
Aspalathin Protects the Heart against Hyperglycemia-Induced Oxidative Damage by Up-Regulating Nrf2 Expression

 ,
,  , ,
, , 
Abstract
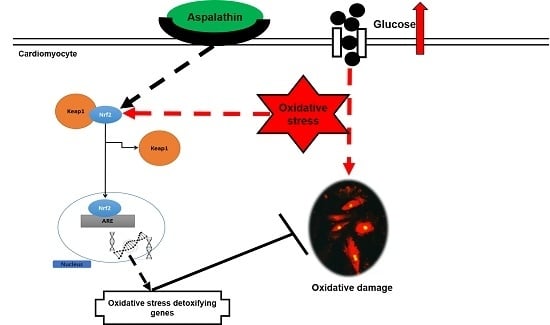
:
1. Introduction
2. Results
2.1. In Vitro Screening of ASP in H9c2 Cardiomyocytes
2.1.1. ASP Maintained Cellular Homeostasis In Vitro
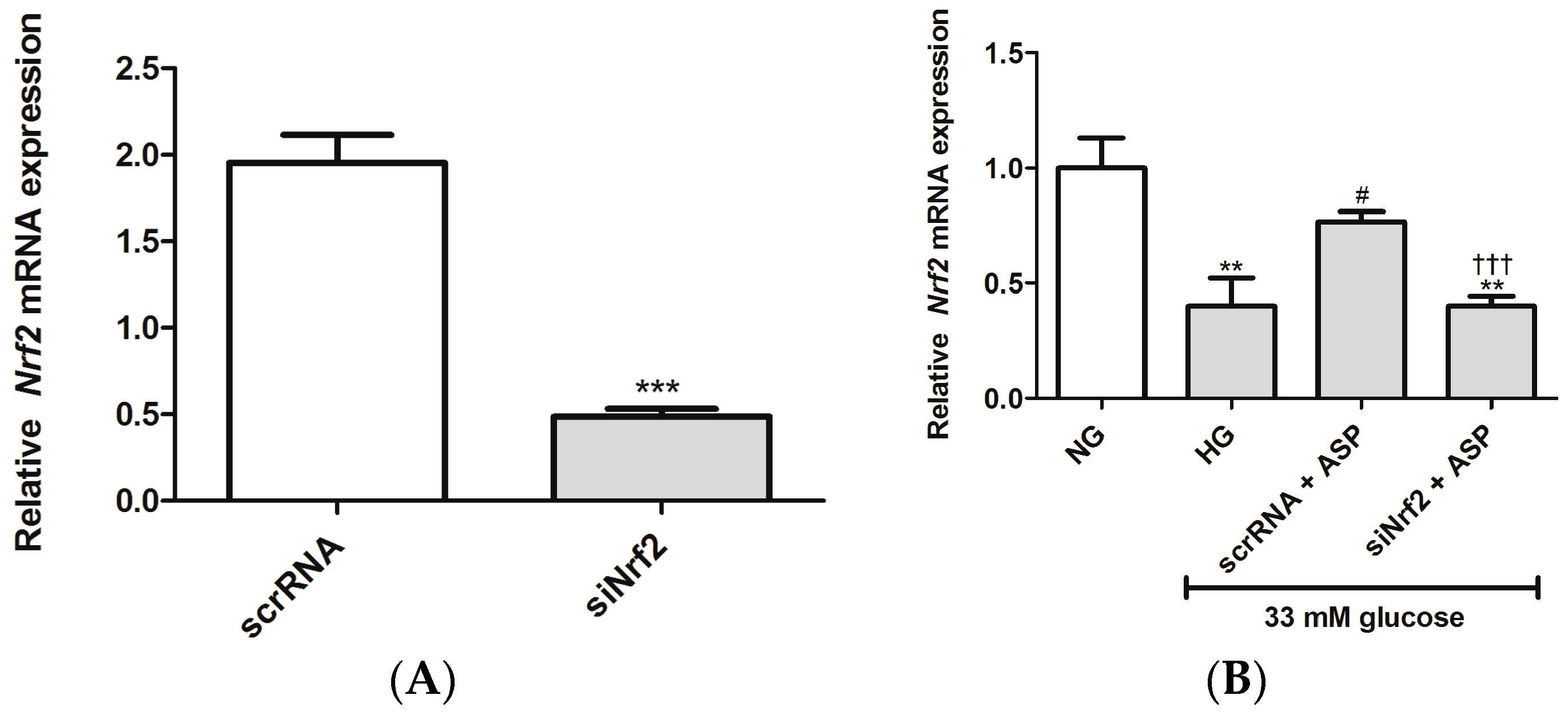
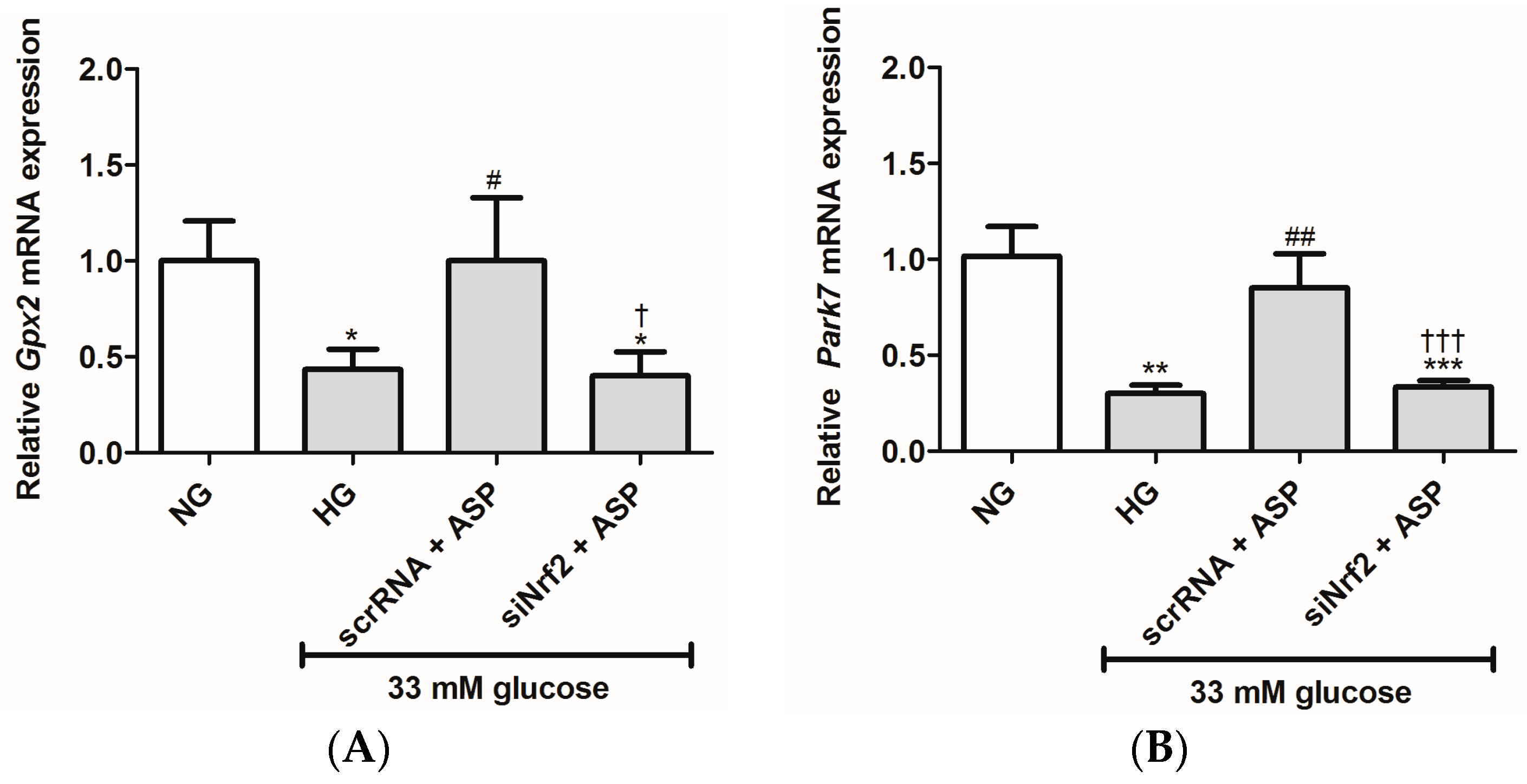
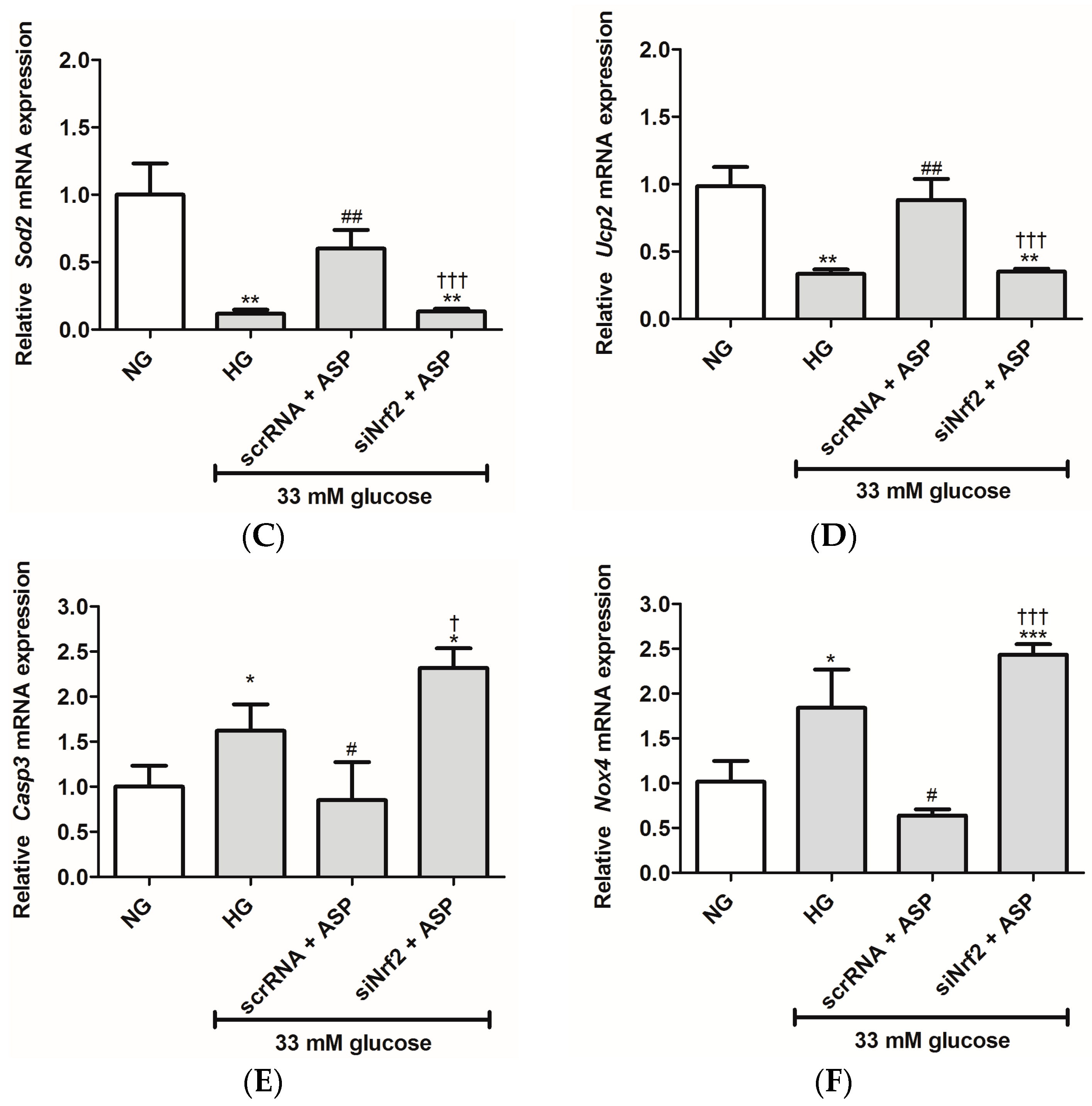
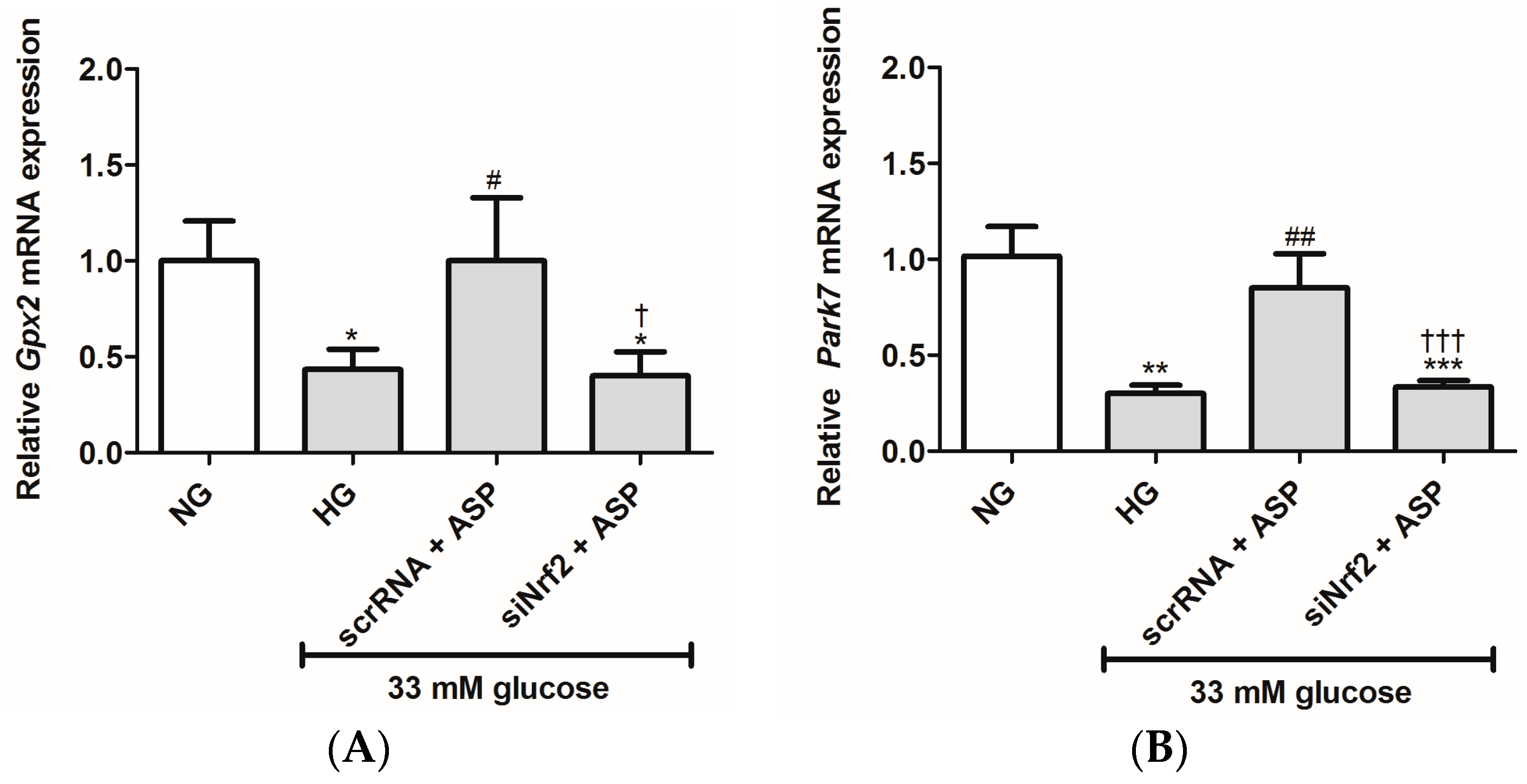
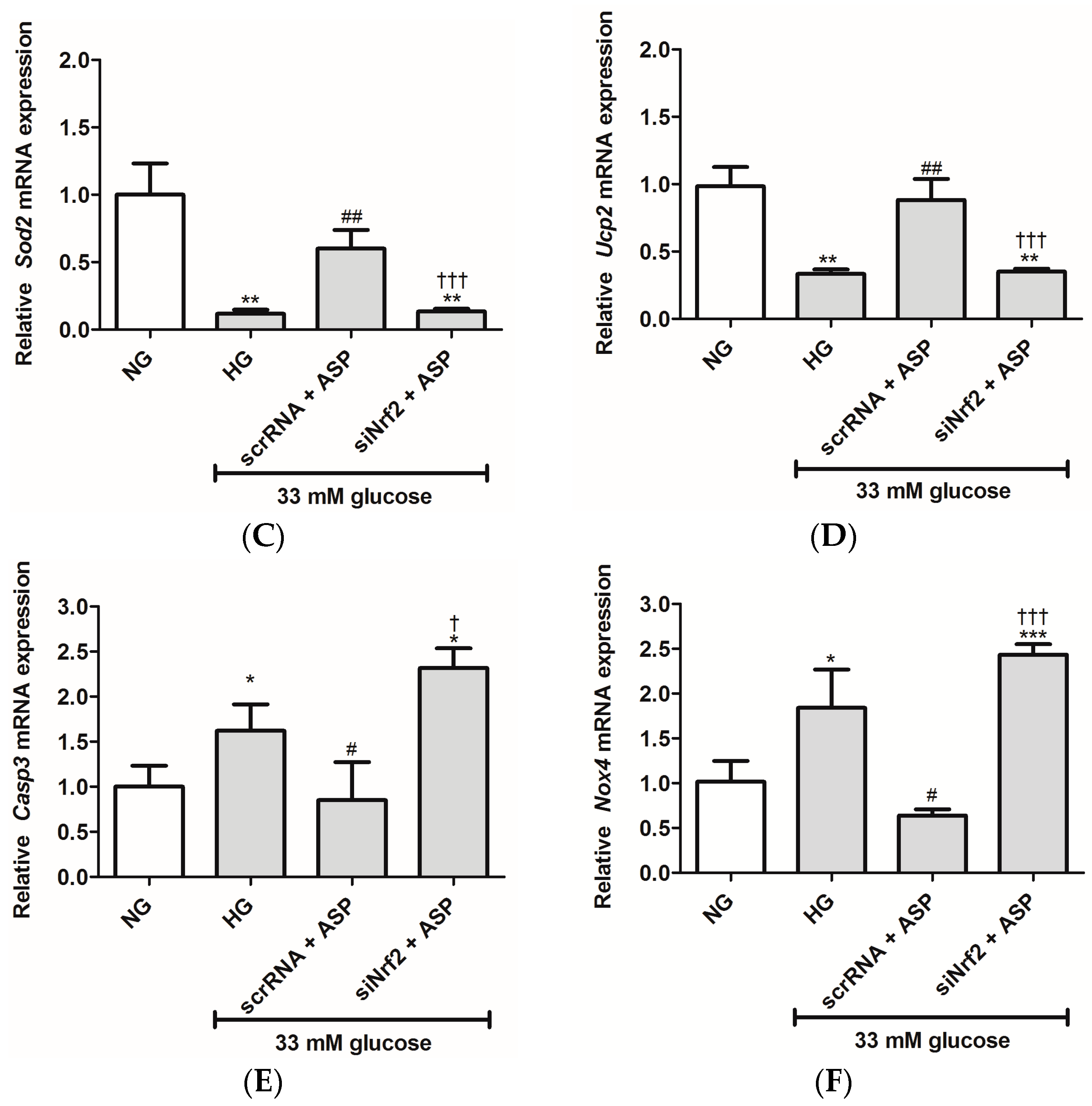
2.1.2. ASP Regulated Expression of Nrf2 and Its Downstream Target Genes In Vitro
2.2. In Vivo Confirmation Studies on db/db Mice
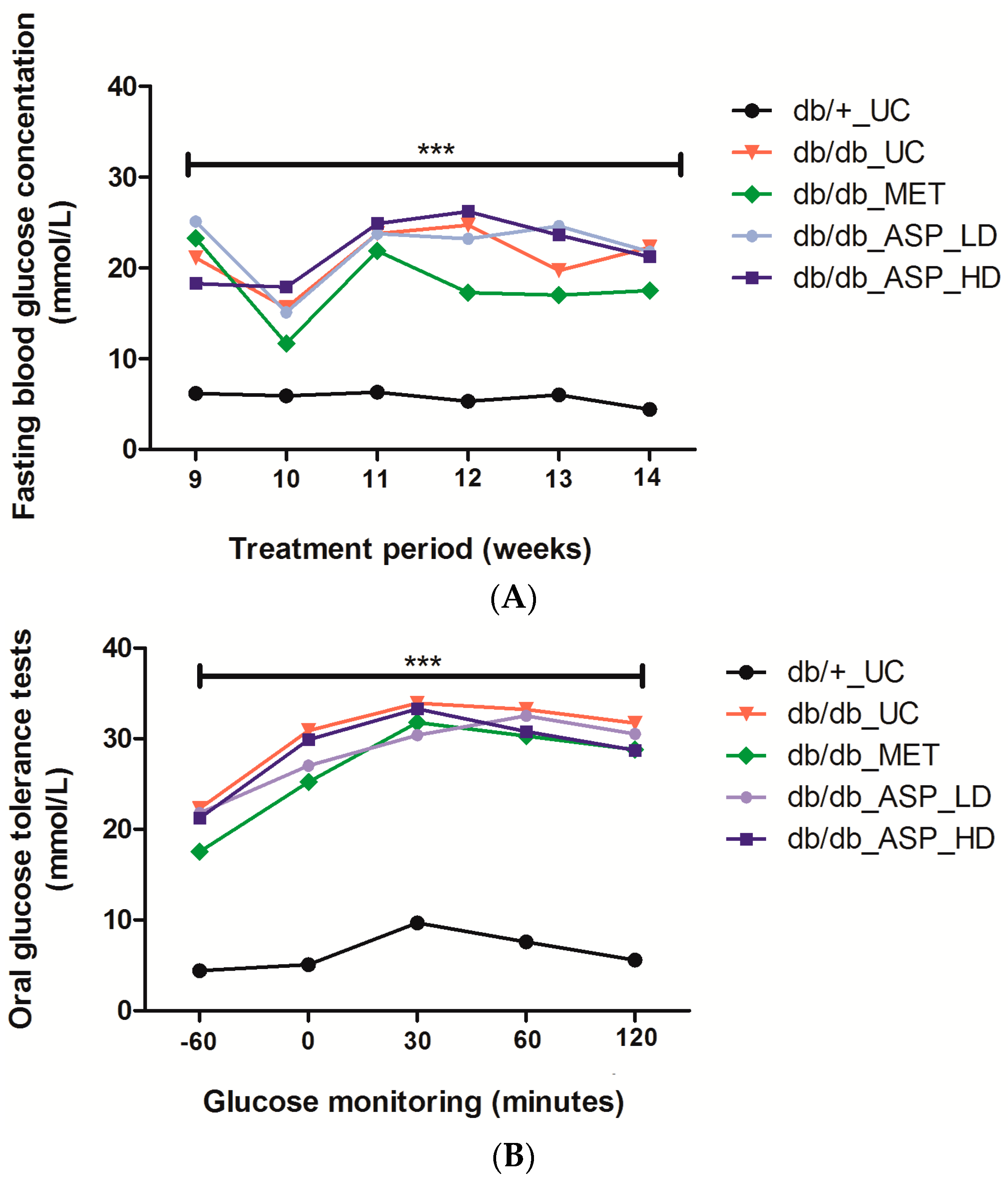
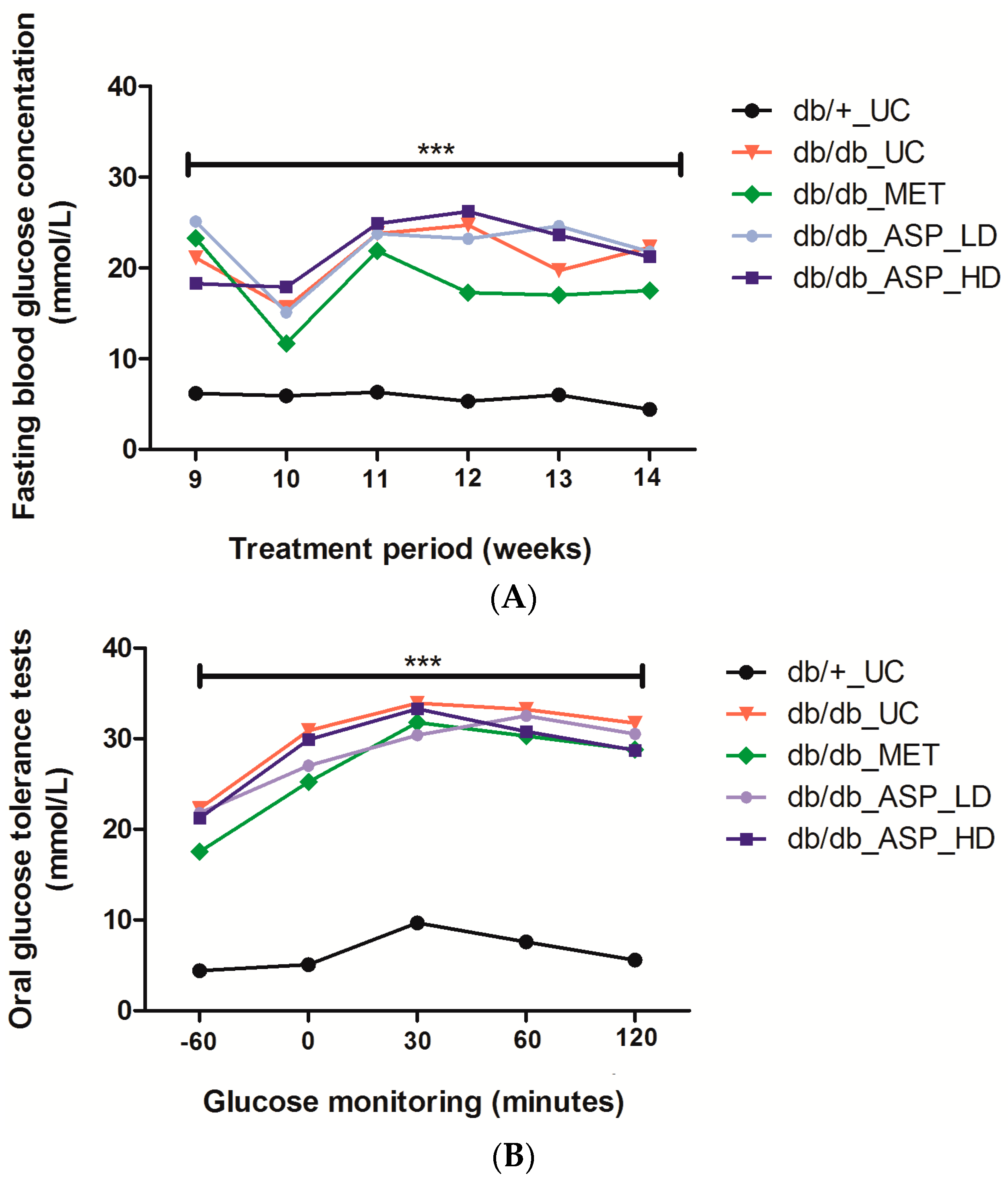
2.2.1. Effect of ASP on Fasting Plasma Glucose (FPG) and Oral Glucose Tolerance Tests (OGTTs)
2.2.2. Assessment of FPG Levels after Administration of a 2 g/kg Glucose Bolus in Mice
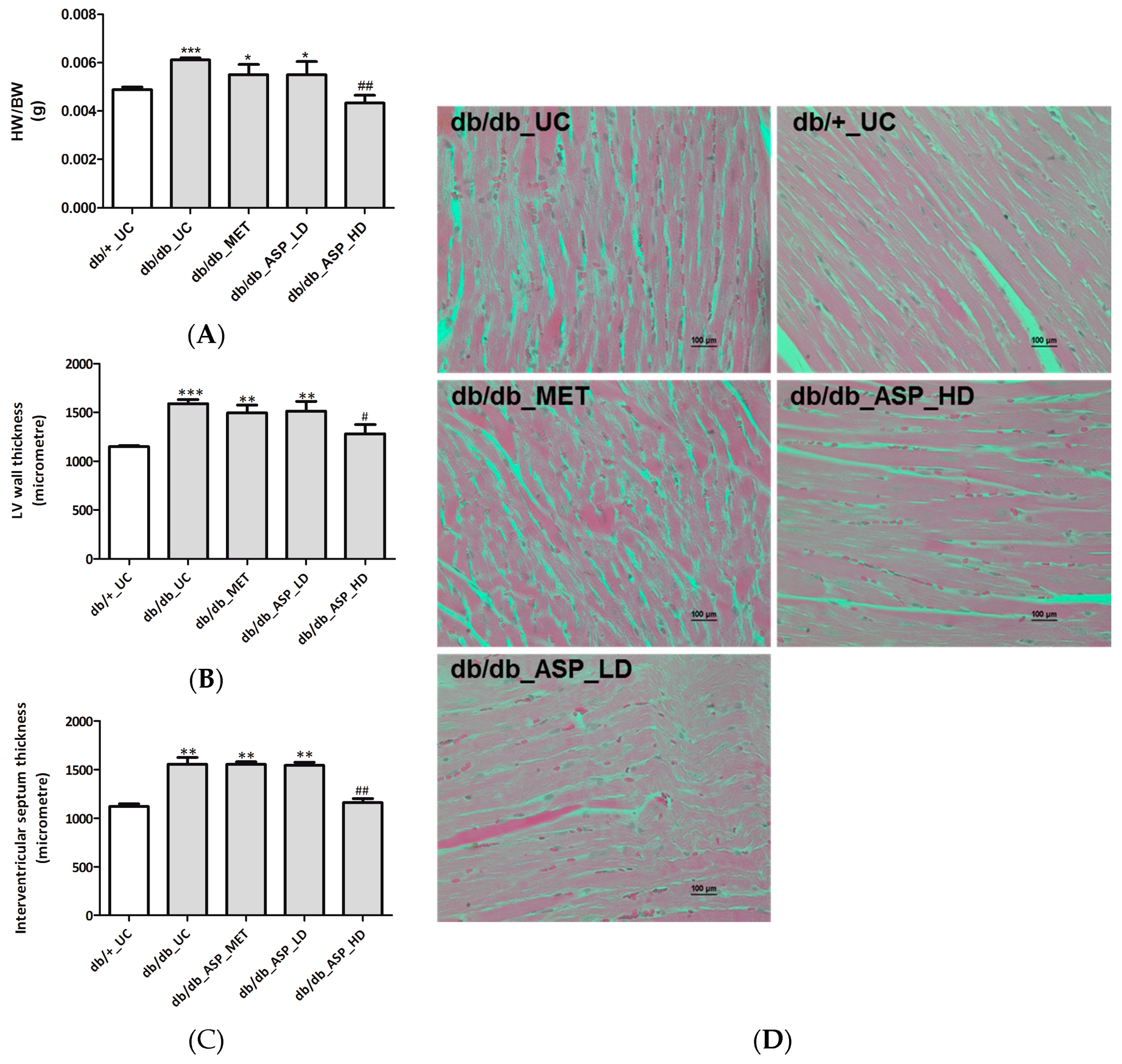
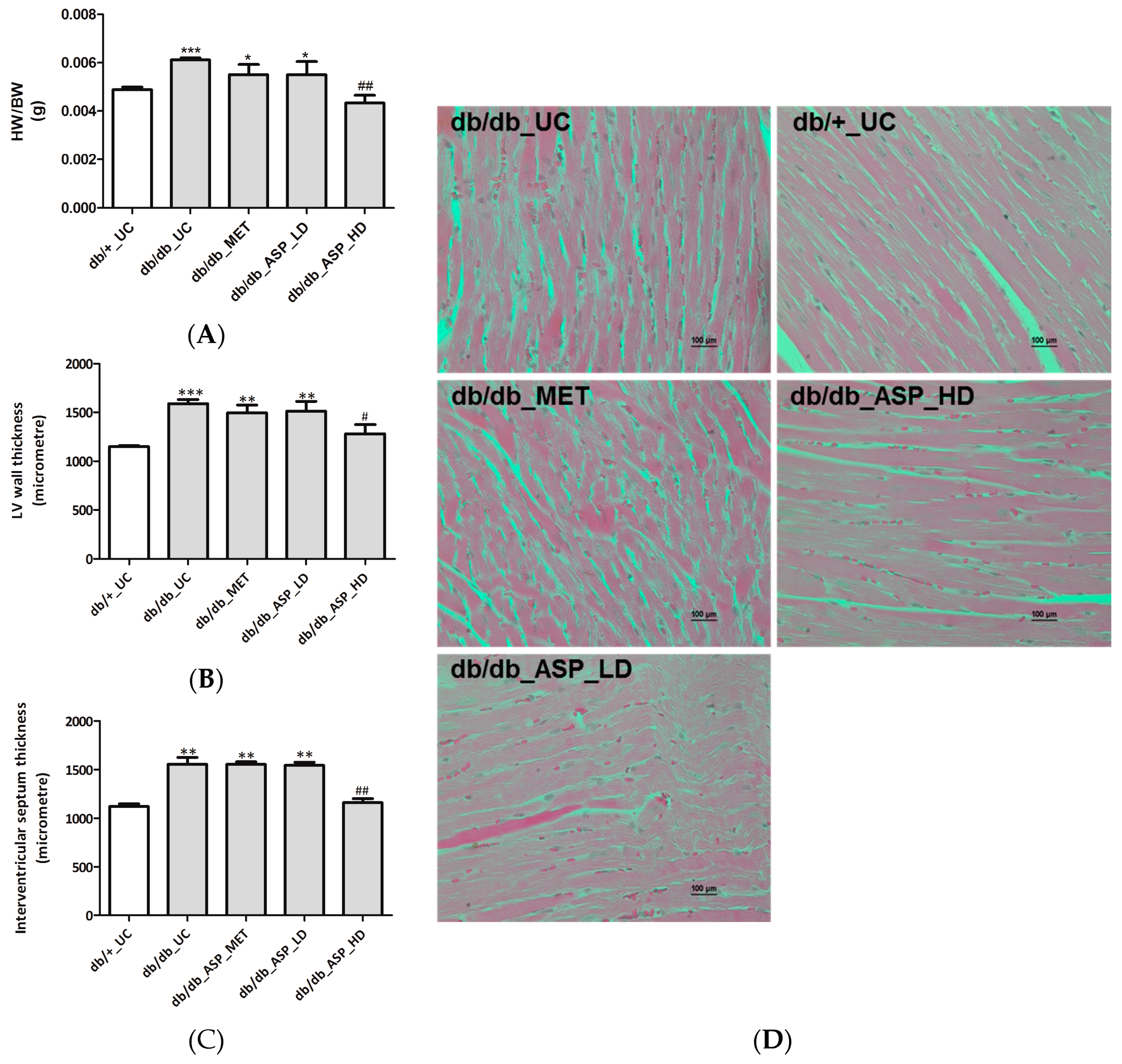
2.2.3. ASP Prevented Hyperglycemia-Induced LV Mass Enlargement In Vivo
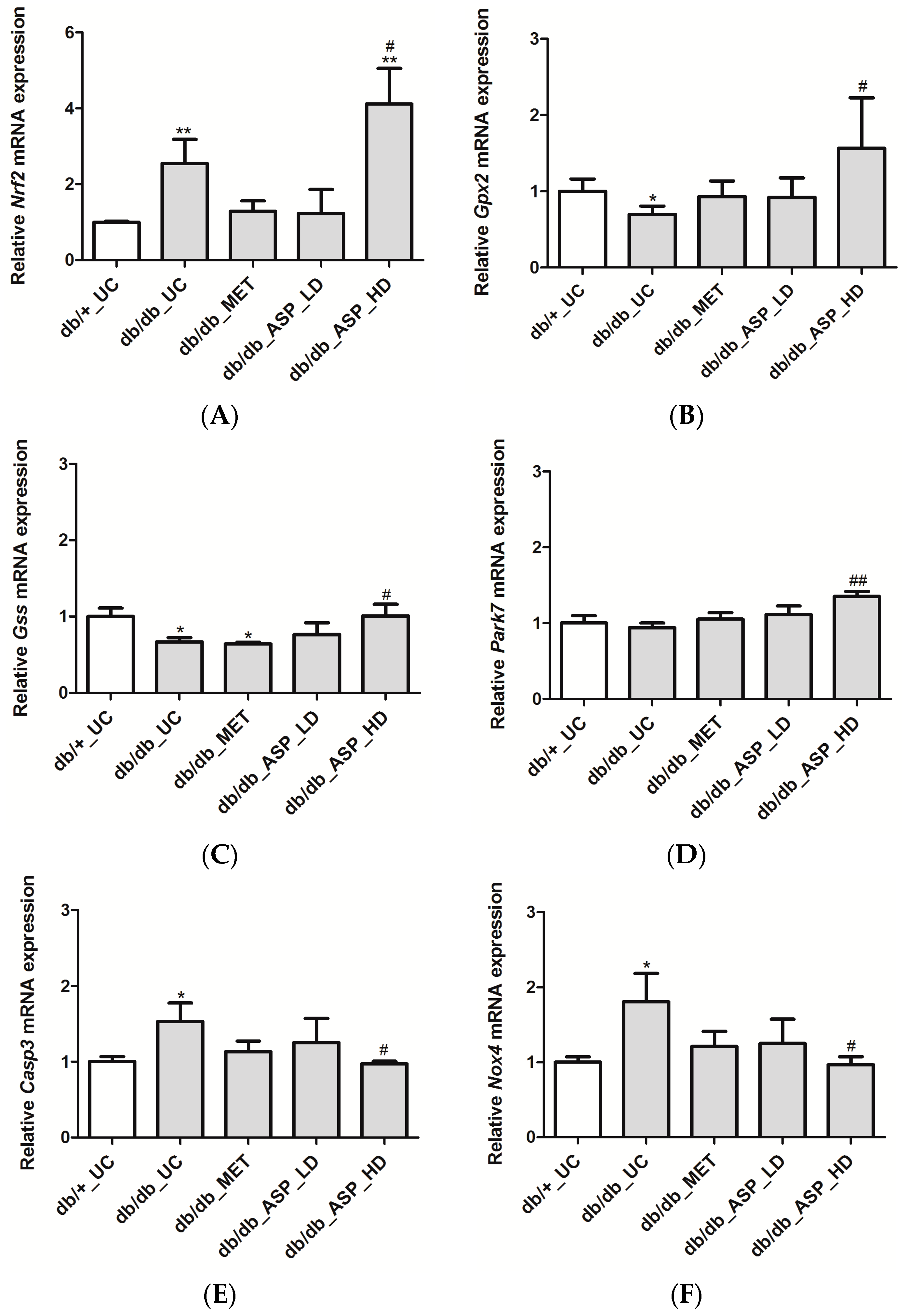
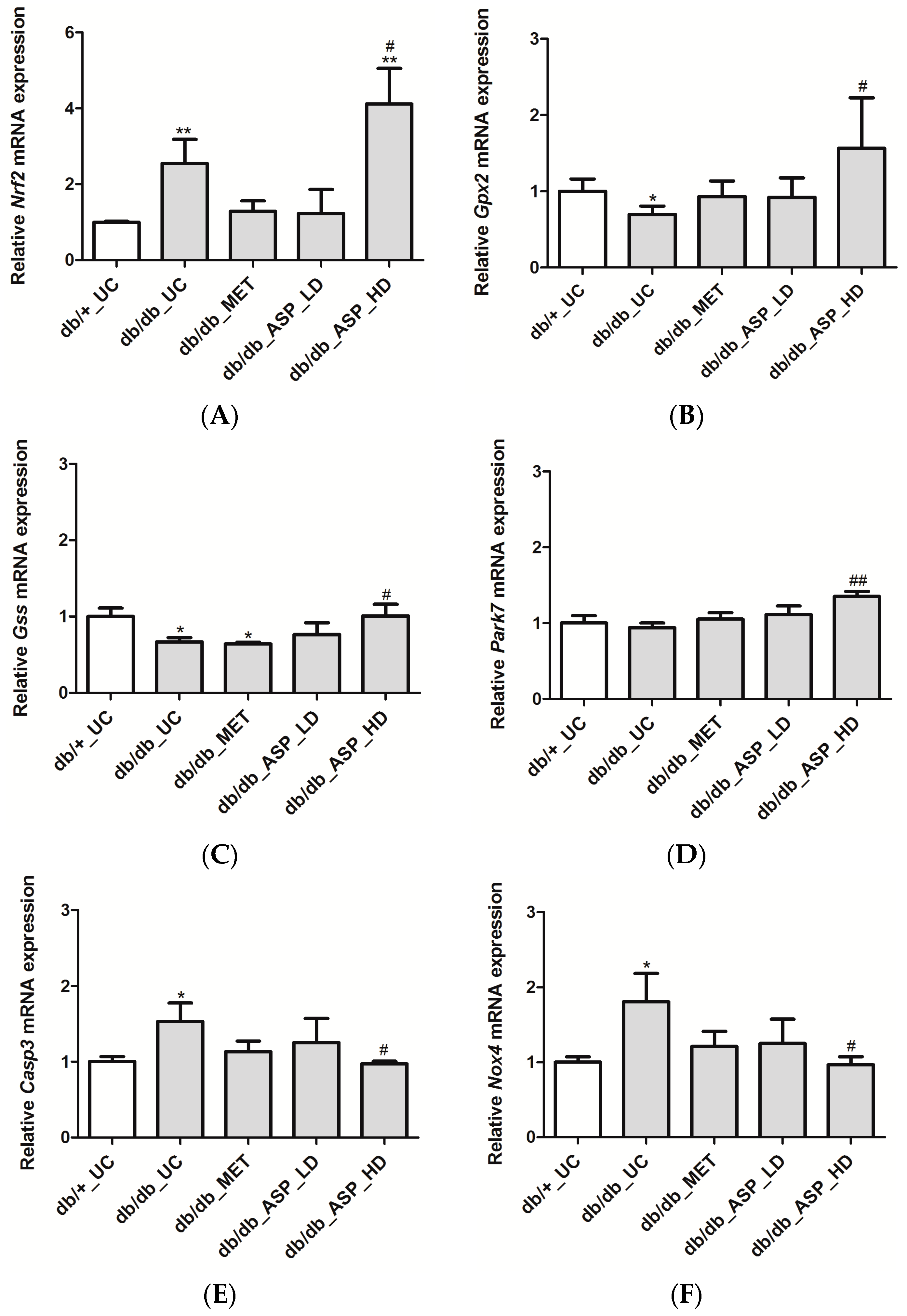
2.2.4. ASP Regulated the Expression of Nrf2 and Its Target Genes In Vivo
3. Discussion
4. Materials and Methods
4.1. Reagents and Kits
4.2. In Vitro Experiments on H9c2 Cardiomyocytes
4.2.1. Cell Culture
4.2.2. Preparation of ASP for Cell Culture Treatment
4.2.3. RNA Isolation and Purification
4.2.4. RT2-PCR Array Analysis
4.2.5. Real-Time PCR to Confirm Oxidative Stress and Apoptosis Markers
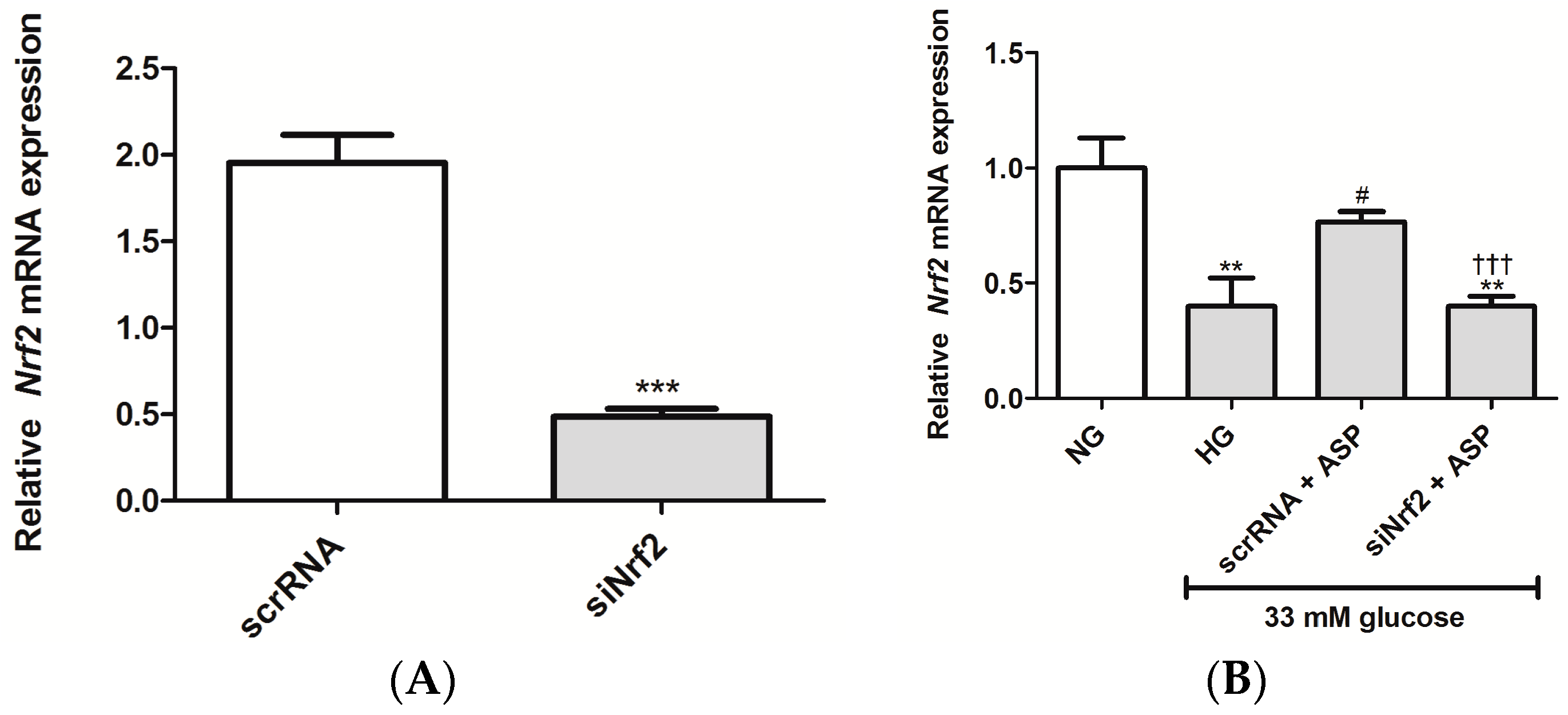
4.2.6. Knockdown of Nrf2 Using Small Interfering RNA
4.3. In Vivo Experiments Using C57BL/KS Mice
4.3.1. Animals
4.3.2. Treatment of Mice with ASP
4.3.3. Heart Tissue Staining and Left Ventricular Hypertrophic Measurements
4.3.4. Measurement of FPG Concentrations
4.3.5. OGTTs
4.4. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- International Diabetes Federation (IDF). IDF Diabetes Atlas, 7th ed. Available online: http://www.diabetesatlas.org/ (accessed on 20 February 2016).
- Fowler, M.J. Microvascular and macrovascular complications of diabetes. Clin. Diabetes 2008, 26, 77–82. [Google Scholar] [CrossRef]
- Inzucchi, S.E.; Bergenstal, R.M.; Buse, J.B.; Diamant, M.; Ferrannini, E.; Nauck, M.; Peters, A.L.; Tsapas, A.; Wender, R.; Matthews, D.R. Management of hyperglycemia in type 2 diabetes, 2015: A patient-centered approach. Update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2015, 38, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef]
- Fonarow, G.C. An approach to heart failure and diabetes mellitus. Am. J. Cardiol. 2005, 96, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy, causes and effects. Rev. Endocr. Metab. Disord. 2010, 11, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Bayeva, M.; Sawicki, K.T.; Ardehali, H. Taking diabetes to heart-deregulation of myocardial lipid metabolism in diabetic cardiomyopathy. J. Am. Heart Assoc. 2013, 2, e000433. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Kang, Y.J. Oxidative stress and diabetic cardiomyopathy: A brief review. Cardiovasc. Toxicol. 2001, 1, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Thandavarayan, R.A.; Giridharan, V.V.; Sari, F.R.; Arumugam, S.; Veeraveedu, P.T.; Pandian, G.N.; Palaniyandi, S.S.; Ma, M.; Suzuki, K.; Gurusamy, N.; et al. Depletion of 14-3-3 protein exacerbates cardiac oxidative stress, inflammation and remodelling process via modulation of MAPK/NF-κB signaling pathways after streptozotocin-induced diabetes mellitus. Cell. Physiol. Biochem. 2011, 28, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, H.Y.; Jiang, Y.N.; Li, N. Protective effect of thymoquinone improves cardiovascular function, and attenuates oxidative stress, inflammation and apoptosis by mediating the PI3K/Akt pathway in diabetic rats. Mol. Med. Rep. 2016, 13, 2836–2842. [Google Scholar] [CrossRef] [PubMed]
- Gupte, A.A.; Lyon, C.J.; Hsueh, W.A. Nuclear factor (erythroid-derived 2)-like-2 factor (Nrf2), a key regulator of the antioxidant response to protect against atherosclerosis and nonalcoholic steatohepatitis. Curr. Diabetes Rep. 2013, 13, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Kan, H.; Cai, L.; Ma, Q. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. J. Mol. Cell. Cardiol. 2009, 46, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Ichikawa, T.; Li, J.; Si, Q.; Yang, H.; Chen, X.; Goldblatt, C.S.; Meyer, C.J.; Li, X.; Cai, L.; et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes 2011, 60, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.S.; Cheng, Y.H.; Lee, C.Y.; Chung, C.Y.; Chang, W.C. Resveratrol protects against methylglyoxal-induced hyperglycemia and pancreatic damage in vivo. Nutrients 2015, 7, 2850–2865. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, J.; Chen, S.Y. Sulforaphane protects against ethanol-induced oxidative stress and apoptosis in neural crest cells by the induction of Nrf2-mediated antioxidant response. Br. J. Pharmacol. 2013, 169, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, Z.; Cai, L. Diabetic cardiomyopathy and its prevention by Nrf2: Current status. Diabetes Metab. J. 2014, 38, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Cui, W.; Xin, Y.; Miao, X.; Barati, M.T.; Zhang, C.; Chen, Q.; Tan, Y.; Cui, T.; Zheng, Y.; et al. Prevention by sulforaphane of diabetic cardiomyopathy is associated with upregulation of Nrf2 expression and transcription activation. J. Mol. Cell. Cardiol. 2013, 57, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Granado-Serrano, A.B.; Martín, M.A.; Bravo, L.; Goya, L.; Ramos, S. Quercetin modulates Nrf2 and glutathione-related defenses in HepG2 cells: Involvement of p38. Chem. Biol. Interact. 2012, 195, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.J.; Joubert, E.; de Beer, D.; Sanderson, M.; Malherbe, C.J.; Fey, S.J.; Louw, J. Acute assessment of an aspalathin-enriched green rooibos (Aspalathus linearis) extract with hypoglycemic potential. Phytomedicine 2012, 20, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Mazibuko, S.E.; Muller, C.J.; Joubert, E.; de Beer, D.; Johnson, R.; Opoku, A.R.; Louw, J. Amelioration of palmitate-induced insulin resistance in C2C12 muscle cells by rooibos (Aspalathus linearis). Phytomedicine 2013, 20, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Dludla, P.V.; Muller, C.J.; Louw, J.; Joubert, E.; Salie, R.; Opoku, A.R.; Johnson, R. The cardioprotective effect of an aqueous extract of fermented rooibos (Aspalathus linearis) on cultured cardiomyocytes derived from diabetic rats. Phytomedicine 2014, 21, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, M.; Mazibuko, S.E.; Joubert, E.; de Beer, D.; Johnson, R.; Pheiffer, C.; Louw, J.; Muller, C.J. Effects of fermented rooibos (Aspalathus linearis) on adipocyte differentiation. Phytomedicine 2014, 21, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.J.; Joubert, E.; Pheiffer, C.; Ghoor, S.; Sanderson, M.; Chellan, N.; Fey, S.J.; Louw, J. Z-2-(β-d-glucopyranosyloxy)-3-phenylpropenoic acid, an α-hydroxy acid from rooibos (Aspalathus linearis) with hypoglycemic activity. Mol. Nutr. Food Res. 2013, 57, 2216–2222. [Google Scholar] [CrossRef] [PubMed]
- Mazibuko, S.E.; Joubert, E.; Johnson, R.; Louw, J.; Opoku, A.R.; Muller, C.J. Aspalathin improves glucose and lipid metabolism in 3T3-L1 adipocytes exposed to palmitate. Mol. Nutr. Food Res. 2015, 59, 2199–2208. [Google Scholar] [CrossRef] [PubMed]
- Von Gadow, A.; Joubert, E.; Hansmann, C.F. Comparison of the antioxidant activity of aspalathin with that of other plant phenols of rooibos tea (Aspalathus linearis), α-tocopherol, BHT, and BHA. J. Agric. Food Chem. 1997, 45, 632–638. [Google Scholar] [CrossRef]
- Snijman, P.W.; Joubert, E.; Ferreira, D.; Li, X.C.; Ding, Y.; Green, I.R.; Gelderblom, W.C. Antioxidant activity of the dihydrochalcones aspalathin and nothofagin and their corresponding flavones in relation to other rooibos (Aspalathus linearis) flavonoids, epigallocatechin gallate, and trolox. J. Agric. Food Chem. 2009, 57, 6678–6684. [Google Scholar] [CrossRef] [PubMed]
- Krafczyk, N.; Woyand, F.; Glomb, M.A. Structure–antioxidant relationship of flavonoids from fermented rooibos. Mol. Nutr. Food Res. 2009, 53, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Kawano, A.; Nakamura, H.; Hata, S.; Minakawa, M.; Miura, Y.; Yagasaki, K. Hypoglycemic effect of aspalathin, a rooibos tea component from Aspalathus linearis, in type 2 diabetic model db/db mice. Phytomedicine 2009, 16, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Son, M.J.; Minakawa, M.; Miura, Y.; Yagasaki, K. Aspalathin improves hyperglycemia and glucose intolerance in obese diabetic ob/ob mice. Eur. J. Nutr. 2013, 52, 1607–1619. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Dludla, P.; Joubert, E.; February, F.; Mazibuko, S.; Ghoor, S.; Muller, C.; Louw, J. Aspalathin, a dihydrochalcone C-glucoside, protects H9c2 cardiomyocytes against high glucose-induced shifts in substrate preference and apoptosis. Mol. Nutr. Food Res. 2016, 60, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, K.; Baldinger, J.; Mayerhofer, B.; Atanasov, A.G.; Dirsch, V.M.; Heiss, E.H. Activated AMPK boosts the Nrf2/HO-1 signaling axis-A role for the unfolded protein response. Free Radic. Biol. Med. 2015, 88, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, P.; Zhao, Y.; Yang, C.; Clark, A.; Leung, T.; Chen, X.; Sang, S. Synthesis, evaluation, and metabolism of novel [6]-shogaol derivatives as potent Nrf2 activators. Free Radic. Biol. Med. 2016, 95, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Xia, X.; Shi, Q.; Song, X.; Fu, J.; Xiao, C.; Chen, H.; Lu, B.; Sun, Z.; Wu, S.; et al. Neohesperidin dihydrochalcone versus CCl₄-induced hepatic injury through different mechanisms: the implication of free radical scavenging and Nrf2 activation. J. Agric. Food Chem. 2015, 63, 5468–5475. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Somaio Neto, F.; Ikejiri, A.T.; Bertoletto, P.R.; Chaves, J.C.; Teruya, R.; Fagundes, D.J.; Taha, M.O. Gene expression related to oxidative stress in the heart of mice after intestinal ischemia. Arg. Bras. Cardiol. 2014, 102, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Antunes, F.; Han, D.; Cadenas, E. Relative contributions of heart mitochondrial glutathione peroxidase and catalase to H2O2 detoxification in in vivo conditions. Free Radic. Biol. Med. 2002, 33, 1260–1267. [Google Scholar] [CrossRef]
- Zheng, A.; Li, H.; Xu, J.; Cao, K.; Li, H.; Pu, W.; Yang, Z.; Peng, Y.; Long, J.; Liu, J.; et al. Hydroxytyrosol improves mitochondrial function and reduces oxidative stress in the brain of db/db mice: Role of AMP-activated protein kinase activation. Br. J. Nutr. 2015, 113, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, R.; Julius, T.; Buxton, K.; Xu, Q.; Kiriazis, H.; McMullen, J.; Forbes, J.; Du, X.; Kaye, D.; Tesch, G. Type 2 diabetic cardiomyopathy in db/db mice is associated with progressive cardiac fibrosis, cardiomyocyte hypertrophy and oxidative stress. Heart Lung Circ. 2008, 17, S231. [Google Scholar] [CrossRef]
- Belke, D.D.; Larsen, T.S.; Gibbs, E.M.; Severson, D.L. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E1104–E1113. [Google Scholar] [PubMed]
- Joseph, D.; Kimar, C.; Symington, B.; Milne, R.; Essop, M.F. The detrimental effects of acute hyperglycemia on myocardial glucose uptake. Life Sci. 2014, 105, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Mapanga, R.F.; Essop, M.F. Damaging effects of hyperglycemia on cardiovascular function: Spotlight on glucose metabolic pathways. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H153–H173. [Google Scholar] [CrossRef] [PubMed]
- Anedda, A.; López-Bernardo, E.; Acosta-Iborra, B.; Saadeh Suleiman, M.; Landázuri, M.O.; Cadenas, S. The transcription factor Nrf2 promotes survival by enhancing the expression of uncoupling protein 3 under conditions of oxidative stress. Free Radic. Biol. Med. 2013, 61, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Quon, M.J.; Kim, J.A. New insights into the mechanisms of polyphenols beyond antioxidant properties; lessons from the green tea polyphenol, epigallocatechin 3-gallate. Redox Biol. 2014, 2, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Dahiru, T. P-value, a true test of statistical significance? A cautionary note. Ann. Ib. Postgrad. Med. 2008, 6, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Du Prel, J.B.; Hommel, G.; Rohrig, B.; Blettner, M. Confidence interval or p-value? Part 4 of a series on evaluation of scientific publications. Dtsch. Arzteblatt Int. 2009, 106, 335–339. [Google Scholar]
- Dludla, P.V.; Muller, C.J.; Joubert, E.; Louw, J.; Gabuza, K.B.; Huisamen, B.; Essop, M.F.; Johnson, R. Phenylpyruvic acid-2-O-β-d-glucoside attenuates high glucose-induced apoptosis in H9c2 cardiomyocytes. Planta Med. 2016, 82, 1468–1474. [Google Scholar] [CrossRef] [PubMed]
- Rios, J.L.; Francini, F.; Schinella, G.R. Natural products for the treatment of type 2 diabetes mellitus. Planta Med. 2015, 81, 975–994. [Google Scholar] [CrossRef] [PubMed]
- Patel, O.; Muller, C.; Joubert, E.; Louw, J.; Rosenkranz, B.; Awortwe, C. Inhibitory interactions of Aspalathus linearis (Rooibos) extracts and compounds, aspalathin and z-2-(β-d-glucopyranosyloxy)-3-phenylpropenoic acid, on cytochromes metabolizing hypoglycemic and hypolipidemic drugs. Molecules 2016, 21, 1515. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Achilonu, M.C.; Kendrekar, P.S.; Joubert, E.; Ferreira, D.; Bonnet, S.L.; van der Westhuizen, J.H. Concise and scalable synthesis of aspalathin, a powerful plasma sugar-lowering natural product. J. Nat. Prod. 2014, 77, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, T.C.; Lin, A.J.; Ververis, K.; Chang, L.; Tang, M.M.; Okabe, J.; El-Osta, A. Trichostatin A accentuates doxorubicin-induced hypertrophy in cardiac myocytes. Aging 2010, 2, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Smit, S.E. An Investigation into the Effects of Aspalathin on Myocardial Glucose Transport Using Cardiomyocytes from Control and Obesity-Induced Insulin Resistant Rats, and Terminally Differentiated H9c2 Cells. Master’s Thesis, Stellenbosch University, South Africa, March 2016. Available online: http://scholar.sun.ac.za/handle/10019.1/98490 (accessed on 10 December 2016). [Google Scholar]
- Page, B.J.; du Toit, D.F.; Muller, C.J.; Mattysen, J.; Lyners, R.; Arends, E. Autogenous transplantation of a duct ligated pancreas: A functional and histological study. J. Oncol. Pract. 2004, 5, 71–80. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Fold Regulation after High Glucose Exposure | Fold Regulation After Post-Treatment with Aspalathin |
|---|---|---|
| Antioxidant genes | ||
| Catalase (Cat) | −1.63 | 11.80 |
| Glutathione peroxidase 2 (Gpx2) | −1.20 | 15.86 |
| Peroxiredoxin 1 (Prdx1) | −1.33 | 2.49 |
| Peroxiredoxin 3 (Prdx3) | −1.21 | 3.09 |
| Peroxiredoxin 4 (Prdx4) | −1.72 | 2.14 |
| Peroxiredoxin 6 (Prdx6) | −2.65 | 2.89 |
| Superoxide dismutase 1 (Sod1) | −1.25 | 2.18 |
| Superoxide dismutase 2 (Sod2) | −1.17 | 1.22 |
| Glutathione synthesis genes | ||
| Glutamate-cysteine ligase catalytic subunit (Gclc) | −1.84 | 6.96 |
| Glutamate-cysteine ligase, modifier subunit (Gclm) | −1.40 | 5.89 |
| Glutathione reductase (Gsr) | −1.50 | 3.29 |
| Reducing agent genes | ||
| Sulfiredoxin 1 (Srxn1) | −3.43 | 6.34 |
| Thioredoxin 1 (Txn1) | −1.08 | 2.05 |
| Thioredoxin reductase 1 (Txnrd1) | −2.88 | 13.70 |
| Thioredoxin reductase 2 (Txnrd2) | −3.25 | 1.08 |
| Cytoprotective genes | ||
| Heme oxygenase 1 (Hmox1) | −2.77 | 3.98 |
| NAD(P)H dehydrogenase (quinone 1) (Nqo1) | −4.57 | 11.45 |
| Uncoupling protein 2 (UCP2) | −3.95 | 2.83 |
| Uncoupling protein 3 (UCP3) | −2.01 | −1.61 |
| Apoptotic genes | ||
| B-cell lymphoma 2 (Bcl2) | −1.8 | 2.6 |
| Caspase 8 (Casp8) | 3.9 | −1.3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dludla, P.V.; Muller, C.J.F.; Joubert, E.; Louw, J.; Essop, M.F.; Gabuza, K.B.; Ghoor, S.; Huisamen, B.; Johnson, R. Aspalathin Protects the Heart against Hyperglycemia-Induced Oxidative Damage by Up-Regulating Nrf2 Expression. Molecules 2017, 22, 129. https://doi.org/10.3390/molecules22010129
Dludla PV, Muller CJF, Joubert E, Louw J, Essop MF, Gabuza KB, Ghoor S, Huisamen B, Johnson R. Aspalathin Protects the Heart against Hyperglycemia-Induced Oxidative Damage by Up-Regulating Nrf2 Expression. Molecules. 2017; 22(1):129. https://doi.org/10.3390/molecules22010129
Chicago/Turabian StyleDludla, Phiwayinkosi V., Christo J. F. Muller, Elizabeth Joubert, Johan Louw, M. Faadiel Essop, Kwazi B. Gabuza, Samira Ghoor, Barbara Huisamen, and Rabia Johnson. 2017. "Aspalathin Protects the Heart against Hyperglycemia-Induced Oxidative Damage by Up-Regulating Nrf2 Expression" Molecules 22, no. 1: 129. https://doi.org/10.3390/molecules22010129
APA StyleDludla, P. V., Muller, C. J. F., Joubert, E., Louw, J., Essop, M. F., Gabuza, K. B., Ghoor, S., Huisamen, B., & Johnson, R. (2017). Aspalathin Protects the Heart against Hyperglycemia-Induced Oxidative Damage by Up-Regulating Nrf2 Expression. Molecules, 22(1), 129. https://doi.org/10.3390/molecules22010129