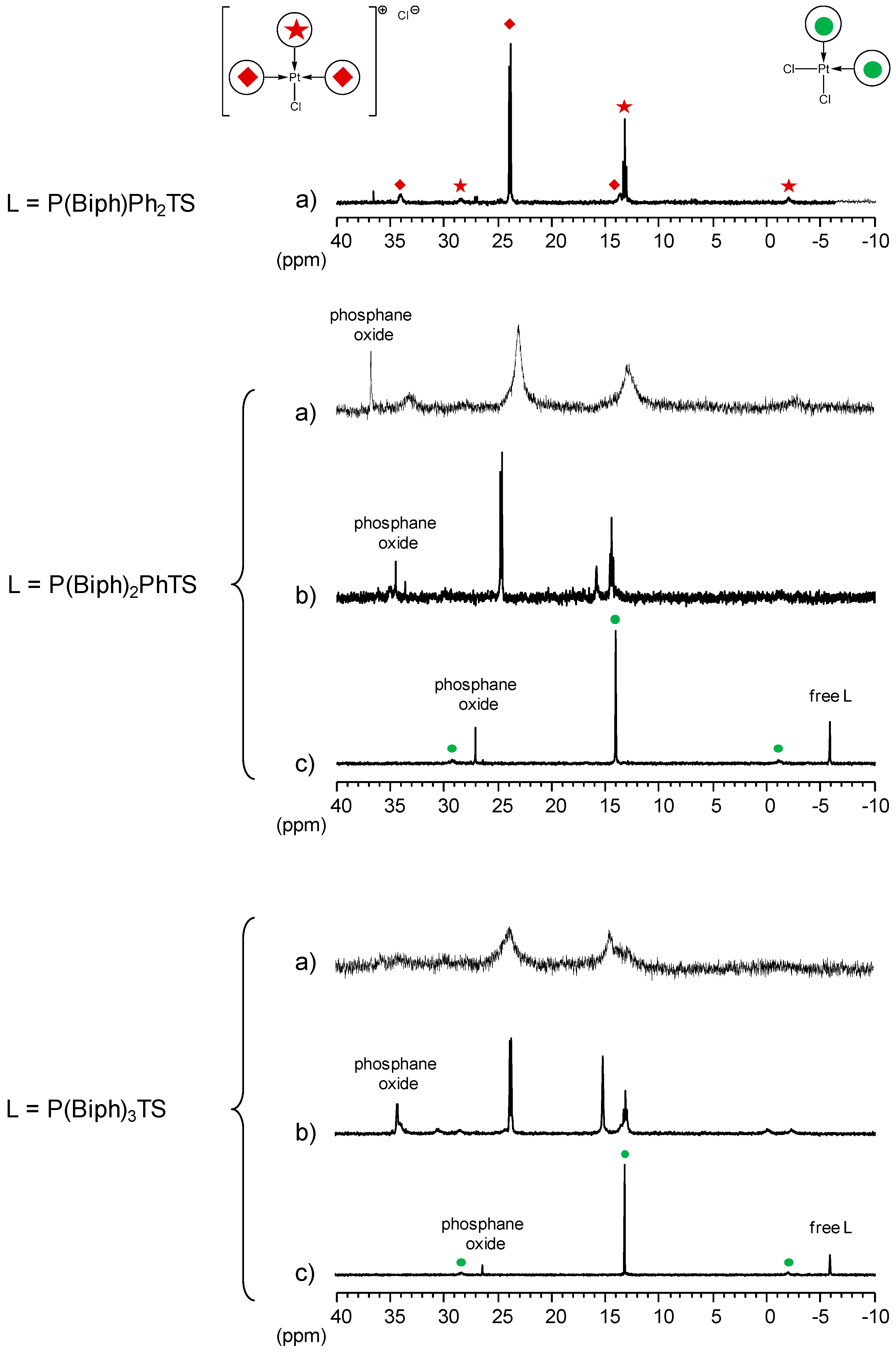
2.2.3. Biphenylphosphane Ligands
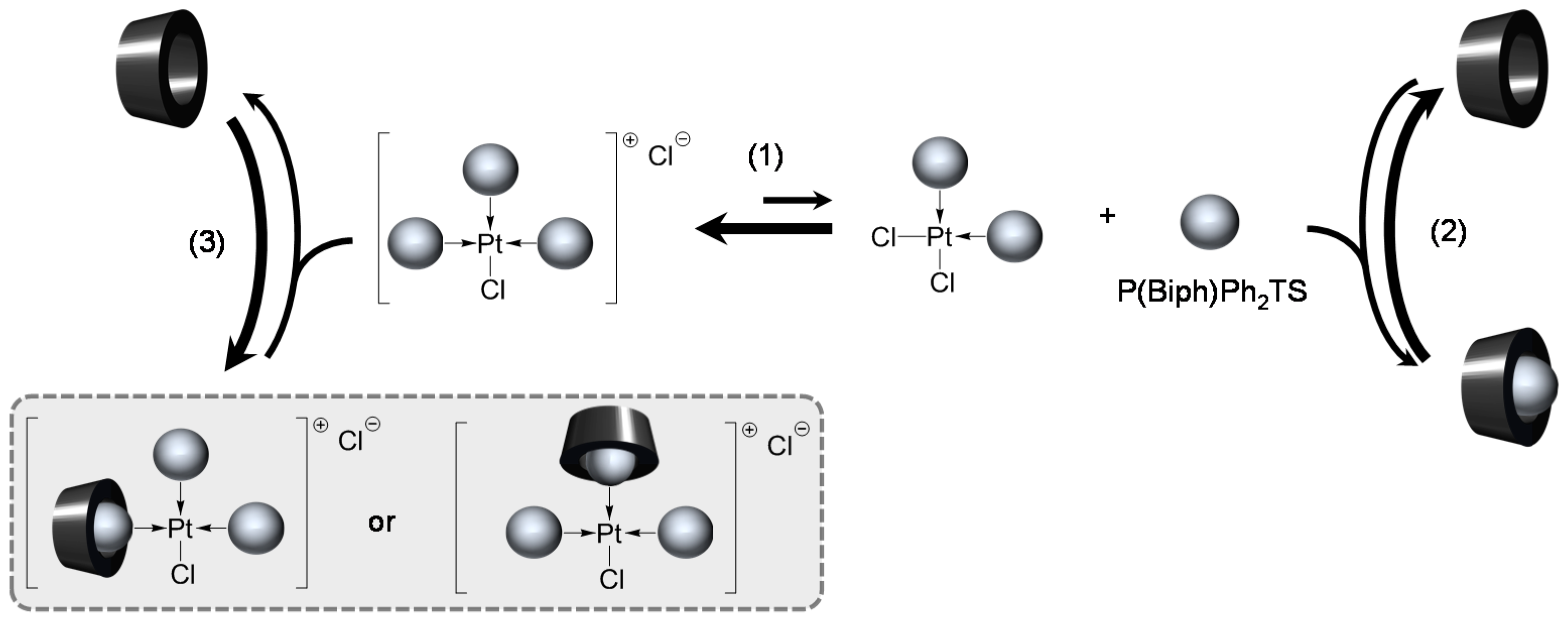
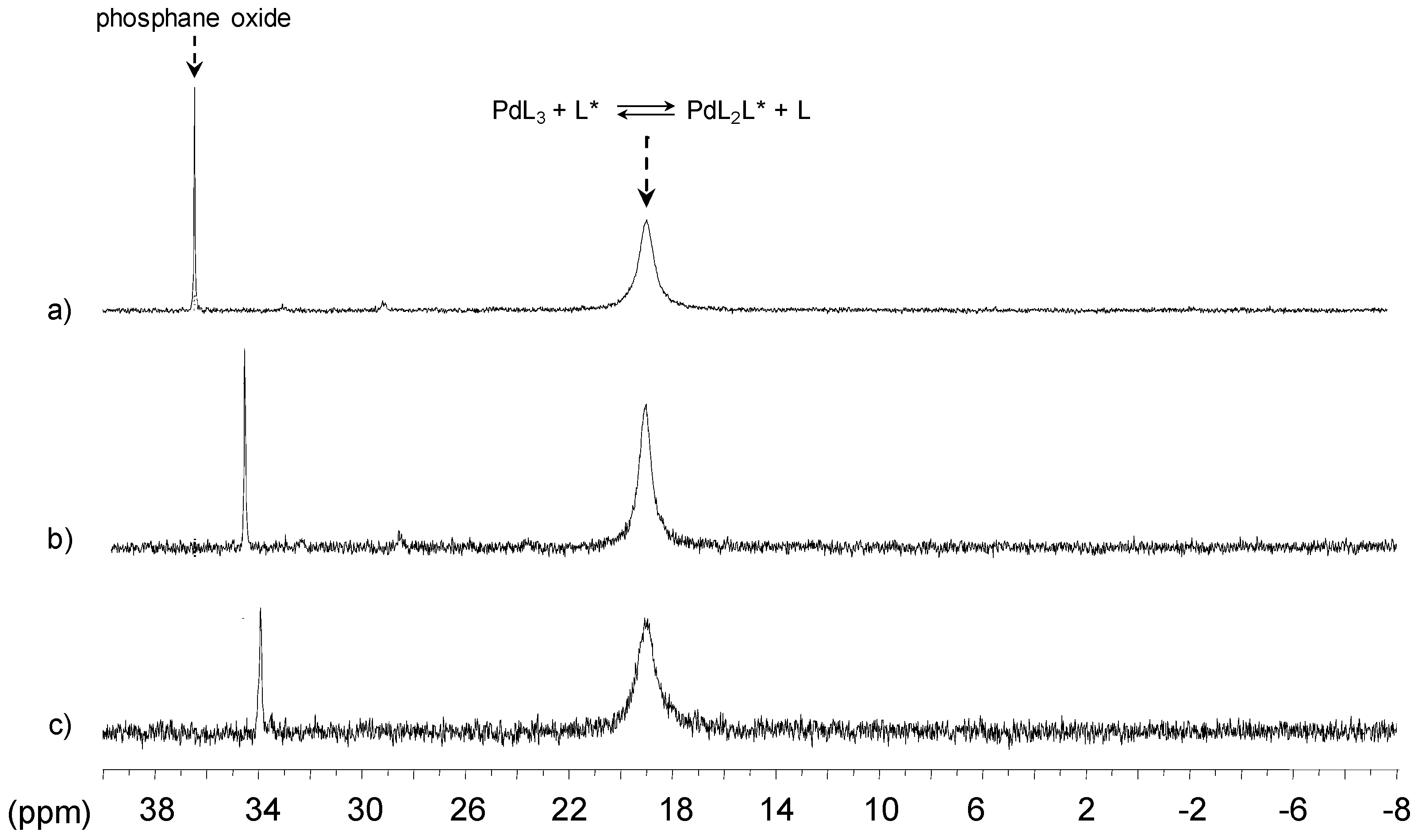
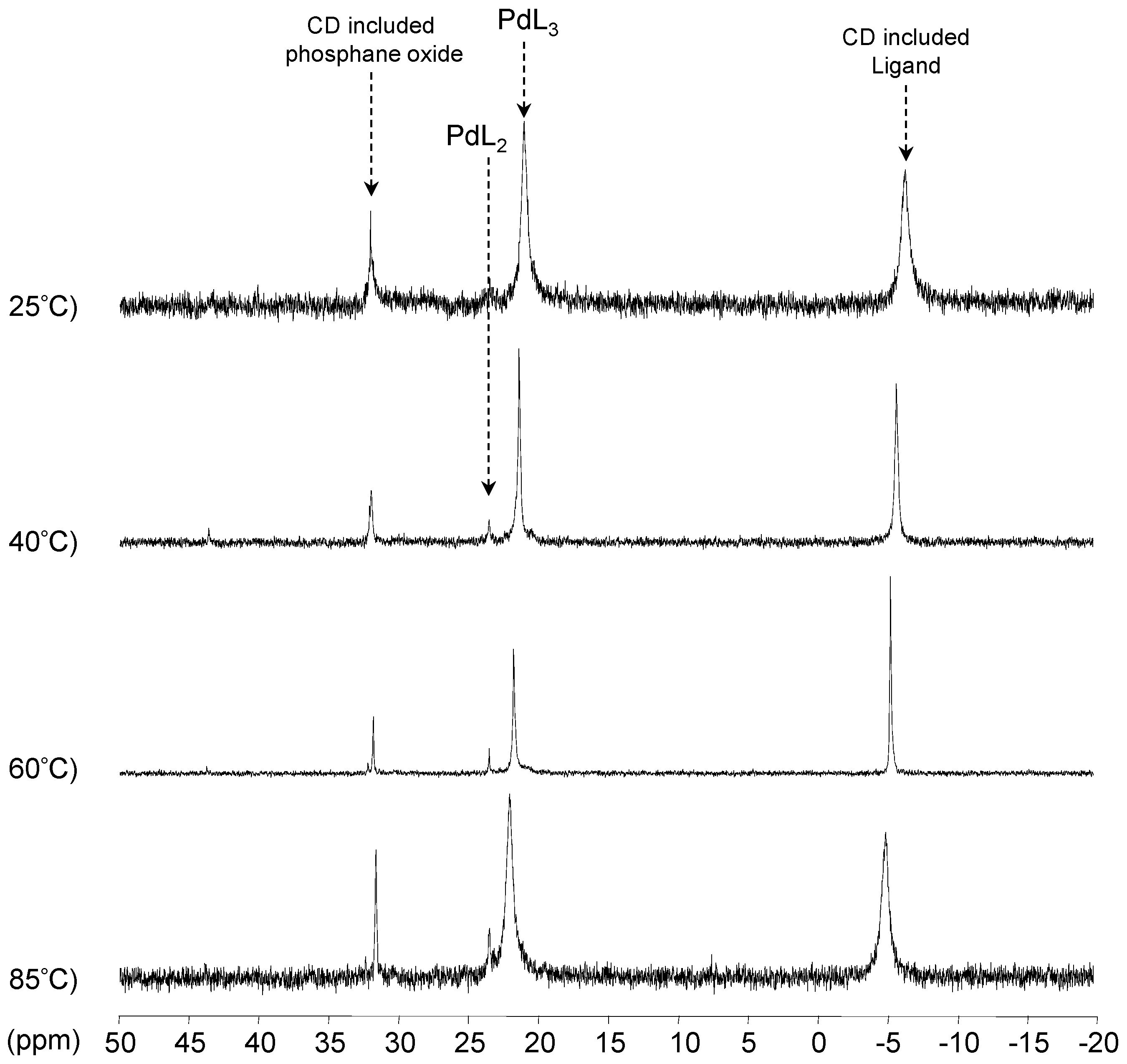
[Pd(0)/P(Biph)Ph2TS] Complex
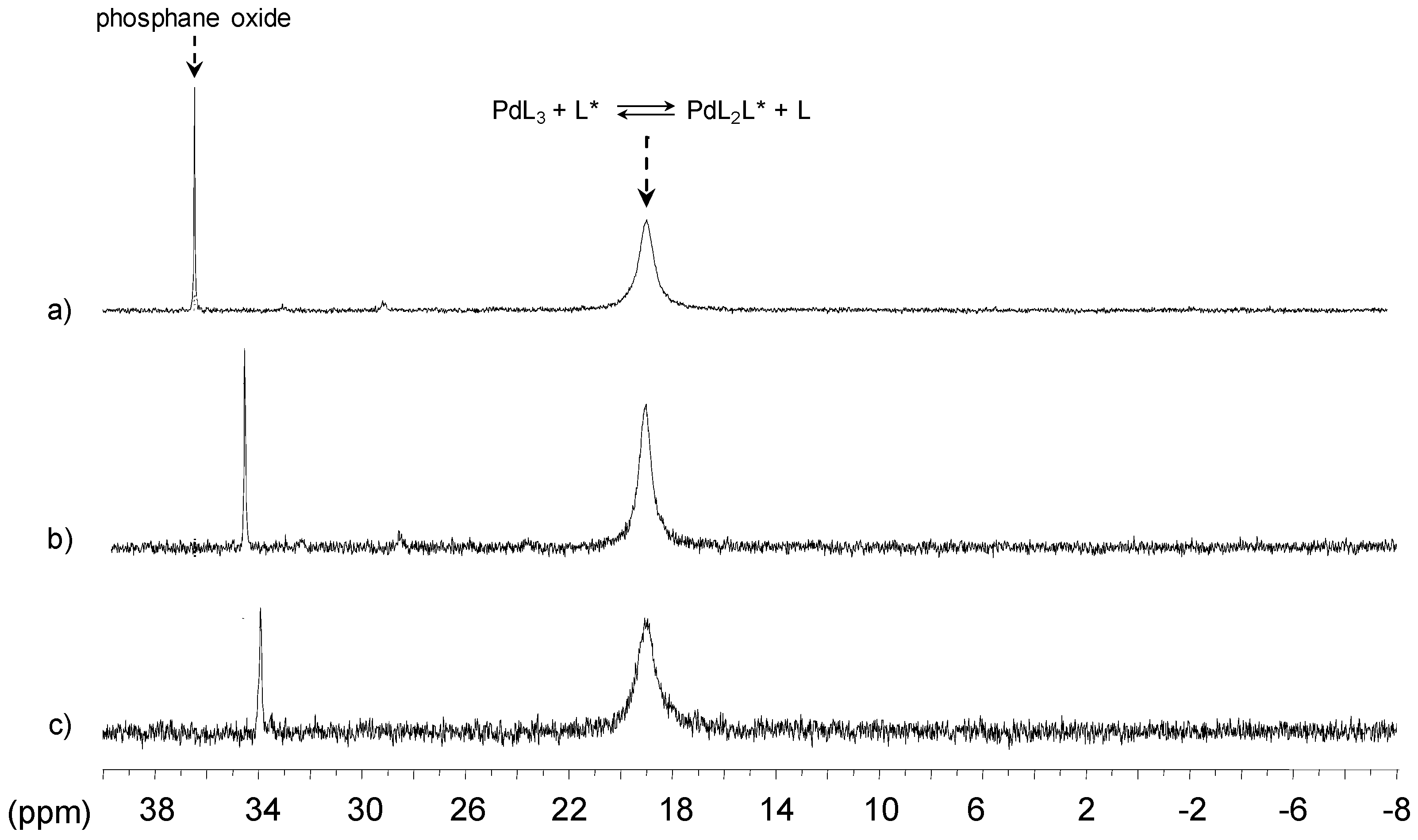
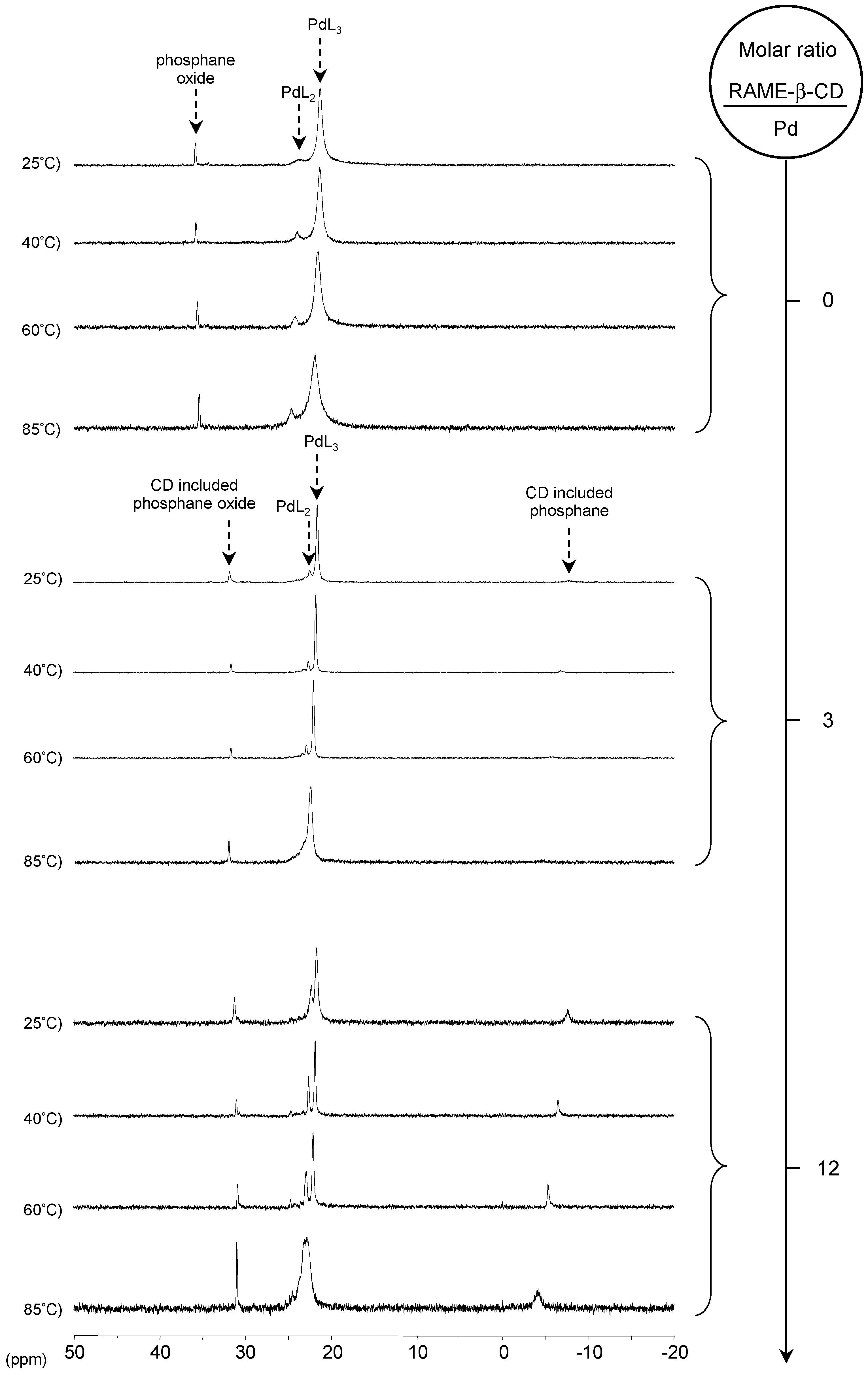
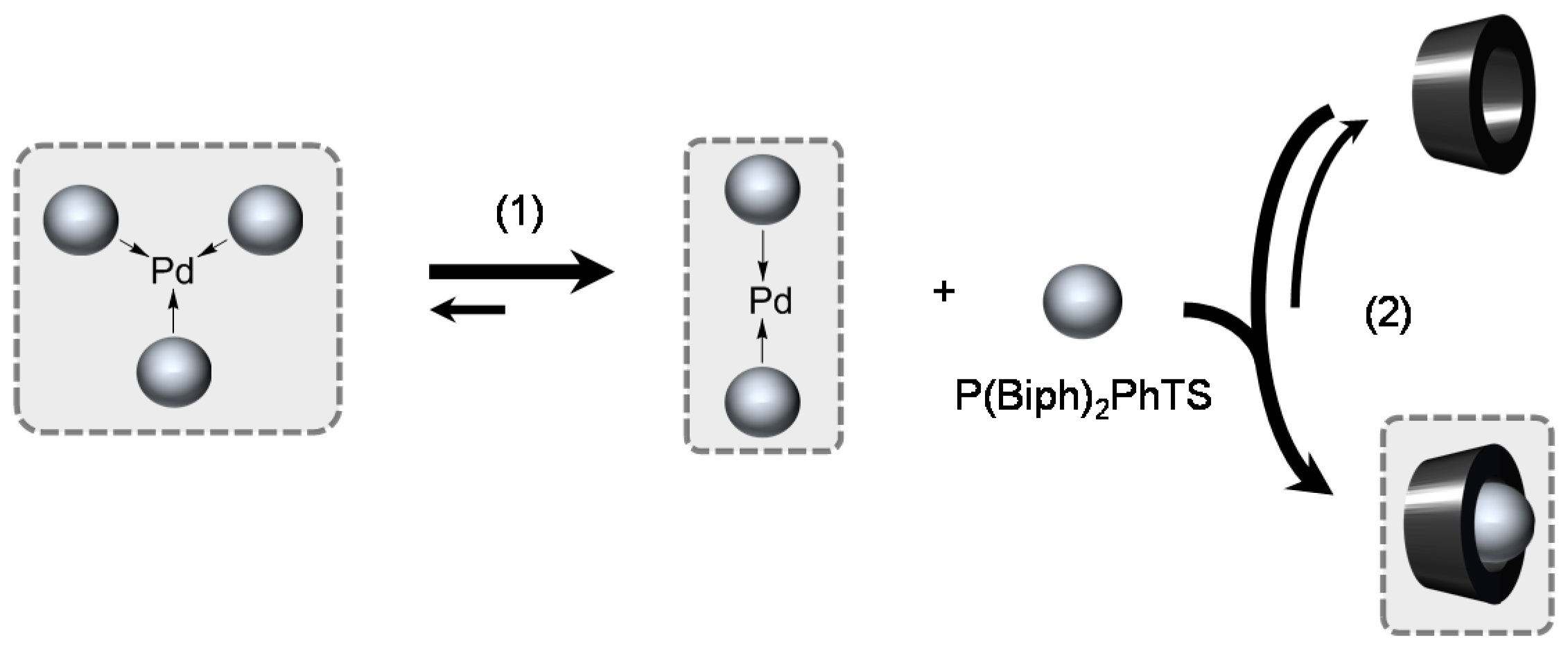
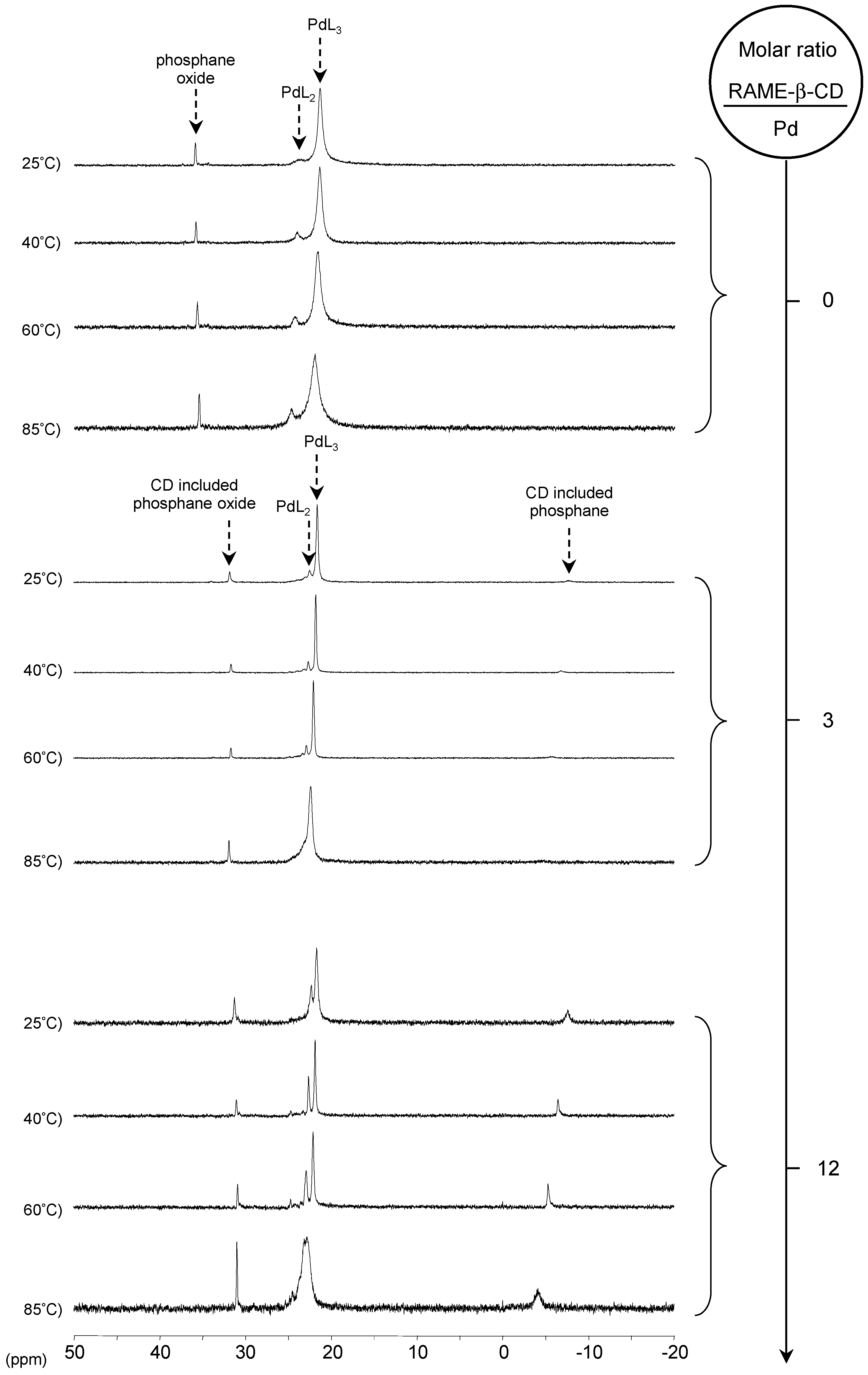
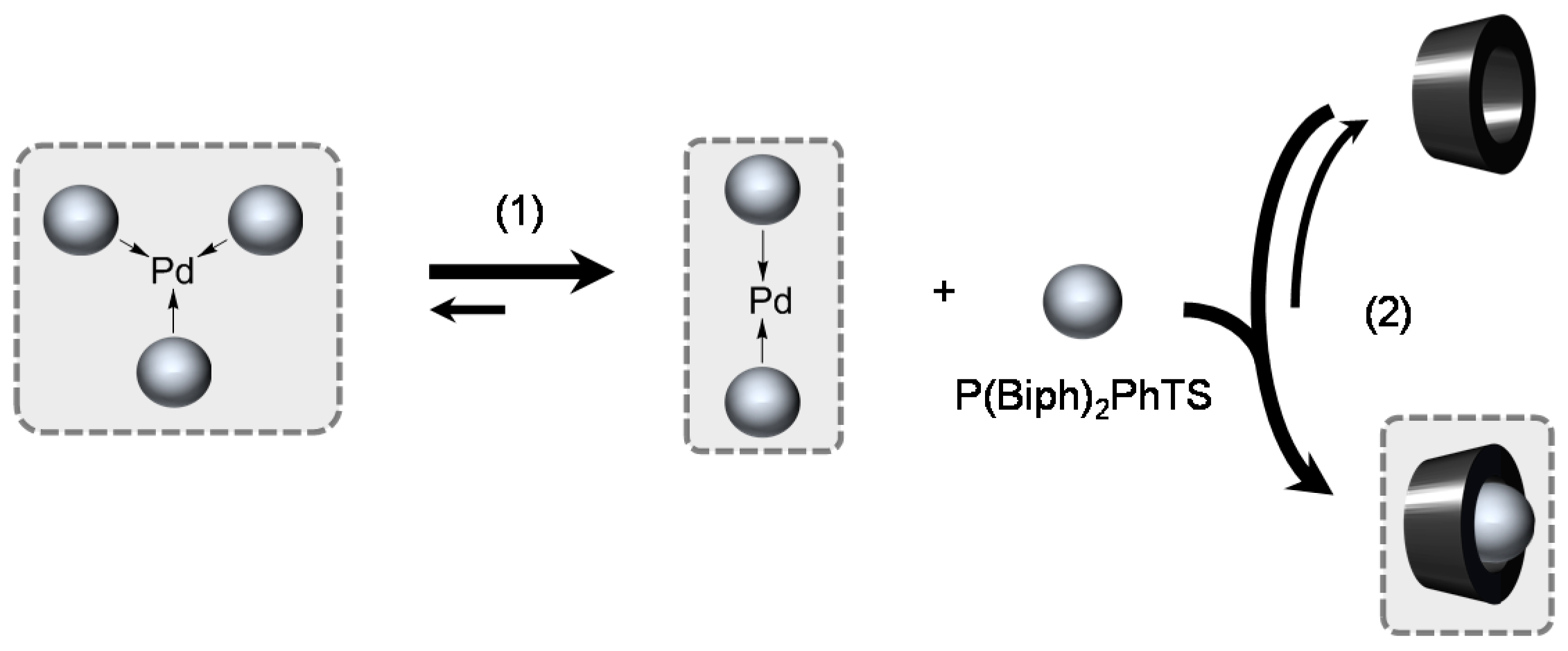
[Pd(0)/P(Biph)2PhTS] Complex
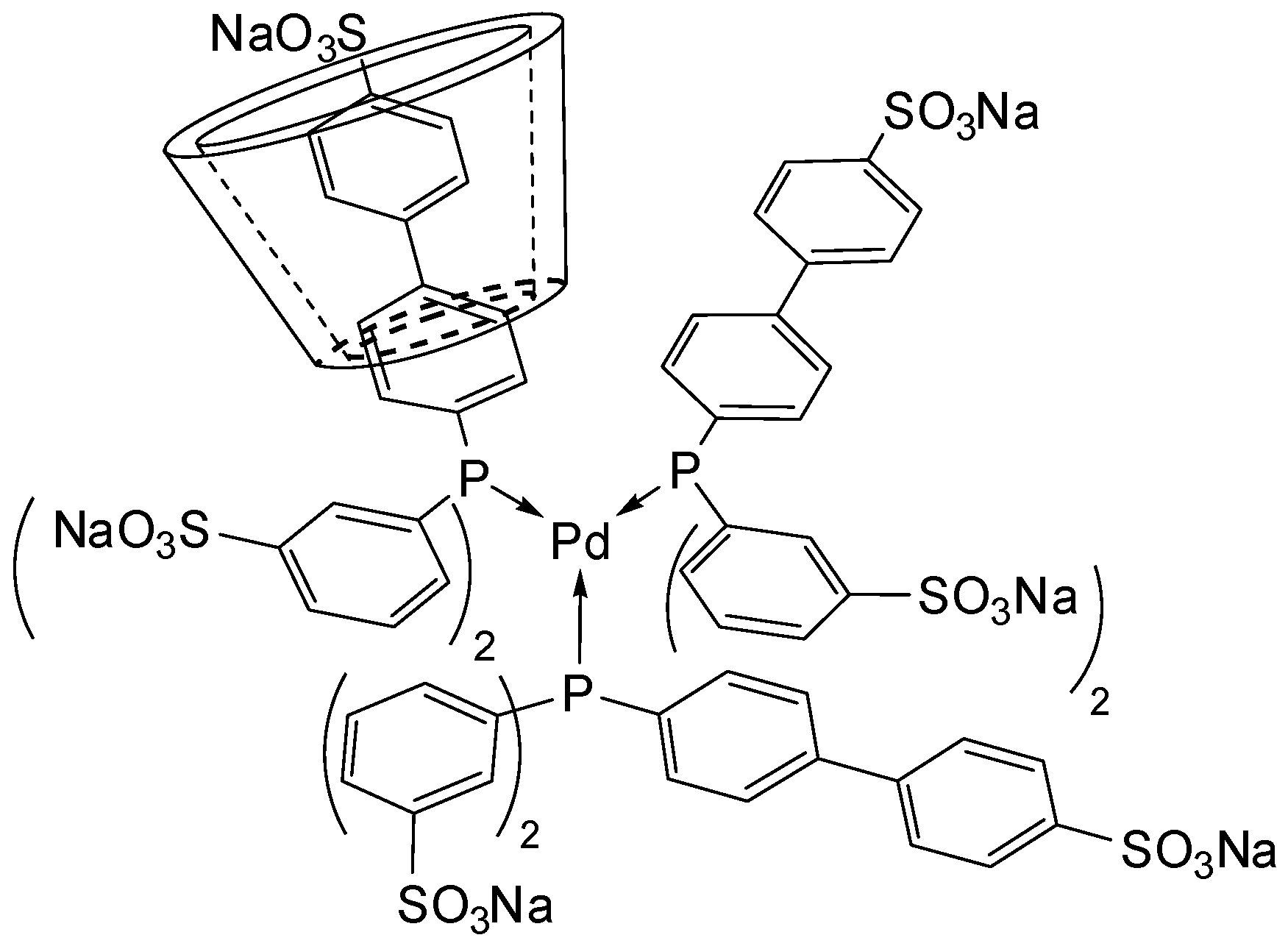
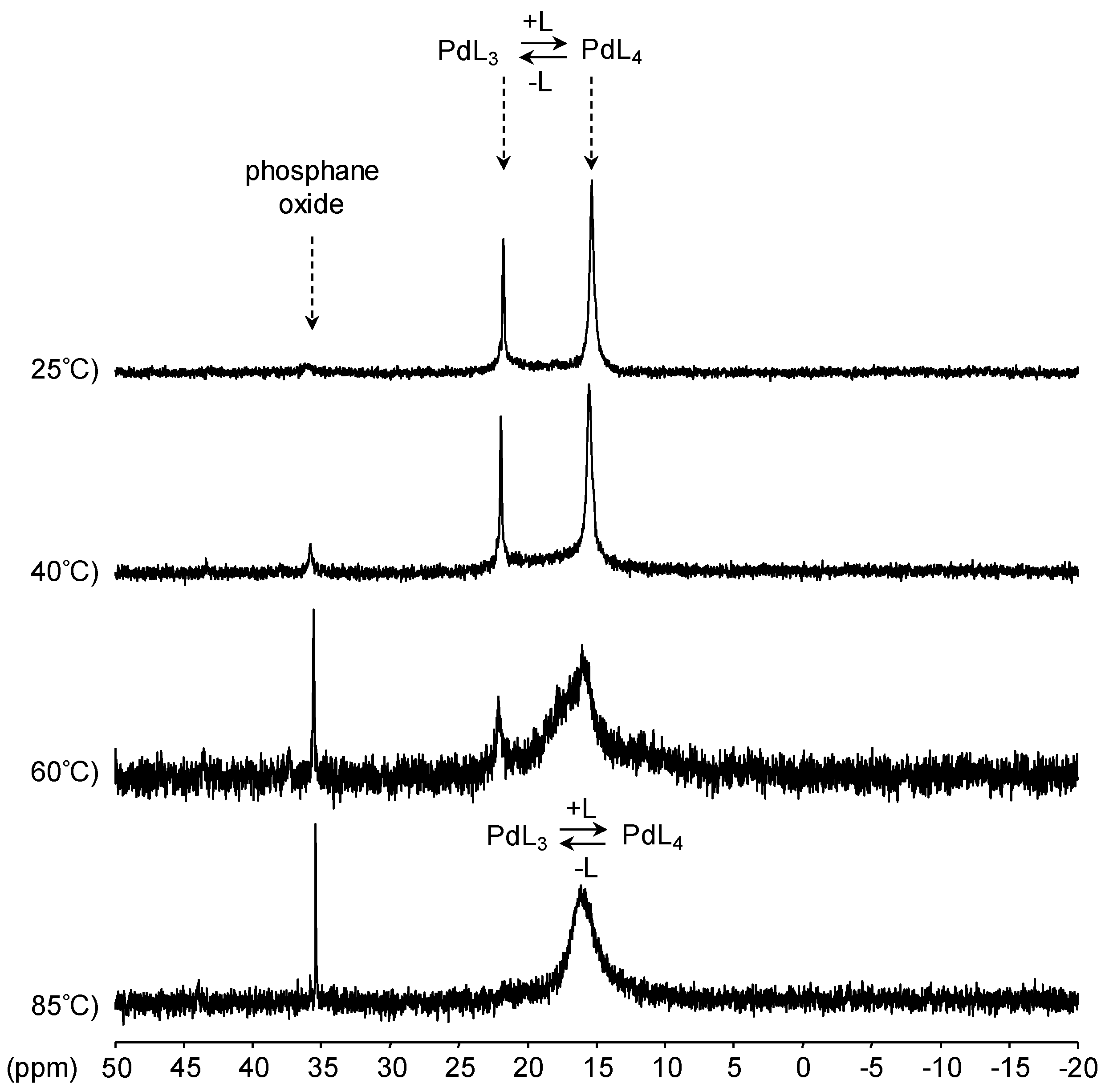
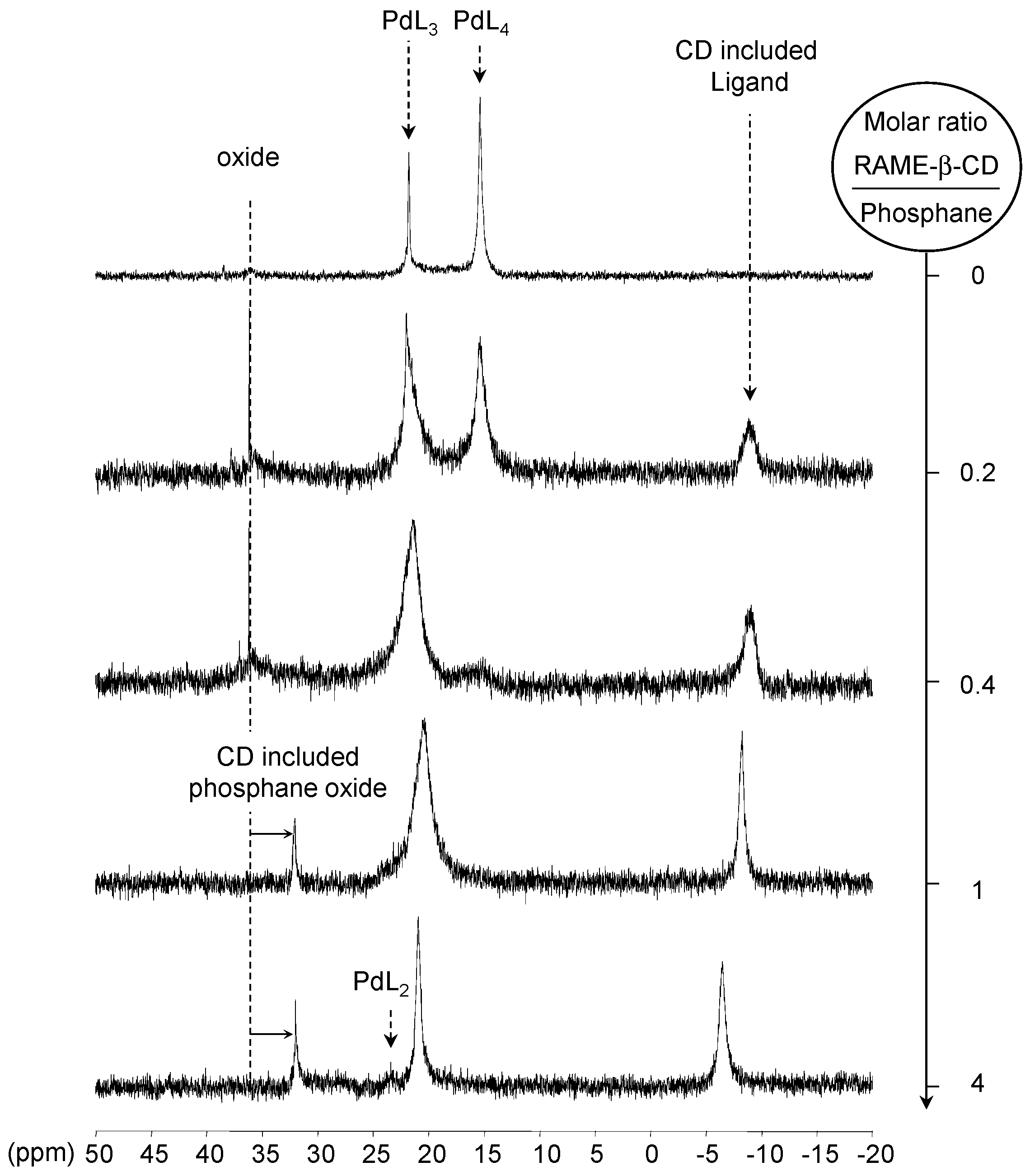
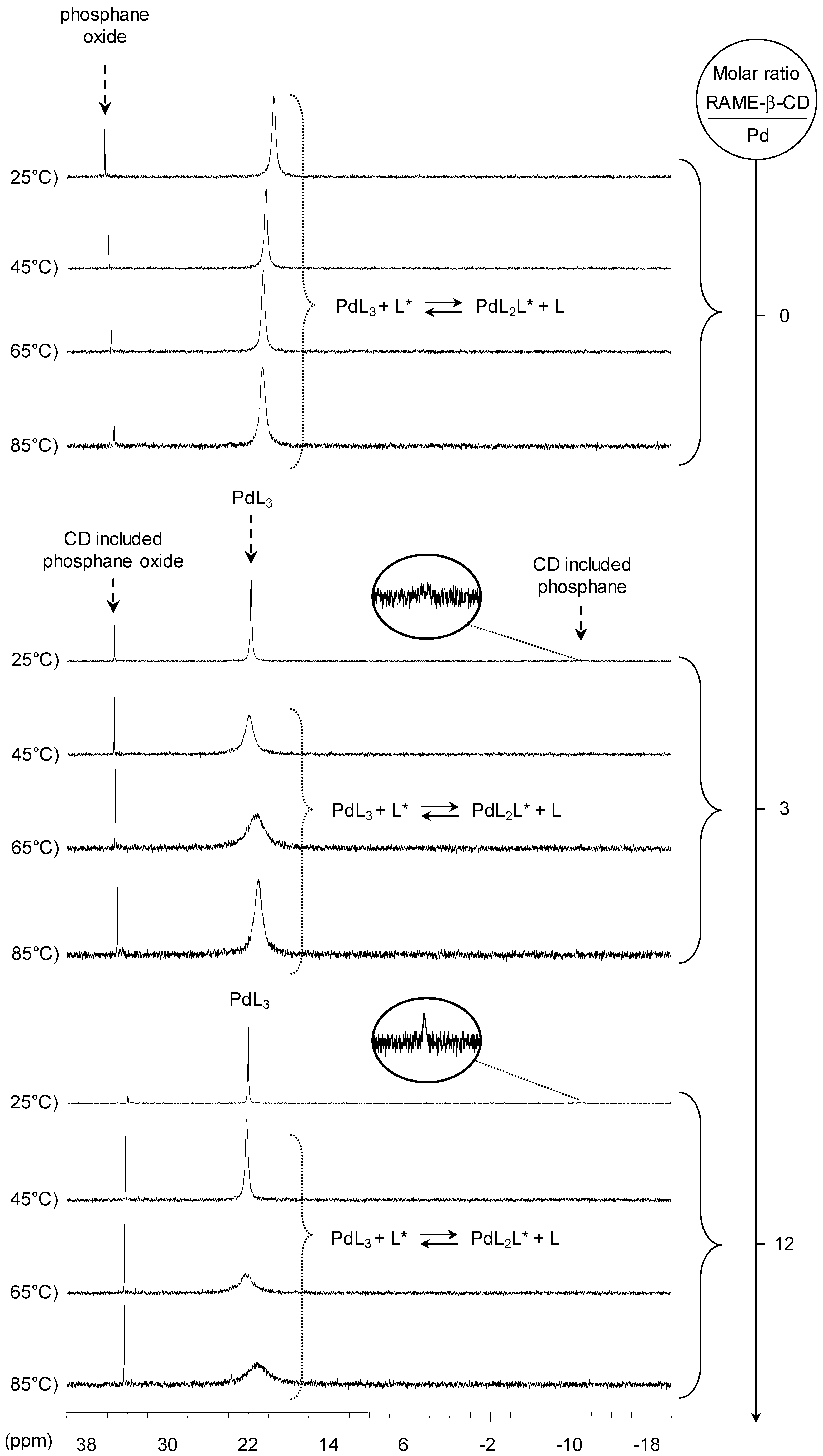
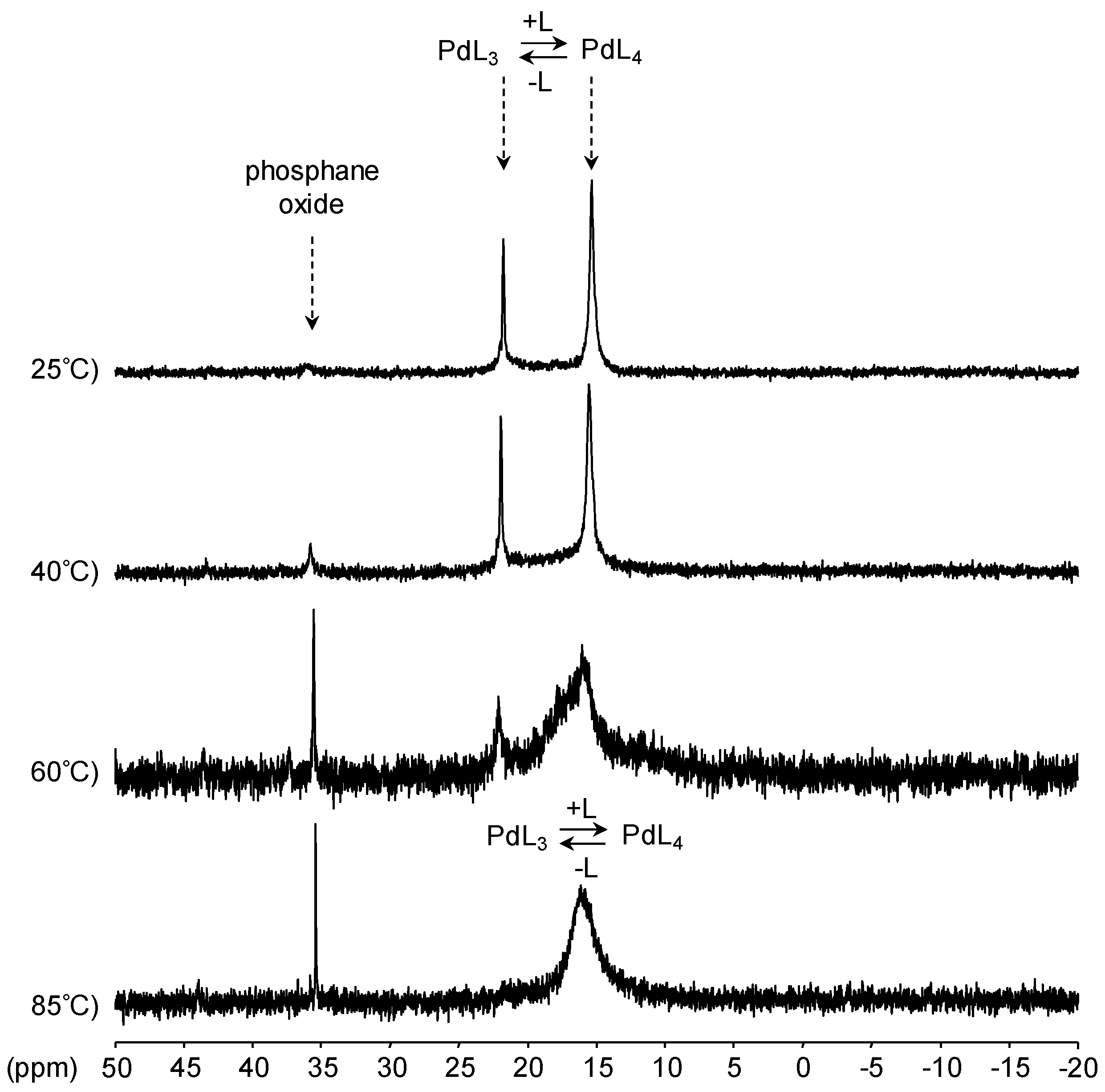
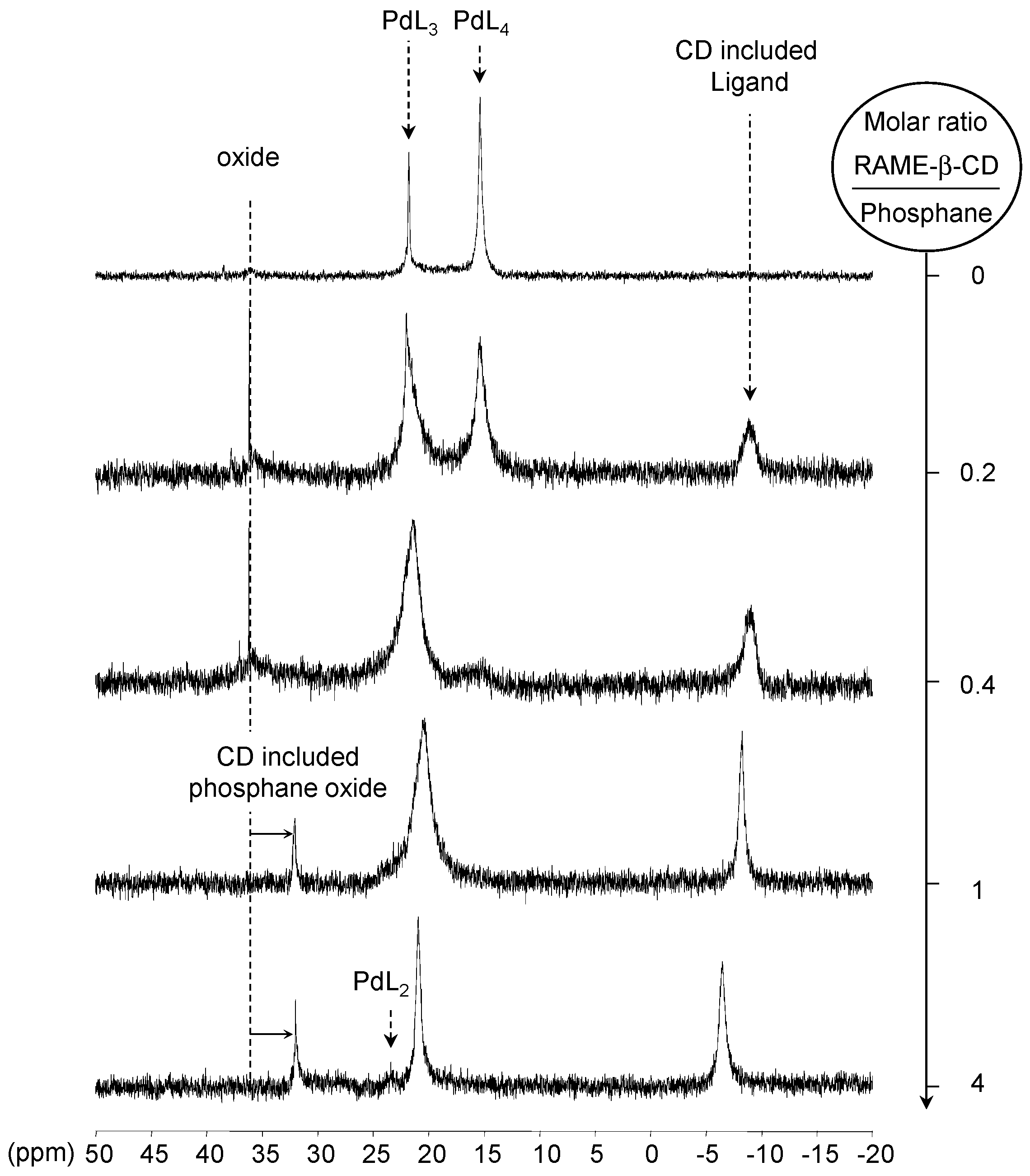
[Pd(0)/P(Biph)3TS] Complex
- (1)
- ligands forming palladium complexes not affected by the RAME-β-CD addition (TPPTS, tris(p-Me)TPPTS and tris(p-OMe)TPPTS)
- (2)
- ligands forming palladium complexes where the CD could play the role of coordination second sphere ligand (P(Biph)Ph2TS)
- (3)
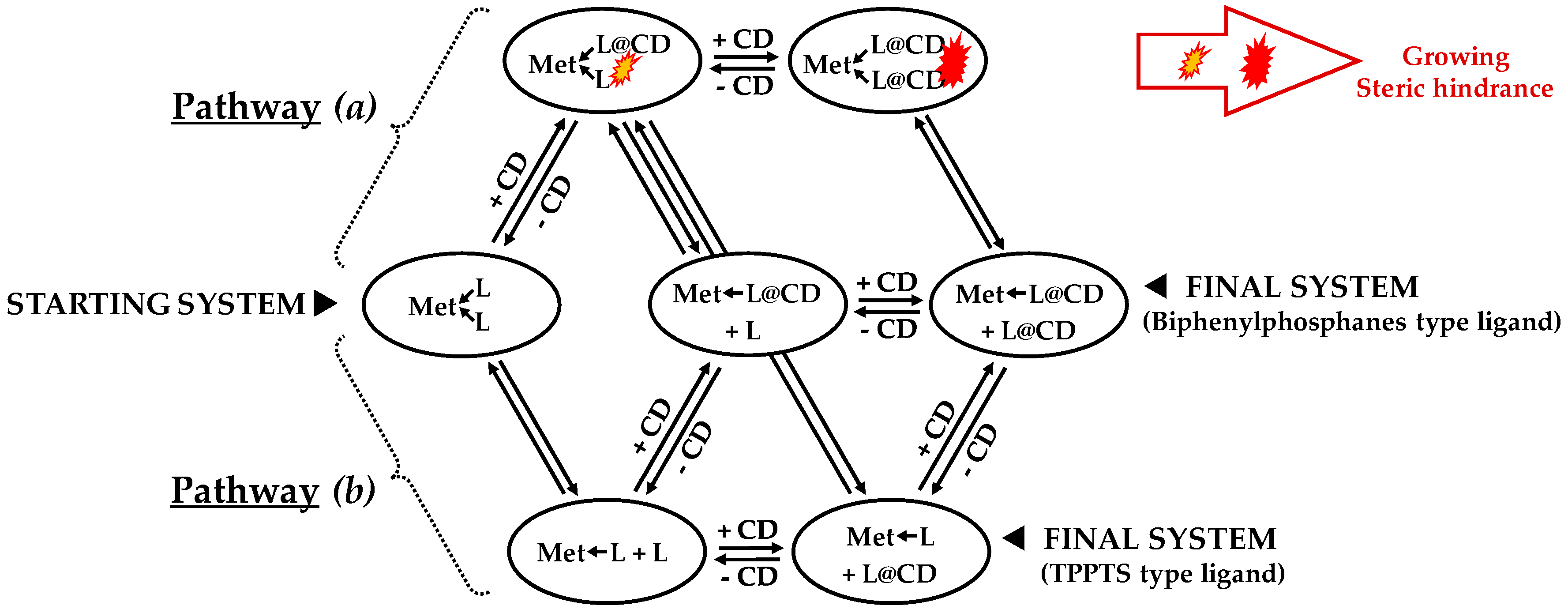
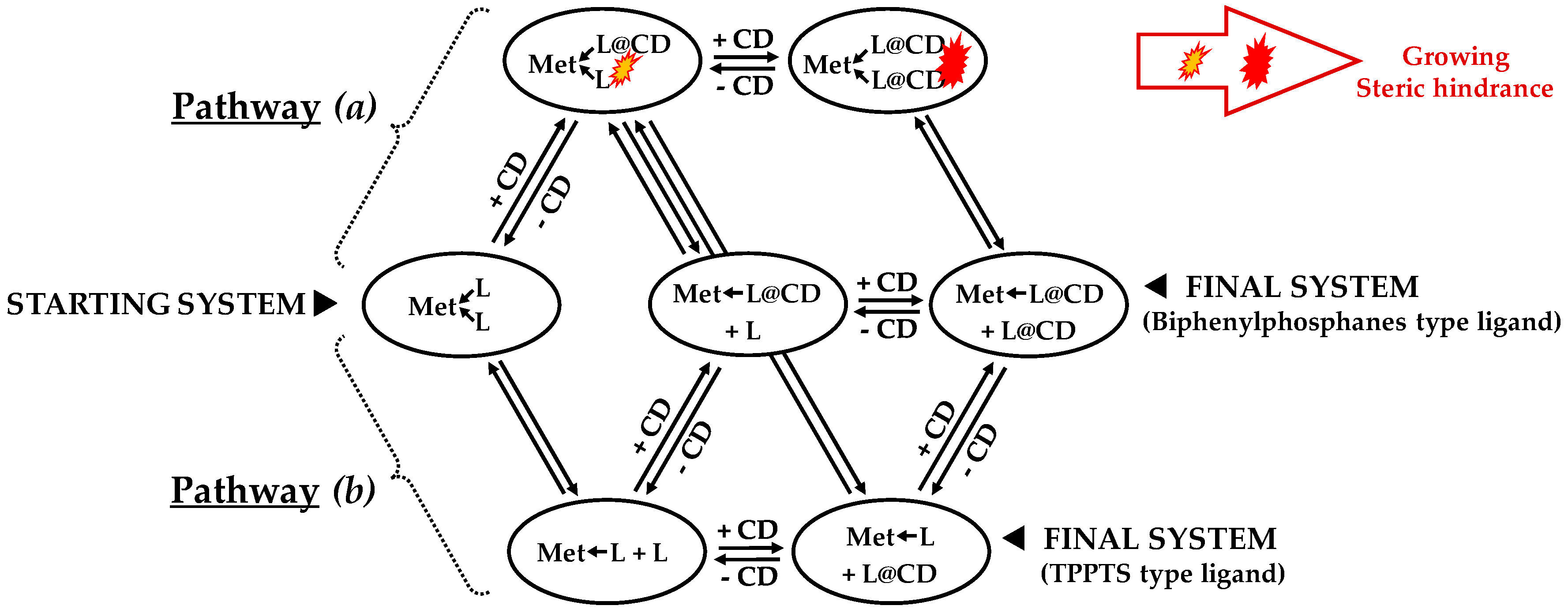
- ligands forming palladium complexes which are transformed by the presence of RAME-β-CD. Indeed, P(Biph)2PhTS and P(Biph)3TS spontaneously form respectively PdL3/PdL2 and PdL4/PdL3 organometallic complexes which are converted in PdL2 complexes when CD is added due to shift equilibria between the different organometallic species. However, formation of species where the CD acts as coordination second sphere ligand could not be avoided as the biphenyl moiety is well recognised by its cavity. In these cases, the organometallic complexes formed would naturally evolve to low-coordinated species through steric decompression by ligand decoordination.
2.3. Comparison of the Results Obtained with Palladium and Platinum Complexes
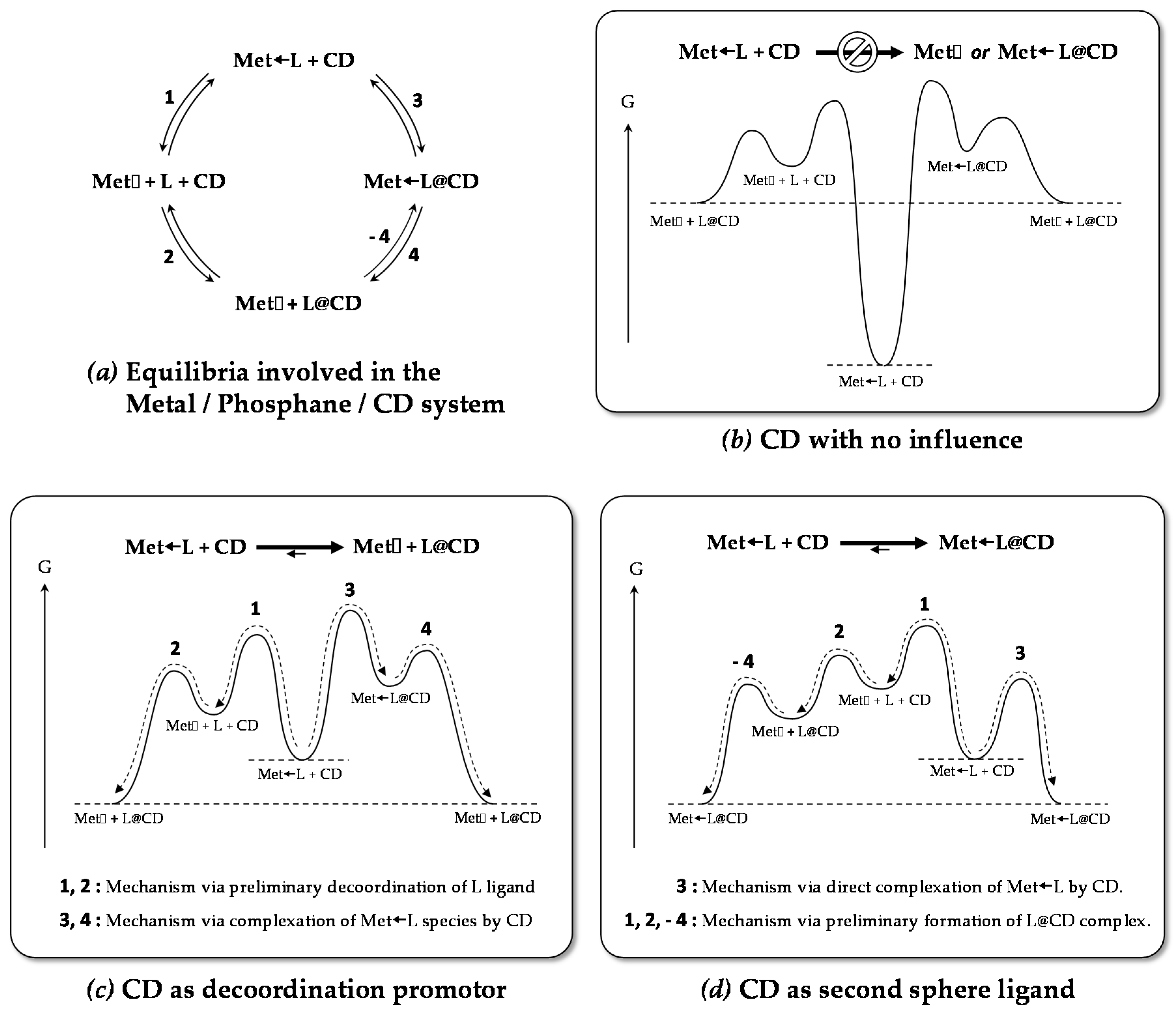
- The CD can have no influence on the organometallic species. That is what is observed with para-substituted hydrosoluble phosphanes for both palladium and platinum complexes (entry 2) and also with TPPTS, only in the case of palladium (entry 1). For the para-substituted hydrosoluble phosphanes, this behaviour could be explained by the lack of interaction between the CD and the phosphane (tris(p-OMe)TPPTS), or by a too strong metal-phosphorus bond coming from a high σ-donor ligand (tris(p-Me)TPPTS). However, the fact that the PdL2 species is not formed after CD addition on the PdL3 complex not only for tris(p-OMe)TPPTS and tris(p-Me)TPPTS but also for TPPTS probably comes from the too unstable PdL2 (14e− species) in the case of TPPTS derivatives.
- The CD can act as a phosphane decoordination promotor. This property was emphasised with TPPTS as ligand in the [PtL3Cl]Cl complex (entry 1), and with P(Biph)2PhTS and P(Biph)3TS ligands in the case of PdLn type complexes (entries 5 and 6). The fact that TPPTS can be removed from Pt by addition of CD, contrary to tris(p-OMe)TPPTS and tris(p-Me)TPPTS, comes not only from its ability to be included in the CD cavity but also to its lower coordination power (less basic ligand).
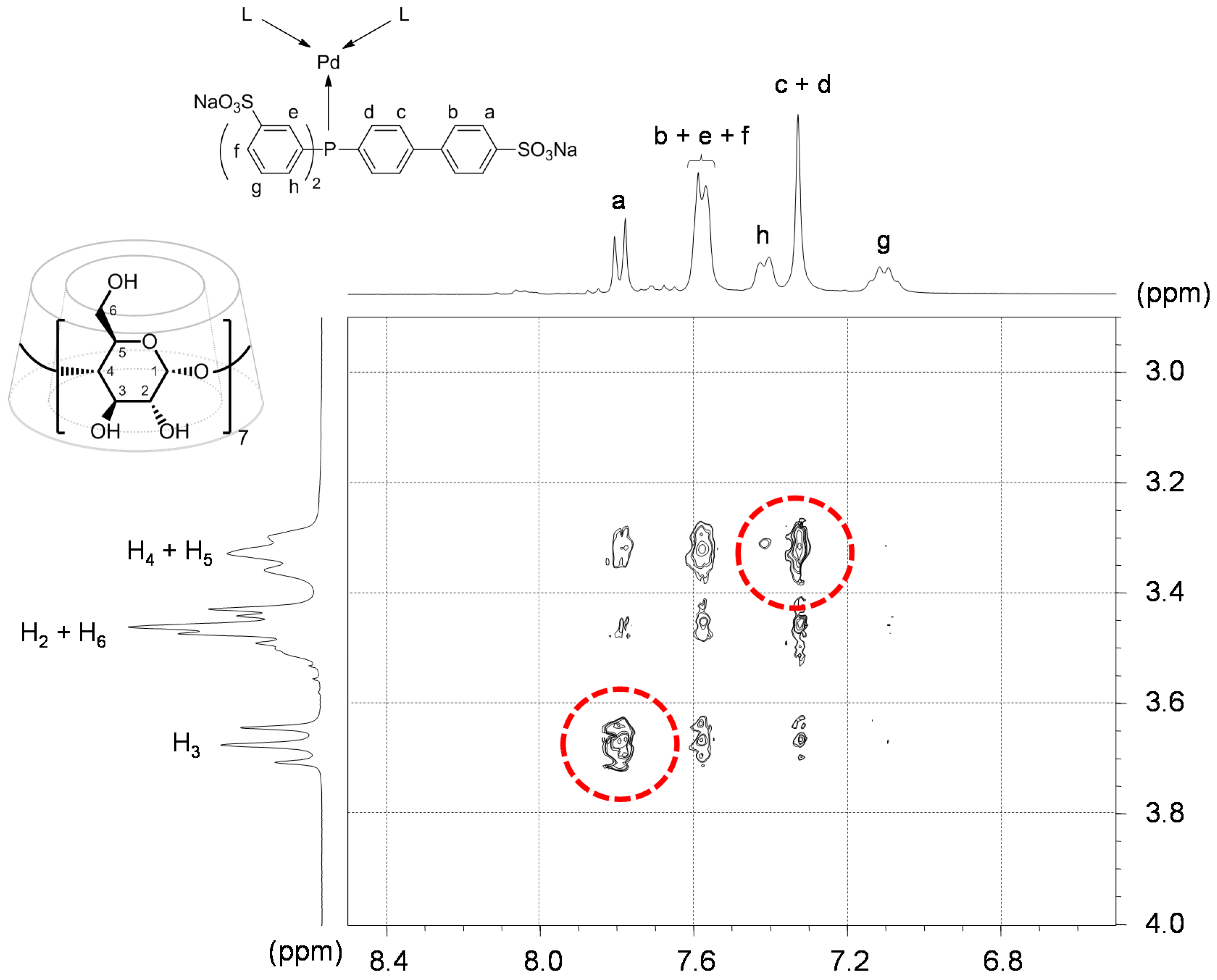
- The CD can act as a second sphere ligand by including the metal-bound phosphane. This property was undoubtedly highlighted for both platinum and palladium complexes with the P(Biph)Ph2TS ligand whose p-sodiosulfobiphenyl group is well recognized by the CD. Because of this identical group present in their structure, making them accessible for a cyclodextrin even when bound to a metal, it is also important to say that P(Biph)2PhTS and P(Biph)3TS based organometallic species form also surely supramolecular complexes with CD. This property plays probably a role in the observed decoordination of P(Biph)2PhTS and P(Biph)3TS from palladium, the CD being able to stabilize the low-coordinated resulting organometallic species. More concretely, this property can explain the observation of PdL2 type species when P(Biph)3TS ligand was used, species not formed with TPPTS ligand. Indeed, TPPTS structure doesn’t permit complexation when bound to the metal.
3. Materials and Methods
3.1. General Remarks
3.2. 31P{1H} NMR Study on Platinum and Palladium Complexes
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Dixneuf, P.H.; Cadierno, V. Metal-Catalyzed Reactions in Water; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Cornils, B.; Hermann, W.A. Aqueous-Phase Organometallic Catalysis; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Shaughnessy, K.H. Hydrophilic ligands and their application in aqueous-phase metal-catalyzed reactions. Chem. Rev. 2009, 109, 643–710. [Google Scholar] [CrossRef] [PubMed]
- Cornils, B.; Kuntz, E.G. Introducing TPPTS and related ligands for industrial biphasic processes. J. Organomet. Chem. 1995, 502, 177–186. [Google Scholar] [CrossRef]
- Obrecht, L.; Kamer, P.C.J.; Laan, W. Alternative approaches for the aqueous–organic biphasic hydroformylation of higher alkenes. Catal. Sci. Technol. 2013, 3, 541–551. [Google Scholar] [CrossRef]
- Sharma, S.K.; Jasra, R.V. Aqueous phase catalytic hydroformylation reactions of alkenes. Catal. Today 2015, 247, 70–81. [Google Scholar] [CrossRef]
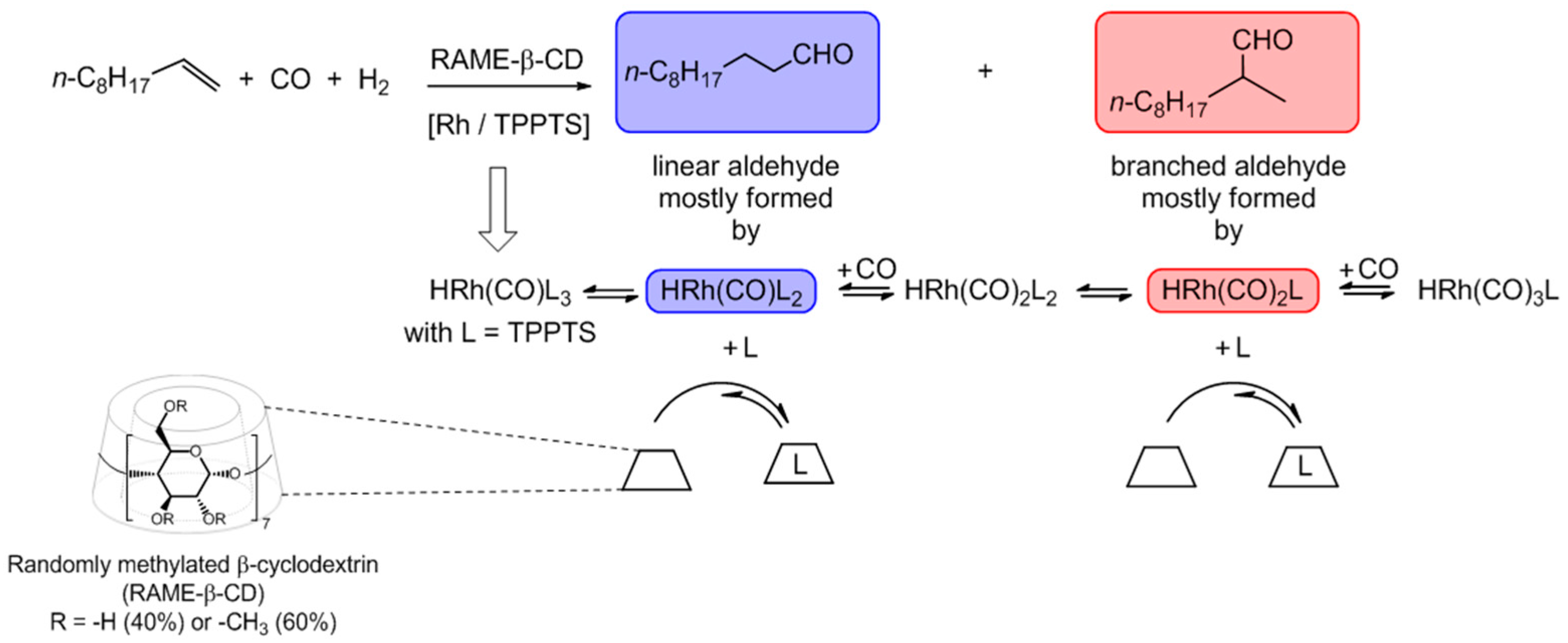
- Hapiot, F.; Ponchel, A.; Tilloy, S.; Monflier, E. Cyclodextrins and their applications in aqueous-phase metal-catalyzed reactions. C. R. Chim. 2011, 14, 149–166. [Google Scholar] [CrossRef]
- Hapiot, F.; Bricout, H.; Menuel, S.; Tilloy, S.; Monflier, E. Recent breakthroughs in aqueous cyclodextrin-assisted supramolecular catalysis. Catal. Sci. Technol. 2014, 4, 1899–1908. [Google Scholar] [CrossRef]
- Mathivet, T.; Méliet, C.; Castanet, Y.; Mortreux, A.; Caron, L.; Tilloy, S.; Monflier, E. Rhodium catalyzed hydroformylation of water insoluble olefins in the presence of chemically modified β-cyclodextrins: Evidence for ligand-cyclodextrin interactions and effect of various parameters on the activity and the aldehydes selectivity. J. Mol. Catal. A Chem. 2001, 176, 105–116. [Google Scholar] [CrossRef]
- Monflier, E.; Bricout, H.; Hapiot, F.; Tilloy, S.; Aghmiz, A.; Masdeu-bultó, A.M. High-Pressure31P{1H} NMR Studies of RhH(CO)(TPPTS)3 in the presence of methylated cyclodextrins: New light on rhodium-catalyzed hydroformylation reaction assisted by cyclodextrins. Adv. Synth. Catal. 2004, 346, 425–431. [Google Scholar] [CrossRef]
- Caron, L.; Canipelle, M.; Tilloy, S.; Bricout, H.; Monflier, E. Unexpected effect of cyclodextrins on water-soluble rhodium complexes. Eur. J. Inorg. Chem. 2003, 4, 595–599. [Google Scholar] [CrossRef]
- Binkowski-Machut, C.; Canipelle, M.; Bricout, H.; Tilloy, S.; Hapiot, F.; Monflier, E. Supramolecular trapping of phosphanes by cyclodextrins: A general approach to generate phosphane coordinatively unsaturated organometallic complexes. Eur. J. Inorg. Chem. 2006, 2006, 1611–1619. [Google Scholar] [CrossRef]
- Caron, L.; Bricout, H.; Tilloy, S.; Ponchel, A.; Landy, D.; Fourmentin, S.; Monflier, E. Molecular recognition between a water-soluble organometallic complex and a β-cyclodextrin: First example of second-sphere coordination adducts possessing a catalytic activity. Adv. Synth. Catal. 2004, 346, 1449–1456. [Google Scholar] [CrossRef]
- Leclercq, L.; Schmitzer, A.R. Assembly of tunable supramolecular organometallic catalysts with cyclodextrins. Organometallics 2010, 29, 3442–3449. [Google Scholar] [CrossRef]
- Ferreira, M.; Bricout, H.; Sayede, A.; Ponchel, A.; Fourmentin, S.; Tilloy, S.; Monflier, E. Biphasic aqueous organometallic catalysis promoted by cyclodextrins: How to design the water-soluble phenylphosphane to avoid interaction with cyclodextrin. Adv. Synth. Catal. 2008, 350, 609–618. [Google Scholar] [CrossRef]
- Ferreira, M.; Bricout, H.; Hapiot, F.; Sayede, A.; Tilloy, S.; Monflier, E. A property-matched water-soluble analogue of the benchmark ligand PPh3. ChemSusChem 2008, 1, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.W.; Moreno, D.A.; Atwood, J.D. Synthesis, characterization, and reaction chemistry of PtCl2[P(m-C6H4SO3Na)3]2, an Alkyne hydration catalyst. Organometallics 2001, 20, 4237–4245. [Google Scholar] [CrossRef]
- Snelders, D.J.M.; van Koten, G.; Klein Gebbink, R.J.M. Steric, electronic, and secondary effects on the coordination chemistry of ionic phosphine ligands and the catalytic behavior of their metal complexes. Chemistry 2011, 17, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Binkowski, C.; Cabou, J.; Bricout, H.; Hapiot, F.; Monflier, E. Cleavage of water-insoluble alkylallylcarbonates catalysed by a palladium/TPPTS/cyclodextrin system: Effect of phosphine/cyclodextrin interactions on the reaction rate. J. Mol. Catal. A Chem. 2004, 215, 23–32. [Google Scholar] [CrossRef]
- Sayede, A.; Ferreira, M.; Bricout, H.; Tilloy, S.; Monflier, E. Interaction of water-soluble triphenylphosphines with β-cyclodextrin: A quantum chemistry study. J. Phys. Org. Chem. 2011, 24, 1129–1135. [Google Scholar] [CrossRef]
- In preliminary experiments, the formation of cis-PtCl2(tris(p-Me)TPPTS)2 and cis-PtCl2(tris(p-OMe)TPPTS)2 was evidenced by mixing 2 equiv. of the corresponding ligand to a K2PtCl4 aqueous solution (see ESI).
- Native β-CD was used for the convenience of understanding the 1H-NMR signals compared to RAME-β-CD which shows overlapping non-interpretable resonances.
- Monteil, F.; Kalck, P. Carbonylation of bromobenzene in a biphasic medium catalysed by water-soluble palladium complexes derived from tris(3-sulphophenyl)phosphine. J. Organomet. Chem. 1994, 482, 45–51. [Google Scholar] [CrossRef]
- Gärtner, R.; Cornils, B.; Springer, H.; Lappe, P. Process for the Preparation of Sulfonated Aryl Phosphine. U.S. Patent 4,483,802, 20 November 1984. [Google Scholar]
- Herrmann, W.A.; Albanese, G.P.; Manetsberger, R.B.; Lappe, P.; Bahrmann, H. New process for the sulfonation of phosphane ligands for catalysts. Angew. Chem. Int. Ed. Engl. 1995, 34, 811–813. [Google Scholar] [CrossRef]
- Gulyás, H.; Szöllősy, Á.; Szabó, P.; Halmos, P.; Bakos, J. Preparation of new sulfonated triarylphosphanes: Control of the selectivity by structural assistance. Eur. J. Org. Chem. 2003, 2003, 2775–2781. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Kellner, J.; Riepl, H. Wasserlösliche Metallkomplexe und Katalysatoren III. Neue wasserlösliche Metallkomplexe des sulfonierten Triphenylphosphans (TPPTS): Mn, Fe, Ru, Co, Rh, Ir, Ni, Pd, Pt, Ag, Au. J. Organomet. Chem. 1990, 389, 103–128. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the water-soluble phosphanes are available from the authors.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Structure | Name Abbreviation | K (M−1) a,b |
|---|---|---|---|
| 1 |  | tris(3-sodium-sulfonatophenyl)phosphane TPPTS | 840 [15] |
| 2 | 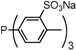 | tris(4-methyl-3-sodium-sulfonatophenyl)phosphane tris(p-Me)TPPTS | 960 [15] |
| 3 | 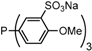 | tris(4-methoxy-3-sodium-sulfonatophenyl)phosphane tris(p-OMe)TPPTS | 0 [15] |
| 4 | 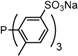 | tris(6-methyl-3-sodium-sulfonatophenyl)phosphane tris(o-Me)TPPTS | 0 [15] |
| 5 | 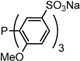 | tris(6-methoxy-3-sodium-sulfonatophenyl)phosphane tris(o-OMe)TPPTS | 0 [15] |
| 6 |  | trisulfonated monobiphenyldiphenylphosphane P(Biph)Ph2TS | 9900 c |
| 7 |  | trisulfonated bisbiphenylmonophenylphosphane P(Biph)2PhTS | >100,000 c |
| 8 | 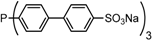 | trisulfonated trisbiphenylphosphane P(Biph)3TS | >100,000 c |
| Ligand | Platinum Complex Obtained without CD | New Species Obtained after RAME-β-CD Addition |
|---|---|---|
| TPPTS | [PtL3Cl]Cl = 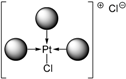 | cis-PtCl2L2 = 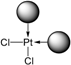 |
| tris(p-Me)TPPTS | [PtL3Cl]Cl | - |
| tris(p-OMe)TPPTS | [PtL3Cl]Cl | - |
| tris(o-Me)TPPTS | trans-PtCl2L2 = 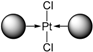 | - |
| tris(o-OMe)TPPTS | trans-PtCl2L2 | - |
| P(Biph)Ph2TS | [PtL3Cl]Cl | 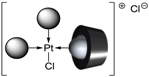 or or 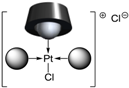 |
| P(Biph)2PhTS | n.d (broad signals) * | n.d (broad signals) |
| P(Biph)3TS | n.d (broad signals) * | n.d (broad signals) |
| Ligand | Palladium Complex Obtained without CD | New Species Obtained after RAME-β-CD Addition |
|---|---|---|
| TPPTS | PdL3 | - |
| tris(p-Me)TPPTS | PdL3 | - |
| tris(p-OMe)TPPTS | PdL3 | - |
| tris(o-Me)TPPTS | No palladium extraction | - |
| tris(o-OMe)TPPTS | No palladium extraction | - |
| P(Biph)Ph2TS | PdL3 |  |
| P(Biph)2PhTS | PdL3 + PdL2 | [PdL2] increase |
| P(Biph)3TS | PdL4 + PdL3 | [PdL3] increase + PdL2 formed |
| Entry | Ligand (L) | Influence of CD on Pt Complexes | Influence of CD on Pd Complexes |
|---|---|---|---|
| 1 | TPPTS | [PtL3Cl]Cl + CD → cis-PtCl2L2 + L@CD CD = decoordination promotor | PdL3 unchanged with CD |
| 2 | tris(p-Me)TPPTS tris(p-OMe)TPPTS | [PtL3Cl]Cl unchanged with CD | PdL3 unchanged with CD |
| 3 | tris(o-Me)TPPTS tris(o-OMe)TPPTS | [PtL3Cl]Cl impossible to obtain; trans-PtCl2L2 was formed with or without CD | Ligand unable to extract Pd from the organic layer with or without CD |
| 4 | P(Biph)Ph2TS | [PtL3Cl]Cl + CD → [(L@CD)PtL2Cl]Cl CD = second sphere ligand | PdL3 + CD → (L@CD)PdL2 CD = second sphere ligand |
| 5 | P(Biph)2PhTS | n.d. (broad signals with or without CD) | PdL3 + CD → PdL*2 + L@CD L* = L or L@CD CD = decoordination promotor and second sphere ligand |
| 6 | P(Biph)3TS | n.d. (broad signals with or without CD) | PdL4 + CD → PdL*3 + L@CD PdL*3 + CD → PdL*2 + L@CD L* = L or L@CD CD = decoordination promotor and second sphere ligand |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, M.; Bricout, H.; Tilloy, S.; Monflier, E. Transition Metal Complexes Coordinated by Water Soluble Phosphane Ligands: How Cyclodextrins Can Alter the Coordination Sphere? Molecules 2017, 22, 140. https://doi.org/10.3390/molecules22010140
Ferreira M, Bricout H, Tilloy S, Monflier E. Transition Metal Complexes Coordinated by Water Soluble Phosphane Ligands: How Cyclodextrins Can Alter the Coordination Sphere? Molecules. 2017; 22(1):140. https://doi.org/10.3390/molecules22010140
Chicago/Turabian StyleFerreira, Michel, Hervé Bricout, Sébastien Tilloy, and Eric Monflier. 2017. "Transition Metal Complexes Coordinated by Water Soluble Phosphane Ligands: How Cyclodextrins Can Alter the Coordination Sphere?" Molecules 22, no. 1: 140. https://doi.org/10.3390/molecules22010140
APA StyleFerreira, M., Bricout, H., Tilloy, S., & Monflier, E. (2017). Transition Metal Complexes Coordinated by Water Soluble Phosphane Ligands: How Cyclodextrins Can Alter the Coordination Sphere? Molecules, 22(1), 140. https://doi.org/10.3390/molecules22010140